

Summary
A 58-year-old woman with debilitating ankylosing spondylitis who was born to consanguineous parents was found to have an apparent severe vitamin D deficiency that did not respond to supplementation. Liquid chromatography–tandem mass spectrometry showed the absence of circulating vitamin D–binding protein, and chromosomal microarray confirmed a homozygous deletion of the group-specific component (GC) gene that encodes the protein. Congenital absence of vitamin D–binding protein resulted in normocalcemia and a relatively mild disruption of bone metabolism, in this case complicated by severe autoimmune disease. (Funded by the National Institutes of Health and the University of Washington.)
Vitamin D is produced by photochemical conversion of 7-dehydrocholesterol to cholecalciferol (vitamin D3) in the skin or is acquired through the diet as either cholecalciferol or ergocalciferol (vitamin D2). After stepwise hydroxylation in the liver and kidneys, the active 1,25-dihydroxyvitamin D (1,25[OH]2D, also known as calcitriol) metabolite participates in calcium and phosphate homeostasis. Severe vitamin D deficiency leads to rickets in children and osteomalacia in adults, both of which can be corrected with supplementation. Epidemiologic studies have proposed that vitamin D deficiency is associated with common health problems such as cancer, cardiovascular disease, diabetes, and various autoimmune conditions.1
The free-hormone hypothesis, as it pertains to lipophilic steroid hormones, states that only molecules that are unbound from their respective binding proteins are active or able to pass through the membrane of target cells. Most vitamin D metabolites are bound to vitamin D–binding protein and are biologically inactive. Free vitamin D metabolites, which are in equilibrium with bound vitamin D metabolites, are available for cellular functions, as is the case with 1,25(OH)2D binding the vitamin D receptor.2–4 Vitamin D–binding protein has an apparent affinity for vitamin D metabolites that is 1000 times as great as that of albumin and thus binds up to 90% of vitamin D metabolites in plasma.2,5,6 The fraction more weakly bound to albumin may contribute to the bioavailable pool of vitamin D metabolites.2 A contrasting hypothesis is that vitamin D–binding protein delivers vitamin D metabolites to target cells — for example, those in the renal proximal tubule — and facilitates their endocytosis through interaction with megalin, a multi-functional receptor.2 Vitamin D–binding protein also serves a number of minor roles, including actin scavenging in tissue injury, C5a-mediated chemotaxis, T-cell response, and macrophage activation.4,7,8 Certain physiological states, such as pregnancy and chronic liver disease, are associated with variations in the level of vitamin D–binding protein and the level of 25-hydroxyvitamin D (25[OH]D), which is currently the most important biomarker of vitamin D stores.2,5,7
Vitamin D–binding protein is encoded by group-specific component (GC), a highly polymorphic gene that belongs to the albuminoid superfamily on chromosome 4q13.3, along with albumin, alpha-fetoprotein, and afamin. More than 120 rare GC variants have been described, although two single-nucleotide polymorphisms (SNPs), which give rise to three common haplo-types, have skewed geographic distributions that appear to correlate with skin pigmentation, sun exposure, and possibly vitamin D metabolite–binding affinity.7,9–11
No GC polymorphisms are known to abolish vitamin D binding. Early studies evaluating thousands of people, in total, noted the absence of deletions or gross alterations in the GC genes,12 and more recent analyses of vitamin D–binding protein and vitamin D metabolites have not identified any person in whom this protein was absent.7,10 Knockout mice lacking vitamin D–binding protein are not only viable and fertile, but when fed a vitamin D–replete diet maintain normal calcium levels and bone structure, despite having significantly lower plasma concentrations of 25(OH)D and 1,25(OH)2D.13,14 In this report, we describe a patient with complete vitamin D–binding protein deficiency caused by homozygous deletion of the GC gene. We compare the patient with her normal and heterozygous siblings.
Case Report
The patient is a 58-year-old nulliparous woman who came to our attention when she was 33 years of age, after she had recently emigrated from Lebanon. She reported that starting in early adolescence, she had had chronic, progressive musculoskeletal pain that involved the lower back, neck, shoulders, and hips. The initial examination revealed marked kyphosis and reduced range of motion in the lumbar and cervical spine, shoulders, and hips. A clinical diagnosis of extensive ankylosing spondylitis was confirmed on radiography that showed advanced bilateral sacroiliitis, bridging syndesmophytes throughout the spine, and evidence of diffuse arthritis and enthesopathy. Her disability worsened over the years, despite treatment with antiinflammatory medications and physiotherapy. The medical history included type 2 diabetes, hypertension, dyslipidemia, nonalcoholic fatty liver disease, gastro-esophageal reflux, and uterine fibroids. Negative duodenal biopsies and HLA typing ruled out celiac disease, although when she was 52 years of age, serologic testing for antibodies to tissue transglutaminase IgA, which had shown normal results 3 years earlier, was positive at 91.5 kU per liter (reference range, 0 to 20). The patient’s parents are second cousins (Fig. S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org). She has seven siblings, none of whom have documented osteopenia, fragility fractures, or rheumatologic disease; three siblings were available for voluntary evaluation at our center.
The patient sustained low-trauma rib fractures at 41 years of age and subsequently had fragility fractures of the feet, left distal radius, and right hip (in separate events). She reported no substantial trauma or tissue injury before these fractures occurred. The results of bone densitometry were in the osteopenic range; T scores when she was 56 years of age were −1.4 in the left femur and −1.3 in the forearm. Lumbar spine T score at L1–L4 was 3.3, which reflects paraspinal ossification due to ankylosing spondylitis. Her height when she was 56 years of age was 137 cm, her weight was 72 kg, and her body-mass index (BMI; the weight in kilograms divided by the square of the height in meters) was 38.4. She had lost at least 20 cm in stature owing to increasing kyphosis over several decades.
Serum 25(OH)D concentrations were persistently undetectable by standard hospital-based methods (<4.0 ng per milliliter), whereas concentrations of 1,25(OH)2D ranged from undetectable (<5 pg per milliliter) to as high as 11.7 pg per milliliter (reference range, 21.2 to 73.1) (Table S1 in the Supplementary Appendix). Concentrations of 25(OH)D and 1,25(OH)2D did not respond to any form of supplementation or ultraviolet B phototherapy. Treatments included, but were not limited to, oral ergocalciferol at a dose of up to 50,000 IU per week, intramuscular ergocalciferol at a dose of up to 600,000 IU per week, a one-time oral dose of 250,000 IU of vitamin D, and calcitriol at a dose of up to 1.25 μg per day (Fig. 1). Serum calcium, albumin, and magnesium levels were normal. Hypophosphatemia and parathyroid hormone (PTH) elevations responded to therapy over time. The alkaline phosphatase level was normal apart from a single elevation before treatment. Urinary calcium excretion was elevated. Normal measurements of fibroblast growth factor 23 (78 RU per milliliter; normal range, <180 RU per milliliter) and 24-hour urine phosphate excretion ruled out renal phosphate wasting. The results of all tests are summarized in Figure 1 and Table S1 in the Supplementary Appendix.
Figure 1. Laboratory Data after Onset of Fragility Fractures.
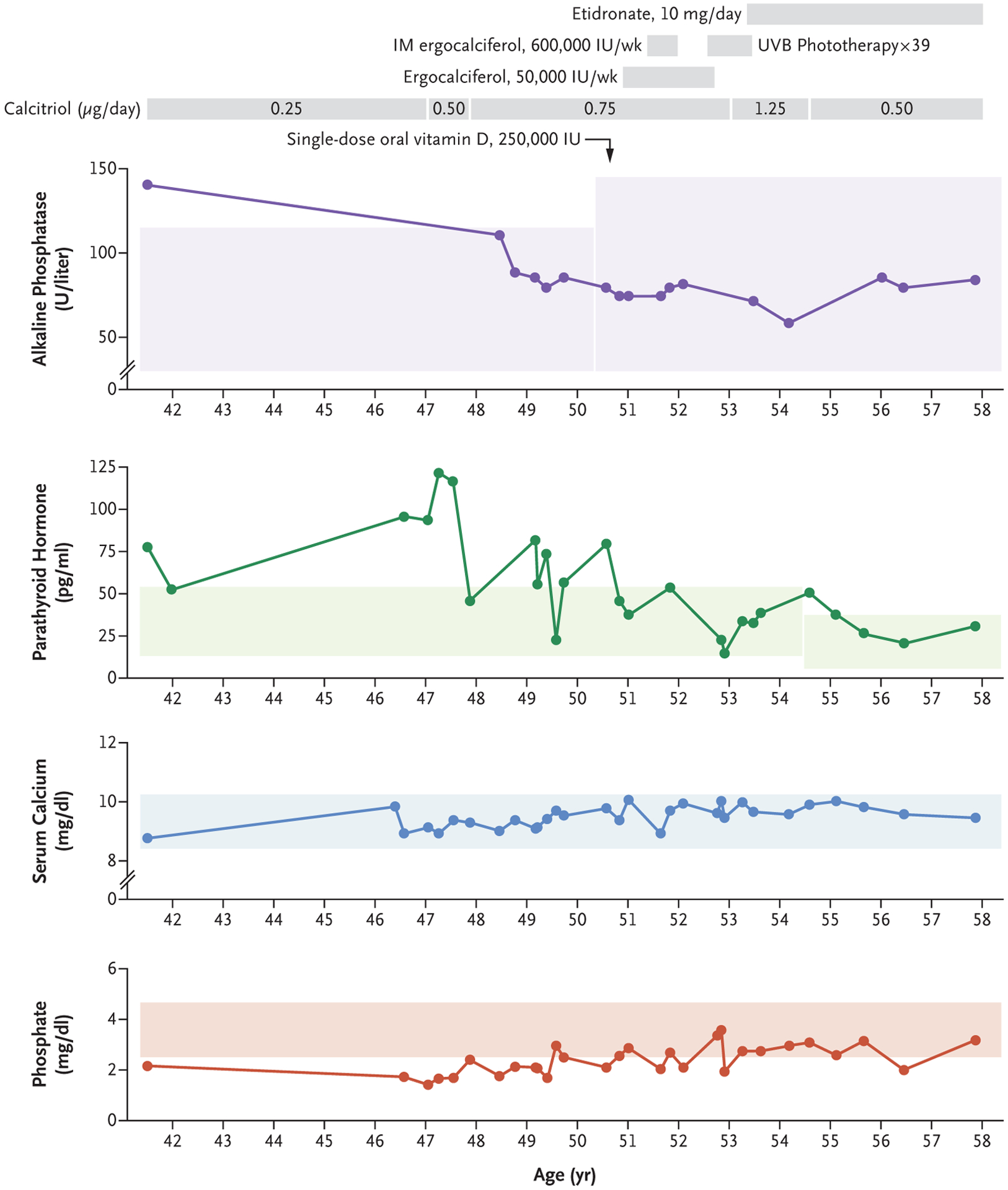
Shown are clinical laboratory results for alkaline phosphatase, parathyroid hormone, serum calcium, and phosphate after the onset of fragility fractures. To convert the values for calcium to millimoles per liter, multiply by 0.250. To convert the values for phosphate to millimoles per liter, multiply by 0.3229. Shaded areas show reference ranges: alkaline phosphatase, 30 to 115 U per liter and 30 to 145 U per liter; parathyroid hormone, 13 to 54 pg per milliliter and 7 to 37 pg per milliliter; calcium, 8.4 to 10.2 mg per deciliter; and phosphate, 2.5 to 4.6 mg per deciliter. The change in reference ranges for alkaline phosphatase and parathyroid hormone are the result of changes in instrumentation in the clinical laboratory during the time the measurements were obtained. Gray bars at the top of the figure provide a chronologic timeline of vitamin D supplementation. IM denotes intramuscular, and UVB ultraviolet B.
Methods
Details on the laboratory methods are provided in the Supplementary Appendix. As we have previously reported, serum concentrations of vitamin D–binding protein were measured with the use of trypsin digestion and liquid chromatography–tandem mass spectrometry,15 and plasma and intracellular concentrations of total 25(OH)D and 1,25(OH)2D were measured with the use of immunoextraction and liquid chromatography–tandem mass spectrometry.16 We performed SNP microarray analysis (Gene Expression Omnibus accession GSE112111) using the CytoScan HD platform (Affymetrix). Metaphase fluorescence in situ hybridization (FISH) was performed with standard methods with the use of fosmid FISH probes G248P83578B9 and RP11–980G14. Fibroblasts from healthy volunteers and the patient were prepared from 2-mm skin-punch biopsy specimens. Serum concentrations of free 25(OH)D were determined by enzyme-linked immunosorbent assay (ELISA; DIASource), according to manufacturer specifications. All the authors vouch for the completeness and accuracy of the data.
Results
Homozygous Deletion of the GC Gene and Absence of Vitamin D–Binding Protein
The patient’s circulating vitamin D–binding protein concentrations were below the limit of detection on the mass spectrometric assay (<3 μg per milliliter) (Fig. S2 in the Supplementary Appendix).15 SNP microarray detected a 139-kb deletion at chromosome 4q13.3 containing the entire GC gene and an adjacent homozygous 144-kb deletion of part of the neuropeptide FF receptor 2 NPFFR2 gene, both nested in the proximal segment of a 67-Mb region of homozygosity (Fig. 2, and Fig. S3 in the Supplementary Appendix). Metaphase FISH confirmed the homozygous GC deletion in the patient, and testing of available siblings identified two who were heterozygotes and one who was homozygous normal. Vitamin D–binding protein concentrations in one carrier and one noncarrier sibling were 95.2 μg per milliliter and 224.5 μg per milliliter, respectively (Table 1); the latter value is similar to concentrations found in normal adults (median, 248.8 μg per milliliter [interquartile range, 224.6 to 275.6]).17 Both siblings were found to have the Gc1S haplotype, as assessed by liquid chromatography–tandem mass spectrometry.15
Figure 2. SNP and FISH Analyses of GC Deletion.
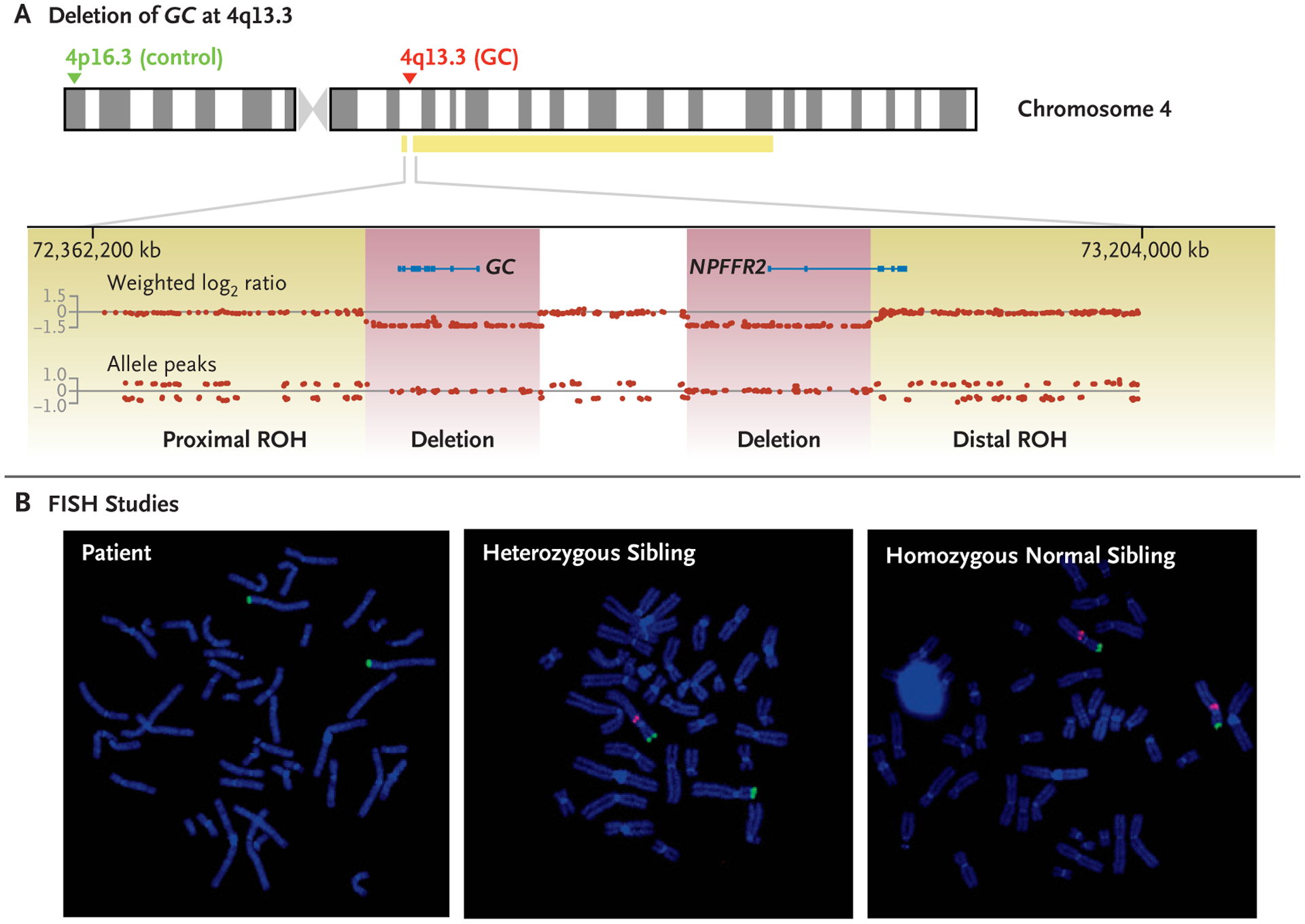
Panel A shows results of single-nucleotide polymorphism (SNP) microarray for the patient. With the use of human genome assembly hg19, homozygous deletions of the group-specific component (GC) gene and part of the neuropeptide FF receptor 2 (NPFFR2) gene are shown on chromosome 4 at 4q13.3, spanning base pairs 72,581,969 to 72,720,986 and 72,839,592 to 72,984,080, respectively. The deletions, shown in purple and separated by an undeleted segment, are flanked by a small proximal 1.13-Mb region of homozygosity (71,449,909 to 72,575,787) and a large distal 66-Mb region of homozygosity (72,991,367 to 139,320,224), shown in yellow and denoted as ROH. The absence of allele peaks is consistent with homozygous deletions. The probes used for metaphase fluorescence in situ hybridization (FISH) are shown with the 4p16.3 control probe RP11–980G14 in green and the 4q13.3 GC probe G24P83578B9 in red. Panel B shows the results of FISH for the patient (left), a heterozygous sibling (middle), and a homozygous normal sibling (right).
Table 1.
Characteristics of the Proband and Her Siblings in 2017.*
| Characteristic | Patient | Carrier Sibling† | Normal Sibling |
|---|---|---|---|
| Age (yr) | 58 | 59 | 60 |
| Sex | Female | Male | Female |
| Calcium (mg/dl) | 9.58 | 9.66 | 9.90 |
| Phosphate (mg/dl) | 2.45 | 2.23 | 2.76 |
| Parathyroid hormone (pg/ml) | 57 | 42 | 43 |
| 25(OH)D2(ng/ml) | 0.060 | 0.085 | 0.172 |
| 25(OH)D3 (ng/ml) | 0.019 | 13.054 | 19.483 |
| Total 25(OH)D (ng/ml) | 0.079 | 13.139 | 19.655 |
| 24,25(OH)2D3 (ng/ml) | 0.000 | 0.312 | 0.918 |
| 1,25(OH)2D2 (pg/ml) | 3.6 | <1.0 | <1.0 |
| 1,25(OH)2D3 (pg/ml) | 2.0 | 19.5 | 55.7 |
| Total 1,25(OH)2D (pg/ml) | 5.6 | 19.5 | 55.7 |
| Free25(OH)D (pg/ml) | |||
| Measured | 1.8 | 3.4 | 3.2 |
| Calculated | <0.2 | 7.6 | 5.5 |
| Vitamin D–binding protein (μg/ml) | <3.0 | 95.3 | 224.5 |
To convert the values for calcium to millimoles per liter, multiply by 0.250. To convert the values for phosphate to millimoles per liter, multiply by 0.3229. Clinical reference ranges are as follows: calcium, 8.9 to 10.2 mg per deciliter; phosphate, 2.5 to 4.5 mg per deciliter; parathyroid hormone, 12 to 88 pg per milliliter; total 25-hydroxyvitamin D (25[OH]D), 20 to 50 ng per milliliter; and total 1,25-dihydroxyvitamin D (1,25[OH]2D, also known as calcitriol), 17 to 72 pg per milliliter. No clinical reference ranges are available for free 25(OH)D or for vitamin D–binding protein. 1,25(OH)2D2 denotes 1,25-dihydroxyvitamin D2, 1,25(OH)2D3 1,25-dihydroxyvitamin D3, 24,25(OH)2D3 24,25-dihydroxyvitamin D3 (also known as secalciferol), 25(OH)D2 25-hydroxyvitamin D2, and 25(OH)D3 25-hydroxyvitamin D3 (also known as calcifediol).
Measurements were performed in one carrier sibling.
Quantification of Vitamin D Metabolites
Vitamin D–binding protein and plasma vitamin D metabolite concentrations were positively correlated (Table 1, and Fig. S4 in the Supplementary Appendix). The patient had reductions in all measured metabolites and undetectable 24,25-dihydroxycholecalciferol (24,25[OH]2D3, also known as secalciferol). Vitamin D metabolites in the noncarrier sibling were at or near the normal range, whereas those in the carrier sibling were intermediate. All three were deficient in 25(OH)D (<20 ng per milliliter), although the proband had markedly lower 25(OH)D (0.079 ng per milliliter) than her siblings. Only the proband had detectable levels of 1,25(OH)2D2 (3.6 pg per milliliter), a finding consistent with ergocalciferol supplementation, whereas her siblings had higher levels of 1,25-dihydroxycholecalciferol (1,25[OH]D3). Measured free 25(OH)D concentrations were within normal limits in all family members but differed from calculated values.
Cellular Vitamin D Uptake and Gene Expression
To determine the effects of vitamin D–binding protein deficiency on cellular uptake of vitamin D metabolites, we measured 25(OH)D concentrations in fibroblasts from the patient and from a volunteer, as well as intracellular expression of CYP24A1, which encodes the vitamin D–responsive 24-hydroxylase involved in 1,25(OH)D3 degradation. Exposure of the fibroblasts to normal serum replete with vitamin D–binding protein and supplemented with exogenous 25-hydroxychole-calciferol (25[OH]D3, also known as calcifediol) resulted in minor increases in intracellular 25(OH)D3 concentrations and induction of CYP24A1 expression (Figs. S5 through S7 in the Supplementary Appendix). The relative increases in intracellular 25(OH)D3 and CYP24A1 induction were larger when fibroblasts were exposed to serum deficient in vitamin D–binding protein and supplemented with exogenous 25(OH)D3. Relative CYP24A1 expression was lower in the patient’s fibroblasts than in the volunteer’s fibroblasts. The differences between plasma replete with vitamin D–binding protein and plasma deficient in vitamin D–binding protein may be greater in vivo, because 10% serum was used in these experiments.
Discussion
Absence of vitamin D–binding protein results in apparent severe vitamin D deficiency, as shown by nearly undetectable plasma concentrations of 25(OH)D. The haploinsufficient sibling had approximately half the normal concentration of circulating vitamin D–binding protein and no appreciable clinical manifestations. The patient’s plasma 25(OH)D levels were approximately 0.4% of those in the unaffected sibling, rather than the approximately 10% predicted from in vitro estimates of albumin affinity, which raises the possibility that albumin does not bind a meaningful amount of 25(OH)D in vivo. This possibility is supported by the observation that estimated free 25(OH)D differed between the proband and her siblings, whereas measured concentrations appeared similar. However, measured concentrations of total 25(OH)D were higher than those of free 25(OH)D, which potentially represents minor amounts of 25(OH)D weakly bound to albumin or other plasma proteins. The finding of low concentrations of bioavailable vitamin D metabolites in the context of vitamin D–binding protein deficiency would seem to support the free-hormone hypothesis and argue against the necessity of megalin-mediated uptake of vitamin D metabolites.
Despite a lifelong deficiency of vitamin D–binding protein, limited sun exposure (for religious reasons), and a diet that was probably lacking sufficient vitamin D, our patient did not have rickets or osteomalacia but rather osteopenia and fragility fractures that occurred in the fifth decade of life. The extent to which her immobility affected her bone densitometry findings is unclear. The disconnect between low plasma 25(OH)D concentration and her relatively mild bone disease highlights the controversy surrounding the use of total 25(OH)D to define vitamin D status2 and provides the strongest support to date for the hypothesis that cholecalciferol is activated locally in a paracrine or intracrine manner.18 Her clinical course and laboratory values were also similar to those of mice deficient in vitamin D–binding protein.13,14 When the mice were fed a vitamin D–replete diet, they showed significant reductions in serum 25(OH)D but maintained normal calcium, phosphate, and PTH concentrations. However, when they were fed a vitamin D–deficient diet, they remained normocalcemic while developing more pronounced secondary hyperparathyroidism, hypophosphatemia, and bone histomorphometric changes than their normal littermates. Correspondingly, although our patient achieved normal phosphate and PTH concentrations after years of supplementation, 25(OH)D and 1,25(OH)2D levels never normalized. Tissue studies in our patient and in knockout mice both showed the inability of plasma vitamin D–binding protein to greatly influence either the accumulation of biologically active vitamin D metabolites within the cell or the expression of vitamin D–responsive CYP24A1. As shown in mice, the absence of vitamin D–binding protein potentially reduces the half-life of plasma vitamin D metabolites by allowing for accelerated distribution to target cells14 and shunting toward an uncharacterized hepatic inactivating pathway.13 Given the relatively lower CYP24A1 expression in the patient’s cells than in fibroblasts from a healthy volunteer, one might anticipate further regulatory adaptations at the genomic level to compensate for vitamin D–binding protein deficiency.
Cross-sectional studies have shown that low 25(OH)D concentrations are associated with chronic inflammation and autoimmune disease19,20 and have noted ubiquitous vitamin D–receptor expression in certain immune cells (e.g., T cells and B cells).21,22 Vitamin D–binding protein also modulates neutrophil chemotaxis and macrophage activation in vitro.7,10 Our patient has severe ankylosing spondylitis, a chronic inflammatory disease that is typically more extensive in affected males. Given that meta-analyses have shown a strong inverse relationship between plasma concentrations of vitamin D metabolites and both disease risk and activity,23 our patient’s clinical association of extensive spondyloarthropathy with lifelong deficiency of vitamin D–binding protein raises the possibility of an important pathophysiological interrelationship.
We are unaware of reports describing homozygous deletion of the GC gene. We have not found similar or identical deletions in our internal copy number variant database or those publicly available (Database of Genomic Variants, DECIPHER, and ClinVar). Only larger heterozygous deletions on the order of megabases have been reported. The Genome Aggregation Database (gnomAD), comprising 125,748 exomes and 15,708 genomes from unrelated persons free of severe pediatric disease, lists 23 high-quality loss-of-function variants in the GC gene, making up 51 alleles in total, of which none are in the homozygous state.24 That our patient carries identical deletions within a long stretch of homozygosity appears to be explained by parental consanguinity. Without further study, we cannot speculate whether it is a private familial deletion or a rare deletion of unknown carrier frequency in the region from which this family originates (Lebanon and surrounding areas). The homozygous partial NPFFR2 deletion is of uncertain significance at this time, since the gene has yet to be associated with human disease. NPFFR2 encodes a G-protein–coupled receptor that in rodents is involved in the hypothalamic–pituitary axis and nociception.25
In conclusion, homozygous deletion of the GC gene in our patient was associated with a relatively mild phenotype of undetectable plasma 25(OH)D concentrations resistant to therapy, normocalcemic osteopenia, and fragility fractures. Delayed recognition of vitamin D–binding protein deficiency and delayed initiation of aggressive vitamin D supplementation, along with the confounding effects of ankylosing spondylitis, may have contributed to adverse outcomes in this case.
Supplementary Material
Acknowledgments
Supported by grants from the National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases (P30DK035816, to Dr. Hoofnagle), and from the NIH National Institute of Allergy and Infectious Diseases (K08AI119142, to Dr. Fink), and the University of Washington Department of Laboratory Medicine.
We thank Hannah Pflaum, M.L.S.(A.S.C.P.), for her technical assistance and Cathy Huculak (genetic counselor) for facilitating intersite sample transfers.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Holick MF. Vitamin D deficiency. N Engl J Med 2007; 357: 266–81. [DOI] [PubMed] [Google Scholar]
- 2.Bikle D, Bouillon R, Thadhani R, Schoenmakers I. Vitamin D metabolites in captivity? Should we measure free or total 25(OH)D to assess vitamin D status? J Steroid Biochem Mol Biol 2017; 173: 105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chun RF, Peercy BE, Orwoll ES, Nielson CM, Adams JS, Hewison M. Vitamin D and DBP: the free hormone hypothesis revisited. J Steroid Biochem Mol Biol 2014; 144: 132–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daiger SP, Schanfield MS, Cavalli-Sforza LL. Group-specific component (Gc) proteins bind vitamin D and 25-hydroxyvitamin D. Proc Natl Acad Sci U S A 1975; 72: 2076–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bikle DD, Gee E, Halloran B, Haddad JG. Free 1,25-dihydroxyvitamin D levels in serum from normal subjects, pregnant subjects, and subjects with liver disease. J Clin Invest 1984; 74: 1966–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouillon R, van Baelen H, de Moor P. Comparative study of the affinity of the serum vitamin D-binding protein. J Steroid Biochem 1980; 13: 1029–34. [DOI] [PubMed] [Google Scholar]
- 7.Bouillon R, Pauwels S. The vitamin D-binding protein In: Feldman D, ed. Vitamin D. 4th ed Vol. 1 Biochemistry, physiology and diagnostic. London: Academic Press, 2018: 97–115. [Google Scholar]
- 8.Trujillo G, Habiel DM, Ge L, Ramadass M, Cooke NE, Kew RR. Neutrophil recruitment to the lung in both C5a- and CXCL1-induced alveolitis is impaired in vitamin D-binding protein-deficient mice. J Immunol 2013; 191: 848–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnaud J, Constans J. Affinity differences for vitamin D metabolites associated with the genetic isoforms of the human serum carrier protein (DBP). Hum Genet 1993; 92: 183–8. [DOI] [PubMed] [Google Scholar]
- 10.Chun RF. New perspectives on the vitamin D binding protein. Cell Biochem Funct 2012; 30: 445–56. [DOI] [PubMed] [Google Scholar]
- 11.Kawakami M, Imawari M, Goodman DS. Quantitative studies of the interaction of cholecalciferol (vitamin D3) and its metabolites with different genetic variants of the serum binding protein for these sterols. Biochem J 1979; 179: 413–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooke NE, Haddad JG. Vitamin D binding protein (Gc-globulin). Endocr Rev 1989; 10: 294–307. [DOI] [PubMed] [Google Scholar]
- 13.Safadi FF, Thornton P, Magiera H, et al. Osteopathy and resistance to vitamin D toxicity in mice null for vitamin D binding protein. J Clin Invest 1999; 103: 239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zella LA, Shevde NK, Hollis BW, Cooke NE, Pike JW. Vitamin D-binding protein influences total circulating levels of 1,25-dihydroxyvitamin D3 but does not directly modulate the bioactive levels of the hormone in vivo. Endocrinology 2008; 149: 3656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henderson CM, Lutsey PL, Misialek JR, et al. Measurement by a novel LC-MS/MS methodology reveals similar serum concentrations of vitamin D-binding protein in blacks and whites. Clin Chem 2016; 62: 179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laha TJ, Strathmann FG, Wang Z, de Boer IH, Thummel KE, Hoofnagle AN. Characterizing antibody cross-reactivity for immunoaffinity purification of analytes prior to multiplexed liquid chromatography-tandem mass spectrometry. Clin Chem 2012; 58: 1711–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson-Cohen C, Zelnick LR, Hoofnagle AN, et al. Associations of vitamin D-binding globulin and bioavailable vitamin D concentrations with coronary heart disease events: the Multi-Ethnic Study of Atherosclerosis (MESA). J Clin Endocrinol Metab 2017; 102: 3075–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollis BW, Wagner CL. The role of the parent compound vitamin D with respect to metabolism and function: why clinical dose intervals can affect clinical outcomes. J Clin Endocrinol Metab 2013; 98: 4619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeffery LE, Raza K, Hewison M. Vitamin D in rheumatoid arthritis — towards clinical application. Nat Rev Rheumatol 2016; 12: 201–10. [DOI] [PubMed] [Google Scholar]
- 20.Robinson-Cohen C, Hoofnagle AN, Ix JH, et al. Racial differences in the association of serum 25-hydroxyvitamin D concentration with coronary heart disease events. JAMA 2013; 310: 179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aranow C Vitamin D and the immune system. J Investig Med 2011; 59: 881–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prietl B, Treiber G, Pieber TR, Amrein K. Vitamin D and immune function. Nutrients 2013; 5: 2502–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai G, Wang L, Fan D, et al. Vitamin D in ankylosing spondylitis: review and meta-analysis. Clin Chim Acta 2015; 438: 316–22. [DOI] [PubMed] [Google Scholar]
- 24.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536: 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin YT, Liu HL, Day YJ, Chang CC, Hsu PH, Chen JC. Activation of NPFFR2 leads to hyperalgesia through the spinal inflammatory mediator CGRP in mice. Exp Neurol 2017; 291: 62–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.