

Abstract
Closthioamide (CTA) is a symmetric nonribosomal peptide (NRP) comprised of two diaminopropane‐linked polythioamidated monomers. CTA is biosynthesized by Ruminiclostridium cellulolyticum via an atypical NRP synthetase (NRPS)‐independent biosynthetic pathway. Although the logic for monomer assembly was recently elucidated, the strategy for the biosynthesis and incorporation of the diamine linker remained a mystery. By means of genome editing, synthesis, and in vitro biochemical assays, we demonstrate that the final steps in CTA maturation proceed through a surprising split‐merge pathway involving the dual use of a thiotemplated intermediate. This pathway includes the first examples of an aldo‐keto reductase catalyzing the reductive release of a thiotemplated product, and of a transthioamidating transglutaminase. In addition to clarifying the remaining steps in CTA assembly, our data shed light on largely unexplored pathways for NRPS‐independent peptide biosynthesis.
Keywords: antibiotics, enzymes, natural products, nonribosomal peptides, thioamide
Closthioamide is a symmetric perthioamidated nonribosomal peptide (NRP) produced by an atypical NRP‐synthetase‐independent biosynthetic pathway. Using a combination of genome editing and in vitro biochemical assays, we elucidated the last steps of closthioamide biosynthesis and found an unexpected asymmetrical route involving novel enzyme functions for members of the aldo‐keto reductase and transglutaminase protein families.
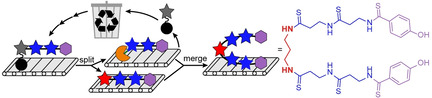
Introduction
Nonribosomal peptides (NRPs) are a diverse class of ecologically and medicinally important secondary metabolites. The majority of NRPs are assembled by either multi‐modular (Type I) or stand‐alone (Type II) NRP synthetases (NRPSs). [1] In both cases, the NRPSs function in a thiotemplated manner on substrates covalently attached to carrier proteins and assemble peptides in linear assembly lines. In addition to these canonical systems, NRPS‐independent biosynthetic pathways are known. [2] NRPS‐independent systems often rely on amide synthases that are homologs of enzymes involved in primary metabolic pathways including, but not limited to, members of the ATP‐grasp, cysteine protease, and acyl‐CoA ligase protein families. [2] In contrast to NRPSs, these atypical amide synthases typically do not follow the canonical thiotemplated program for NRP assembly, but rather act on free substrates. [2] A notable deviation from this trend is exemplified by the NRPS‐independent, yet thiotemplated biosynthesis of the DNA gyrase‐targeting antibiotic closthioamide (CTA; Figure 1 A and B). [3]
Figure 1.
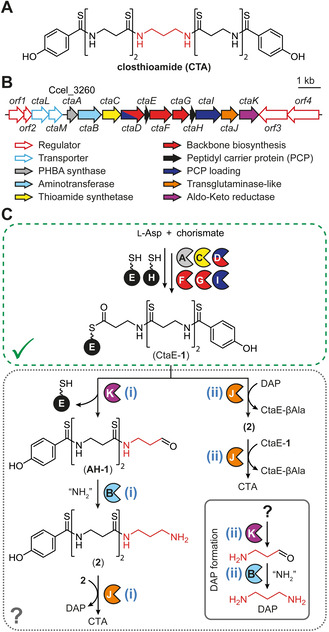
Closthioamide biosynthesis gene cluster and proposed routes to diaminopropane incorporation. A) Structure of closthioamide (CTA) with diaminopropane (DAP) core colored red. B) CTA biosynthesis gene cluster. PHBA; p‐hydroxybenzoic acid. C) Proposed CTA biosynthetic pathway. Steps that have been previously characterized are outlined in a green box while the uncharacterized portion of the pathway is outlined in a gray box. Two possible routes (i and ii) for DAP formation and incorporation are displayed.
CTA is a symmetric perthioamidated NRP produced by the obligate anaerobic bacterium Ruminiclostridium cellulolyticum.[ 3c , 4 ] This potent copper chelator [5] is a rare example of both a secondary metabolite isolated from an obligate anaerobe [6] and a thioamidated natural product. [7] CTA is comprised of two p‐hydroxybenzoic acid (PHBA)‐end‐capped β‐alanine (βAla) polymers linked through diaminopropane (DAP, Figure 1 A).[ 3c , 4 ] Recently, a combination of knockout studies and in vitro biochemical assays illuminated the type II peptidyl carrier protein (PCP)‐dependent assembly of the polythioamidated backbone of CTA from l‐aspartate and chorismate (1; Figure 1 C).[ 3a , 8 ] However, the biosynthetic steps for the synthesis and incorporation of the DAP linker remained unclear. Here, we elucidate the final steps in CTA maturation using a combination of genome editing, in vitro biochemical assays, and chemical synthesis.
Results and Discussion
Of the eleven biosynthetic proteins encoded in the CTA biosynthesis gene cluster, three remained to be functionally characterized: a putative pyridoxal phosphate (PLP)‐dependent aminotransferase (CtaB), an aldo‐keto reductase (AKR; CtaK), and a transglutaminase (CtaJ; Figure 1 B and C). [8b] Based on previous knockout studies [3a] and the activities of characterized transglutaminases, [9] we predicted that CtaJ would be responsible for incorporating the DAP linker into the CTA backbone. However, the source of this diamine core was unknown. In bacteria, DAP is biosynthesized from l‐aspartate semialdehyde by the sequential function of a PLP‐dependent aminotransferase and a decarboxylase. [10] Although the presence of CtaB and a PLP‐dependent decarboxylase (CtaF) in the genetic locus is consistent with such a pathway, we recently demonstrated that CtaF is involved in the assembly of the poly‐βAla backbone of CTA (Figure 1 C). [8a] Thus, we predicted that the diamine core of CTA would originate from a novel pathway involving CtaB and the AKR CtaK.
Based on the bioinformatically predicted activities of CtaB (Figure S1 in the Supporting Information) and CtaK (Figure S2), we envisioned two possible linear pathways for DAP production and incorporation into CTA (Figure 1 C). In the first pathway (i in Figure 1 C), the terminal βAla of PCP‐linked 1 (CtaE‐1; Figure 1 C) would be converted to DAP by the sequential action of CtaK and CtaB to afford 2. CtaJ would then merge two molecules of 2 into CTA, releasing DAP in the process. In the second pathway (ii in Figure 1 C), DAP could be generated as a free diamine by the CtaK‐dependent reduction and CtaB‐dependent transamination of an unknown primary metabolite. CtaJ would cleave the terminal thioamide CtaE‐1, releasing CtaE‐linked βAla (CtaE‐βAla), and transfer the PHBA‐containing fragment to DAP to produce 2. CtaJ would then further process this new intermediate using a second equivalent of CtaE‐1 to produce CTA and CtaE‐βAla.
Although 2 is a predicted intermediate of both pathways (Figure 1 C), it has not previously been detected from R. cellulolyticum cultures.[ 3a , 3c , 4 ] Upon growth of R. cellulolyticum under CTA‐producing conditions, a metabolite with an exact mass (calc. m/z 385.1185 [M+H]+; found m/z 385.1193 [M+H]+) and isotope pattern consistent with 2 was detected (Figure 2 and Figures S3 and S4). Due to low production titers, attempts to isolate sufficient quantities of the compound for structure elucidation by NMR were unsuccessful. Therefore, we synthesized 2 (Supporting Information) and used this characterized compound as a reference. The unknown metabolite has the same retention time and MS2 fragmentation pattern as the reference (Figure 2 and Figures S3 and S5), demonstrating that 2 is indeed produced by R. cellulolyticum.
Figure 2.
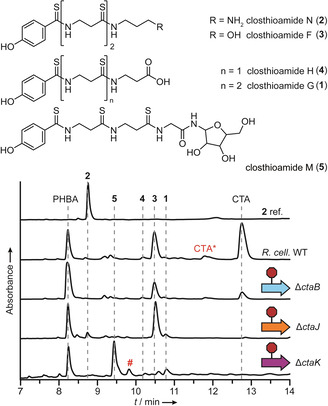
In vivo dissection of the final steps in CTA maturation. HPLC profiles (270 nm absorbance) of extracts from cultures of R. cellulolyticum wild‐type (WT) and mutant strains and the structures of the CTA congeners identified in the extracts. An HPLC profile of a synthetic reference compound of 2 is displayed for comparison. PHBA, p‐hydroxybenzoic acid; CTA*, CTA degradation product lacking a single thioamide; #, unrelated metabolite.
The origin of 2 differs between the two putative biosynthetic routes for DAP formation and incorporation (Figure 1 C). While both pathways predict that CtaB and CtaK are required for the formation of 2, its production is only dependent on CtaJ in pathway ii. To interrogate the roles of CtaB, CtaJ, and CtaK in the production of 2 and CTA, we used CRISPR‐Cas9‐mediated genome editing [11] to generate mutant R. cellulolyticum strains harboring inactivated versions of the corresponding genes (R. cellulolyticum ΔctaB, ΔctaJ, [3a] and ΔctaK; Figures S6 and S7) and analyzed their metabolite profiles by HPLC‐HRMS (Figure 2 and Figure S3). R. cellulolyticum ΔctaB produces low levels of CTA in addition to a suite of known derivatives and intermediates (compounds 1–5; Figure 2), demonstrating that the aminotransferase is not essential for CTA production, likely due to genetic redundancy. In contrast, CTA biosynthesis is abrogated in R. cellulolyticum ΔctaJ [3a] and R. cellulolyticum ΔctaK (Figure 2). R. cellulolyticum ΔctaK also lost the ability to produce 2 and closthioamide F (3), and instead produces increased levels of 1 (closthioamide G), closthioamide H (4), and closthioamide M (5) (Figure 2 and Figure S3). The CtaJ‐deficient strain no longer assembles 4 and 5 but produces increased levels of 1, 2, and 3 (Figure 2 and Figure S3). The metabolite profiles of the mutant strains, in particular the continued production of 2 by R. cellulolyticum ΔctaJ, strongly support pathway i. Furthermore, CtaK is implicated in the reductive release of 1 from CtaE. Although common in secondary metabolite pathways, the reductive release of thiotemplated products is performed by members of the short‐chain dehydrogenase protein family, [12] making CtaK the first AKR protein family member identified to date that catalyzes the reduction of a thioester. [13]
With a putative pathway identified for the assembly and introduction of the DAP linker, we sought to reconstitute the activities of the implicated enzymes in vitro. As such, CtaB, CtaK, and CtaJ were heterologously produced as an N‐terminal His6‐tagged fusion protein in Escherichia coli. Although CtaK was not soluble under any of the production conditions tested, we were able to obtain sufficient quantities of CtaB and CtaJ (Figure S8) for in vitro biochemical assays.
CtaB is predicted to be a PLP‐dependent aminotransferase responsible for the conversion of AH‐1 to 2 (Figure 1 C). Since the poor solubility of CtaK precluded the in situ enzymatic generation of the native substrate from 1, we performed in vitro assays with a surrogate of AH‐1 (Figure 3 A). Assays were supplemented with PLP, since CtaB did not co‐purify with high levels of the cofactor (Figure S9). Phylogenetic analysis (Figure S1) indicated that CtaB is most closely related to class‐III aminotransferases that use diamines, β‐amino acids, and γ‐amino acids as amine donors. [14] Therefore, we assayed a diverse set of potential amine donors and monitored reaction progress by HPLC‐HRMS. In all assays performed with CtaB and a diamine‐containing co‐substrate, we detected a new compound with an exact mass (calc. m/z 209.1285 [M+H]+; found m/z 209.1284 [M+H]+), MS2 fragmentation pattern, and elution time consistent with a synthetic reference compound of the expected aminated product (6; Figure 3 A and Figures S10–S12). To ascertain the preferred amine donor for CtaB, we first performed reaction time courses with all substrates that were accepted in the initial screen (Figure S13) and then subjected the top three amine donors—lysine, putrescine, and cadaverine—to a detailed kinetic investigation (Figure S14). Although the kinetic profiles obtained for each substrate were quite similar, the specificity constant for lysine was 5‐fold and 10‐fold greater than the constants obtained for cadaverine and putrescine, respectively. Combined with the observation that cadaverine and putrescine titers are low when R. cellulolyticum (Figure S15) is grown under CTA production conditions, we concluded that lysine is the preferred amine donor for CtaB.
Figure 3.
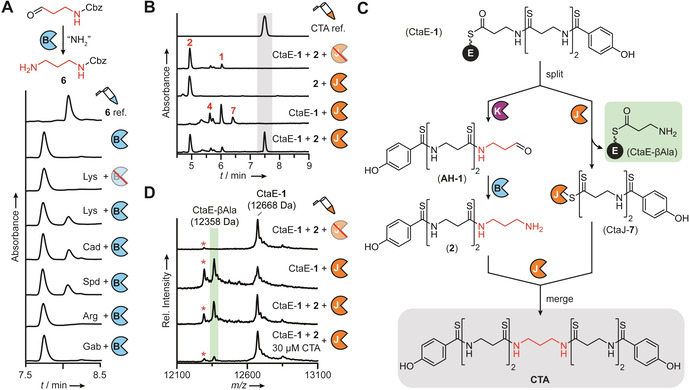
CtaB, CtaK, and CtaJ work together to convert CtaE‐1 to CTA in a split‐merge pathway. A) HPLC profile (255 nm absorbance) of CtaB reactions performed with Cbz‐propionaldehyde and various amine donors. A synthetic reference compound of the expected product, 6, is displayed for comparison. Lys, lysine; Cad, cadaverine; Spd, spermidine; Arg, arginine; Gab, γ‐aminobutyric acid. B) HPLC profiles (270 nm absorbance) of CtaJ reactions. A synthetic reference compound of CTA is displayed for comparison. C) Split‐merge pathway for the conversion of CtaE‐1 to CTA. D) MALDI‐TOF‐MS spectral overlay of CtaJ reactions. The average masses of CtaE‐1 and CtaE‐βAla are indicated. The red asterisk denotes holo‐CtaE formed from thioester hydrolysis. A,B,D) Red strikethrough=heat‐inactivated enzyme.
With the aminotransferase function of CtaB assigned, we proceeded to characterize the activity of CtaJ. We expected that CtaJ would convert two molecules of 2 into CTA, producing DAP as a byproduct. Therefore, reactions were performed with CtaJ and synthetically prepared 2, and CTA production was monitored by HPLC‐HRMS. Although the metabolite profiles of the knockout strains (Figure 2 and Figure S3) provided compelling evidence that 2 is an intermediate in CTA biosynthesis and the substrate for CtaJ, no processing of 2 was detected in the enzyme assays (Figure 3 B and Figure S16). While bacterial transglutaminases are not known to require any cofactors or additives for activity, [9c] the activity of some mammalian transglutaminases is allosterically regulated by calcium and guanosine triphosphate. [9a] As such, we also performed assays with CaCl2 and GTP supplementation; however, regardless of the reaction conditions, the processing of 2 was not observed (Figure S16).
Surprised by the inability of CtaJ to convert 2 to CTA, we reevaluated our biosynthetic model. Our initial assumption was that the pathway proceeded in a linear fashion, akin to canonical thiotemplated systems. [1] However, we realized that the data are also consistent with an atypical branching pathway where CtaE‐1 is a substrate for both CtaK and CtaJ (Figure 3 C). In the first branch, CtaE‐1 would be converted to 2 by the sequential action of CtaK and CtaB as initially proposed. In the second branch, CtaJ would cleave the terminal thioamide of CtaE‐1 to generate an enzyme‐bound intermediate (CtaJ‐7) and CtaE‐βAla. The pathways would then merge with the CtaJ‐dependent transfer of 7 to 2, affording CTA without the production of DAP.
As an initial test for the feasibility of this branching biosynthetic route, we grew R. cellulolyticum under CTA‐producing conditions and checked for the accumulation of DAP. Consistent with this new proposal, no DAP was detected (Figure S15). Encouraged by this result, we synthesized 1 and an N‐acetyl cysteamine thioester of 1 (SNAC‐1) and repeated the CtaJ assays with the addition of these potential substrates. HPLC‐HRMS analysis of the reactions demonstrated that low levels of CTA were only formed in assays containing SNAC‐1, 2, and CtaJ (Figure S17). The removal of any of these components or the substitution of SNAC‐1 with 1 led to a complete loss of CTA production. Furthermore, in addition to CTA, a new peak was observed in the chromatogram of the SNAC‐1 reaction. The exact mass (calc. m/z 764.1679 [M−H]−; found m/z 764.1682 [M−H]−), isotope pattern, and MS2 fragmentation pattern of the corresponding molecule suggested that it is a CTA congener bearing an additional βAla residue (Figures S18 and S19). We expect that this is formed by the non‐specific cleavage of the thioester of SNAC‐1 by CtaJ and attachment of the corresponding intermediate to 2. Attempts to increase SNAC‐1 turnover by the addition of CaCl2 or GTP were unsuccessful (Figure S17), demonstrating that the activity of CtaJ, like other bacterial transglutaminases, [9c] is calcium‐ and GTP‐independent.
As turnover was very low in the assays performed with SNAC‐1, we sought to improve turnover by using the predicted in vivo substrate, CtaE‐1. Therefore, we synthesized coenzyme A‐linked 1 (CoA‐1), purified His6‐tagged apo‐CtaE from E. coli (Figure S8), and loaded the substrate onto apo‐CtaE using the promiscuous phosphopantetheinyl transferase Sfp. [15] CtaJ reactions were then performed with CtaE‐1 and 2, and reaction progress was monitored by HPLC‐HRMS. In contrast to the low level of CTA detected in assays containing the SNAC thioester (Figure S17), we observed robust production in assays supplemented with CtaE‐1 (Figure 3 B). When CtaJ or either substrate was omitted, CTA production was lost. Furthermore, consistent with the split‐merge biosynthetic model, substituting 2 with DAP also abrogated CTA production (Figure S20). However, in reactions performed with CtaE‐1 alone, 4 and a derivative of 4 harboring a terminal thioacid (7) were produced (Figure 3 B and Figures S21 and S22). This result is consistent with a mechanism where CtaJ cleaves the terminal thioamide of CtaE‐1 and attaches the PHBA‐containing fragment to 2 (Figure 3 C). To corroborate this model, we monitored the fate of CtaE‐1 by matrix‐assisted laser desorption/ionization‐time‐of‐flight‐mass spectrometry (MALDI‐TOF‐MS) and found that, regardless of the presence or absence of 2, CtaJ converts CtaE‐1 to a new species with a mass consistent with the expected βAla adduct (Figure 3 D). Taken together, these results demonstrate that CtaJ is a thiotemplated transthioamidase that synthesizes CTA by coupling CtaE‐1 and 2.
Over the course of our studies, we noticed that—irrespective of the reaction time used—we never achieved complete consumption of the CtaE‐1 precursor. Furthermore, the processing of CtaE‐1 decreased in reactions performed with 2 relative to assays lacking 2 (Figure 3 D). We therefore suspected that the activity of CtaJ might be strongly product inhibited. Indeed, when we added CTA to CtaJ assays we observed a substantial decrease in CtaE‐1 processing by MALDI‐TOF‐MS (Figure 3 D and Figure S23). From a biological perspective, this pronounced product inhibition is unsurprising, as a buildup of CTA in the cell would be indicative of a failure in the transport machinery. Under such conditions, the negative regulation of CtaJ activity by CTA could prevent the self‐poisoning that would presumably arise from the continued synthesis of CTA.
Transglutaminases typically catalyze the inter‐ and intra‐molecular crosslinking of glutamine and lysine residues on proteins and peptides.[ 9a , 9c ] Apart from CtaJ, only one other transglutaminase has been reported that catalyzes the thiotemplated formation of an amide bond, an enzyme involved in the biosynthesis of the acylated NRP andrimid, AdmF. [9b] However, AdmF cleaves a thioester, not a thioamide linkage. Given the unprecedented nature of the transthioamidation reaction catalyzed by CtaJ, we next sought to gain deeper mechanistic insights. Transglutaminases are members of the CA clan of proteases and, accordingly, typically have a Cys‐His‐Asp catalytic triad.[ 9a , 9c ] For canonical transglutaminases, the transamidation reaction proceeds via a covalent intermediate with the catalytic cysteine residue (Figure 4 A). Despite the large sequence divergence between CtaJ and characterized members of the transglutaminase protein family, a putative catalytic triad is present in CtaJ (Cys114, His148, Asp165; Figure S24). We generated mutant versions of CtaJ bearing alanine substitutions to the residues of the putative catalytic triad (CtaJC114A, CtaJH148A, and CtaJD165A; Figure S8) and tested their catalytic competency. Consistent with the bioinformatic prediction, all CtaJ mutants were impaired in CTA production and CtaE‐1 processing (Figure 4 B and C). These data demonstrate that the CtaJ uses the standard catalytic triad of the transglutaminase protein family to catalyze the transthioamidation reaction.
Figure 4.
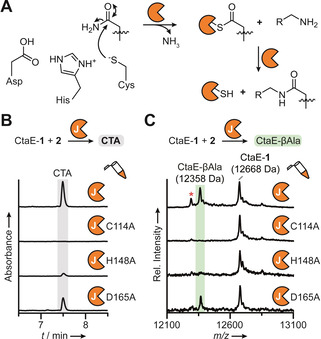
CtaJ uses the catalytic triad of canonical transglutaminases. A) General mechanism of canonical transglutaminases. B) HPLC profiles (270 nm absorbance) of CtaJ reactions performed with wild‐type or mutant CtaJ. C) MALDI‐TOF‐MS spectral overlay of CtaJ reactions performed with wild‐type or mutant CtaJ. The red asterisk denotes holo‐CtaE formed from thioester hydrolysis.
Based on the results described herein, we propose a model for the complete biosynthetic pathway to CTA (Scheme 1). The biosynthesis begins with the conversion of chorismate to PHBA by CtaA and the assembly of the amide backbone on CtaE (CtaE‐amido‐1) by CtaD and CtaF–G.[ 3a , 8a ] Next, CtaE‐amido‐1, is iteratively thioamidated by CtaC to afford CtaE‐1. [8b] The pathway branches at this point. In one branch, the AKR CtaK reductively releases the perthioamidated intermediate from CtaE, and CtaB converts the corresponding aldehyde, AH‐1, to 2 in a diamine‐dependent reaction. In a separate branch, CtaJ cleaves the terminal thioamide of CtaE‐1 forming an enzyme‐bound intermediate (CtaJ‐7) and CtaE‐βAla. The two pathways converge with the transfer of 7 from the active site cysteine of CtaJ to 2, affording CTA. The CtaE‐βAla byproduct is then recycled by the upstream enzymes to form CtaE‐1. This unexpected split‐merge pathway relies on the conversion of the terminal βAla residue to DAP in one branch, and its use as a recyclable priming residue in the other (Scheme 1). In this way, an elegant solution is provided for the biosynthesis of a symmetric molecule from an asymmetric pathway without the production of unused byproducts. Notably, the deconvolution of these final steps in CTA assembly not only assigns roles to each of the biosynthetic enzymes encoded in the biosynthetic operon but also accounts for all known intermediates and congeners (Scheme S1).
Scheme 1.
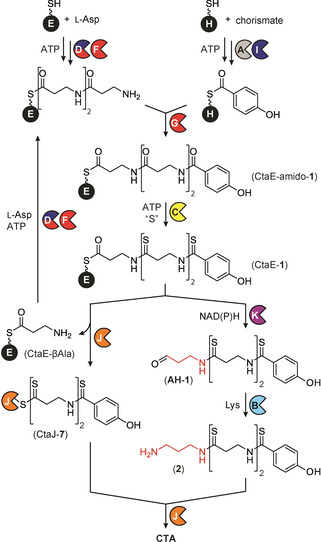
Complete pathway for CTA biosynthesis, featuring an unusual split‐merge route.
In type I thiotemplated biosynthetic pathways the carrier protein is covalently linked to the biosynthetic machinery and, as a consequence, each intermediate is typically passed directly to a single downstream enzyme in a linear fashion.[ 1a , 16 ] Biosynthetic pathways utilizing stand‐alone type II carrier proteins do not face the same structural constraints and often a single carrier protein can be recognized by multiple enzyme partners.[ 1b , 17 ] This behavior is effectively illustrated by the association of CtaE with six biosynthetic enzymes over the course of CTA maturation. Despite this inherent flexibility, type II thiotemplated pathways typically proceed in a linear fashion, with split‐merge pathways, like the one described here, being remarkably rare.[ 1a , 16 , 18 ] The discovery of split‐merge logic in the CTA maturation pathway highlights the flexibility of type II thiotemplated biosynthetic machinery and presents novel opportunities for pathway engineering.
Conclusion
In summary, we have unraveled the final steps of CTA maturation and discovered an unusual split‐merge assembly strategy for the synthesis and incorporation of the DAP linker. In the process, we have not only uncovered the first example of a transthioamidase but also implicated an AKR protein family member in the reductive release of a thiotemplated product. The identification of these non‐canonical members of well‐characterized protein families highlights novel enzyme functions that can be found through the investigation of atypical biosynthetic pathways.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank A. Perner, T. Kindel and M. García‐Altares Peréz for MS measurements and H. Heinecke for NMR measurements. K. Dunbar was supported by the Humboldt Research Fellowship for Postdoctoral Researchers. Financial support by the DFG (Leibniz Award to C. Hertweck) is gratefully acknowledged. Open access funding enabled and organized by Projekt DEAL.
K. L. Dunbar, M. Dell, E. M. Molloy, H. Büttner, J. Kumpfmüller, C. Hertweck, Angew. Chem. Int. Ed. 2021, 60, 4104.
References
- 1.
- 1a. Finking R., Marahiel M. A., Annu. Rev. Microbiol. 2004, 58, 453–488; [DOI] [PubMed] [Google Scholar]
- 1b. Jaremko M. J., Davis T. D., Corpuz J. C., Burkart M. D., Nat. Prod. Rep. 2020, 37, 355–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Giessen T. W., Marahiel M. A., Int. J. Mol. Sci. 2014, 15, 14610–14631; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Goswami A., Van Lanen S. G., Mol. Biosyst. 2015, 11, 338–353; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Carroll C. S., Moore M. M., Crit. Rev. Biochem. Mol. Biol. 2018, 53, 356–381. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Dunbar K. L., Büttner H., Molloy E. M., Dell M., Kumpfmüller J., Hertweck C., Angew. Chem. Int. Ed. 2018, 57, 14080–14084; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14276–14280; [Google Scholar]
- 3b. Chiriac A. I., Kloss F., Kramer J., Vuong C., Hertweck C., Sahl H. G., J. Antimicrob. Chemother. 2015, 70, 2576–2588; [DOI] [PubMed] [Google Scholar]
- 3c. Lincke T., Behnken S., Ishida K., Roth M., Hertweck C., Angew. Chem. Int. Ed. 2010, 49, 2011–2013; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2055–2057; [Google Scholar]
- 3d. Kloss F., Lincke T., Hertweck C., Eur. J. Org. Chem. 2011, 1429–1431; [Google Scholar]
- 3e. Kloss F., Chiriac A. I., Hertweck C., Chem. Eur. J. 2014, 20, 15451–15458; [DOI] [PubMed] [Google Scholar]
- 3f. Miari V. F., Solanki P., Hleba Y., Stabler R. A., Heap J. T., Antimicrob. Agents Chemother. 2017, 61, e00929-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Behnken S., Lincke T., Kloss F., Ishida K., Hertweck C., Angew. Chem. Int. Ed. 2012, 51, 2425–2428; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2475–2478. [Google Scholar]
- 5. Kloss F., Pidot S., Goerls H., Friedrich T., Hertweck C., Angew. Chem. Int. Ed. 2013, 52, 10745–10748; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10945–10948. [Google Scholar]
- 6.
- 6a. Li J. S., Barber C. C., Zhang W., J. Ind. Microbiol. Biotechnol. 2019, 46, 375–383; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Schieferdecker S., Shabuer G., Knuepfer U., Hertweck C., Org. Biomol. Chem. 2019, 17, 6119–6121; [DOI] [PubMed] [Google Scholar]
- 6c. Schieferdecker S., Shabuer G., Letzel A. C., Urbansky B., Ishida-Ito M., Ishida K., Cyrulies M., Dahse H. M., Pidot S., Hertweck C., ACS Chem. Biol. 2019, 14, 1490–1497; [DOI] [PubMed] [Google Scholar]
- 6d. Ishida K., Shabuer G., Schieferdecker S., Pidot S. J., Stinear T. P., Knuepfer U., Cyrulies M., Hertweck C., Chem. Eur. J. 2020, 26, 13147–13151; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Li J. S., Barber C. C., Herman N. A., Cai W., Zafrir E., Du Y., Zhu X., Skyrud W., Zhang W., J. Ind. Microbiol. Biotechnol. 2020, 47, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Dunbar K. L., Scharf D. H., Litomska A., Hertweck C., Chem. Rev. 2017, 117, 5521–5577; [DOI] [PubMed] [Google Scholar]
- 7b. Mahanta N., Szantai-Kis D. M., Petersson E. J., Mitchell D. A., ACS Chem. Biol. 2019, 14, 142–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Dunbar K. L., Dell M., Gude F., Hertweck C., Proc. Natl. Acad. Sci. USA 2020, 117, 8850–8858; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Dunbar K. L., Dell M., Molloy E. M., Kloss F., Hertweck C., Angew. Chem. Int. Ed. 2019, 58, 13014–13018; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13148–13152. [Google Scholar]
- 9.
- 9a. Griffin M., Casadio R., Bergamini C. M., Biochem. J. 2002, 368, 377–396; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Fortin P. D., Walsh C. T., Magarvey N. A., Nature 2007, 448, 824–827; [DOI] [PubMed] [Google Scholar]
- 9c. Strop P., Bioconjugate Chem. 2014, 25, 855–862. [DOI] [PubMed] [Google Scholar]
- 10. Lee J., Sperandio V., Frantz D. E., Longgood J., Camilli A., Phillips M. A., Michael A. J., J. Biol. Chem. 2009, 284, 9899–9907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang H., La Russa M., Qi L. S., Annu. Rev. Biochem. 2016, 85, 227–264. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Du L., Lou L., Nat. Prod. Rep. 2010, 27, 255–278; [DOI] [PubMed] [Google Scholar]
- 12b. Mullowney M. W., McClure R. A., Robey M. T., Kelleher N. L., Thomson R. J., Nat. Prod. Rep. 2018, 35, 847–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Ellis E. M., FEMS Microbiol. Lett. 2002, 216, 123–131; [DOI] [PubMed] [Google Scholar]
- 13b. Penning T. M., Chem.-Biol. Interact. 2015, 234, 236–246; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Mindnich R. D., Penning T. M., Hum. Genomics 2009, 3, 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schiroli D., Peracchi A., Biochim. Biophys. Acta Proteins Proteomics 2015, 1854, 1200–1211. [DOI] [PubMed] [Google Scholar]
- 15. Beld J., Sonnenschein E. C., Vickery C. R., Noel J. P., Burkart M. D., Nat. Prod. Rep. 2014, 31, 61–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hertweck C., Angew. Chem. Int. Ed. 2009, 48, 4688–4716; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4782–4811. [Google Scholar]
- 17. Chen A., Re R. N., Burkart M. D., Nat. Prod. Rep. 2018, 35, 1029–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Keating T. A., Marshall C. G., Walsh C. T., Biochemistry 2000, 39, 15522–15530; [DOI] [PubMed] [Google Scholar]
- 18b. Keating T. A., Marshall C. G., Walsh C. T., Biochemistry 2000, 39, 15513–15521; [DOI] [PubMed] [Google Scholar]
- 18c. Lautru S., Deeth R. J., Bailey L. M., Challis G. L., Nat. Chem. Biol. 2005, 1, 265–269; [DOI] [PubMed] [Google Scholar]
- 18d. Dimise E. J., Widboom P. F., Bruner S. D., Proc. Natl. Acad. Sci. USA 2008, 105, 15311–15316; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18e. Giessen T. W., Franke K. B., Knappe T. A., Kraas F. I., Bosello M., Xie X., Linne U., Marahiel M. A., J. Nat. Prod. 2012, 75, 905–914; [DOI] [PubMed] [Google Scholar]
- 18f. Mantovani S. M., Moore B. S., J. Am. Chem. Soc. 2013, 135, 18032–18035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary