

Abstract
Background and Aims
Hepatitis B virus (HBV) integrations are common in hepatocellular carcinoma (HCC). In particular, alterations of the telomerase reverse transcriptase (TERT) gene by HBV integrations are frequent; however, the molecular mechanism and functional consequence underlying TERT HBV integration are unclear.
Approach and Results
We adopted a targeted sequencing strategy to survey HBV integrations in human HBV‐associated HCCs (n = 95). HBV integration at the TERT promoter was frequent (35.8%, n = 34/95) in HCC tumors and was associated with increased TERT mRNA expression and more aggressive tumor behavior. To investigate the functional importance of various integrated HBV components, we employed different luciferase reporter constructs and found that HBV enhancer I (EnhI) was the key viral component leading to TERT activation on integration at the TERT promoter. In addition, the orientation of the HBV integration at the TERT promoter further modulated the degree of TERT transcription activation in HCC cell lines and patients’ HCCs. Furthermore, we performed array‐based small interfering RNA library functional screening to interrogate the potential major transcription factors that physically interacted with HBV and investigated the cis‐activation of host TERT gene transcription on viral integration. We identified a molecular mechanism of TERT activation through the E74 like ETS transcription factor 4 (ELF4), which normally could drive HBV gene transcription. ELF4 bound to the chimeric HBV EnhI at the TERT promoter, resulting in telomerase activation. Stable knockdown of ELF4 significantly reduced the TERT expression and sphere‐forming ability in HCC cells.
Conclusions
Our results reveal a cis‐activating mechanism harnessing host ELF4 and HBV integrated at the TERT promoter and uncover how TERT HBV‐integrated HCCs may achieve TERT activation in hepatocarcinogenesis.
Abbreviations
- ABI
Applied Biosystems Inc
- ChIP
chromatin immunoprecipitation
- DMEM
Dulbecco’s modified Eagle’s medium
- ELF4
E74‐like ETS transcription factor 4
- EnhI
enhancer I
- EnhII
enhancer II
- HBsAg
hepatitis B surface antigen
- HBV
hepatitis B virus
- HBx
hepatitis B x protein
- HBxC1
C‐terminal truncated hepatitis B virus X protein
- HCC
hepatocellular carcinoma
- MT
mitochondrial
- NTC
nontarget control
- qPCR
quantitative real‐time PCR
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- TERT
telomerase reverse transcriptase
Hepatocellular carcinoma (HCC) is a leading cause of cancer deaths worldwide.( 1 , 2 ) Chronic hepatitis B virus (HBV) infection is a major etiological factor of HCC. HBV frequently integrates into the human genome, with the telomerase reverse transcriptase (TERT) gene being the most frequent target of HBV integration in HBV‐associated HCCs.( 3 ) Interestingly, TERT promoter mutations were recently identified as another frequent genetic alteration in HCC.( 4 ) Therefore, telomerase activation and its maintenance of telomere length are critical events driving hepatocarcinogenesis.( 4 , 5 , 6 ) However, the molecular mechanism of how HBV integration into the TERT gene leads to TERT activation is unclear. Here, we present our targeted DNA sequencing approach to systematically determine HBV integrations in a cohort of HBV‐associated HCCs. We also identified a mechanism of TERT HBV integration on TERT activation through recruitment of host transcription factor to the viral enhancer I (EnhI) leading to cis‐activation consequence. Furthermore, using small interfering RNA (siRNA) library screening, we discovered that the key transcription factor, E74‐like ETS transcription factor 4 (ELF4), plays a crucial role in driving TERT activation in HCC with TERT HBV integration. Knockdown of ELF4 not only reduced TERT expression but also diminished the sphere‐forming ability of HCC cells. This suggests the potential additional involvement of TERT in self‐renewal ability apart from its better‐known role in maintaining telomere stability. Interestingly, with the use of two HCC cell models that naturally harbor two different forms of TERT HBV integration, we demonstrated that the orientation of HBV integration could modulate the degree of TERT transcription activation. Our data have provided insight into the molecular mechanism of TERT HBV integration in HBV‐associated HCC.
Patients and Methods
Patient Samples
Ninety‐five HCC cases from patients who underwent surgical resection of tumors at Queen Mary Hospital, Queen Elizabeth Hospital, and Pamela Youde Hospital, Hong Kong, were randomly selected. The patients aged from 24 to 74 years (mean: 52.5 years) with male predominance (74.7%, 71/95 were male). All patients had chronic HBV infection and were serum positive for hepatitis B surface antigen (HBsAg). All specimens were obtained immediately after surgical resection, snap frozen in liquid nitrogen, and kept at ‐80°C. Frozen sections were cut from tumor blocks and stained for histological examination to ensure a homogenous cell population of tissues. The demographic data are summarized in Supporting Table S1. This study was approved by the institutional review board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster and requirement for informed consent was waived by IRB.
DNA Extraction, Library Preparation, and Targeted Sequencing on HBV‐Associated HCCs
Total DNA was isolated from the HCC tissues using a described method.( 7 ) Library preparation and custom target enrichment were performed by Illumina TruSeq DNA Library Preparation Kit (San Diego, CA) and NimbleGen EZ Developer Library Kit (Roche, Basel, Switzerland), respectively, as according to the manufacturers’ protocol. Target‐enriched libraries were then PCR amplified and sequenced using Illumina HiSeq 2000 system. Paired‐end reads of 101 base pairs were generated. Library preparation and massively parallel sequencing for targeted sequencing were performed by the Centre for Genomic Sciences, The University of Hong Kong. In the assay design for the targeted sequencing, the whole HBV genome (National Center for Biotechnology Information GenBank: DQ089769.1) was incorporated. By doing so, probes were designed to target the HBV genome and were able to extract chimeric genomic fragments (containing both human and HBV genomic sequences) at HBV integration junctions. Such fragments were able to generate soft‐clipped sequencing reads to detect HBV integrations and determine their exact genomic locations.
HBV Integration Detection
We developed our tool Virus‐Clip( 8 ) to detect viral integration events. It relies on soft‐clipped sequencing reads that represent chimeric fusion of human and virus genomic sequences. With the targeted sequencing data, Virus‐Clip identified HBV integration events and determined the exact positions of integration junctions at single‐base resolution. HBV integration events were supported by at least three soft‐clipped sequencing reads that showed chimeric fusions. Recurrent HBV‐integrated genes were defined as having integration events located at the same gene for >1 sample, i.e., sample recurrence.
TERT Expression Assay by Quantitative Real‐Time PCR
To confirm the TERT activating events, TERT expression level was subjected to quantitative real‐time PCR (qPCR) by TaqMan qPCR assay (Hs00972656_m1; Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. Total RNA was extracted by Trizol (Invitrogen, Carlsbad, CA), and complementary DNA was synthesized by a reverse transcription kit (Invitrogen) on 81 randomly selected human HCC cases (both HCC and the corresponding nontumorous liver tissues) that had targeted sequencing performed on HCC cell lines.
Cell Lines
Human HCC cell lines PLC/PRF/5, SNU449, and HepG2 were obtained from American Type Culture Collection, whereas MHCC‐97L was a gift from Dr. Z. Y. Tang of Fudan University in Shanghai. All HCC cell lines used were authenticated by short tandem repeat assays and a computation algorithm( 9 ) and confirmed to have no HeLa cell and mouse cell contamination. PLC/PRF/5 and HepG2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) high glucose supplemented with 10% fetal bovine serum, SNU449 cells in RPMI‐1640 medium supplemented with 10% fetal bovine serum and 1 mM sodium pyruvate, and MHCC‐97L cells in DMEM high glucose supplemented with 10% fetal bovine serum and 1 mM sodium pyruvate. HepG2.2.15 cells were obtained from Dr. Se‐Jong Kim (Yonsei University College of Medicine, Seoul) and cultured in minimum essential medium supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, and 400 μg/mL G418.
Dual Luciferase Reporter Assay
HepG2 and SNU449 cells were transfected with different combinations of plasmids using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s protocol. The plasmids used included TERT–wild type/pGL3‐Basic, various forms of HBV‐TERT promoter fusion DNA/pGL3‐Basic and mutated HBV‐TERT promoter fusion DNA/pGL3‐Basic reporter constructs, and an internal control pRL‐PGK. Twenty‐four hours after transfection, luciferase and Renilla luciferase activities were measured by a Dual Luciferase Reporter assay system (Promega, Madison, WI) according to the manufacturer’s protocol. Transfection efficiency was normalized with the Renilla luciferase activity. Experiments were done three times independently.
PCR Confirmation
To confirm the HBV‐TERT promoter integration events within 1 kb upstream of the TERT transcriptional start site, a set of PCR primers (HBV‐1174‐F: 5′‐TGCCAAGTGTTTGCTGACGC‐3′ and chr5:1295168F: 5′‐CAGCGCTGCCTGAAACTC‐3′) was used to amplify the HBV‐TERT promoter integration events from human HCC tissue DNA and subjected to Sanger sequencing for confirmation of HBV‐TERT promoter integration events. To validate the HBV‐TERT promoter integration in PLC/PRF/5 and MHCC‐97L, three sets of primers (HBV‐1174‐F: 5′‐TGCCAAGTGTTTGCTGACGC‐3′ and chr5:1295168F: 5′‐CAGCGCTGCCTGAAACTC‐3′; HBV‐1024R: 5′‐GCAGCAAAACCCAAAAGACCC‐3′ and chr5:1295540F: 5′‐GTAACCCGAGGGAGGGGCCA‐3′ and HBV‐1174F: 5′‐TGCCAAGTGTTTGCTGACGC‐3′ and HBV‐1572R: 5′‐GCAGATGAGAAGGCACAGAC‐3′) were used to amplify HBV‐TERT promoter integration events from HCC cell line DNA and subjected to Sanger sequencing for confirmation of HBV‐TERT promoter integration events. All the PCR reactions were carried out using Applied Biosystems Inc. (ABI) AmpliTaq Gold 360 master mix (Applied Biosystems) with supplement of 360 GC enhancer solution according to the manufacturer’s protocol.
siRNA Library Screening
Human ON‐TARGETplus SMARTpool siRNA Library‐Transcription Factor panel and Human ON‐TARGETplus SMARTpool siRNA Library‐Nuclear Receptor panel were purchased from Dharmacon (Lafayette, CO). HepG2.2.15 and PLC/PRF/5 were transfected with siRNA duplexes using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Four days after siRNA transfection, cultured medium was removed and the wells were washed with phosphate‐buffered saline 3 times, and 100 μL of fresh medium was added and incubated for 12 hours. Cultured medium was collected and subjected to HBsAg protein detection using a Bio‐Rad Monalisa HBsAg ELISA detection kit (Bio‐Rad, Hercules, CA) according to the manufacturer’s protocol.
Cell Transfection and Establishment of Stable Knockdown Cells
Short hairpin RNAs (shRNAs) incorporated with sequences targeting ELF4 mRNA were subcloned into pLKO.1‐Puro shRNA expression vector. The shRNA vectors were transfected using Lipofectamine 2000 (Invitrogen) into 293FT according to the MISSION Lentiviral Packaging System (Sigma‐Aldrich, St. Louis, MO) manufacturer’s protocol. For viral packaging, the viral particles containing shRNA were transduced into PLC/PRF/5 and MHCC‐97L to establish the shRNA stably expressing cells, and the transfected cells were kept under 3 μg/mL puromycin selection for 7 days.
Western Blot Analysis
Cells were lyzed in sodium dodecyl sulfate (SDS)‐containing buffer and equal amounts of protein were separated in SDS‐polyacrylamide gel electrophoresis gel for western blot analysis. Immunodetection was performed using anti‐ELF4 (Sigma‐Aldrich) and antitubulin (Sigma‐Aldrich) antibodies.
Chromatin Immunoprecipitation Assay
The detailed protocol has been described.( 10 ) The antibody against ELF4 protein (Sigma‐Aldrich) was used, and the primer sets (HBV‐1000F: 5′‐TGTGGGTCTTTTGGGTTTTGCTGCC‐3′, HBV‐1120R: 5′‐AAGGCCTTGTAAGTTGGCGAG‐3′, HBV‐1240F: 5′‐GCGCGTGCGTGGAAGATCTGTGTCTCCTCTGC‐3′, and HBV‐1300R: 5′‐TTGCTCCAGACCTGCTGCGAGC‐3′) covering the putative ELF4 binding site on HBV EnhI DNA region were employed for standard qPCR using ABI Power SYBR Green master mix supplements of AmpliTaq Gold 360 master mix (Applied Biosystems) and detected by ABI QuantStudio 5 Real‐Time PCR System (Applied Biosystems) in the chromatin immunoprecipitation (ChIP) assay.
Sphere Formation Assay
Briefly, cells were suspended in 0.25% methyl cellulose in serum‐free DMEM/F12 in wells precoated with 1% Poly(2‐hydroxyethyl methacrylate) to form spheres in the presence of the B‐27 supplement. At the end point, the numbers of spheres greater than 100 μm in diameter were counted.
Immunohistochemistry
Immunohistochemistry was performed on formalin‐fixed, paraffin‐embedded sections using rabbit polyclonal antibody against ELF4 (Sigma‐Aldrich) at 1:500 dilution.
Results
Landscape of HBV DNA Integrations Using Targeted DNA Sequencing
With targeted DNA sequencing, we surveyed HBV integrations on a cohort of 95 human HBV‐associated HCCs (demographic and clinicopathologic data summarized in Supporting Table S1) and detected HBV integrations in 85 (89.5%) of the 95 cases (Supporting Fig. S1). Among our cases with identified HBV integrations, the median number of integration events was 4 per case, with a range from 1 to 636 (Supporting Fig. S1). The targeted sequencing strategy achieved a sensitivity in identifying HBV integration that is fairly comparable with other strategies, including whole genome sequencing and whole transcriptome sequencing,( 3 , 11 , 12 , 13 , 14 , 15 , 16 ) in terms of the detection rate and number of integration events detected (Supporting Table S2).
Among all the HBV integration events detected, HBV integration breakpoints (at which circular HBV genome linearized and joined with the human genome) frequently occurred at an interval centered at around position 1,820 (Fig. 1A). A similarly high frequency at this breakpoint was seen after stratifying the events according to the genic locations (Fig. 1B,E). Intriguingly, these integration breakpoints located mainly near the 3' end or C‐terminus of the HBV X protein (HBx) gene (position 1,374‐1,838; Fig. 1F). Indeed, HBV integration breakpoints at around position 1,820 likely resulted in C‐terminal truncated HBx protein, which we recently demonstrated to be a common natural mutant (46%) in HCCs and associated with metastasis.( 10 ) We then determined the HBx C‐terminal truncation status of these 95 cases, as identified by sequence alignments to the HBV genome. We found that 41 (43.2%) of these 95 cases had HBx C‐terminal truncation, 45 (47.4%) had no C‐terminal truncated HBx, and the remaining 9 had undetermined results (because of low read coverage). On clinicopathologic correlation, the presence of integrated HBx C‐terminal truncation in the tumors was significantly associated with more aggressive tumor behavior, namely the presence of venous invasion (P = 0.017), poorer cellular differentiation (P = 0.028), larger tumor size (P = 0.046), and liver cirrhosis (P = 0.017), whereas there was also a trend for more advanced tumor stage (P = 0.075; Fig. 1G).
Fig. 1.
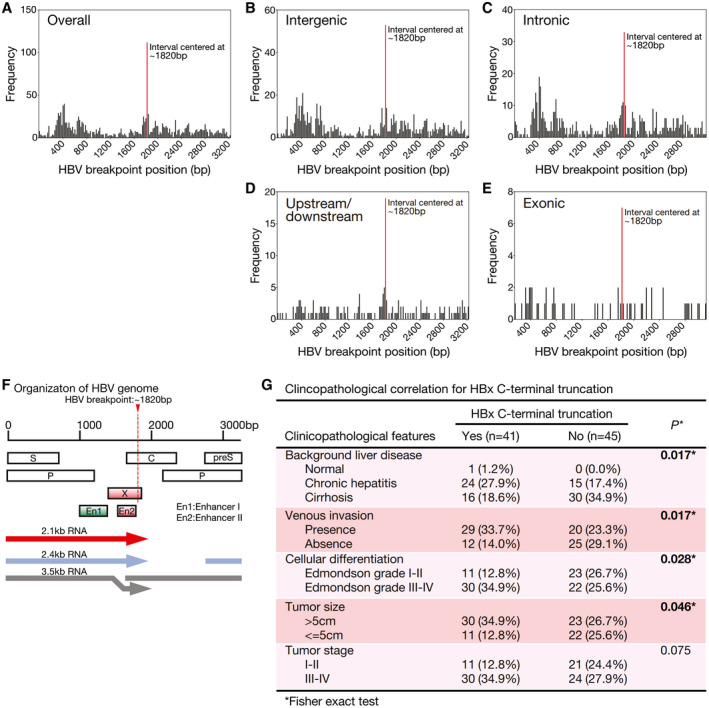
Frequency distribution of HBV breakpoint positions. (A) Overall distribution or (B‐E) stratified by genic location demonstrated hotspot breakpoint position at around position 1,820 (indicated with red line), which is located at the 3' end or C‐terminal region of the HBx gene. (F) Organization of HBV viral genes in HBV genome. (G) Clinicopathologic correlation of HBx C‐terminal truncation in human HCCs. There were nine cases with undetermined HBx C‐terminal truncation status because of low read coverage, and they were excluded.
HBV Integration at TERT Promoter and TERT Promoter Mutations
HBV integration was most frequently seen at the TERT gene (35.8%, n = 34; Fig. 2A). Apart from the TERT gene, lysine methyltransferase 2B (KMT2B, also known as MLL4), cyclin E (CCNE1), cyclin A2 (CCNA2), and Rho associated coiled‐coil containing protein kinase 1 (ROCK1) were also recurrently affected by HBV integration (11.6%, 3.2%, 2.1%, and 2.1%, respectively; Fig. 2A). In addition, genes located at the mitochondrial (MT) genome (MT‐CO1, MT‐CO3, MT‐CYB, MT‐ND1, MT‐ND2, MT‐ND4, MT‐ND5, and MT‐ND6) were recurrently affected, and this has very recently been reported.( 17 ) Besides, we also identified some integrated genes such as eyes shut homolog, microRNA 4457, and CLPTM1 like (CLPTM1L). Collectively, a total of 46 different TERT HBV integration events were identified in these 34 HCC cases (Fig. 2B and Supporting Table S3). We randomly selected 10 HBV integration events at the TERT gene, and they were successfully validated by PCR and Sanger sequencing (Fig. 2B).
Fig. 2.
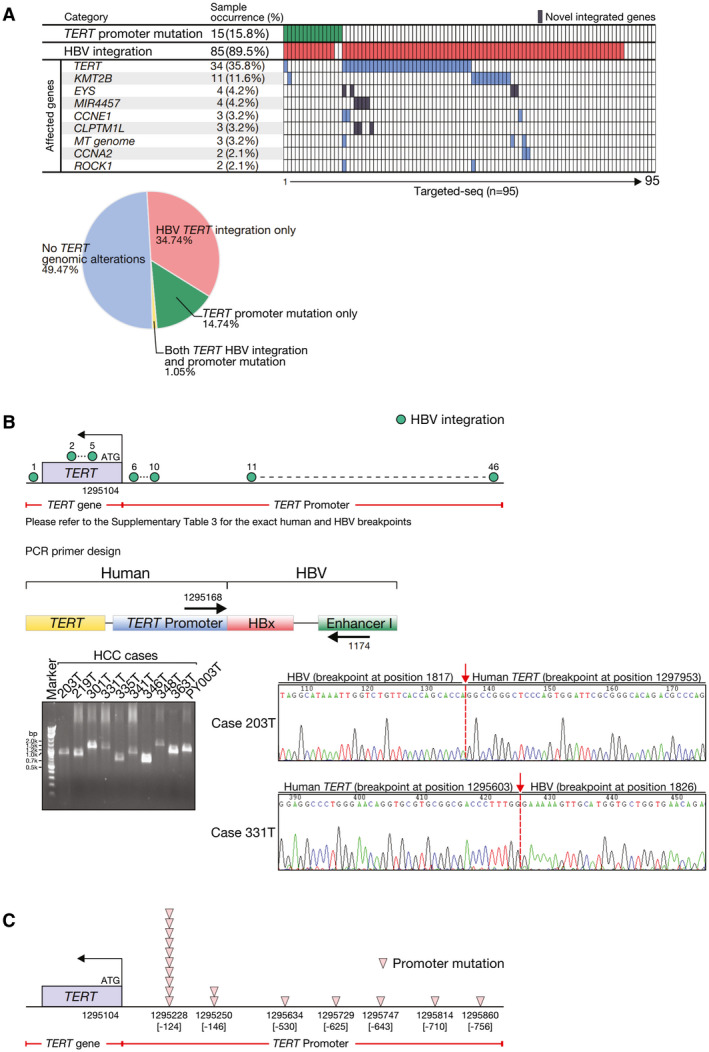
Frequent TERT genomic alterations detected in HBV‐associated HCCs. (A) TERT promoter mutations and recurrently HBV‐integrated human genes. The pie chart shows the distribution of TERT genomic alterations detected in the HCC cohort. (B) Schematic diagram showing the frequent TERT HBV integrations and their genomic locations in human HCC tissues. The direct joining of HBV EnhI and HBx DNA sequences to human TERT promoter on HBV integration in human HCCs was confirmed using PCR primers flanking human TERT promoter and HBV EnhI and subsequent Sanger sequencing. Representative Sanger sequencing results are shown. (C) Schematic diagram showing frequent TERT promoter mutations and their genomic locations in HCC tissue. CCNE1, cyclin E1; CLPTM1L, CLPTM1 like; EYS, eyes shut homolog; MIR4457, microRNA 4457; ROCK1, Rho‐associated coiled‐coil containing protein kinase 1.
By analyzing the same set of targeted sequencing data, we also identified frequent TERT promoter mutations (15.8%, n = 15; Fig. 2A). Of note, they displayed a mutually exclusive pattern with TERT HBV integrations (Fig. 2A), suggesting the acquisition of either type of the TERT genomic alterations might be sufficient to result in activation of TERT in HCC. For the recurrent mutations of the TERT promoter, we detected 2 hot spots (‐124 and ‐146 bp from the ATG start site; Fig. 2C), as described.( 4 ) The former one (‐124G>A) was found in 10 cases (10.5%), whereas the latter one (‐146G>A) was found in 2 (2.1%). In addition, there were 5 other putatively novel TERT promoter mutations (‐530C>T, ‐625C>T, ‐643C>T, ‐710G>C, and ‐756G>T) detected in 4 cases. Regarding the other 5 putatively novel mutations outside the hot spots, 2 of them (‐625C>T and ‐710G>C) were confirmed to be somatic, whereas the remaining ones (‐530C>T, ‐643C>T, and ‐756G>T) were nonsomatic and could also be detected from the corresponding nontumorous tissue. Collectively, these 17 TERT promoter mutations were found in 15 cases (15.8%) and confirmed with Sanger sequencing (Fig. 2C and Supporting Fig. S2). These TERT promoter mutations were taken as a group in later analysis without further stratification.
To assess the effect of TERT promoter alterations (including TERT HBV integration and TERT promoter mutations), we compared the TERT mRNA expression levels with the alterations in patients with HCC. Tumors with TERT HBV integration had significantly up‐regulated TERT mRNA expression levels as compared with those without TERT genomic alterations (P = 0.0019), whereas tumors with TERT promoter mutations had TERT mRNA expression similar to those without TERT genomic alterations (P = 0.1417; Fig. 3A). On clinicopathologic correlation, the presence of TERT HBV integration alone was associated with more frequent venous invasion (P = 0.041) and had a trend of more advanced tumor stage (P = 0.055; Fig. 3B). There was also a trend of association with shorter overall survival rates (P = 0.065), although it did not reach statistical significance (Fig. 3B). In addition, the presence of TERT promoter mutation alone was associated with more advanced tumor stage (P = 0.047) and had a trend of presence of venous invasion (P = 0.051) but was not associated with shorter overall survival rates (P = 0.184; Fig. 3C). Collectively, the presence of TERT genomic alterations, consisting of either TERT HBV integration or TERT promoter mutations, was associated with more frequent venous invasion (P = 0.023) and more advanced tumor stage (P = 0.021). There was also a trend of association with shorter overall survival rates (P = 0.054; Fig. 3D). Notably, TERT genomic alterations collectively achieved greater statistically significant clinicopathologic correlations than either form of TERT genomic alterations alone, suggesting there was an overall concordance in associating with more aggressive tumor behavior (Fig. 3B‐D).
Fig. 3.
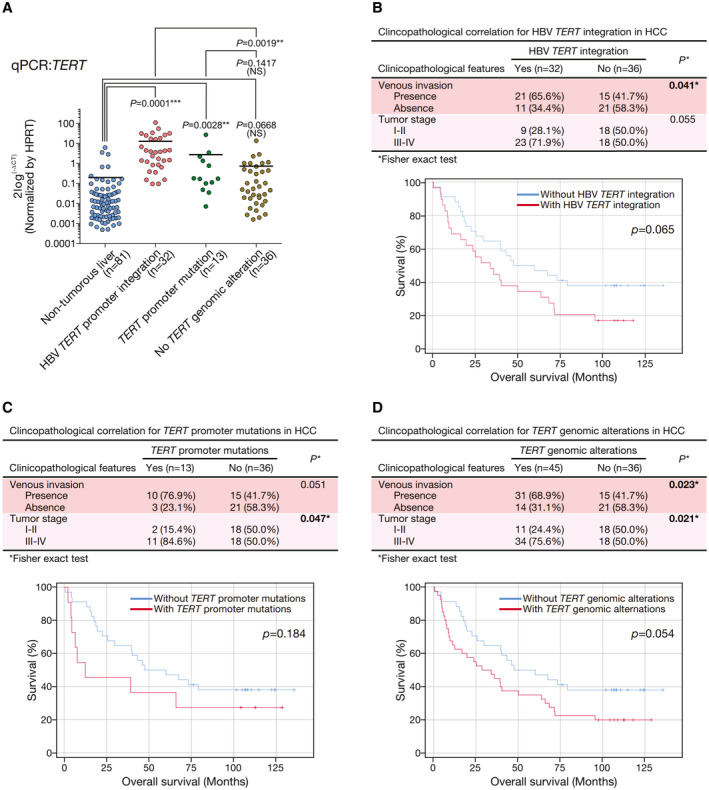
HBV‐TERT‐promoter integration resulted in TERT activation and with poor clinical outcome. (A) TERT mRNA expression was significantly up‐regulated in HCCs with HBV integration at TERT locus, as compared with those without HBV integration. (B) Clinicopathologic correlation of TERT HBV integrations in human HCCs. Tumors having TERT HBV integration were associated with more frequent venous invasion and had a trend of association with more advanced tumor stage and poorer overall survival. (C) Clinicopathologic correlation of TERT promoter mutations in human HCCs. (D) Clinicopathologic correlation of collective TERT genomic alterations in human HCCs. *P < 0.05; **P < 0.01; ***P < 0.001. HPRT, hypoxanthine‐guanine phosphoribosyltransferase; NS, not significant.
A Key Role of HBV EnhI in Driving TERT Activation
It is known that, in HCCs not associated with HBV, TERT promoter mutations create a typical ETS/TCF‐binding sequence leading to TERT activation.( 4 ) However, the corresponding mechanism of activation with TERT HBV integration remains elusive, except that early studies suggested recurrent HBV integrations at the TERT promoter( 18 ) and TERT HBV integrations resulted in the juxtaposition of viral enhancers near the TERT gene.( 19 ) We observed that TERT HBV integration events preferentially had breakpoints at the HBx region (P < 0.0001; Supporting Fig. S3). We postulated that the HBx genomic sequence might carry specific element(s) that, on chimeric fusion to the TERT upstream region during HBV integration, led to TERT activation. To this end, we used a panel of luciferase reporter constructs consisting of different combinations of human TERT promoter and HBV genomic elements (Fig. 4) based on the knowledge that a typical HBV integration event at a human TERT promoter contains a partial TERT promoter fused with an HBV genome having a breakpoint at the C‐terminus of HBx (Fig. 2B). We transfected HepG2 cells with different plasmids of human TERT promoter fused with different HBx sequences (Fig. 4A,B). We found that the 4 plasmids carrying the HBV EnhI region (position 830‐1,366) with or without various lengths of HBx (plasmids B‐E, TERTpshortHBxFLEnhI+, TERTpshortHBx‐14aaEnhI+, TERTpshortHBxC1EnhI+, and TERTpshortHBx‐EnhI+, respectively) had significantly higher transcription (22 to 51 folds relative to wild‐type TERT promoter) as compared with the TERTpWTHBx‐EnhI‐ control (plasmid A) in HepG2 cells (Fig. 4B) . Of note, EnhI was found to be the predominant activator for gene expression, as the plasmids carrying no EnhI but various lengths of HBx (plasmids F‐H, TERTpshortHBxFLEnhI‐, TERTpshortHBx‐14aaEnhI‐, and TERTpshortHBxC1EnhI‐, respectively) had significantly reduced transcription compared with those EnhI‐containing plasmids (plasmids B‐E; Fig. 4B). Similarly, EnhI (plasmid E, TERTpshortHBx‐EnhI+) was found to be the predominant activator for gene expression, as compared with enhancer II (EnhII; plasmid F, TERTpshortHBxFLEnhI‐) or wild‐type TERT promoter (plasmid A, TERTpWTHBx‐EnhI‐) control in SNU449 cells (Supporting Fig. S4). Taken altogether, our data indicate that HBV EnhI plays a key role in driving TERT promoter transcription.
Fig. 4.
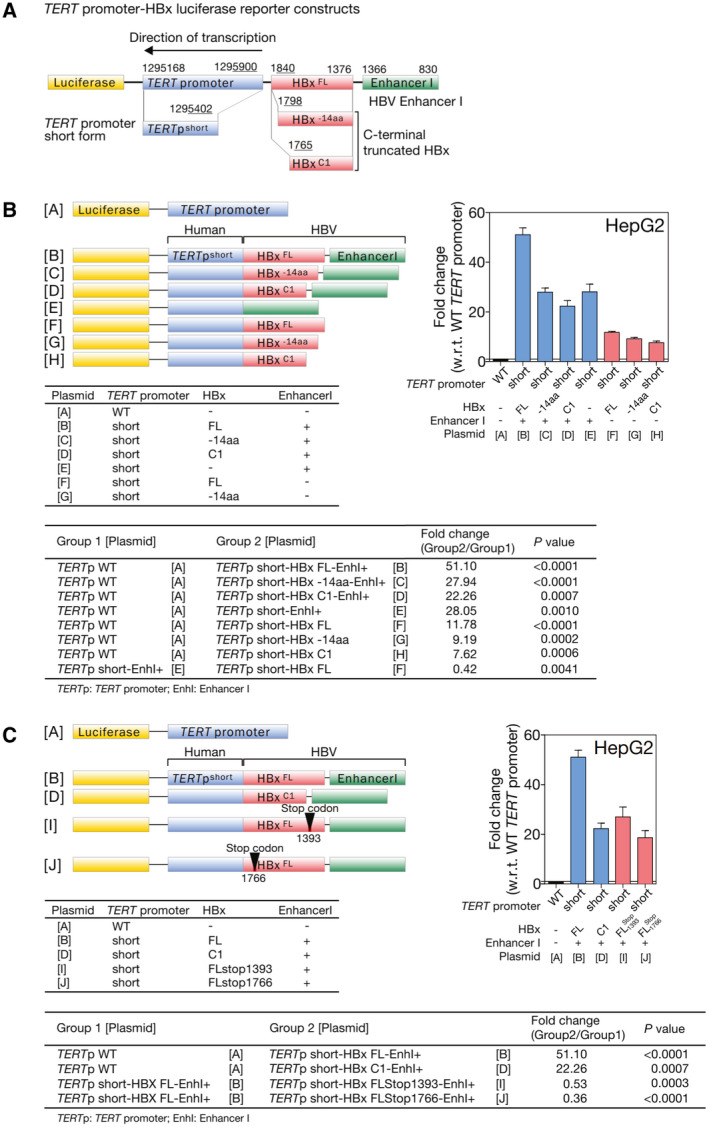
TERT transcription activation was driven by EnhI of integrated HBV. (A) Typical HBV integration event at human TERT promoter consists of a partial TERT promoter fused with HBV genome having a breakpoint at C‐terminal of HBx. (B,C) Mechanism of transcription activation by TERT HBV integration was studied by luciferase reporter assay using plasmids containing different combinations of the TERT promoter sequence (TERTp), HBx coding sequence (HBx), and HBV EnhI mimicking the actual TERT HBV integration events detected (human TERT is transcribed by reverse strand, with positive strand of integrated HBx joining to the reverse strand of TERT upstream/promoter region forming chimeric fusion). All the numbers refer to the genomic location of the forward strand. TERTpWT and TERTpshort represent wild‐type or partial TERT promoter detected in TERT HBV integration events. As EnhII is located within HBx, it was included in all plasmids B‐J except E. HBxFL, HBx‐14aa, HBxC1, HBxFLsg1393, and HBxFLsg1766 represent different lengths of HBx coding sequences with or without specific single‐nucleotide stop‐gain mutation at positions 1,393 and 1,766. EnhI+ and EnhI‐ denote the presence or absence of HBV EnhI. Investigation of the transcriptional activation effects of HBV enhancers and HBx protein in HepG2 cells using luciferase reporter assay. Fl, full length; w.r.t., with regard to; WT, wild type.
Full‐Length Form of HBx Partially Contributed to TERT Transcription Activation
Furthermore, we tested the potential effect of the HBx protein by introducing single‐nucleotide stop‐gain mutation to full‐length HBx at 2 positions—position 1,393 (stop1393) and position 1,766 (stop1766; plasmid I, TERTpshortHBxFLstop1393EnhI+ and plasmid J, TERTpshortHBxFLstop1766EnhI+; Fig. 4C). Although these two plasmids essentially consisted of almost the same construct sequence as TERTpshortHBxFLEnhI+ (plasmid B), they resulted in protein translation stopping at the 5th and 130th amino acid of HBx, respectively, and the resultant HBx protein was either negligibly short or resembled C‐terminal truncated HBx protein (HBxC1). Essentially, they did not produce the full‐length form of HBx. The transcription levels of these two constructs with stop‐gain mutations (plasmids I and J) were consistently much higher than the TERTpWTHBx‐EnhI‐ control (plasmid A) but significantly reduced when compared with that of TERTpshortHBxFLEnhI+ (plasmid B, producing full‐length form or HBx) in HepG2 cells (Fig. 4C). In contrast to the full‐length form of HBx, the presence or absence of C‐terminal truncated HBx protein did not affect the HBV DNA‐driven TERT promoter activity in these cell lines (plasmids D, I, and J of Fig. 4C). The results suggested that the full‐length form of HBx might partially contribute to TERT transcription activation.
Orientation of HBV Integration Into TERT Promoter Modulates the Degree of TERT Transcription Activation
HBV integration with different orientations might result in creation of different transcription factor‐binding sites and hence affect the degree of TERT transcription activation. We identified from whole transcriptome sequencing that two human HCC cell lines (PLC/PRF/5 and MHCC‐97L) had HBV integration at the TERT promoter but with opposite integration orientation, with HBV integration in the same direction of TERT promoter in PLC/PRF/5 and reverse direction in MHCC‐97L (Fig. 5A). We validated these TERT HBV integrations with conventional PCR followed by Sanger sequencing using DNA as template (Fig. 5B,C). Moreover, the apparent difference in the TERT promoter activity as a result of the different orientations of HBV integration was recapitulated in terms of TERT transcription levels in these cell lines (same orientation: PLC/PRF/5; opposite orientation: MHCC‐97L; Fig. 5D). Of note, in patients with HCC, the tumors having HBV integration parallel to the TERT promoter orientation had significantly higher TERT mRNA expression levels as compared with those having HBV integration opposite to the TERT promoter orientation (P = 0.0338; Fig. 5E). Those HCC tumors having HBV integration opposite to the TERT promoter orientation also had higher TERT mRNA expression levels as compared with the nontumorous livers (P < 0.0001), suggesting that both orientations could recruit host transcription factors to activate the gene transcription on HBV integration but to different degrees. We further constructed a TERTpshortHBx‐EnhI+(reverse) promoter (plasmid E.1), which had the TERT promoter join to inverted HBV EnhI DNA (Fig. 5F). We found that the promoter activity of TERTpshortHBx‐EnhI+(reverse) was reduced by 35.7% in HepG2 (P < 0.0001) as compared with the TERTpshortHBx‐EnhI+ promoter. Collectively, the results suggest that TERT transcription activation could be modulated by the orientation of HBV integration into TERT promoter.
Fig. 5.
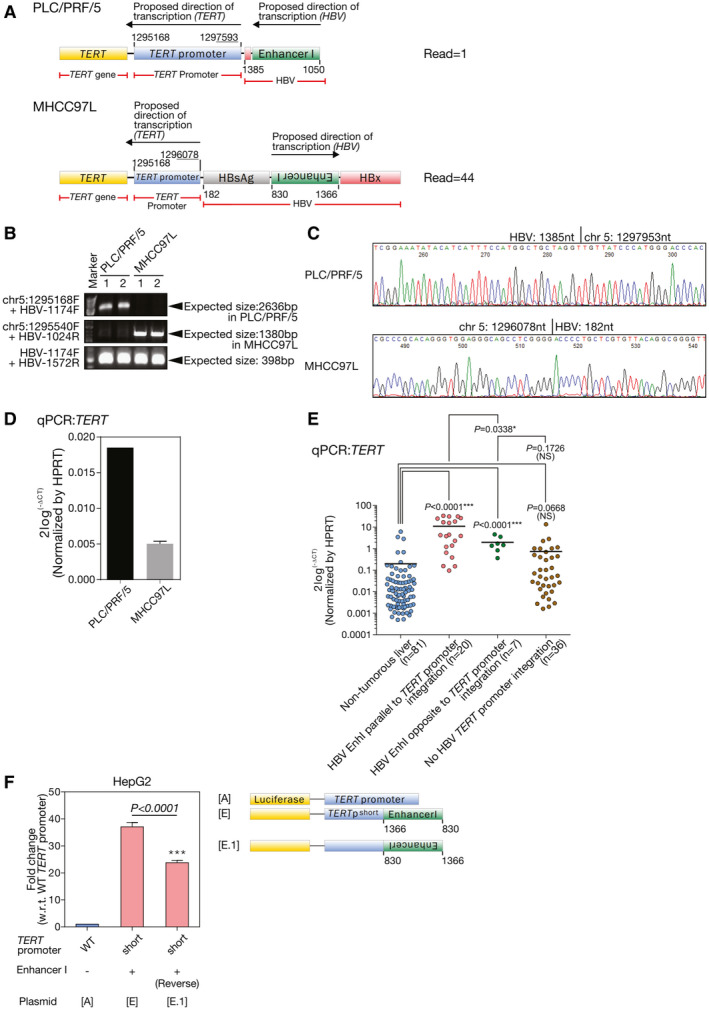
Orientation of the HBV‐TERT promoter integration significantly influenced the degree of TERT promoter activation in HCC cells. (A) Schematic diagram showing the HBV DNA integration event at the TERT promoter region in PLC/PRF/5 and MHCC‐97L as informed by transcriptome sequencing. (B) Confirmation of HBV DNA integration events at TERT promoter region in PLC/PRF/5 and MHCC‐97L cells with conventional PCR using DNA from PLC/PRF/5 and MHCC‐97L. The HBV‐TERT chimeric fusion DNA in PLC/PRF/5 was amplified using primer set chr5:1295168F and HBV‐1174F, whereas chimeric fusion DNA in MHCC‐97L was amplified using chr5:1295540F and HBV‐1024R. The primer set flanking the region 1174‐1572 of HBV DNA was successfully amplified from PLC/PRF/5 and MHCC‐97L, suggesting the presence of integrated HBV DNA in both cell lines. (C) The TERT HBV integration breakpoints in the PLC/PRF/5 and MHCC‐97L were confirmed by Sanger sequencing. (D) mRNA expression levels of TERT in PLC/PRF/5 and MHCC‐97L were quantified by qPCR. (E) Comparison between the two forms of HBV‐TERT promoter integration in human HCC tumors. HCC tumor samples having HBV integration parallel to TERT promoter orientation had significantly higher TERT mRNA expression levels by qPCR as compared with those with HBV integration opposite to the TERT promoter orientation (P = 0.0338). (F) The promoter activity of TERTpshortHBx‐EnhI+(reverse) with reverse orientation of HBV EnhI integration (plasmid E.1) was reduced by 40% as compared with TERTpshortHBx‐EnhI+ promoter in HepG2 cells (P = 0.0003). ***P < 0.001. chr, chromosome; HPRT, hypoxanthine‐guanine phosphoribosyltransferase; NS, not significant; w.r.t., with regard to; WT, wild type.
Transcription Factor ELF4 Mediates TERT Promoter Activation on HBV DNA Integration
We were intrigued to find out the host transcription factor(s) that might physically interact with HBV DNA to drive the transcription of host genes on HBV DNA integration. First, we screened for host transcription factors and transcription‐associated proteins that might be involved in physically interacting with HBV DNA and resulting in HBV viral mRNA and protein production. As suppression of viral mRNA production results in suppression of HBsAg and/or HBV secretion in vitro,( 20 ) we employed an array‐based loss‐of‐function siRNA library screening approach to identify key proteins that were involved in the HBV viral mRNA transcription process. To achieve this goal, we made use of commercially available array‐based siRNA library for transfection into the HepG2.2.15 HCC cells, which have integrated HBV DNA and secrete HBsAg and HBV viral particles, and then measured the HBsAg level (representing both HBsAg and HBV production) as a read‐out of HBV viral gene transcription. The array‐based loss‐of‐function siRNA library targeted a total of 1,527 human transcription‐related proteins for screening (Supporting Table S4). The percentage of inhibition of secretion of HBV viral particles and/or HBsAg in HepG2.2.15 cells (with HBV genotype B) on transfection of individual siRNAs was measured, and the top 58 genes with most remarkable inhibition of HBsAg secretion (as compared with the corresponding nontarget control [NTC] siRNA transfected cells) were subjected to a second round of screening using human PLC/PRF/5 HCC cells (with HBV genotype A) to mimic HBV genotype‐specific effects. Among the gene list, ELF4 was found to result in the greatest reduction of HBsAg secretion on siRNA transfection in both HepG2.2.15 (43.32%) and PLC/PRF/5 (93.09%) cells (Table 1). We then tested if ELF4 might interact with the integrated HBV DNA and in turn regulate TERT transcription. Ectopic expression of ELF4 open reading frame in SNU449 cells, which had relatively lower ELF4 protein expression as compared with other HCC cells (Supporting Fig. S5A), resulted in significant up‐regulation of HBV EnhI TERT fusion promoter activity (plasmid E) as compared with the empty vector control (Supporting Fig. S5B). Ectopic expression of other previously suggested transcription factors such as androgen receptor (AR), forkhead box A1 (FOXA1), forkhead box A2 (FOXA2), Jun proto‐oncogene (JUN), and hepatocyte nuclear factor 4 alpha (HNF4A) did not show significant up‐regulation of HBV EnhI TERT fusion promoter activity (Supporting Fig. S5B).
TABLE 1.
The Top 20 Genes With Highest Reduction in HBsAg Level on siRNA Knockdown as Compared With the Corresponding NTC in HepG2.2.15 and PLC/PRF/5 Cells
| HepG2 2.15 | PLC/PRF/5 | ||
|---|---|---|---|
| Gene Name | Reduction (%) | Gene Name | Reduction (%) |
| ELF4 | 43.32 | ELF4 | 93.09 |
| HOXD13 | 35.62 | PPARA | 67.27 |
| ZFP36L2 | 34.44 | ESRRB | 65.86 |
| TFAP4 | 33.68 | PPP1R27 | 58.49 |
| ERF | 32.35 | CERS2 | 57.61 |
| POLR3E | 31.42 | POLR3E | 51.05 |
| HOXA1 | 30.20 | VDR | 45.09 |
| NKX3‐2 | 29.94 | FOXA1 | 42.09 |
| GTF2A1L | 27.77 | LITAF | 41.98 |
| GATAD2A | 27.34 | NOTCH4 | 39.02 |
| PPP1R27 | 27.00 | SHOX2 | 31.10 |
| KDM5B | 26.53 | TCEB2 | 27.66 |
| BRCA1 | 26.08 | MEF2BNB‐MEF2B | 27.20 |
| HOXB9 | 24.47 | E2F5 | 26.78 |
| E2F5 | 22.99 | ZNF326 | 24.59 |
| ELK1 | 22.29 | TFAP4 | 21.50 |
| FOXA1 | 22.28 | TCEB1 | 20.65 |
| GCDH | 21.90 | ERF | 19.65 |
| KLF9 | 21.85 | DMRTA2 | 15.21 |
| SSX2 | 21.83 | GATAD2A | 13.18 |
BRCA1, BRCA1 DNA repair associated; CERS2, ceramide synthase 2; DMRTA2, DMRT like family A2; E2F5, E2F transcription factor 5; ELK1, ETS transcription factor ELK1; ERF, ETS2 repressor factor; ESRRB, estrogen related receptor beta; FOXA1, forkhead box A1; GATAD2A, GATA zinc finger domain containing 2A; GCDH, glutaryl‐CoA dehydrogenase; GTF2A1L, general transcription factor IIA subunit 1 like; HOXA1, homeobox A1; HOXB9, homeobox B9; HOXD13, homeobox D13; KDM5B, lysine demethylase 5B; KLF9, Kruppel like factor 9; LITAF, lipopolysaccharide induced TNF factor; MEF2BNB‐MEF2B, MEF2BNB‐MEF2B Readthrough; NKX3‐2, NK3 homeobox 2; NOTCH4, notch receptor 4; POLR3E, RNA polymerase III subunit E, PPARA, peroxisome proliferator activated receptor alpha; PPP1R27, protein phosphatase 1 regulatory subunit 27; SHOX2, short stature homeobox 2; SSX2, SSX family member 2; TCEB1, elongin C; TCEB2, elongin B; TFAP4, transcription factor AP‐4; VDR, vitamin D receptor; ZFP36L2, ZFP36 ring finger protein like 2; ZNF326, zinc finger protein 326.
ELF4 Physically Binds to HBV EnhI
Because we observed that HBV EnhI was critical in driving TERT activation, to this end, we attempted to define the physical binding site of ELF4 on HBV EnhI region. A series of plasmids with N‐terminal deletion (plasmid E.2‐E.6; Fig. 6A) and mutations (plasmid E.7‐E.10; Fig. 6B) on the putative ELF4 binding sites between positions 1,050 and 1,300 of the HBV EnhI region were created and subjected to luciferase reporter assays. The results suggested that the putative ELF4 binding site on HBV EnhI region was likely located at positions 1,111, 1,240, and 1,274 (Fig. 6A,B).
Fig. 6.
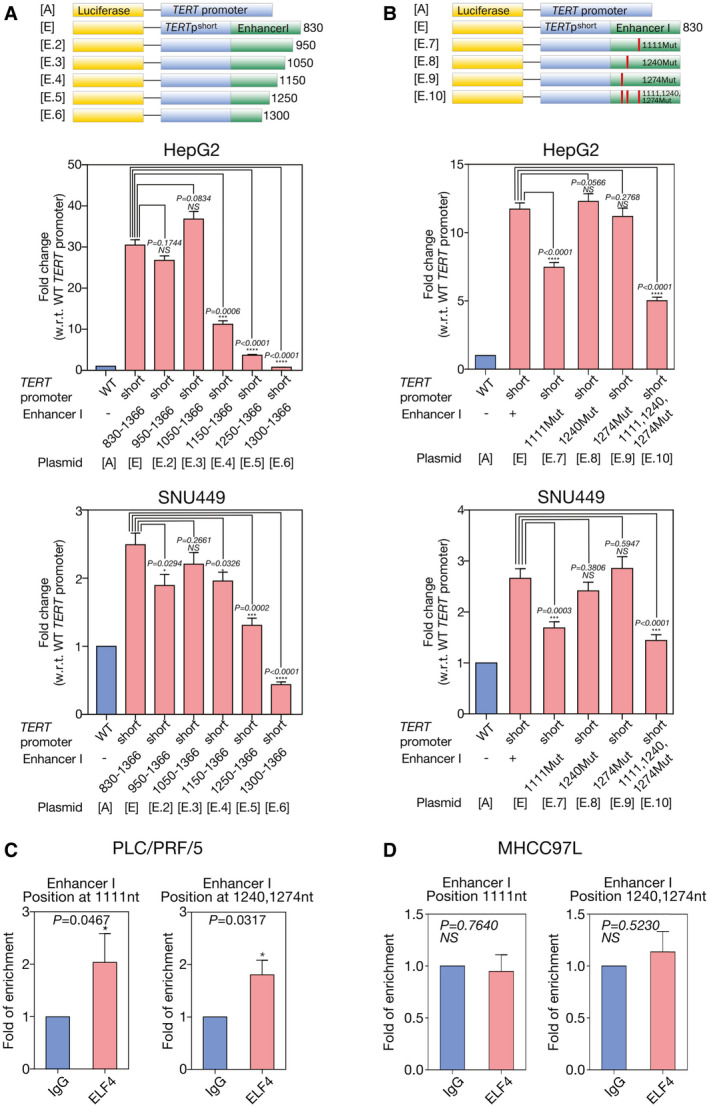
Binding of ELF4 at HBV EnhI region resulted in enhanced TERT HBV chimeric fusion promoter activity. (A) The promoter activity of TERT HBV chimeric fusion promoter was significantly reduced on deletion of the 830‐1150 HBV EnhI region (TERTpshortHBx‐EnhI(Δ830‐1150), plasmid E.4; 63.2% reduction, P < 0.0001), 830‐1250 region (TERTpshortHBx‐EnhI(Δ830‐1250), plasmid E.5; 87.9% reduction, P < 0.0001), and 830‐1300 region (TERTpshortHBx‐EnhI(Δ830‐1300), plasmid E.6; 97.5% reduction, P < 0.0001) but not for deletion of the 830‐950 (TERTpshortHBx‐EnhI(Δ830‐950), plasmid E.2) and 830‐1050 (TERTpshortHBx‐EnhI(Δ830‐1050), plasmid E.3) regions (both P > 0.05) in HepG2 cells. In SNU449 cells, the promoter activity of TERT HBV chimeric fusion promoter was significantly reduced on deletion of the 830‐1250 region (TERTpshortHBx‐EnhI(Δ830‐1250), plasmid E.5; 47.4% reduction, P = 0.0002) and 830‐1300 region (TERTpshortHBx‐EnhI(Δ830‐1300), plasmid E.6; 82.4% reduction, P < 0.0001). (B) The promoter activity of TERT HBV chimeric fusion promoter was significantly reduced on single mutation of the putative ELF4 binding site at position 1,111 (TERTpshortHBx‐EnhI+(1111Mut), plasmid E.7; 36.2% reduction, P < 0.0001) and triple putative ELF4 binding site mutations at positions 1,111, 1,240, and 1,274 (TERTpshortHBx‐EnhI+(1111,1240,1274Mut), plasmid E.10; 57.3% reduction, P < 0.0001) although not on mutating single putative ELF4 binding sites at positions 1,240 (TERTpshortHBx‐EnhI+(1240Mut), plasmid E.8) and 1,274 (TERTpshortHBx‐EnhI+(1274Mut), plasmid E.9) of the HBV DNA region as compared with the wild‐type TERT HBV chimeric fusion (TERT shortHBx‐EnhI+, plasmid E) promoter in HepG2 cells. In SNU449 cells, the promoter activity of TERT HBV chimeric fusion promoter was significantly reduced on single mutation of the putative ELF4 binding site at position 1,111 (TERTpshortHBx‐EnhI+(1111Mut), plasmid E.7; 36.6% reduction, P < 0.0001) and triple putative ELF4 binding site mutations at positions 1,111, 1,240, and 1,274 (TERTpshortHBx‐EnhI+(1111,1240,1274Mut), plasmid E.10; 45.8% reduction, P < 0.0001). (C) ChIP assay using primers flanking positions 1,000‐1,120 and 1,240‐1,300 of HBV DNA region showed ELF4 physically interacted with HBV EnhI region in PLC/PRF/5. (D) There was no significant interaction of ELF4 with HBV EnhI region at positions between 1,000‐1,120 and 1,240‐1,300 in MHCC‐97L on ChIP assay. ***P < 0.001; ****P < 0.0001. Mut, mutation; NS, not significant; w.r.t., with regard to; WT, wild type.
Furthermore, ChIP assay showed that ELF4 physically interacted with HBV EnhI region at positions 1,000‐1,120 and 1,240‐1,300 in PLC/PRF/5 HCC cells (Fig. 6C). In contrast, ChIP assay showed insignificant ELF4 binding to the integrated HBV EnhI in MHCC‐97L cells (Fig. 6D).
Stable Knockdown of ELF4 Reduces TERT mRNA Expression and Sphere‐Forming Ability of HCC Cells
We further investigated the functional significance of ELF4 using stable knockdown approach. Because the siRNA library we adopted used a pool of four siRNA sequence duplexes predicted to specifically target the genes of interest, we established stable knockdown shELF4 PLC/PRF/5 cells using individual specific shRNA sequences. Successful depletion of ELF4 at protein levels by shRNA (shELF4 #1 and shELF4 #4) in PLC/PRF/5 showed a significant reduction of TERT mRNA expression (P = 0.0093 and P = 0.0023, respectively; Fig. 7A). These stable knockdown shELF4 clones also had reduced sphere‐forming ability, which is a test for self‐renewal ability in vitro,( 21 ) (P = 0.0082 and P = 0.0058, respectively) as compared with NTC (Fig. 7B). In spite of the diminished sphere‐forming ability on knockdown of ELF4 in PLC/PRF/5, there was no significant alteration of the cell proliferation rate as compared with NTC with or without serum supplements, suggesting that shRNA was not toxic to the HCC cells (Supporting Fig. S6). On the other hand, in MHCC‐97L with opposite orientation of HBV integration, there was no significant alteration of the TERT mRNA expression and sphere‐forming ability on knockdown of ELF4 (Fig. 7C,D). These observations further supported our finding that different orientations of HBV integration might result in differential ELF4 binding, leading to different degrees of TERT activation and sphere‐forming ability of HCC cells.
Fig. 7.
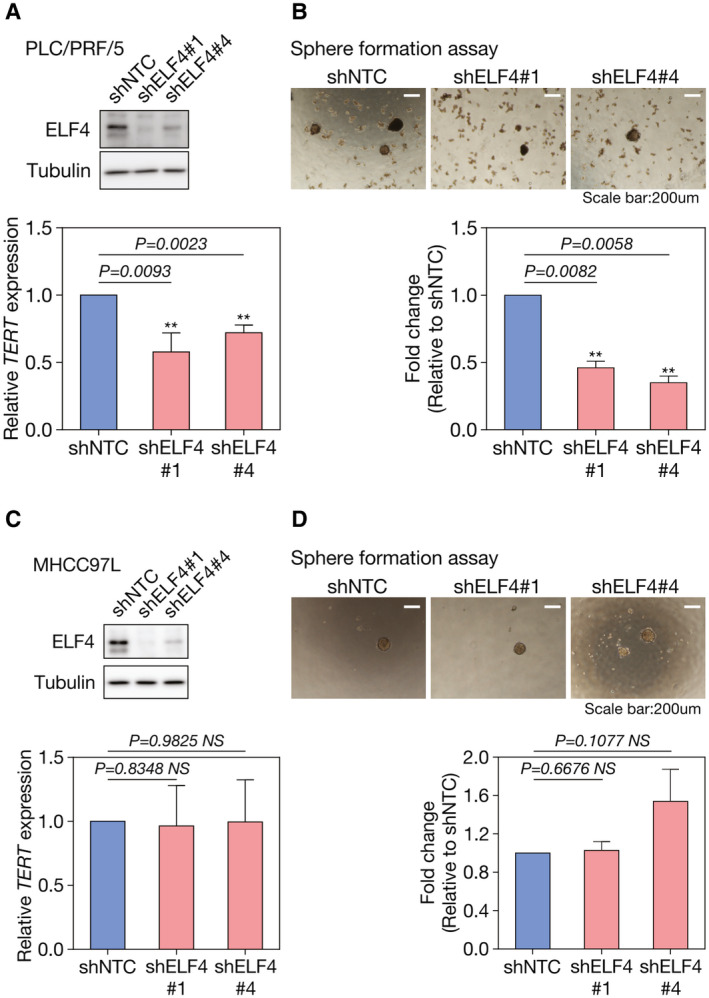
Knockdown of ELF4 suppressed TERT mRNA expression and sphere‐forming ability in PLC/PRF/5 but not in MHCC‐97L. (A) Successful depletion of ELF4 at protein level by shRNA (shELF4 #1 and shELF4 #4) suppressed TERT mRNA expression (P = 0.0093 and P = 0.0023, respectively) and (B) sphere‐forming ability (P = 0.0082 and P = 0.0058, respectively), as compared with non‐targeting control shRNA (shNTC) cells in PLC/PRF/5. (C, D) Knockdown of ELF4 in MHCC‐97L, which has the opposite orientation of HBV DNA integration at the TERT promoter region, did not alter the TERT mRNA expression and sphere‐forming ability. **P < 0.01.
Discussion
Based on the targeted sequencing on a sizeable cohort of clinical HCC cases, we identified preferential HBV‐human chimeric fusions with breakpoints at the C‐terminus of HBx and TERT promoter, respectively. HBV integration at the TERT promoter was frequent (35.8%) in HCCs and was associated with increased TERT mRNA expression and more aggressive tumor behavior.
We further investigated the functional importance of the various integrated HBV components in HBV‐TERT promoter integration using luciferase reporter assays with multiple constructs. We found that HBV EnhI was the key HBV enhancer leading to telomerase activation on integration at the TERT promoter. It is known that the two HBV enhancers, EnhI and EnhII (EnhII is located within HBx [Figs. 1F and 4A]), can cis‐activate both HBV promoters and other heterologous promoters.( 22 ) However, the relative strength of the cis‐activation effect of HBV EnhI and EnhII on TERT promoter remains elusive. Although HBV EnhII is strictly liver specific, HBV EnhI is less restricted to liver cells.( 23 ) We observed that HBV EnhI had a far stronger effect in driving TERT transcription than HBV EnhII. This might have implications for the possible adaptive utilization of non‐liver‐specific HBV EnhI in driving oncogenic TERT expression in less differentiated hepatocyte HCC cells during hepatocarcinogenesis, and further investigation is warranted.
Importantly, we demonstrated that the orientation of HBV integration could result in differential transcription activation in both HCC cell lines and patients’ HCC samples. In general, enhancers are able to exert their functions irrespective of their directions. However, the three‐dimensional DNA loop conformation in controlling the gene expression may be changed on HBV DNA integration into the human genome. The orientation‐dependent binding of CCCTC‐binding factor (CTCF) in altering enhancer‐promoter loop formation( 24 ) and subsequently modulating gene expression is an example.( 25 ) Notably, both HBV EnhI and CTCF are involved in the formation of DNA loop structure to regulate enhancer‐promoter interactions. Our finding that the orientation of HBV integration resulted in differential transcription activation might imply different degrees of affinity with the transcription factor at the binding site with different orientations. Because the TERT promoter region does not contain any TATA box or CAAT box to serve as the transcription initiation site for TERT gene transcription,( 26 , 27 ) HBV EnhI and EnhII DNA fragments might serve as both enhancer and promoter elements in driving TERT gene transcription. Taken together, our study revisited the classical cis‐activation of gene targets on HBV integration and identified mechanisms of HBV integration at TERT promoter.
We observed that the full‐length form of HBx protein might partially contribute to the transcription activation of HBV‐TERT promoter activity as demonstrated by the reporter assays. This might be due to the transactivating ability of HBx protein in gene transcription, as exemplified by stemness‐related markers in Wnt/β‐catenin pathway.( 28 ) However, most of the HBV integration breakpoints in our human HCCs were within the HBx gene, thus resulting in HBx C‐terminal truncation; the full‐length HBx was observed only in a minority of our patients’ HCCs in this study (Supporting Table S3). Therefore, the integrated full‐length form of HBx protein contributing to further enhanced levels of TERT mRNA transcription might not be significant in our HCCs.
Little is known on the transcriptional regulation after HBV DNA is integrated into a particular gene locus. It is known that the liver‐specific surface receptor solute carrier family 10 member 1 and NTCP can capture HBV viral particles and import HBV viral DNA into hepatocytes.( 29 ) This further suggests that the host transcription factors in hepatocytes might participate in HBV viral mRNA transcription. In this study, we hypothesized that host transcription‐associated proteins that might physically interact with HBV DNA were recruited to the integrated HBV DNA and drove the transcription of host genes. Making use of an array‐based siRNA library that targets human transcription‐associated proteins and an HCC cell line that constitutively produces HBV viral particles, we were able to shortlist candidate genes that might be involved in the transcription regulation of HCC with TERT HBV integration.
There have been several reports using a siRNA library screening approach to study the underlying mechanisms of HBV replication.( 30 , 31 , 32 , 33 ) To this end, our present study focused specifically on the TERT transcriptional activation on HBV integration in the TERT promoter of the hepatocyte genome. We identified a molecular mechanism of TERT activation through ELF4, which normally could drive HBV gene transcription. ELF4 is a DNA‐binding transcription factor that is ubiquitously expressed in various tissues, including nontumorous livers and HCCs (Supporting Fig. S7). It is involved in the p53‐dependent senescence and development of nature‐killer cells in ELF4 knockout mice.( 34 , 35 ) We showed that ELF4 was able to bind to the chimeric HBV EnhI at the TERT promoter, resulting in telomerase activation. Furthermore, stable knockdown of ELF4 significantly reduced TERT expression and sphere‐forming ability in HCC cells. Altogether, the results suggest that ELF4 may be a major transcription factor in controlling HBV gene transcription, including HBx.
In summary, our results have provided evidence demonstrating a molecular mechanism of telomerase activation in TERT HBV‐integrated HCC. It involves a cis‐activating mechanism harnessing host ELF4 and HBV integrated at the TERT promoter, uncovering how TERT HBV‐integrated HCCs may achieve TERT activation in hepatocarcinogenesis.
Author Contributions
K.M.‐F.S., D.W.‐H.H., and I.O.‐L.N. conceived the study. K.M.‐F.S., D.W.‐H.H, Y.‐T.C., Y.‐M.T., L.‐K.C., and J.M.‐F.L. performed the experiments. K.M.‐F.S., D.W.‐H.H., and I.O.‐L.N. analyzed the data. K.S.‐H.C., A.C.‐Y.C., C.‐N.T., V.W.‐L.T., I.L.‐O.L., D.T.‐W.Y., and T.‐T.C. contributed to patient recruitment and sample collection. K.M.‐F.S., D.W.‐H.H., and I.O.‐L.N. wrote the manuscript.
Supporting information
Supplementary Material
The study was supported by the Hong Kong Research Grants Council Theme‐based Research Scheme (T12‐704/16‐R), Innovation and Technology Commission grant for State Key Laboratory of Liver Research, National Natural Science Foundation of China (81872222), University Development Fund of The University of Hong Kong, and Lee Shiu Family Foundation. I.O.L. Ng is Loke Yew Professor in Pathology.
Potential conflict of interest: Nothing to report.
References
- 1. El‐Serag HB. Hepatocellular carcinoma. N Engl J Med 2011;365:1118‐1127. [DOI] [PubMed] [Google Scholar]
- 2. Villanueva A, Llovet JM. Liver cancer in 2013: mutational landscape of HCC–the end of the beginning. Nat Rev Clin Oncol 2014;11:73‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, et al. Genome‐wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet 2012;44:765‐769. [DOI] [PubMed] [Google Scholar]
- 4. Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac‐Sage P, Laurent C, et al. High frequency of telomerase reverse‐transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun 2013;4:2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Borah S, Xi L, Zaug AJ, Powell NM, Dancik GM, Cohen SB, et al. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science 2015;347:1006‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barthel FP, Wei W, Tang M, Martinez‐Ledesma E, Hu X, Amin SB, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet 2017;49:349‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wong CM, Lee JM, Ching YP, Jin DY, Ng IO. Genetic and epigenetic alterations of DLC‐1 gene in hepatocellular carcinoma. Cancer Res 2003;63:7646‐7651. [PubMed] [Google Scholar]
- 8. Ho DW, Sze KM, Ng IO. Virus‐Clip: a fast and memory‐efficient viral integration site detection tool at single‐base resolution with annotation capability. Oncotarget 2015;6:20959‐20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Conway T, Wazny J, Bromage A, Tymms M, Sooraj D, Williams ED, et al. Xenome—a tool for classifying reads from xenograft samples. Bioinformatics 2012;28:i172‐i178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sze KM, Chu GK, Lee JM, Ng IO. C‐terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by C‐Jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology 2013;57:131‐139. [DOI] [PubMed] [Google Scholar]
- 11. Jiang Z, Jhunjhunwala S, Liu J, Haverty PM, Kennemer MI, Guan Y, et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res 2012;22:593‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, et al. Whole‐genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet 2012;44:760‐764. [DOI] [PubMed] [Google Scholar]
- 13. Ding D, Lou X, Hua D, Yu W, Li L, Wang J, et al. Recurrent targeted genes of hepatitis B virus in the liver cancer genomes identified by a next‐generation sequencing‐based approach. PLoS Genet 2012;8:e1003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li W, Zeng X, Lee NP, Liu X, Chen S, Guo B, et al. HIVID: an efficient method to detect HBV integration using low coverage sequencing. Genomics 2013;102:338‐344. [DOI] [PubMed] [Google Scholar]
- 15. Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, et al. Trans‐ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet 2014;46:1267‐1273. [DOI] [PubMed] [Google Scholar]
- 16. Lau CC, Sun T, Ching AK, He M, Li JW, Wong AM, et al. Viral‐human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 2014;25:335‐349. [DOI] [PubMed] [Google Scholar]
- 17. Berger AW, Schwerdel D, Ettrich TJ, Hann A, Schmidt SA, Kleger A, et al. Targeted deep sequencing of circulating tumor DNA in metastatic pancreatic cancer. Oncotarget 2017;9:2076‐2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paterlini‐Bréchot P, Saigo K, Murakami Y, Chami M, Gozuacik D, Mugnier C, et al. Hepatitis B virus‐related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 2003;22:3911‐3916. [DOI] [PubMed] [Google Scholar]
- 19. Ferber MJ, Montoya DP, Yu C, Aderca I, McGee A, Thorland EC, et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene 2003;22:3813‐3820. [DOI] [PubMed] [Google Scholar]
- 20. Tang HM, Gao WW, Chan CP, Cheng Y, Chaudhary V, Deng JJ, et al. Requirement of CRTC1 coactivator for hepatitis B virus transcription. Nucleic Acids Res 2014;42:12455‐12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. CD24(+) liver tumor‐initiating cells drive self‐renewal and tumor initiation through STAT3‐mediated NANOG regulation. Cell Stem Cell 2011;9:50‐63. [DOI] [PubMed] [Google Scholar]
- 22. Guo WT, Bell KD, Ou JH. Characterization of the hepatitis B virus EnhI enhancer and X promoter complex. J Virol 1991;65:6686‐6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Su H, Yee JK. Regulation of hepatitis B virus gene expression by its two enhancers. Proc Natl Acad Sci U S A 1992;89:2708‐2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Wit E, Vos ES, Holwerda SJ, Valdes‐Quezada C, Verstegen MJ, Teunissen H, et al. CTCF binding polarity determines chromatin looping. Mol Cell 2015;60:676‐684. [DOI] [PubMed] [Google Scholar]
- 25. Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, et al. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell 2015;162:900‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cong YS, Wen J, Bacchetti S. The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum Mol Genet 1999;8:137‐142. [DOI] [PubMed] [Google Scholar]
- 27. Takakura M, Kyo S, Kanaya T, Hirano H, Takeda J, Yutsudo M, et al. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res 1999;59:551‐557. [PubMed] [Google Scholar]
- 28. Arzumanyan A, Friedman T, Ng IO, Clayton MM, Lian Z, Feitelson MA. Does the hepatitis B antigen HBx promote the appearance of liver cancer stem cells? Cancer Res 2011;71:3701‐3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012;1:e00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang WH, Studach LL, Andrisani OM. Proteins ZNF198 and SUZ12 are down‐regulated in hepatitis B virus (HBV) X protein‐mediated hepatocyte transformation and in HBV replication. Hepatology 2011;53:1137‐1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ghosh S, Kaushik A, Khurana S, Varshney A, Singh AK, Dahiya P, et al. An RNAi‐based high‐throughput screening assay to identify small molecule inhibitors of hepatitis B virus replication. J Biol Chem 2017;292:12577‐12588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang H, Liu K, Fang BAM, Wu H, Li F, Xiang X, et al. Identification of acetyltransferase genes (HAT1 and KAT8) regulating HBV replication by RNAi screening. Cell Biosci 2015;5:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wooddell CI, Yuen MF, Chan HL, Gish RG, Locarnini SA, Chavez D, et al. RNAi‐based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci Transl Med 2017;9:eaan0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. You F, Wang P, Yang L, Yang G, Zhao YO, Qian F, et al. ELF4 is critical for induction of type I interferon and the host antiviral response. Nat Immunol 2013;14:1237‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sashida G, Liu Y, Elf S, Miyata Y, Ohyashiki K, Izumi M, et al. ELF4/MEF activates MDM2 expression and blocks oncogene‐induced p16 activation to promote transformation. Mol Cell Biol 2009;29:3687‐3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material