

Abstract
The role of IL-21, produced mainly by T helper (Th) 17 cells and T follicular helper cells, has been intensively investigated in B cell differentiation and antibody class switch. However, how IL-21 regulates memory IgA+ B cell development and memory IgA responses in the intestines is still not completely understood. In this study, we found the total IgA+ B cells as well as CD38+CD138−IgA+ memory B cells were significantly increased in intestinal lamina propria (LP) of TCRβxδ−/− mice after transfer of microbiota antigen-specific Th17 cells but not Th1 cells. Although IL-21R−/− mice or IL-17R−/− mice showed decreased antigen-specific memory IgA production in the intestines upon infection with Citrobacter rodentium (C. rodentium), the percentage of IgA+CD38+CD138− memory B cells in Peyer’s patches (PP) and LP was decreased only in IL-21R−/− mice, but not in IL-17R−/− mice, after re-infection with C. rodentium compared with WT mice. Blockade IL-21 in vivo suppressed intestinal C. rodentium-specific IgA production as well as IgA+CD38+CD138− memory B cells in PP and LP. Furthermore, IL-21 significantly induced B cell IgA production in vitro, with the increased expression of genes related with class-switching and memory B cell development, including Aicda, Ski, Bmi1, and Klf2. Consistently, Acida and Ski expression was decreased in B cells of IL-21R−/− mice after C. rodentium re-infection. In conclusion, our study demonstrated that IL-21 promotes intestinal memory IgA B cell development, possibly through up-regulating differentiation-related and class switching-related genes, indicating a potential role of IL-21 in memory IgA+ B cell responses in the intestines.
Introduction
Secreted antibodies are essential for providing immune protection in the intestine. The enrichment and maintenance of antibody production after antigen encounter have been considered as the result of the activation of limited memory B cells that are generated and sustained after the primary responses to the antigen (1, 2). B cells normally undergo somatic hypermutation and differentiation in the germinal center (GC), and emerge out as memory B cells or plasma cells. The selection into memory B cell fate is regulated by several mechanisms, one prominent of which is extrinsic signals from T cells, including cytokines and cell-contact-dependent signals (3). Several transcription regulators, which are essential for T cell development and differentiation, have been showed to be critical for the survival of immunoglobulin class-specific memory B cells, such as T-bet and retinoic acid in IgG2a+ memory B cells, and receptor-related orphan receptor-α (RORα) in IgA+ memory B cells, probably by controlling cell-surface B cell receptor transcription (4). Furthermore, IL-2 and IL-10 cooperated with CD40L play an important role in driving the transition of GC B cells to memory B cells (5).
IgA, one of the most enriched isotypes of antibodies in the intestine, has been crucial in regulation of host-microbiota interaction to maintain the intestinal homeostasis (6). It has been reported that the generation and maintenance of intestinal IgA response are different from that of systemic IgG response (7). The microbiota antigen-specific IgA+ B cells can retain in the intestine for more than 16 weeks, which suggests the unique relationship between IgA+ producing cells and intestinal environments. There are abundant long-lived microbiota antigen-specific Th17 cells in intestinal mucosa, which provide supports for the development of IgA+ B cells (8–10). It has been shown that Th17 cells facilitate B cell proliferation and promote the GC formation, as well as antibody isotype switching to IgG1, IgG2a, IgG2b, and IgG3 (11). IL-17, which predominately produced by Th17 cells, is suggested to promote the antibody production indirectly through inducing the production of B cell activators in other immune cells (12). IL-21, a signature cytokine of Th17 cells and T follicular helper cells, binds to IL-21R, which is widely expressed on T cells, B cells, and DCs, and is upregulated after activation by anti-CD40 mAb in memory B cells (13, 14). IL-21 has been shown to regulate B cell proliferation, class switch recombination, and plasma cell differentiation, as well as IgA production (10, 15, 16, 17). IgA+ memory B cell response can be boosted rapidly upon antigen re-challenge, which is crucial in the host defense. It has been shown that IL-21 deficiency led to reduction of GC B cells (18). However, the roles of Th17 cells as well as IL-17 and IL-21 in regulating intestinal memory IgA+ B cell responses are still unclear.
In this study, we demonstrated that transfer of gut microbiota antigen specific Th17 cells from CBir1 flagellin-specific TCR transgenic (CBir1 Tg) mice, which are specific for an immunodominant gut microbiota antigen CBir1 flagellin, significantly induced the CD19+CD38+CD138−IgA+ memory B cells in TCRβxδ−/− mice. In addition, upon Citrobacter rodentium (C. rodentium) infection, C. rodentium-specific IgA+ memory B cells as well as antigen-specific memory IgA production were impaired in IL-21R−/− mice. Our study thus indicates a critical role of IL-21 produced by Th17 cells in promoting the intestinal memory IgA responses.
Materials and Methods
Mice
Wild-type C57BL/6 (WT), TCRβxδ−/− and IL-21R−/− mice were obtained from Jackson Laboratory. IL-17R−/− mice were kindly provided by Amgen. CBir1 Tg mice were maintained and bred in the Animal Facilities at University of Texas Medical Branch (UTMB). All experiments were reviewed and approved by the Institutional Animal Care and Use Committees of the UTMB. All mice are bred onto the C57BL/6 background. 8–12 weeks-old mice were used for all experiments. All the mice used in this study were gender-matched, and maintained under SPF conditions.
Reagents
The following antibodies were used for flow cytometry: FITC-anti-CD19 (6D5), PE/Cy7-anti-IL-17A (TC11–18H10.1), FITC-anti-B220 (RA3–6B2), Brilliant Violet 421-anti-CD4 (RM4–5), PE/Cy7-anti-CD38 (90), APC-anti-CD138 (281–2), Percp/Cy5.5-anti-GL-7 (29F.1A12), and Percp/Cy5.5-IL-17 (TC11–18H10.1) were purchased from Biolegend. APC-anti-IL-21 (mhalx21), and Brilliant Violet 421-anti-CD95 (Jo2) were from BD Biosciences; PE-IgA (polyclonal) was from Southern Biotechnology. Foxp3 perm/fix kit for intracellular permeabilization and Live/Dead Fixable Dead Cell stain kit for gating live cells were from Thermo Scientific. Phorbolmyristate acetate was purchased from Sigma-Aldrich, and ionomycin was purchased from Invitrogen. Golgistop was purchased from BD Biosciences. Mouse recombinant IL-6, IL-17 and IL-21, and human recombinant TGFβ1 were purchased from Biolegend. Anti-IFNγ (XMG1.2), anti-IL-4 (11B11), and anti-IL21R (4A9) were from BioXCell. Antibody against IgD was purchased from Southern Biotechnology. Anti-μ was purchased from Jackson ImmunoResearch Laboratories. Anti-biotin microbeads from Miltenyi Biotec were used to isolate naïve IgD+ B cells. Anti-mouse phosphorylated Erk1/2, anti-mouse total Erk1/2, anti-mouse phosphorylated stat3, and anti-mouse total phosphorylated stat3 antibodies were purchased from Cell Signaling Technology. Anti-CD4 antibody was purchased from Abcam.
Polarization of Th17 cells and Th1 cells
CD4+ T cells were isolated from spleens of CBir1 Tg mice using anti-mouse CD4 magnetic beads (GK1.5, BD Biosciences) as previously described (19). To polarize Th17 cells, CBir1-Tg CD4+ T cells were cultured for 5 days with 15 ng/ml TGFβ, 20 ng/ml IL-6, 10 μg/ml anti-IFNγ, 10 μg/ml anti-IL-4 and 1 μg/ml CBir1 peptide with irradiated splenic antigen presenting cells (APC) (20). To polarize Th1 cells, CBir1-Tg CD4+ T cells were cultured with 10 ng/ml IL-12, 1 μg/ml CBir1 peptide, and irradiated antigen-presenting cells (APCs).
Fecal pellet preparation
Fecal pellets were collected every week from individual mouse, weighted and homogenized in cold PBS containing 0.04 mg/ml soybean trypsin inhibitor, 20 mM EDTA and 2 mM PMSF, and centrifuged at 15000 rpm for 15 mins to remove bacteria and insoluble debris as described previously (21). The supernatants were kept in −20°C for further ELISA processing.
ELISA
96-well plates were coated with 1 μg/ml anti-IgA (Kirkegaard & Perry Labs, KPL) or 1 μg/ml of C. rodentium bacterial lysate overnight at 4 degree. The plates were washed with PBS/Tween and blocked with 1% BSA/PBS at room temperature for 2 hrs, following with incubation of fecal pellets supernatant for 2 hrs. After washing, adherent antibodies were detected by biotinylated anti-IgA (KPL) and HRP-labeled streptavidin. Plates were developed using a two-component TMB substrate (KPL) and read at 450 nm. Results were quantified by normalizing to standard concentrations of IgA (Southern Biotechnology).
Intestinal lamina propria cell isolation
Intestines were removed from individual mouse and the Peyer’s patches (PP) were excised as previously described (19). Intestines were sliced and incubated with 0.5 mM EDTA-PBS at 37 degrees for 40 mins to remove epithelial cells. Intestines were then digested by collagenase IV for 40 min. After went through 100 μm filter, the cell lysis was resuspended in 40% Percoll and overload onto 70 % Percoll, and centrifuged at 2000 rpm for 20 mins at room temperature. The interface containing lymphocytes was collected. Lymphocytes were directly stained for T cell and B cell analysis.
Flow cytometry staining
For CD4+ T cell staining, cells were activated with phorbolmyristate acetate (50 ng/ml) and ionomycin (750 ng/ml) for 2 hrs, followed by adding 0.7μl/ml Golgistop for another 3 hrs. Cells were then stained with live/dead and antibody against CD4. Subsequently, cells were permeabilized and fixed by Foxp3 perm/fix kit. Finally, cells were stained with intracellular antibodies against IL-17 and IL-21.
For B cell staining, cells were stained with live/dead as well as surface markers of CD19. B220, CD38, CD38, CD95, and GL7. After permeabilization, cells were stained with antibody against IgA.
Citrobacter rodentium infection
The infection method was descripted previously (22). Briefly, C. rodentium strain DBS100 (ATCC) was grown overnight in lysogeny broth at 37°C and subcultured (1:100 dilution) in fresh LB for another 5 to 6 hrs. Bacteria were harvested and re-suspended in sterile PBS at 1 × 107 colony-forming unit (CFU)/ml. Adult mice (6–8 weeks old) were given 200 μl of bacterial suspension (2 × 106 CFU) by gavage. Fecal pellets were collected from individual mice and weighed, one part was homogenized in PBS and serial diluted onto MacConkey’s agar (BD Biosciences) for CFU counting; the other part was processed for fecal pellets supernatant. Mice were re-infected with same amount of C. rodentium at 5 weeks after initial infection, the fecal pellets were collected and processed for bacteria quantification and supernatant preparation. During the experiment period, all the mice were monitored and weighted.
Quantitative Real-Time/Reverse transcription PCR
RNA was extracted with TRIzol (Invitrogen) and followed by cDNA synthesis with Revertaid reverse transcriptase (Fermentas). Quantitative PCR was performed using SYBR Gene Expression Assays. Primers were synthesized from Integrated DNA Technologies, Inc., and data were normalized to Gapdh mRNA expression. Primer sequences as follows: Aicda: ©, 5’- AGAAAGTCACGCTGGAGACC-3′, reverse, 5’-CTCCTCTTCACCACGTAGCA-3’; Bmi1: forward, 5’-ATGAGTCACCAGAGGGATGG-3’, reverse, 5’-AAGAGGTGGAGGGAACACCT-3’; KLF2: forward, 5’-GCCTGTGGGTTCGCTATAAA-3’, reverse, 5’-TTTCCCACTTGGGATACAGG-3’; Ski: forward, 5’-AAAAGCCCTCCGCTCTAGTC-3’, reverse, 5’-GACGTCAGGGCTTAGCAGTC-3’; Il17r: forward 5’-AGTGTTTCCTCTACCCAGCAC-3’, reverse 5’-GAAAACCGCCACCGCTTAC-3’; Il21r: forward 5’-GGCTGCCTTACTCCTGCTG-3’, reverse 5’-TCATCTTGCCAGGTGAGACTG-3’; GAPDH: forward, 5’-CCATGGAGAAGGCTGGGG-3’, reverse, 5’-CAAAGTTGTCATGGATGACC-3. Aliquots of PCR products were visualized by electrophoresis on 1.5% agarose gels.
Western Blot
Cellular protein was extracted using radio-immunoprecipitation assay buffer, and the protein concentration was determined with a BCA Protein Assay kit (Thermo Fisher Scientific). Six microgram total protein/ sample was loaded and separated by TGX gels (Bio-rad), and then transferred to polyvinylidene difluoride membranes. After blocking, membranes were incubated with primary antibodies (1:1000), followed by incubation with a secondary antibody (1:2000). Protein bands were developed using enhanced chemiluminescence assay.
Statistical analysis
For comparisons between samples, levels of significance were determined by Student’s t test or One-way ANOVA in Prism 8.0 (Graphpad). Mean ± SD is represented. *p < 0.05 was considered as significance.
Results
1. Gut microbiota specific Th17 cells promote intestinal memory IgA+ B cell development and IgA production.
To investigate how gut microbiota antigen specific Th17 cells regulate intestinal memory IgA responses to microbiota, we generated Th17 cells from CBir1 Tg mice, which are specific for an immunodominant gut microbiota antigen CBir1 flagellin (23), in vitro by culture under Th17 polarization condition with TGFβ and IL-6, and transferred them into TCRβxδ−/− mice, which lack T cells. To address the specific role of Th17, but not other effector T cells, in memory IgA responses, we transferred the in vitro generated Th1 cells from CBir1 Tg mice into TCRβxδ−/− mice. We also gave another group of mice PBS to serve as controls. The fecal pellets were collected for measuring IgA on day 21. Transfer of Th17 cells significantly increased fecal IgA production in TCRβxδ−/− mice compared with transfer of Th1 cells and the controls with PBS (Fig. 1A). To investigate whether transferred T cells localize to PP, the recipient mice were sacrificed 21 post T cell transfer and PPs were stained with anti-CD4 antibody. As shown in Supplementary Fig. 1A, CD4+ T cells presented in PPs from the mice received either Th1 or Th17 cells. Spleen, PP, and Lamina propria (LP) cells from these mice were harvested for analysis of B cell phenotypes by flow cytometry (FACS). The percentage of CD95+GL7+ B cells in PP, which represent the GC B cells, was significantly higher in mice which received Th17 cells than that in control mice with PBS (Fig. 1B), and also higher than that in mice received Th1 cells although not reaching statistical significance (Fig. 1B). There were no significant differences of CD95+GL7+ B cells in PP between mice transferred with Th1 cells and control mice with PBS (Fig. 1B). The IgA+ B cells were significantly increased in spleen, PP, and LP in the recipient mice with Th17 cells but not with Th1 cells (Fig. 1C), and significantly higher in PP of the mice received Th17 cells compared to the mice with Th1 cells (Fig. C). Importantly, total CD138−CD38+IgA+ memory B cells were significantly expanded in spleen, PP, and LP in the recipient mice with Th17 cells compared with control mice, whereas transfer of Th1 cells did not increase memory IgA+ B cells (Fig. 1D). In addition, CD138−CD38+IgA+ memory B cells in PP and LP were significantly increased in mice received Th17 cells compared with those in mice received Th1 cells (Fig. 1D). Similar results were observed on CD19−CD38−CD138+IgA+ plasma cells among spleen, PP, and LP (Supplementary Fig. 1B). Taken together, these data indicated that Th17 cells promoted IgA memory B cell development in the intestine. The levels of IL-17 and IL-21, which are signature cytokines of Th17 cells, in those Th17 cells before and after transfer were shown in Supplementary Fig. 1C and D.
Figure 1. Transfer of microbiota specific Th17 cells promotes the memory IgA production in TCRβxδ−/− mice.
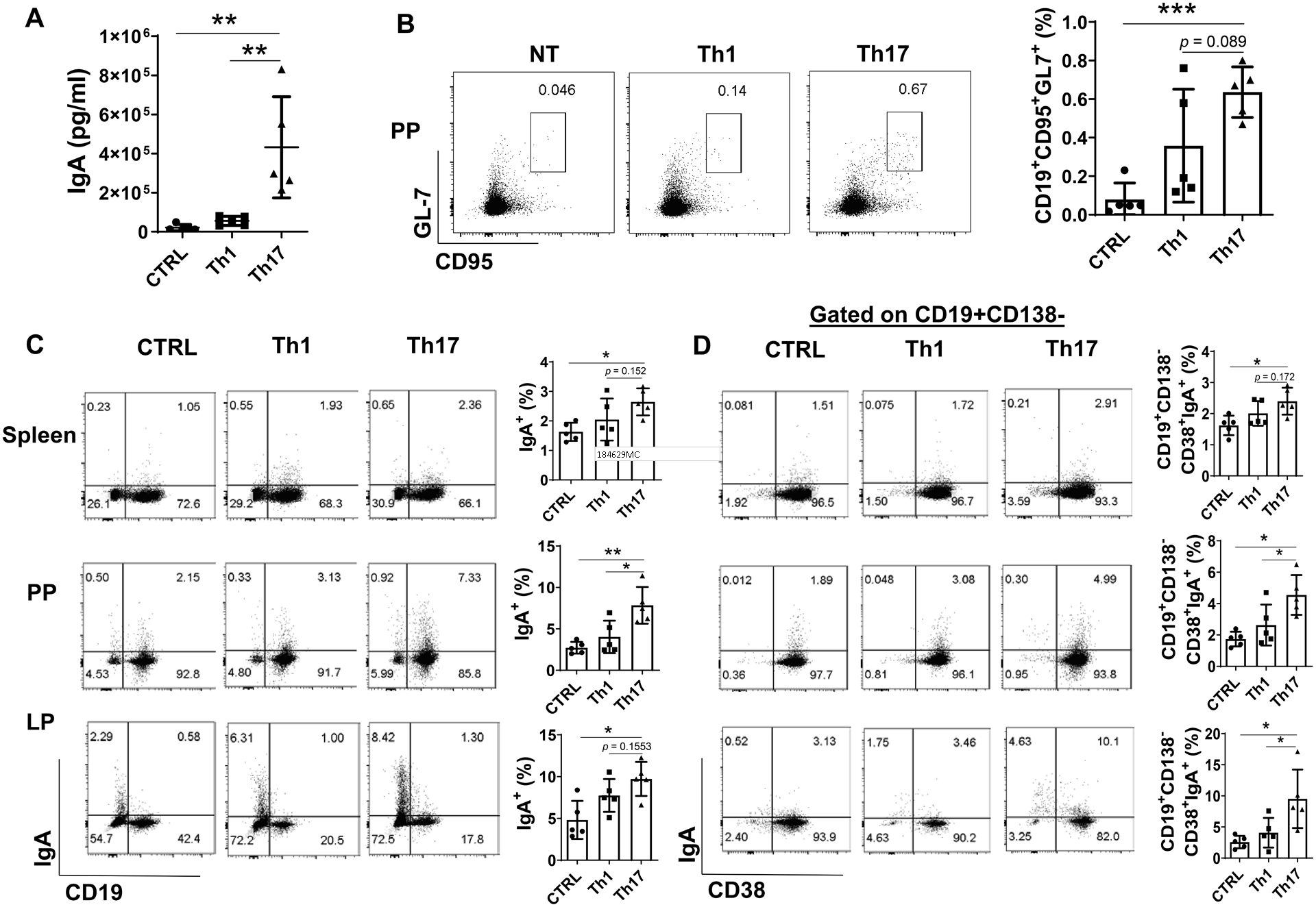
Splenic CBir1 Tg CD4+ T cells were activated with irradiated APCs and CBir1 peptide under Th1 or Th17 polarization condition for 5 days. Th1 (1 × 106/mice) or Th17 (1 × 106/mice) cells were transferred to TCRβxδ−/− mice (n = 5/group) by intravenous injection. TCRβxδ−/− mice receiving PBS served as controls (n = 5). (A) Mice were sacrificed on day 21. Fecal IgA levels were quantified by ELISA and normalized to total protein. The CD95+GL7+ B cells in Peyer’s patches (PP) (B), total IgA+ B cells (C), and the CD19+CD138−CD38+IgA+ B cells in the spleen, PP, and LP were analyzed by FACS. One representative of 2–3 experiments with similar results was shown. *p < 0.05, **p < 0.01, ***p < 0.001. One-way ANOVA.
2. IL-21R−/− mice and IL-17R−/− mice demonstrate impaired intestinal antigen-specific IgA production and reduced capacity of bacteria clearance upon enteric infection.
As Th17 cells produce their signature cytokines IL-17 and IL-21, we asked whether IL-17 or IL-21 is involved in the regulation of intestinal memory IgA+ B cell development. We first infected WT mice with a low dose of C. rodentium (2 × 106 CFU/mouse) by oral gavage to avoid induction of intestinal inflammation, which could confound data interpretation, and re-infected with the same dose of C. rodentium on day 35. Mice were sacrificed 7 days post re-infection. We found that CD4+ T cells produced both IL-17 and IL-21 in PP and LP (Fig. 2A). We then infected WT, IL-17R−/−, and IL-21R−/− mice with a low dose of C. rodentium (2 × 106 CFU/mouse) by oral gavage. Fecal pellets were collected weekly for analysis of intestinal IgA secretion. We found that C. rodentium-specific IgA levels were dramatically increased in feces of WT mice from day 21 post initial infection, while intestinal C. rodentium-specific IgA secretion in IL-17R−/− mice and IL-21R−/− mice were significantly lower than that in WT mice (Fig. 2B and 2C). Once the specific antibody secretion dropped back to the relatively low levels in feces at 5 weeks after initial infection (Fig. 2B and C), these mice were re-infected with same amount of C.rodentium for analysis of the memory B cell responses. Seven days after re-infection, the intestinal C. rodentium-specific IgA production was induced rapidly in WT mice but not in IL-17R−/− mice and IL-21R−/− mice (Fig. 2B and 2C), indicating the importance of IL-17 and IL-21 in the intestine memory IgA responses. Additionally, we found that IL-17R−/− and IL-21R−/− mice showed higher levels of fecal C. rodentium upon re-infection compared to WT mice (Fig. 2D and E).
Figure 2. IL-21R−/− and IL-17R−/− mice showed impaired bacteria specific IgA production upon Citrobacter rodentium infection.
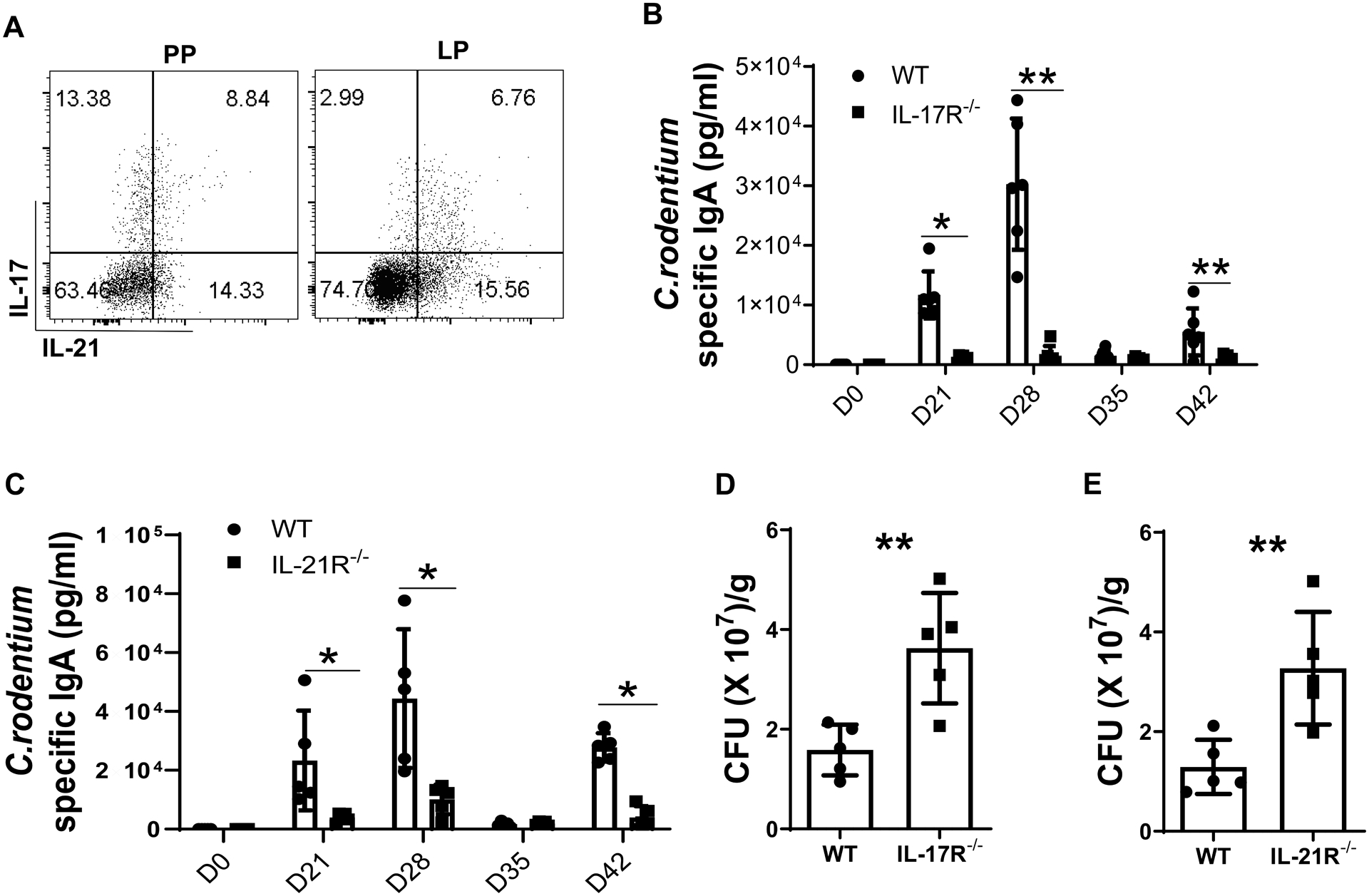
(A) WT mice (n = 4) were orally infected with C. rodentium (2 × 106 CFU/mouse) on day 0, and re-infected with the same amount of C. rodentium on day 35 after initial infection. Mice were sacrificed 7 days post re-infection, and IL-17 and IL-21 production in CD4 T cells from PP and LP was measured by FACS. (B-E) WT, IL-17R−/−, and IL-21R−/− mice (n = 5/group) were orally infected with C. rodentium (2 × 106 CFU/mouse) on day 0, and re-infected with the same amount of C. rodentium on day 35 after initial infection. The fecal pellets were collected on day 21, 28, 35, and 42 post initial infection, and C. rodentium-specific IgA production in the feces of WT and IL-17R−/− mice (B) or IL-21R−/− mice (C) were measured by ELISA. CFUs were measured in WT and IL-17R−/− mice (D) or IL-21R−/− mice (E). One representative of 2–3 experiments with similar results was shown. *p < 0.05. Unpaired Student t test.
3. IL-21R−/− mice but not IL-17R−/− mice show the impaired intestinal memory IgA+ B cells.
To investigate the role of IL-17 in memory B cell development, we analyzed B cell phenotypes of WT and IL-17R−/− mice which were infected with C. rodentium (2 × 106 CFU/mouse) by oral gavage, and re-infected mice 5 weeks post initial infection. One week after re-infection, WT and IL-17R−/− mice developed similar levels of GL7+CD95+ GC B cells in PP (Fig. 3A), with comparable levels of the total IgA+ B cells (Fig. 3B), B220−CD38−CD138+IgA+ plasma cells (Supplementary Fig. 2A), and B220+CD138−CD38+IgA+ memory B cells (Fig. 3C), in spleen, PP, and LP. Moreover, serum level of C. rodentium-specific IgA was similar between WT and IL-17R−/− mice (Supplementary Fig. 2B). These data indicated that decreased antigen specific memory IgA secretion in IL-17R−/− mice is not due to the impaired memory IgA+ B cell development, which could be explained by our previous study showing the lower polymeric immunoglobulin receptor (pIgR) expression, which facilitates the secretion of the IgA into lumen, in intestinal epithelial cells in IL-17R−/− mice (24).
Figure 3. Intact intestinal IgA+ B cell development in IL-17R−/− mice.
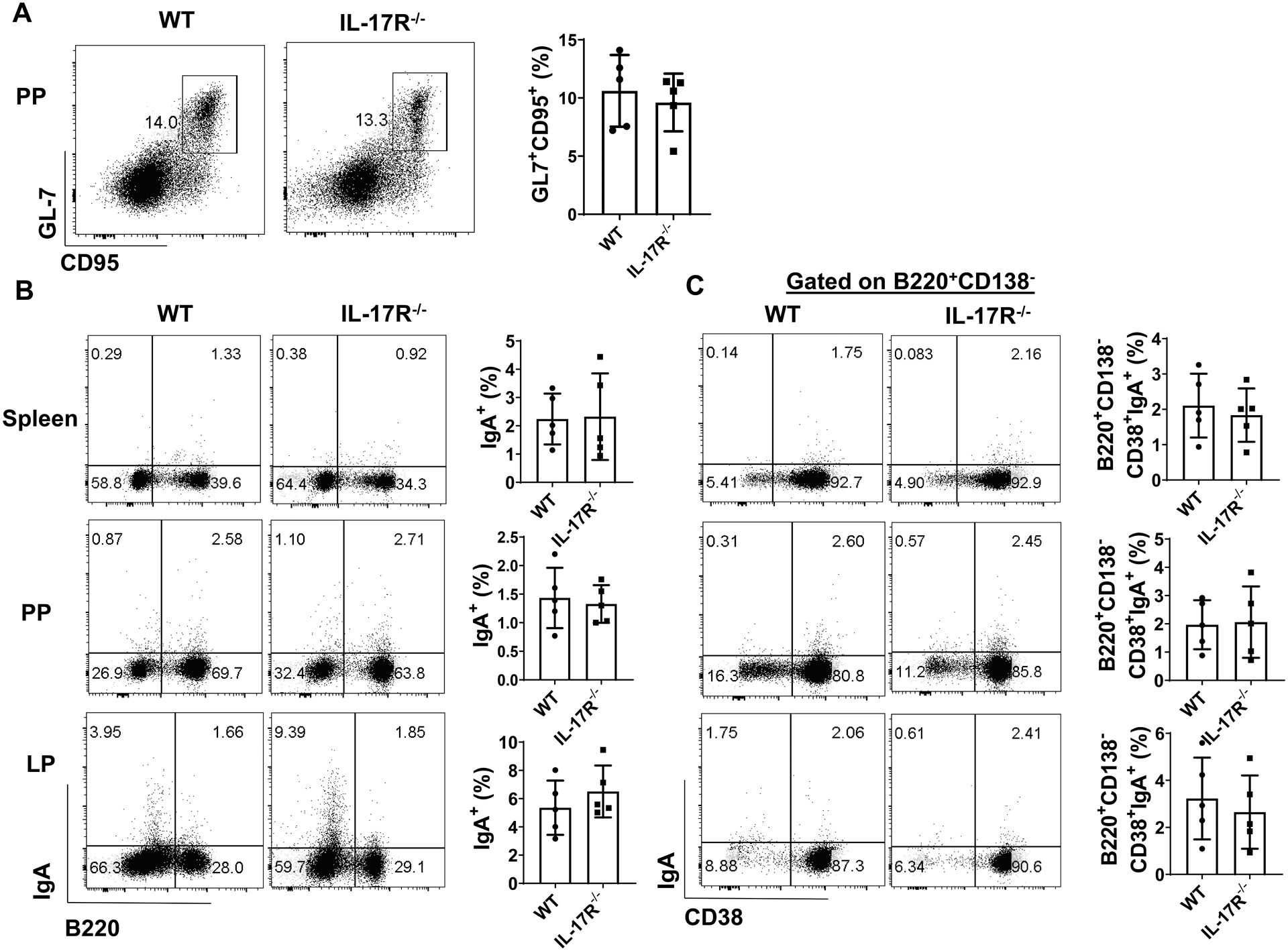
WT and IL-17R−/− mice (n = 5/group) were orally infected with C. rodentium (2 × 106 CFU/mouse) on day 0, and re-infected with the same amount of C. rodentium on day 35 after initial infection. Mice were sacrificed one week post re-infection. The CD95+GL7+ B cells in PP (A), total IgA+ B cells (B), and the CD19+CD138−CD38+IgA+ B cells (C) in the spleen, PP, and LP were analyzed by FACs. One representative of three experiments with similar results was shown. Unpaired Student t test.
Next, we investigated whether IL-21 is responsible for Th17 induction of memory IgA+ B cell responses. WT and IL-21R−/− mice were infected with C. rodentium (2 × 106 CFU/mouse) by oral gavage, and re-infected mice 5 weeks post initial infection. One week after re-infection, the levels of CD95+GL7+ GC B cells were significantly lower in PP of IL-21R−/− mice than those of WT mice (Fig. 4A). The levels of IgA+ B cells and B220−CD38−CD138+IgA+ plasma cells were also decreased in spleen, PP, and LP of IL-21R−/− mice compared with WT mice (Fig. 4B, supplementary Fig. 2D). Interestingly, IL-21R−/− mice showed reduced levels of B220+CD138−CD38+IgA+ memory B cells compared with WT mice (Fig. 4C). Taken together, these data demonstrated a crucial role of IL-21 in development of intestinal memory IgA+ B cells. Additionally, there was no difference of serum IL-21 levels between WT and IL-17R−/− mice (Supplementary Fig. 2C), indicating that reduced fecal IgA in IL-17R−/− was not attributed to a decreased IL-21 production.
Figure 4. IL-21R−/− mice demonstrated impaired memory IgA B cell development.
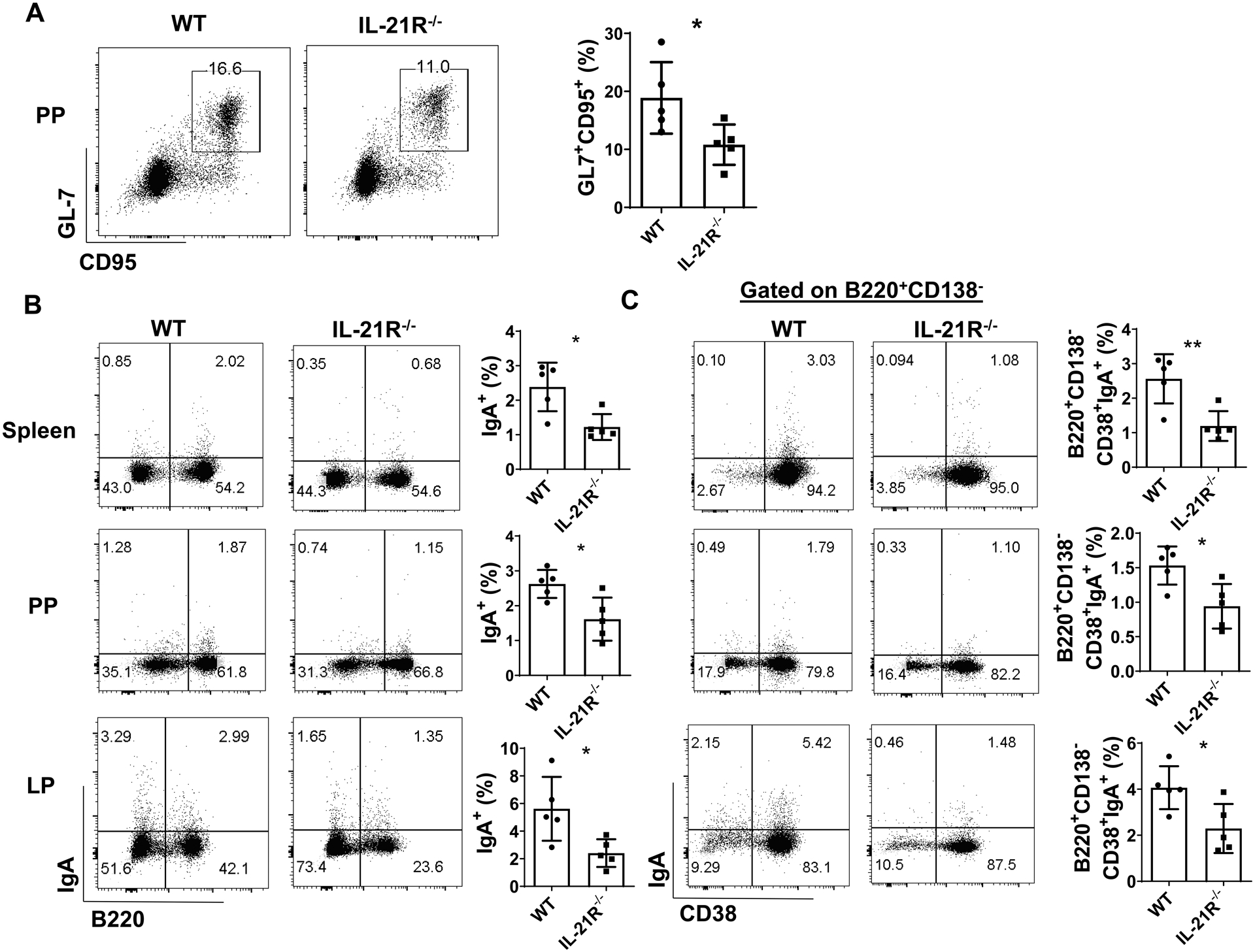
WT and IL-21R−/− mice (n = 5/group) were orally infected with C. rodentium (2 × 106 CFU/mouse) on day 0, and re-infected with the same amount of C. rodentium on day 35 after initial infection. Mice were sacrificed one week post re-infection. The CD95+GL7+ B cells in PP (A), total IgA+ B cells (B), and the CD19+CD138−CD38+IgA+ B cells (C) in spleen, PP, and LP were analyzed by FACS. One representative of two experiments with similar results. *p < 0.05. Unpaired Student’s t test.
To further confirm the roles of IL-17 and IL-21 for development of intestinal IgA memory B cells, we analyzed the B cell phenotypes in IL-17R−/− and IL-21R−/− mice under steady condition. Interestingly, IL-21R−/− mice showed less B220+CD138−CD38+IgA+ memory B cells in PP and LP under steady condition (Supplementary Fig. 3B). Furthermore, PP CD95+GL7+ GC B cells as well as B220−CD38−CD138+IgA+ plasma cells in PP and LP were also decreased in IL-21R−/− mice (Supplementary Fig. 3A and 3C). However, there were no differences of PP CD95+GL7+ GC B cells, B220+CD138−CD38+IgA+ memory B cells, and B220−CD38−CD138+IgA+ plasma cells between WT and IL-17R−/− mice (Supplementary Fig. 3A–C).
4. Blockade of IL-21 suppresses intestinal memory IgA+ B cell development.
To further investigate the role of IL-21 in regulating memory IgA+ B cell development, we infected WT mice orally with C. rodentium (2 × 106 CFU/mouse) on day 0, and re-infected on day 35. Group of mice were given anti-IL-21R antibody or control anti-IgG antibody i.p. every the other day from the day of re-infection. The mice were sacrificed one week post re-infection. Administration of anti-IL-21R antibody suppressed fecal C. rodentium-specific IgA (Fig. 5A) and inhibited bacterial clearance (Fig. 5B). Treatment of anti-IL-21R antibody decreased GL7+CD95+ GC B cells in PP (Fig. 5C), and suppressed total IgA+ B cells and B220+CD138−CD38+IgA+ memory B cells in spleen, PP, and LP (Fig. 5D and E). In addition, B220−CD38−CD138+IgA+ plasma cells were also decreased after treatment of anti-IL-21R antibody (Supplementary Fig. 4).
Figure 5. Blockade of IL-21-IL-21R pathway suppressed memory IgA B cell development.
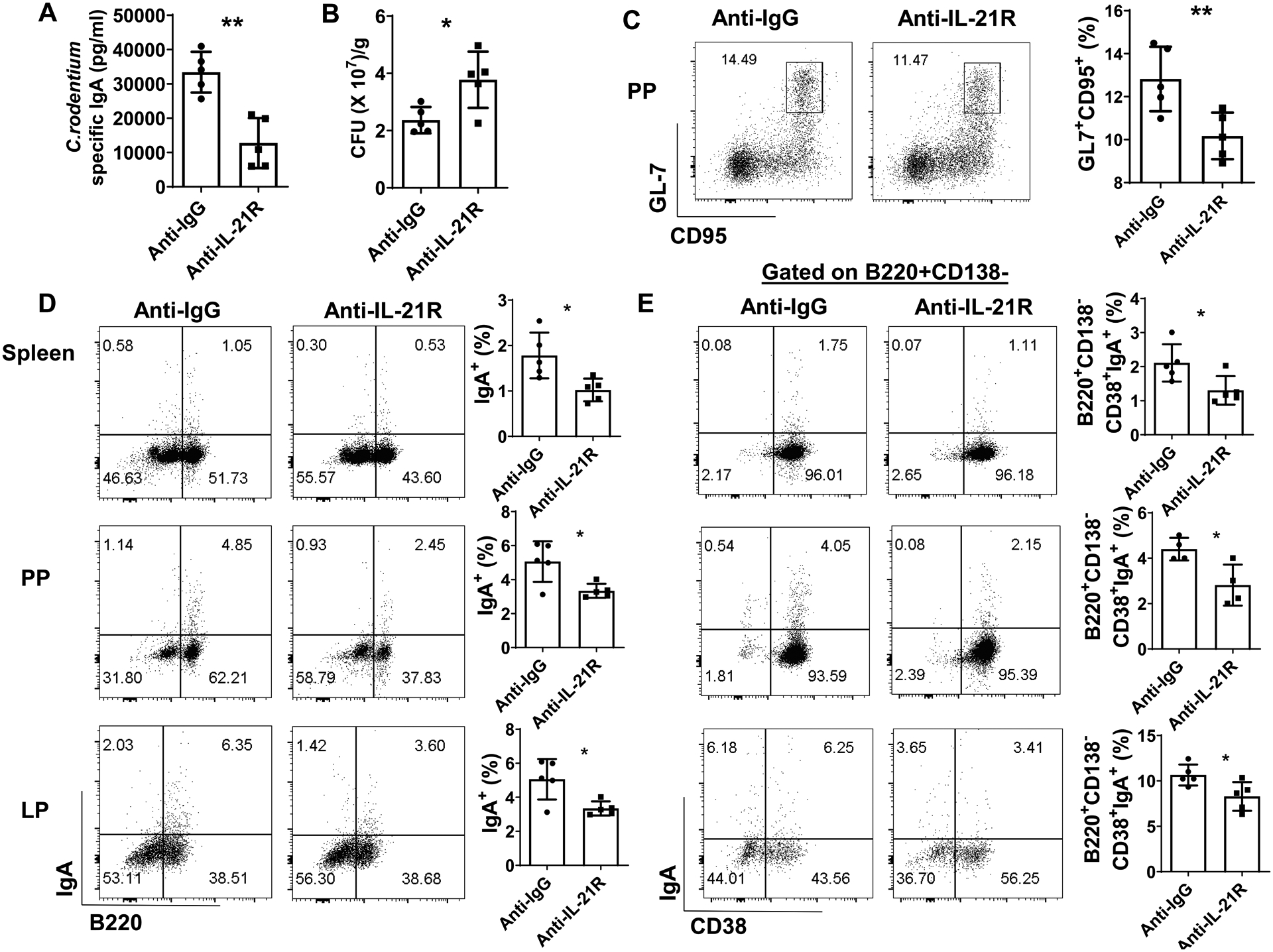
WT mice were orally infected with C. rodentium (2 × 106 CFU/mouse) on day 0, and re-infected with the same amount of C. rodentium on day 35 after initial infection. One group of mice (n = 5/group) were administrated with anti-IL-21R antibody (25mg/kg) i.p every the other day from day 35, and another group of mice (n = 5/group) were given anti-IgG antibody as control. Mice were sacrificed one week post re-infection. Fecal C. rodentium-specific IgA (A) and CFU (B) were measured. CD95+GL7+ B cells in PP (C), total IgA+ B cells (D), and CD19+CD138−CD38+IgA+ B cells (E) in spleen, PP, and LP were measured by FACS. One representative of two experiments with similar results. *p < 0.05, **p < 0.01. Unpaired Student’s t test.
5. IL-21 promote the memory B cell development related gene expression.
To investigate the mechanisms by which IL-21 promotes IgA+ memory B cell development, we treated splenic naïve IgD+ B cells in vitro with IL-21 and/or IL-17. We first confirmed the B cell expression of IL-17 receptor and IL-21 receptor (Fig. 6A). IL-17 and IL-21 activated their downstream Erk1/2 (Fig. 6B) and Stat3 (Fig. 6C) in B cells, respectively, indicating that IL-17 and IL-21 at the tested dose of 10 ng/ml stimulates their pathway. Interestingly, IL-21, but not IL-17, promoted IgA production (Fig. 6D), which is consistent with our previous reports (10). IL-21 treatment also increased IgA+ B cells in in vitro cultures (Fig. 6E). We then analyzed the genes, which have been associated with development of memory B cells, Krüppel-like factor 2 (Klf-2), Ski, Bmi1, as well as class switch related gene, activation-induced cytidine deaminase (Aicda). IL-21 treatment promoted the expression of those genes in B cells (Fig. 6F). Consistently, the expression of Ski and Aicda in B cells in PP and spleen of IL-21R−/− mice infected with C. rodentium was significantly decreased compared with C. rodentium infected WT mice (Fig. 6G–H).
Figure 6. IL-21 promoted the expression of the genes related to memory B cell development.
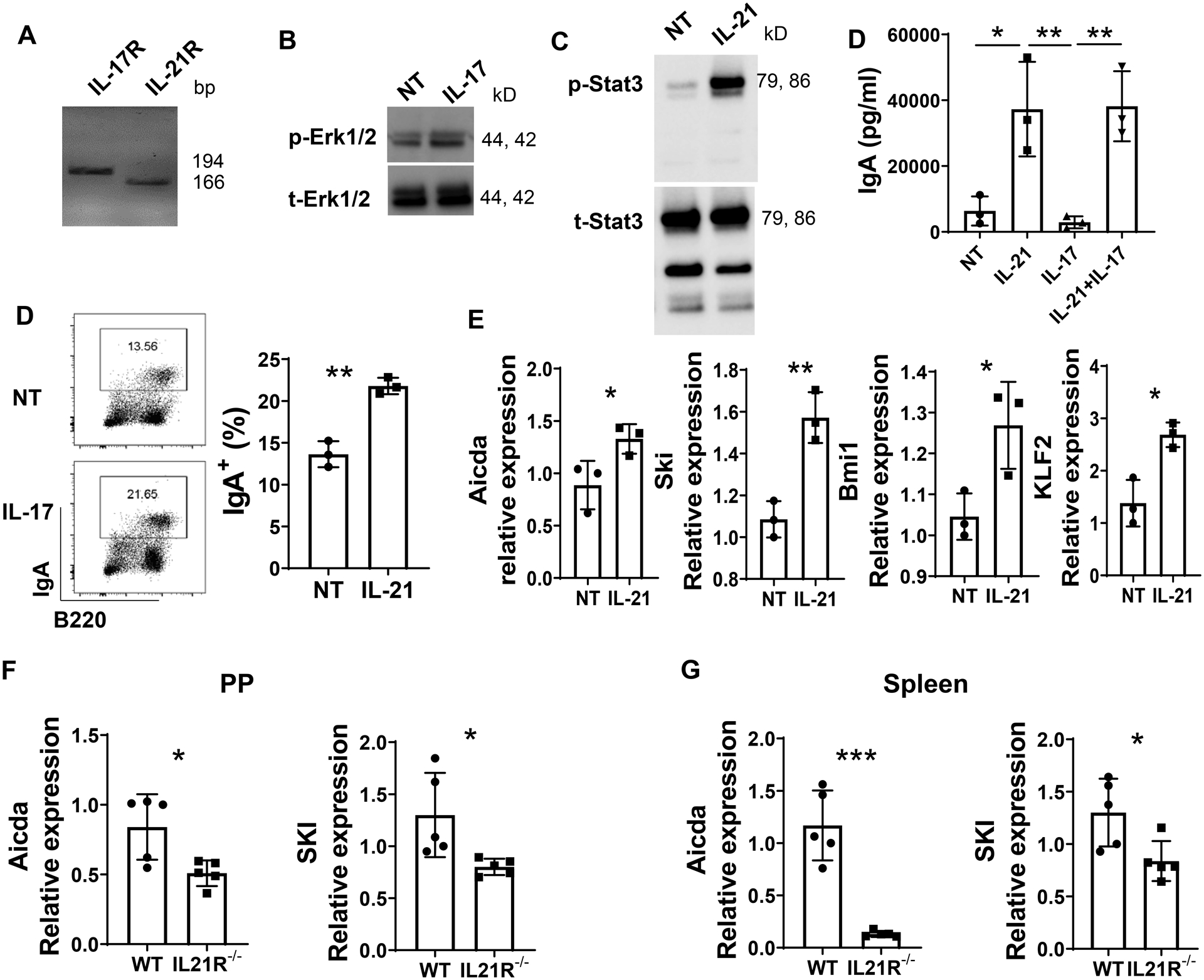
(A) IL-17R and IL-21R mRNA expression in splenic IgD+ B cells was measured by RT-PCR and visualized by electrophoresis on 1.5% agarose gels. (B) IgD+ B cells were activated with anti-μ (5 μg/ml) and CD40L (5 μg/ml) in the presence or absence of IL-17 (10 ng/ml) for 30 min, and phosphorylated Erk1/2 (p-Erk1/2) and total Erk1/2 (t-Erk1/2) protein levels were determined by western blot. (C) IgD+ B cells were activated with anti-μ (5 μg/ml) and CD40L (5 μg/ml) in the presence or absence of IL-21 (10 ng/ml) for 30 min, and phosphorylated stat3 (p-stat3) and total stat3 (t-stat3) protein levels were determined by western blot. (D) IgD+ B cells (n = 3/group) were activated with anti-μ (5 μg/ml) and CD40L (5 μg/ml) in the presence or absence of IL-21 (10 ng/ml), and/or IL-17 (10 ng/ml) for 5 days, the IgA concentration in the supernatant was measured by ELISA. (E and F) IgD+ B cells (n = 3/group) were activated with anti-μ (5 μg/ml) and CD40L (5 μg/ml) in the presence or absence of IL-21 (10 ng/ml) for 3 days, and IgA production was determined by FACS (E), and the expression of Aicda, SKI, Bmi1, KLF2 was analyzed by real-time PCR (F). (G-H) WT and IL-21R−/− mice (n = 5/group) were orally infected with C. rodentium (2 × 106 CFU/mouse) on day 0, and re-infected with the same amount of C. rodentium on day 35 after initial infection. Mice were sacrificed one week post re-infection. B220+ B cells were isolated from the PP (G) and spleen (H). The expression of Acida and SKI was analyzed with real-time PCR. *p < 0.05, **p < 0.01, ***p < 0.001. (D) One-way ANOVA. (E-H) Unpaired Student t test.
Discussion
The secretory IgA production in the intestine is pivotal to keep microbiota symbiosis and maintain intestine hemostasis. In this report, we demonstrated that IL-21, a signature cytokine of Th17 cells, promotes intestinal memory IgA+ B cell development and memory IgA production. Our results thus revealed a potential aspect of Th17 cells on the development of intestinal memory IgA+ B cells, in addition of the impacts on total IgA production in the intestines.
Interaction of Th17 cells and B cells have been well studied previously on promoting the antibody class switching and B cell differentiation. However, it is still not completely clear whether and how Th17 cells influence the development and proliferation of different B cell populations in the intestines. Unlike Th1 and Th2 cells, which are known to provide helps on B cells through their signature cytokines IFN-γ and IL-4 to induce class switch recombination to IgG2a or IgG1 and IgE, respectively (25, 26), Th17 cells have distinct roles in triggering B cell proliferation and promoting the formation of GC (11). IgA in the intestine is essential in maintenance of intestinal homeostasis and mucosal host defense (27). Besides, IgA plays a role in promoting specific bacteria colonization in the gut, such as B. fragilis, beyond its role in pathogen clearance (28). In this study, we used gut microbiota antigen specific T cells from CBir1 Tg mice, which are specific for an immunodominant gut microbiota antigen CBir1 flagellin, to investigate how Th17 cells regulate intestinal memory the IgA+ B cells. Although CBir1 Tg T cells do not respond to luminal CBir1 antigen when transferred into WT mice (19, 29), which is attributed to the presence of intestinal IgA blocking direct contact between T cells and gut microbiota/gut microbiota antigens, these T cells do respond robustly when transferred into IgA deficient mice, or immunocompromised mice whose intestinal IgA response was impaired completely or partially, such as RAG−/− mice and TCRβxδ−/− mice (19, 24, 30). This also indicates a crucial role of intestinal IgA response in regulation of intestinal T cell response to gut microbiota. We reported here that in the TCRβxδ−/− mice, transfer of gut microbiota antigen specific Th17 cells increased fecal IgA production as well as the IgA+ B cells in the spleen, PP, and LP compared with control mice. Th17 cells promoted GC formation in PP. Interestingly, the CD19+CD138−CD38+IgA+ memory B cells were significantly induced by Th17 cells in spleen, PP, and LP. Similar effects were demonstrated on CD19−CD38−CD138+IgA+ plasma cells as well. CBir1 antigen is just one of the gut microbiota antigens. Although we showed here that CBir1 specific Th17 cells induce intestinal IgA responses once activated, our results do not exclude the possibility that Th17 cells reactive to other gut microbiota antigens could also induce IgA responses. Our results thus suggest that gut microbiota antigen specific Th17 cells have potential roles to promoting the development of intestinal IgA+ B cells, more specifically the memory IgA+ B cells.
It has been shown that IL-17 promotes B cell class switch recombination to IgG2a and IgG3, while IL-21 promotes the switch recombination to IgG2b and IgG1 (11). In addition, IL-17 regulates the expression of pIgR on intestinal epithelial cells, which mediates the translocation of IgA into intestinal lumen, thus indirectly induces the intestine IgA production (24, 31, 32). IL-21, in cooperation of either IL-4 and IL-10 or TGF-β and retinoid acid (RA), regulates antibody isotype switching to subclasses of IgG and IgA in B cells (10, 16). In this study, we showed that IL-21 alone increased B cell IgA production in vitro, while the IL-17 alone showed limited effect. The combination of IL-21 and IL-17 showed similar effects on promoting IgA production as IL-21 alone. These results suggest that the induction of B cell IgA production by Th17 cells may mainly rely on the IL-21. Moreover, IL-21R−/− mice demonstrated significant lower GC B cells in PP compare with WT (P < 0.05). Interestingly, the B220+CD138−CD38+IgA+ memory B cells were lower in IL-21R−/− mice compared with WT mice. IL-21 was previously found to be involved in plasma cell differentiation from both naïve and memory B cells, and was believed to play a role in the maintenance of serologic memory (33, 34). Nevertheless, our data suggested that IL-21 plays a crucial role in development of intestinal memory IgA responses. Interestingly, it has been reported that through promoting IgA responses to atypical commensals intestine, IL-21 also encounters for altering the microbiota composition and mucosal immune response (35).
Upon re-exposure to same antigen, gut memory B cell migrate into multiple sites in the gut-associated lymphoid tissue, synchronize the response, and exhibit strong cross-protection against related pathogens (36, 37). Our previous studies showed that the deficiency of IL-17 or IL-21 decreased intestinal total IgA production. To further elucidate the roles of IL-17 and IL-21 in development of antigen specific memory IgA+ B cell development and IgA production, we infected IL-17R−/− and IL-21R−/− mice as well as WT mice with low dose of C. rodentium, both IL-21R−/− and IL-17R−/− mice showed impaired IgA responses in the gut. Moreover, the IL-21R−/− mice did not acquire the specific IgA responses after reinfection, while the WT mice showed a rapid elevated IgA response against C. rodentium, and no changes were observed on the IgA levels in IL-17R−/− mice as time progresses. Previous study has indicated that the intestine may adapt its memory IgA response to the predominant commensal species dominant in the lumen (7). Upon re-challenge of C. rodentium, total IgA+ B cells and B220+CD138−CD38+IgA+ memory B cells in the spleen, PP, and LP were lower in IL-21R−/− mice when compared with WT mice. In contrast, IgA+ B cells in spleen, PP, and LP were similar between WT and IL-17R−/− mice, which suggested that IL-17 did not directly induce IgA+ B cell differentiation. The lower secretory IgA production in the intestinal lumen is mainly due to lower pIgR expression in IL-17R−/− mice, which limits the IgA translocation across intestinal epithelial cells into intestinal lumen (24, 32, 38). Previous studies also suggested that IL-21 plays an important role in driving B cell differentiation into memory cells and plasma cells in both murine and human (15, 39). Thus, our data provided new evidence of IL-21 in promoting the memory IgA responses in the intestine.
Up-regulation of genes involved in activation, co-stimulation, and survival of memory B cells has been showed in increasing frequencies of antigen-specific precursor cells, which rapidly acquire T cell helps upon antigen re-encounter (40, 41). The constitutive expression of Aicda in memory B cells indicates that they remain poised to undergo additional rounds of antibody isotype switching and somatic hypermutation upon antigen re-exposure. In our study, transfer of microbiota antigen specific Th17 cells significantly increased Aicda expression in both PP and spleen B cells compared with those in control mice. The Aicda expression in B cells isolated from the PP and spleen of WT mice after re-exposure to C. rodentium were significantly higher compared with IL-21R−/− mice. Moreover, the IL-21 treatment promoted the Aicda expression in B cells in vitro. Polycomb group gene Bmi-1 and Krüppel-like factor 2 (Klf2) have been suggested to function in memory B cells, which is important for controlling the differentiation of immune-stimulation after pathogen re-enter (40, 42). Our data demonstrated the IL-21 promoted the Bmi-1 and Klf2 expression in B cells, which provided further support for signifying the role of IL-21 in driving the development of memory B cells. Ski, a transcriptional factor, is highly expressed in memory B cells compared with naïve B cells, suggesting it may play a role in memory B cell differentiation (40). In our study, IL-21 treatment increased Ski expression in B cells. Furthermore, re-infection with C. rodentium induced Ski expression in PP and spleen B cells, which is positively correlated with the memory IgA production in WT mice. In contrast, the Ski expression as well as C. rodentium-specific memory IgA response were defected in IL-21R−/− mice, suggesting that Ski may play a role in IL-21 induction of memory IgA+ B cell development.
Supplementary Material
Key points.
Th17 cells promote intestinal memory IgA+ B cell development and IgA response.
IL-21 is critical in regulating intestinal memory IgA+ B cell development.
Grant Support:
This work was supported by NIH grants DK105585, DK112436, DK125011, AI150210 and DK124132, University of Texas System STARs award (YC), and McLaughlin postdoctoral Fellowship, UTMB (XH).
Abbreviations:
- Th
T helper
- GC
Germinal center
- PP
Peyer’s patch
- LP
Lamina Propria
- RORα
receptor-related orphan receptor-α
- APCs
antigen-presenting cells
- Klf-2
Krüppel-like factor 2
- Aicda
activation-induced cytidine deaminase
- CBir1 Tg
CBir1 flagellin-specific TCR transgenic
- WT
Wild-type
- C. rodentium
Citrobacter rodentium
- CFU
colony-forming unit
Footnotes
Disclosures: The authors declare no competing interests.
Reference
- 1.Ahmed R, and Gray D. 1996. Immunological memory and protective immunity: understanding their relation. Science 272: 54–60. [DOI] [PubMed] [Google Scholar]
- 2.Kurosaki T, Kometani K, and Ise W. 2015. Memory B cells. Nat Rev Immunol 15: 149–159. [DOI] [PubMed] [Google Scholar]
- 3.Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, Takahashi Y, Fukuyama H, Okada T, and Kurosaki T. 2016. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol 17: 861–869. [DOI] [PubMed] [Google Scholar]
- 4.Wang NS, McHeyzer-Williams LJ, Okitsu SL, Burris TP, Reiner SL, and McHeyzer-Williams MG. 2012. Divergent transcriptional programming of class-specific B cell memory by T-bet and RORalpha. Nat Immunol 13: 604–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roy MP, Kim CH, and Butcher EC. 2002. Cytokine control of memory B cell homing machinery. J Immunol 169: 1676–1682. [DOI] [PubMed] [Google Scholar]
- 6.Bunker JJ, and Bendelac A. 2018. IgA Responses to Microbiota. Immunity 49: 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hapfelmeier S, Lawson MA, Slack E, Kirundi JK, Stoel M, Heikenwalder M, Cahenzli J, Velykoredko Y, Balmer ML, Endt K, Geuking MB, Curtiss R 3rd, McCoy KD, and Macpherson AJ. 2010. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 328: 1705–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kryczek I, Zhao E, Liu Y, Wang Y, Vatan L, Szeliga W, Moyer J, Klimczak A, Lange A, and Zou W. 2011. Human TH17 cells are long-lived effector memory cells. Sci Transl Med 3: 104ra100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, Sukumar M, Reger RN, Yu Z, Kern SJ, Roychoudhuri R, Ferreyra GA, Shen W, Durum SK, Feigenbaum L, Palmer DC, Antony PA, Chan CC, Laurence A, Danner RL, Gattinoni L, and Restifo NP. 2011. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity 35: 972–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao AT, Yao S, Gong B, Nurieva RI, Elson CO, and Cong Y. 2015. Interleukin (IL)-21 promotes intestinal IgA response to microbiota. Mucosal immunology 8: 1072–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitsdoerffer M, Lee Y, Jager A, Kim HJ, Korn T, Kolls JK, Cantor H, Bettelli E, and Kuchroo VK. 2010. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci U S A 107: 14292–14297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shibui A, Shimura E, Nambu A, Yamaguchi S, Leonard WJ, Okumura K, Sugano S, Sudo K, and Nakae S. 2012. Th17 cell-derived IL-17 is dispensable for B cell antibody production. Cytokine 59: 108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spolski R, and Leonard WJ. 2008. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol 26: 57–79. [DOI] [PubMed] [Google Scholar]
- 14.Good KL, Bryant VL, and Tangye SG. 2006. Kinetics of human B cell behavior and amplification of proliferative responses following stimulation with IL-21. J Immunol 177: 5236–5247. [DOI] [PubMed] [Google Scholar]
- 15.Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Spolski R, Leonard WJ, and Lipsky PE. 2005. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol 175: 7867–7879. [DOI] [PubMed] [Google Scholar]
- 16.Avery DT, Bryant VL, Ma CS, de Waal Malefyt R, and Tangye SG. 2008. IL-21-induced isotype switching to IgG and IgA by human naive B cells is differentially regulated by IL-4. J Immunol 181: 1767–1779. [DOI] [PubMed] [Google Scholar]
- 17.Lindner C, Thomsen I, Wahl B, Ugur M, Sethi MK, Friedrichsen M, Smoczek A, Ott S, Baumann U, Suerbaum S, Schreiber S, Bleich A, Gaboriau-Routhiau V, Cerf-Bensussan N, Hazanov H, Mehr R, Boysen P, Rosenstiel P, and Pabst O. 2015. Diversification of memory B cells drives the continuous adaptation of secretory antibodies to gut microbiota. Nat Immunol 16: 880–888. [DOI] [PubMed] [Google Scholar]
- 18.Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, Wang YH, Watowich SS, Jetten AM, Tian Q, and Dong C. 2008. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity 29: 138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cong Y, Feng T, Fujihashi K, Schoeb TR, and Elson CO. 2009. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proceedings of the National Academy of Sciences of the United States of America 106: 19256–19261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng T, Qin H, Wang L, Benveniste EN, Elson CO, and Cong Y. 2011. Th17 cells induce colitis and promote Th1 cell responses through IL-17 induction of innate IL-12 and IL-23 production. J Immunol 186: 6313–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cong Y, Brandwein SL, McCabe RP, Lazenby A, Birkenmeier EH, Sundberg JP, and Elson CO. 1998. CD4+ T cells reactive to enteric bacterial antigens in spontaneously colitic C3H/HeJBir mice: increased T helper cell type 1 response and ability to transfer disease. J Exp Med 187: 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dann SM, Le C, Choudhury BK, Liu H, Saldarriaga O, Hanson EM, Cong Y, and Eckmann L. 2014. Attenuation of intestinal inflammation in interleukin-10-deficient mice infected with Citrobacter rodentium. Infect Immun 82: 1949–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, Fort M, and Hershberg RM. 2004. Bacterial flagellin is a dominant antigen in Crohn disease. The Journal of clinical investigation 113: 1296–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao AT, Yao S, Gong B, Elson CO, and Cong Y. 2012. Th17 cells upregulate polymeric Ig receptor and intestinal IgA and contribute to intestinal homeostasis. Journal of immunology 189: 4666–4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snapper CM, and Paul WE. 1987. Interferon-gamma and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science 236: 944–947. [DOI] [PubMed] [Google Scholar]
- 26.Toellner KM, Luther SA, Sze DM, Choy RK, Taylor DR, MacLennan IC, and Acha-Orbea H. 1998. T helper 1 (Th1) and Th2 characteristics start to develop during T cell priming and are associated with an immediate ability to induce immunoglobulin class switching. J Exp Med 187: 1193–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shulzhenko N, Morgun A, Hsiao W, Battle M, Yao M, Gavrilova O, Orandle M, Mayer L, Macpherson AJ, McCoy KD, Fraser-Liggett C, and Matzinger P. 2011. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med 17: 1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donaldson GP, Ladinsky MS, Yu KB, Sanders JG, Yoo BB, Chou WC, Conner ME, Earl AM, Knight R, Bjorkman PJ, and Mazmanian SK. 2018. Gut microbiota utilize immunoglobulin A for mucosal colonization. Science 360: 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hand TW, Dos Santos LM, Bouladoux N, Molloy MJ, Pagan AJ, Pepper M, Maynard CL, Elson CO 3rd, and Belkaid Y. 2012. Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science 337: 1553–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng T, Wang L, Schoeb TR, Elson CO, and Cong Y. 2010. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. The Journal of experimental medicine 207: 1321–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crottet P, and Corthesy B. 1998. Secretory component delays the conversion of secretory IgA into antigen-binding competent F(ab’)2: a possible implication for mucosal defense. J Immunol 161: 5445–5453. [PubMed] [Google Scholar]
- 32.Jie Z, Yang JY, Gu M, Wang H, Xie X, Li Y, Liu T, Zhu L, Shi J, Zhang L, Zhou X, Joo D, Brightbill HD, Cong Y, Lin D, Cheng X, and Sun SC. 2018. NIK signaling axis regulates dendritic cell function in intestinal immunity and homeostasis. Nat Immunol 19: 1224–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuchen S, Robbins R, Sims GP, Sheng C, Phillips TM, Lipsky PE, and Ettinger R. 2007. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. J Immunol 179: 5886–5896. [DOI] [PubMed] [Google Scholar]
- 34.Ettinger R, Sims GP, Robbins R, Withers D, Fischer RT, Grammer AC, Kuchen S, and Lipsky PE. 2007. IL-21 and BAFF/BLyS synergize in stimulating plasma cell differentiation from a unique population of human splenic memory B cells. J Immunol 178: 2872–2882. [DOI] [PubMed] [Google Scholar]
- 35.Cho H, Jaime H, de Oliveira RP, Kang B, Spolski R, Vaziri T, Myers TG, Thovarai V, Shen Z, Fox JG, Leonard WJ, and Kelsall BL. 2019. Defective IgA response to atypical intestinal commensals in IL-21 receptor deficiency reshapes immune cell homeostasis and mucosal immunity. Mucosal Immunol 12: 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clemens JD, Sack DA, Harris JR, Chakraborty J, Neogy PK, Stanton B, Huda N, Khan MU, Kay BA, Khan MR, and et al. 1988. Cross-protection by B subunit-whole cell cholera vaccine against diarrhea associated with heat-labile toxin-producing enterotoxigenic Escherichia coli: results of a large-scale field trial. J Infect Dis 158: 372–377. [DOI] [PubMed] [Google Scholar]
- 37.Dogan I, Bertocci B, Vilmont V, Delbos F, Megret J, Storck S, Reynaud CA, and Weill JC. 2009. Multiple layers of B cell memory with different effector functions. Nat Immunol 10: 1292–1299. [DOI] [PubMed] [Google Scholar]
- 38.Kumar P, Monin L, Castillo P, Elsegeiny W, Horne W, Eddens T, Vikram A, Good M, Schoenborn AA, Bibby K, Montelaro RC, Metzger DW, Gulati AS, and Kolls JK. 2016. Intestinal Interleukin-17 Receptor Signaling Mediates Reciprocal Control of the Gut Microbiota and Autoimmune Inflammation. Immunity 44: 659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ozaki K, Spolski R, Ettinger R, Kim HP, Wang G, Qi CF, Hwu P, Shaffer DJ, Akilesh S, Roopenian DC, Morse HC 3rd, Lipsky PE, and Leonard WJ. 2004. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J Immunol 173: 5361–5371. [DOI] [PubMed] [Google Scholar]
- 40.Bhattacharya D, Cheah MT, Franco CB, Hosen N, Pin CL, Sha WC, and Weissman IL. 2007. Transcriptional profiling of antigen-dependent murine B cell differentiation and memory formation. J Immunol 179: 6808–6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Good KL, Avery DT, and Tangye SG. 2009. Resting human memory B cells are intrinsically programmed for enhanced survival and responsiveness to diverse stimuli compared to naive B cells. J Immunol 182: 890–901. [DOI] [PubMed] [Google Scholar]
- 42.Luckey CJ, Bhattacharya D, Goldrath AW, Weissman IL, Benoist C, and Mathis D. 2006. Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc Natl Acad Sci U S A 103: 3304–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.