

Abstract
Tau protein aggregates are a recognized neuropathological feature in Alzheimer's disease as well as many other neurodegenerative disorders, known as tauopathies. The development of tau‐targeting therapies is therefore extremely important but efficient strategies or protein targets are still unclear. Here, we performed a cell‐based phenotypic screening under endoplasmic reticulum (ER) stress conditions and identified a small molecule, SB1617, capable of suppressing abnormal tau protein aggregation. By applying label‐free target identification technology, we revealed that the transient enhancement of protein kinase‐like endoplasmic reticulum kinase (PERK) signaling pathway through the inhibition of stress‐responsive SB1617 targets, PDIA3 and DNAJC3, is an effective strategy for regulating proteostasis in tauopathies. The molecular mechanism and the promising efficacy of SB1617 were demonstrated in neuronal cells and a mouse model with traumatic brain injury, a tauopathy known to involve ER stress.
Keywords: ER stress response, PERK signaling, proteostasis, target Identification, tauopathies
A novel neuroprotective compound, SB1617, was developed via phenotypic assay by image‐based monitoring of tau protein aggregation. Label‐free target identification study revealed that SB1617 inhibits PDIA3 and DNAJC3, thereby regulates tau proteostasis through the activation of PERK signaling pathway in a stress‐responsive manner. The administration of SB1617 showed neuroprotective effects in mice with tauopathy involving ER stress.
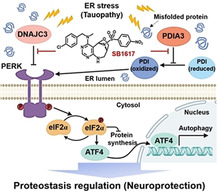
Introduction
Age‐associated neurodegenerative diseases have become a substantial medical and social burden in the 21st century as the human lifespan steadily increases. These diseases, characterized by the accumulation of misfolded proteins, are referred to as proteinopathies. [1] One of the best‐known proteinopathies is Alzheimer's disease (AD) which exhibits characteristic extracellular deposits of amyloid β (Aβ) plaques and intracellular tau protein aggregates known as neurofibrillary tangles (NFTs). [2] Because numerous studies targeting Aβ plaques have failed in the late stages of drug development, the biology of tau aggregation received much attention in the quest for new therapeutic targets. [3] The main therapeutic strategies for tauopathies include the development of tau immunotherapies, modulators of tau post‐translational modifications, inhibitors of tau aggregation, and enhancers of protein clearance mechanisms to promote tau degradation. [3b] Although several tau targeting immunotherapy drugs are currently being evaluated in clinical trials, they are still in the early stages of development, and no approved therapeutics or prophylactics are available for tauopathies, because of the limited knowledge on the mechanisms related to pathological tau protein in tauopathies. [3]
Recently, endoplasmic reticulum (ER) stress has attracted much attention because of its involvement in neurodegenerative disorders, including tauopathies. [4] ER is the structure in which nascent and misfolded proteins are correctly folded by various chaperones, before transport via secretory vesicles. Stressful stimuli to ER, caused by the accumulation of misfolded proteins and changes in intracellular calcium levels, activate an adaptive signaling pathway for cell survival, known as the unfolded protein response (UPR). [5] Various ER stress markers or UPR proteins have been found together with accumulated NFTs in post‐mortem brain tissues from tauopathy patients, and a positive relationship has been observed between the severity of protein aggregation and disease status. [4] Although tauopathies have been reported to be exacerbated by chronic ER stress, not many studies have evaluated the development of tauopathy therapeutics targeting ER stress. [6] In this study, we hypothesized that screening for bioactive small molecules able to suppress tau aggregation in live cells under ER stress conditions might lead to the identification of compounds with the potential to become the first‐in‐class therapeutics for tauopathies. Available target‐based approaches are, however, limited, because the information about functional targets in neurodegenerative diseases associated with ER stress is insufficient. [3b] To address this limitation, we performed a phenotypic screening by monitoring tau assembly in live cells to explore potential targets for tauopathy therapy.
Results and Discussion
To identify bioactive small molecules that can reduce abnormal tau assembly, we carried out a phenotypic screening using HEK293 human embryonic kidney cells with stable expression of Venus‐based bimolecular fluorescence‐complemented (BiFC)‐tau (Table S1, Figure S1). [7] In this system, each N‐terminus Venus (VN173) and C‐terminus Venus (VC155) fragment sequence was fused with the full‐length human tau (hTau441) sequence. Co‐expression of these two vectors (hTau441‐VN173 and hTau441‐VC155) enabled the measurement of soluble tau dimerization by monitoring turn‐on/off signals of Venus fluorescence (Figure 1 A). As a stimulator of tau assembly, we used the ER stress inducer thapsigargin (TG), a direct inhibitor of Sarco/endoplasmic reticulum Ca2+‐ATPase, which perturbs calcium homeostasis. [8] The TG treatment induces tau dimerization as shown by the BiFC‐tau Venus fluorescence turn‐on, which corresponded to the level of phosphorylated tau (p‐tau) measured by western blot analysis, as tau hyper‐phosphorylation is a representative pathological post‐translational modification in tauopathy (Figures 1 B, C). [9] Importantly, the phenotypic screening of our in‐house compound library, generated using a privileged substructure‐based diversity‐oriented synthesis (pDOS) strategy, [10] allowed the identification of an initial hit compound SB1605, that effectively suppressed BiFC‐tau Venus fluorescence with a half‐maximal inhibitory concentration (IC50) of 5.2 μM (Table 1, Figures 1 D, S2). We further conducted a small structure–activity relationship (SAR) study to understand if the observed effect of SB1605 on tau dimerization could be improved. Briefly, we generated more than twenty analogues by adding three different substituents to the bridged pyrimidodiazepine core structure of SB1605 and evaluated their activity in the above‐mentioned phenotypic assay. Interestingly, we observed that as long as the compounds’ core structure contained a para‐substituted phenyl on its sulfonamide group and an N‐alkyl, N‐benzylamine on the pyrimidine moiety, the tau dimerization inhibitory activity was retained. Furthermore, among the synthesized analogues, SB1617 showed improved activity (IC50 1.9 μM) compared to the parent compound (SB1605), and was selected as a lead for further biological evaluations (Figures 1 D,E, S2,3 and Table 1). Of note, SB1617 effect on BiFC‐tau Venus fluorescence intensity was also confirmed by p‐tau level using immunoblot assay (Figure 1 C). On the contrary, the simplification of SB1617 chemical structure by removing an aromatic ring from the sulfonamide group (SB1607) led to a significant loss of activity, suggesting that the tau dimerization inhibitory effect is dependent on specific compound‐target interactions (Figures 1 C–E).
Figure 1.
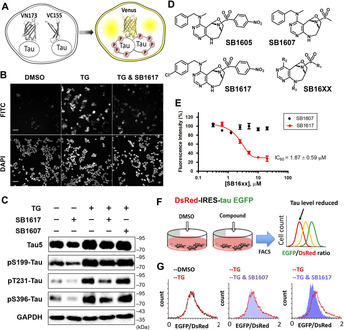
SB1617 reduces tau assembly and regulates proteostasis in tau‐overexpressing cell lines. A) Depiction of BiFC‐tau Venus HEK293 cell system for monitoring tau assembly. B) Representative microscopic data of BiFC‐tau screening via monitoring of Venus fluorescence. Cells were co‐treated with 80 nM TG and 10 μM of screening compounds (current data with SB1617) for 24 h. (FITC channel; BiFC‐Venus fluorescence, DAPI channel; the nuclei of cells from the same areas presented by FITC images) (scale bar=25 μm). C) Immunoblot data investigating total tau (Tau5) and phospho‐tau (S199, T231, S396) levels upon treatment of 80 nM TG with either 5 μM of SB1617 or SB1607 for 20 h in BiFC‐tau Venus HEK293 cells (n ≥3). Quantitative data are shown in Figure S4. D) Chemical structures of SB16XX compounds. E) Dose‐dependency data on Venus fluorescence intensity change upon co‐treatment of 80 nM TG with various concentrations of SB1617 or SB1607 for 20 h in BiFC‐tau Venus HEK293 cells (n=4). See Figure S3 for SB1617 concentration dependency data without TG co‐treatment. F) Illustration of the IRES‐incorporated cell system to measure tau proteostasis. G) Representative flow cytometry data upon treatment with 100 nM TG together with either 5 μM of SB1617 or SB1607 for 20 h in DsRed‐IRES‐EGFP‐tau HEK293 cells. Quantitative data pertaining to Figure 1 G (n≥4) are shown in Figure S6.
Table 1.
Structure‐activity relationship data investigated in BiFC‐tau Venus HEK293 cellls. BiFC‐tau‐Venus fluorescence intensity was presented as % values upon co‐treatment of 10 μM SB16XX series compounds and 80 nM thapsigargin for 24 h in HEK293‐BiFC‐tau‐Venus cells. Parentheses values shows IC50 values. R‐groups are shown in Figure 1 D with the chemical structure.
|
|
R1 |
R2 |
R3 |
BiFC‐tau intensity [%] |
|---|---|---|---|---|
|
SB1601 |
4‐methylphenyl |
N‐methyl, N‐benzyl |
H |
65 (9.1±0.9) |
|
SB1602 |
4‐methoxyphenyl |
N‐methyl, N‐benzyl |
H |
79 |
|
SB1603 |
2‐nitrophenyl |
N‐methyl, N‐benzyl |
H |
102 |
|
SB1604 |
3‐nitrophenyl |
N‐methyl, N‐benzyl |
H |
115 |
|
SB1605 |
4‐nitrophenyl |
N‐methyl, N‐benzyl |
H |
46 (5.2±0.5) |
|
SB1606 |
4‐fluorophenyl |
N‐methyl, N‐benzyl |
H |
81 |
|
SB1607 |
methyl |
N‐methyl, N‐benzyl |
H |
94 |
|
SB1608 |
phenyl |
N‐methyl, N‐benzyl |
H |
105 |
|
SB1609 |
benzyl |
N‐methyl, N‐benzyl |
H |
84 |
|
SB1610 |
pyridyl |
N‐methyl, N‐benzyl |
H |
130 |
|
SB1611 |
thiophenyl |
N‐methyl, N‐benzyl |
H |
110 |
|
SB1612 |
4‐nitrophenyl |
N, N‐dimethyl |
H |
90 |
|
SB1613 |
4‐nitrophenyl |
N‐methyl, N‐(2‐phenylethyl) |
H |
71 |
|
SB1614 |
4‐nitrophenyl |
N‐isopropyl, N‐benzyl |
H |
47 (6.9±0.9) |
|
SB1615 |
4‐nitrophenyl |
isopropenyl |
H |
99 |
|
SB1616 |
4‐nitrophenyl |
4‐methoxylphenyl |
H |
135 |
|
SB1617 |
4‐nitrophenyl |
N‐methyl, N‐(4‐chlorobenzyl) |
H |
36 (1.9±0.5) |
|
SB1618 |
4‐nitrophenyl |
N‐methyl, N‐(4‐fluorobenzyl) |
H |
55 (7.6±1.2) |
|
SB1619 |
4‐nitrophenyl |
N‐methyl, N‐(4‐azidobenzyl) |
H |
56 (4.6±2.1) |
|
SB1620 |
4‐fluorophenyl |
N‐methyl, N‐(4‐chlorobenzyl) |
H |
81 |
|
SB1621 |
4‐methoxyphenyl |
N‐methyl, N‐(4‐chlorobenzyl) |
H |
51 (6.6±0.6) |
|
SB1622 |
4‐nitrophenyl |
N‐methyl, N‐(4‐chlorobenzyl) |
acetyl |
81 |
|
SB1623 |
4‐nitrophenyl |
N‐methyl, N‐(4‐chlorobenzyl) |
cyclopropyl carbonyl |
78 |
|
SB1624 |
4‐ethynylphenyl |
N‐methyl, N‐(4‐azidobenzyl) |
H |
50 (8.1±0.6) |
The immunoblot data indicated that TG treatment increased the levels of total tau as well as p‐tau, whereas SB1617 treatment reduced both levels. To exclude the effect of transcriptional changes upon stress stimulation that might happen in the engineered cell lines, we used a bicistronic cell line containing an internal ribosome entry site (IRES) between DsRed and EGFP‐tau sequences. [11] As shown in Figure 1 F, changes in the EGFP‐to‐DsRed fluorescence ratio upon compound treatment indicate alterations in tau levels via clearance pathways, regardless of any transcriptional alteration. TG treatment showed a slight, if any, increase in the EGFP‐to‐DsRed signal ratio, whereas co‐treatment of SB1617 with TG resulted in a decreased EGFP‐to‐DsRed signal ratio, indicating that SB1617 regulated tau proteostasis by promoting tau clearance (Figures 1 G, S5–S7). Overall, our observations in two independent cellular systems suggest that SB1617 suppresses the assembly of overexpressed, aggregation‐prone tau protein by regulating protein homeostasis.
To investigate the mechanism of action of SB1617, we conducted a target identification study using the thermal stability shift‐based fluorescence difference in two‐dimensional gel electrophoresis (TS‐FITGE), which is based on changes in protein stability to thermal denaturation when the protein engages in a specific interaction with a small molecule. [12] Briefly, soluble lysate fractions of the vehicle‐, SB1607 (partial negative control)‐, and SB1617‐treated cells, were heated at the increasing temperatures, and chemically conjugated with Cy2‐, Cy3‐, and Cy5‐N‐hydroxysuccinimide esters, respectively. Then the three proteomes (fluorescently labeled with three different dyes) from each temperature condition, were combined and analyzed by 2D gel electrophoresis. As shown in Figure 2 A, several red spots appeared in the 2D‐gel images of samples treated at higher temperatures, indicating enhanced protein thermal stability induced by specific interactions with SB1617; however, these proteins were not or less stabilized by vehicle or the compound SB1607 with weak activity, respectively (Figure S11). These red spots ultimately disappeared upon full thermal denaturation. The identities of these reproducibly thermostable proteins (red spots) were investigated by mass spectrometry analyses. The list of target protein candidates for SB1617 was deconvolved by excluding proteins with inherent non‐specific binding, based on the previous chemical proteomics data, and by performing phenocopying tests upon knockdown of the listed proteins in BiFC‐tau cells (Figures S8–S10 and Table S2). [13]
Figure 2.
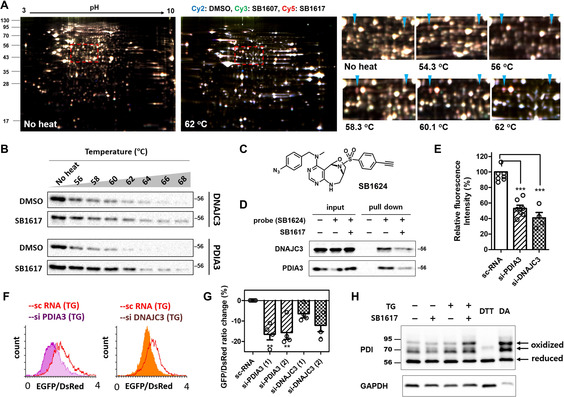
Target identification and target validation of SB1617. A) Representative images of TS‐FITGE 2D gel data. Merged gel images of Cy2‐ (blue, DMSO), Cy3‐ (green, SB1607), and Cy5‐ (red, SB1617) channels. The area in the red dashed boxes is magnified with the marked target candidates. Left arrow‐PDIA3, right arrow‐DNAJC3. B) CETSA data for specific binding of SB1617 towards DNAJC3 and PDIA3. Quantitative data are shown in Figure S11. C) Chemical structure of the photo‐reactive probe SB1624. (D) Pull‐down data by SB1624, the photo‐reactive probe, in the absence and presence of SB1617, towards DNAJC3 and PDIA3 (n≥2). E) BiFC‐tau Venus fluorescence intensity changes upon depletion of either PDIA3 or DNAJC3 using siRNAs. BiFC‐tau cells were transfected with siRNAs for 48 h then were treated with 100 nM TG for 20 h (n≥4). F) Flow cytometry data investigating EGFP‐tau/DsRed ratio alteration upon depletion of either PDIA3 or DNAJC3 using siRNAs. DsRed IRES EGFP‐tau cells were transfected with siRNAs for 48 h and then treated with 100 nM TG for 18 h. G) Quantitative data pertaining to Figure 2 F (n=4). H) PEG‐maleimide modification assay to monitor the oxidation status of PDI via alterations in PDIA3 reductase activity by SB1617. BiFC‐tau HEK293 cells were treated with 200 nM TG and 40 μM SB1617 for 3.5 h. As controls of the reduced and the oxidized forms of PDI, 10 mM 1,4‐dithiothreitol (DTT) and 5 mM tetramethylazodicarboxamide (DA) were added to cells for 15 min, respectively (n≥3). (ns, P>0.05, * P≤0.05, ** P≤0.01, *** P≤0.001).
This target deconvolution procedure led to two possible SB1617 target proteins: protein disulfide isomerase A3 (PDIA3) and DNA J homology subfamily C member 3 (DNAJC3). Interactions of SB1617 with these target protein candidates in cells were confirmed by a cellular thermal stability assay (CETSA; Figures 2 B, S11) and pull‐down experiments using the probe compound SB1624. Based on our SAR study, the chemical probe SB1624 was designed from the structure of SB1617 by introducing a photo‐reactive aryl azido group on the pyrimidine moiety, which can covalently crosslink to adjacent proteins upon target engagement via UV irradiation, and by replacing a nitro group with a terminal alkyne moiety on its sulfonamide group for the subsequent conjugation with biotin as an affinity handle, via a bioorthogonal Click reaction (Figure 2 C). The activity of SB1624 was confirmed in BiFC‐tau HEK293 cells before its use as a chemical probe (Figure S2G). Interactions of SB1624 with PDIA3 and DNAJC3 were competed out by SB1617 treatment in a protein pull‐down assay, confirming the binding of SB1617 to both PDIA3 and DNAJC3 (Figure 2 D). We also biophysically confirmed the direct binding events of SB1617 with PDIA3 and DNAJC3 using surface plasmon resonance spectroscopy (Figure S12).
Depletion of PDIA3 and DNAJC3 in both BiFC‐tau and DsRed‐IRES‐EGFP‐tau HEK293 cells using short interfering RNAs (siRNAs) phenocopied the cellular activity of SB1617 by reducing the tau‐Venus intensity, p‐tau levels, and EGFP‐to‐DsRed intensity ratio (Figures 2 E–G). We then carried out additional functional/enzymatic assays as a validation step. However, because DNAJC3 has not been widely studied enzymatically, we only focused on the biophysical studies for this protein. In the case of PDIA3, its knockdown should promote protein disulfide isomerase (PDI) oxidation, since PDIA3 is a known PDI reductase (Figure S13). [14] To assess whether the interaction of SB1617 with PDIA3 can enhance PDI oxidation, we performed a PEG‐maleimide modification assay. In this assay, free thiols in cysteines were pre‐alkylated with low molecular‐weight maleimide molecules, leaving oxidized, or disulfide‐bonded, cysteines intact. These intact cysteines were then alkylated with high molecular‐weight PEG‐maleimide (5 kDa) after a disulfide reduction step. The two different maleimide molecular weights facilitate the band separation of oxidized and reduced forms of PDI by electrophoresis. Substantial PDI conversion from the reduced to oxidized form was observed upon co‐treatment with SB1617 and TG, compared to that observed upon treatment with TG alone, thus, indirectly demonstrating that SB1617 perturbed the cellular function of PDIA3 (Figure 2 H).
PDIA3 (or ERp57), a member of the protein disulfide isomerase family, has multiple functions. [15] Particularly, PDIA3 has been shown to facilitate peripheral nerve regeneration and was suggested to have crucial roles in cell survival. [16] However, certain aspects of PDIA3 indicate its pathological involvement. PDIA3 upregulated the steady‐state levels of protease‐resistant prion proteins, whereas the PDIA3 knockdown decreased the prion protein levels. [17] Furthermore, the knockdown of PDIA3 or the pharmacological enhancement of PDI oxidation by LOC14 resulted in the suppression of neuronal degeneration induced by mutant huntingtin. [18] On the other hand, DNAJC3 (or P58IPK) has less frequently been reported as a therapeutic target, although it is considered to play a role in ER stress‐associated apoptosis and inflammation. [19]
Interestingly, both PDIA3 and DNAJC3 are stress‐responsive ER chaperones in the UPR signaling pathway and are direct or indirect inhibitors of the protein kinase‐like endoplasmic reticulum kinase (PERK) signaling pathway.[ 14 , 20 ] PERK is one of the three ER stress sensors on the ER membrane, along with inositol‐requiring enzyme 1 alpha (IRE1α) and activating transcription factor 6 (ATF6), and is autophosphorylated and activated upon the accumulation of misfolded proteins. [5a] DNAJC3 forms a negative feedback loop in the ER stress cycle and is upregulated to terminate PERK activation via direct binding. [20] PDIA3 is a PDI reductase. Since the oxidized PDI activates PERK, PDIA3 indirectly suppresses PERK activation via PDI reduction.[ 14 , 18b ] Therefore, the functional inhibition of PDIA3 and DNAJC3 by SB1617 would increase and prolong the activation of PERK signaling.
To confirm the functional roles of DNAJC3 and PDIA3 in the repression of PERK signaling, we performed siRNA knockdown experiments and investigated the alteration of PERK downstream pathways using immunoblot assays. Depletion of either PDIA3 or DNAJC3 suppressed the abnormal increase of p‐tau upon TG treatment in tau‐overexpressing cells and upregulated the signals downstream of PERK, including the phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) and expression of transcription factor 4 (ATF4), which confirms the inhibitory function of PDIA3 and DNAJC3 in the PERK signaling pathway (Figure 3 A). Double knockdown of PDIA3 and DNAJC3 did not exhibit noticeable synergistic effects in tau‐overexpressing cells, implying that both PDIA3 and DNAJC3 exert their regulatory roles in tau aggregation by suppressing the PERK signaling pathway (Figures S15–S17). We predict that SB1617 binds to PDIA3 and DNAJC3, and impairs their PERK repression functions. As shown in Figure 3 B, SB1617 prolonged PERK activation, demonstrated by the sustained levels of p‐PERK and p‐eIF2α, and upregulated ATF4 under stress conditions induced by TG treatment in SH‐SY5Y human neuroblastoma cells (Figure S18). Notably, the activation of downstream PERK signaling by SB1617 was not significant in the absence of cell stress (Figures 3 C, S19). The stress‐responsive efficacy of SB1617 might be due to the upregulation of cellular PDIA3 and DNAJC3 levels (Figure S20) or to changes in redox states of their partner proteins in ER stress conditions.[ 14 , 20 ]
Figure 3.
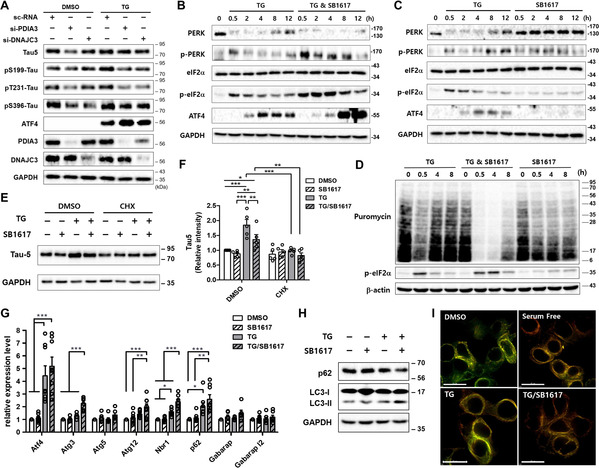
SB1617 stimulates the PERK pathway in a stress‐responsive manner and regulates proteostasis. A) Immunoblot data showing the PDIA3 and DNAJC3 depletion effect on PERK activation and tau reduction in HEK293 BiFC‐tau cells. Cells were transfected with siRNAs for 48 h and then treated with 200 nM TG for 6 h (n≥3). Quantitative data are shown in Figure S14. B, C) Time‐course analysis of PERK activation upon SB1617 treatment in SH‐SY5Y cells. 10 μM SB1617 was added with (B) or without (C) 1 μM TG for the indicated times (n≥3). See also Figures S14 and S15. D) Immunoblot data investigating the translational regulation by SB1617. SH‐SY5Y cells were treated with 10 μM SB1617 for the indicated time, then newly synthesized proteins were labeled with 10 μg mL−1 puromycin for 10 min and visualized by anti‐puromycin antibody staining (n≥2). E) Immunoblot data of total tau upon treatment with 10 μM SB1617 in the absence and presence of 200 nM TG and 20 μg mL−1 cycloheximide (CHX) for 8 h in HEK293 BiFC‐tau cells. F) Quantitative data pertaining to Figure 3 E (n=5). G) Autophagy‐related genes regulated by ATF4 were analyzed by RT‐qPCR (n≥6). SH‐SY5Y cells were treated with 5 μM SB1617 with or without 1 μM TG for 8 h. H) Immunoblot data measuring autophagy: LC3‐I to LC3‐II conversion and p62 levels. HEK293 BiFC‐tau cells were treated with 5 μM SB1617 with or without 500 nM TG for 6 h. Quantitative data are shown in Figure S21 (n=2–4). I) mCherry and GFP fluorescence images of live HEK293 cells transfected with mCherry‐GFP‐tau plasmid and treated with compounds for 6 h (n≥3). Scale bar, 15 μm. (ns, P>0.05, * P≤0.05, ** P≤0.01, *** P≤0.001).
Although a small subset of mRNAs undergoes translation, [21] phosphorylation of eIF2α leads to translational repression of most mRNAs [5b] allowing cells to survive by greatly reducing their ER workload. Upon TG treatment, cellular protein synthesis was transiently suppressed, recovering at later time points, as observed via nascent protein labeling with puromycin (Figure 3 D). [22] When SB1617 was administered together with TG, the suppression of cellular protein synthesis was intensified and prolonged, which was consistent with the level of p‐eIF2α. Without ER stress, SB1617 appeared to only mildly affect protein translation, likely because of the basal PDIA3 and DNAJC3 levels. When BiFC‐tau cells were pre‐treated with a translational inhibitor, cycloheximide, SB1617 treatment did not induce significant changes in tau levels, confirming the effects of SB1617 on protein translation regulation (Figures 3 E,F). ATF4 is a transcription factor, that is elevated upon eIF2α‐phosphorylation and upregulates genes related to autophagy and redox control as a recovery mechanism upon ER stress. [23] SB1617 treatment under ER stress conditions upregulated the transcription levels of autophagy‐related genes, regulated by ATF4, compared to those in cells treated with TG alone, suggesting that SB1617 promoted autophagy (Figure 3 G), which was further validated using immunoblotting to detect microtubule‐associated protein light chain 3 (LC3)‐I to LC3‐II conversion (Figure 3 H). To confirm the induction of entire autophagic flux for tau clearance upon SB1617 treatment, we monitored live HEK293 cells stably overexpressing mCherry‐green fluorescent protein (GFP)‐tau protein, of which fluorescent emission alters by the environmental acidity due to the acid‐sensitive characteristic of GFP. [24] Upon co‐treatment of cells with SB1617 and TG, the GFP signal was reduced while the mCherry signal was unaffected, which is similar to the serum‐free condition (Figure 3 I). Since both fluorescent proteins are tandemly fused to tau protein, this observation suggested that tau is proteolytically processed in autolysosome, of which acidic environment led to the selective reduction of GFP fluorescence. When the autophagic process was blocked via treatment with either 3‐methyladenine or bafilomycin A1 to interfere with the early and late autophagy stages, respectively, the efficacy of SB1617 on the reduction of tau levels was diminished, but not completely abrogated, suggesting that the SB1617 effect on tau clearance is less impactful than its translational regulatory properties for proteostasis control (Figure S22).
Traumatic brain injury (TBI) leads to the development of a tau pathology along with ER stress, resulting in significant injury to the brain that compromises neuronal function and cognitive abilities. [25] To evaluate the suitability of SB1617 for in vivo use, we first investigated its pharmacokinetic and blood–brain barrier penetration properties. Intraperitoneally injected SB1617 showed suitable pharmacokinetic behaviors with a half‐life of approximately 6.6 h and sufficient blood–brain barrier crossing‐at least 50 % of SB1617 was detected in the brain compared to that in the plasma (Figures S23,S24). We then examined the potential therapeutic effects of SB1617 on TBI mice by investigating the levels of tau, p‐tau, oxidative stress, ER stress, neuronal viability, and behavioral improvement (Figures 4, S25–S28). Immunohistochemistry data indicated significant increases in ER stress and oxidative stress upon TBI, suggesting that the TBI pathology was induced (Figures S26,S27). 24 h after TBI, drastic increases in the total tau and p‐tau levels were observed in the hippocampal and cortical regions (Figures 4 A,B, S28), likely a consequence of impaired tau clearance.[ 25 , 26 ] Furthermore, 3 days after TBI, neuronal death was apparent (Figure 4 C). These pathological symptoms were ameliorated when SB1617 was administered to mice with TBI (5 mg kg−1 of body weight, twice per day). Compared to vehicle treatment, SB1617 treatment inhibited the abnormal increase in the tau and p‐tau levels, suppressed cellular stress induction, and increased neuronal viability in mice with TBI (Figures 4 A–C, S26–S28). SB1617 exerted further beneficial effects on motor neuronal behaviors, as measured by the neurological severity score and pole tests up to 7 days post‐TBI (Figures 4 D–F, movie S1). Overall, the administration of SB1617 to mice with TBI lead to neuroprotective effects.
Figure 4.
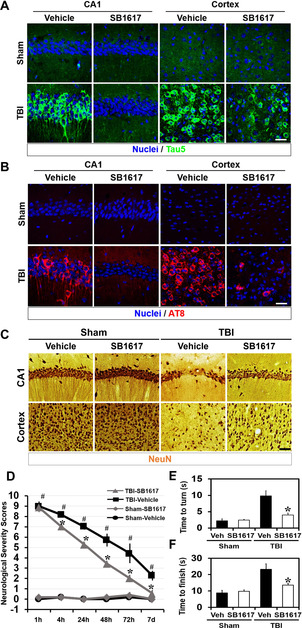
SB1617 is neuroprotective in a TBI mouse model. A, B) Representative immunofluorescence images showing expression of total tau (anti‐Tau5) (A) and p‐tau (AT8) (B) in the ipsilateral hippocampal CA1 and cortex sections from vehicle‐ and SB1617‐treated mice at 24 h after sham surgery or TBI. n=3–6 from each group. Scale bar, 20 μm. C) Representative images showing expression of the neuronal marker NeuN for live neuronal cells in the ipsilateral hippocampal CA1 and cortex sections from vehicle‐ and SB1617‐treated mice at 72 h after sham surgery or TBI. n=5 from each group. Scale bar, 50 μm. Quantitative data pertaining to Figure 4 A–C are shown in Figure S28. D) The mice neurologic severity scores were determined 1, 4, 24, 48, 72 h and 7 days after TBI. A score of 10 means that all tasks were failed; a score of 0 means that all tasks were successfully completed (n=5–7 from each group, * P<0.05 versus vehicle‐treated group; # P<0.05 versus sham‐operated group). E, F) The pole test analyzed both downward orientation (E) and bottom arrival (F) when the mouse was placed head upward on the top of a vertical pole, thus predominantly testing extrapyramidal motor locomotion. Maximal testing time was 60 s (n=5–7 from each group, * P<0.05 versus vehicle‐treated group).
PERK is a controversial target in the context of neurodegenerative diseases. Salubrinal [27] and sephin1 [28] showed neuroprotective effects in various mouse models of neurodegeneration including ALS, [28] Parkinson's disease, [29] and TBI [30] by stimulation of PERK signaling pathway. On the other hand, Walter et al. reported a bioactive small molecule‐integrated stress response inhibitor bisglycolamide (ISRIB)‐discovered by an ATF4‐based reporter gene assay as an inhibitor of PERK signaling, was efficacious in preventing rodent neurodegeneration. [31] These contradictory results regarding PERK signaling may be due to some unknown mechanisms underlying PERK activation and eIF2α phosphorylation, which has been suggested to induce non‐canonical translation of a specific group of mRNAs, that could be critical for cell survival. [21] SB1617, with its stress‐responsive PERK stimulation activity, can be used as a model compound for further mechanistic studies.
Conclusion
The convergent strategy of cell‐based phenotypic screening with label‐free target identification revealed that PDIA3 and DNAJC3 are attractive stress‐responsive targets for regulating proteostasis in tauopathies. A novel compound, SB1617, inhibited the activity of PDIA3 and DNAJC3, leading to conditional stimulations of PERK signaling pathway, and showed beneficial effects on mice with tauopathies. Thus, PDIA3 and DNAJC3 should be considered as novel targets in the development of new therapies for various proteinopathies to overcome pathological states in several neurodegenerative diseases with insufficient induction of innate ER stress. [32]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Acknowledgements
This work was supported by the National Creative Research Initiative Grant (2014R1A3A2030423) and the Bio & Medical Technology Development Program (2012M3A9C4048780) through the National Research Foundation of Korea (NRF) funded by the Korean Government (Ministry of Science & ICT). We thank Dr. Yun Kyung Kim, Korea Institute of Science and Technology (KIST) for kindly providing BiFC‐tau stable HEK293 human embryonic kidney cell line. Y.‐H.S. is grateful for the BK21 Postdoctoral Fellowship Program. J.H. is grateful for the Fostering Core Leaders of the Future Basic Science Program/Global Ph.D. Fellowship Program (NRF‐2016H1A2A1907084).
Y.-H. Shin, H. Cho, B. Y. Choi, J. Kim, J. Ha, S. W. Suh, S. B. Park, Angew. Chem. Int. Ed. 2021, 60, 1831.
References
- 1. Soto C., Nat. Rev. Neurosci. 2003, 4, 49–60. [DOI] [PubMed] [Google Scholar]
- 2. Grundke-Iqbal I., Iqbal K., Tung Y. C., Quinlan M., Wisniewski H. M., Binder L. I., Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Iqbal K., Liu F., Gong C. X., Nat. Rev. Neurol. 2016, 12, 15–27; [DOI] [PubMed] [Google Scholar]
- 3b. Congdon E. E., Sigurdsson E. M., Nat. Rev. Neurol. 2018, 14, 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hetz C., Saxena S., Nat. Rev. Neurol. 2017, 13, 477–491. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Walter P., Ron D., Science 2011, 334, 1081–1086; [DOI] [PubMed] [Google Scholar]
- 5b. Hetz C., Chevet E., Oakes S. A., Nat. Cell Biol. 2015, 17, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bocai N. I., Marcora M. S., Belfiori-Carrasco L. F., Morelli L., Castano E. M., J. Alzheimer′s Dis. 2019, 68, 439–458. [DOI] [PubMed] [Google Scholar]
- 7. Tak H., Haque M. M., Kim M. J., Lee J. H., Baik J. H., Kim Y., Kim D. J., Grailhe R., Kim Y. K., PloS One 2013, 8, e81682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Földi I., Tóth A. M., Szabó Z., Mózesa E., Berkecz R., Datki Z. L., Penke B., Janáky T., Neurochem. Int. 2013, 62, 58–69. [DOI] [PubMed] [Google Scholar]
- 9. Wang Y. P., Mandelkow E., Nat. Rev. Neurosci. 2016, 17, 5–21. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Kim J., Jung J., Koo J., Cho W., Lee W. S., Kim C., Park W., Park S. B., Nat. Commun. 2016, 7, 13196; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Kim J., Kim H., Park S. B., J. Am. Chem. Soc. 2014, 136, 14629–14638. [DOI] [PubMed] [Google Scholar]
- 11. Lasagna-Reeves C. A., de Haro M., Hao S., Park J., Rousseaux M. W. C., Al-Ramahi I., Jafar-Nejad P., Vilanova-Velez L., See L., De Maio A., et al., Neuron 2016, 92, 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Park H., Ha J., Koo J. Y., Park J., Park S. B., Chem. Sci. 2017, 8, 1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Park J., Koh M., Koo J. Y., Lee S., Park S. B., ACS Chem. Biol. 2016, 11, 44–52; [DOI] [PubMed] [Google Scholar]
- 13b. Park H., Koo J. Y., Srikanth Y. V., Oh S., Lee J., Park J., Park S. B., Chem. Commun. 2016, 52, 5828–5831. [DOI] [PubMed] [Google Scholar]
- 14. Kranz P., Neumann F., Wolf A., Classen F., Pompsch M., Ocklenburg T., Baumann J., Janke K., Baumann M., Goepelt K., Riffkin H., Metzen E., Brockmeier U., Cell Death Dis. 2017, 8, e2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hettinghouse A., Liu R. H., Liu C. J., Pharmacol. Ther. 2018, 181, 34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Castillo V., Onate M., Woehlbier U., Rozas P., Andreu C., Medinas D., Valdes P., Osorio F., Mercado G., Vidal R. L., Kerr B., Court F. A., Hetz C., PLoS One 2015, 10, e0136620; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Linz A., Knieper Y., Gronau T., Hansen U., Aszodi A., Garbi N., Hammerling G. J., Pap T., Bruckner P., Dreier R., J. Bone Miner. Res. 2015, 30, 1481–1493. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Sepulveda M., Rozas P., Hetz C., Medinas D. B., Prion 2016, 10, 50–56; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Torres M., Medinas D. B., Matamala J. M., Woehlbier U., Cornejo V. H., Solda T., Andreu C., Rozas P., Matus S., Munoz N., et al., J. Biol. Chem. 2015, 290, 23631–23645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Hoffstrom B. G., Kaplan A., Letso R., Schmid R. S., Turmel G. J., Lo D. C., Stockwell B. R., Nat. Chem. Biol. 2010, 6, 900–906; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Kaplan A., Gaschler M. M., Dunn D. E., Colligan R., Brown L. M., Palmer A. G., Lo D. C., Stockwell B. R., Proc. Natl. Acad. Sci. USA 2015, 112, E2245–E2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boriushkin E., Wang J. J., Zhang S. X., J. Ophthalmic Vis. Res. 2014, 9, 134–143. [PMC free article] [PubMed] [Google Scholar]
- 20. Yan W., Frank C. L., Korth M. J., Sopher B. L., Novoa I., Ron D., Katze M. G., Proc. Natl. Acad. Sci. USA 2002, 99, 15920–15925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Starck S. R., Tsai J. C., Chen K., Shodiya M., Wang L., Yahiro K., Martins-Green M., Shastri N., Walter P., Science 2016, 351, aad3867; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Cagnetta R., Wong H. H., Frese C. K., Mallucci G. R., Krijgsveld J., Holt C. E., Mol. Cell 2019, 73, 474–489,e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schmidt E. K., Clavarino G., Ceppi M., Pierre P., Nat. Methods 2009, 6, 275–277. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. B'Chir W., Maurin A. C., Carraro V., Averous J., Jousse C., Muranishi Y., Parry L., Stepien G., Fafournoux P., Bruhat A., Nucleic Acids Res. 2013, 41, 7683–7699; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Pitale P. M., Gorbatyuk O., Gorbatyuk M., Front. Cell Neurosci. 2017, 11, 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T., J. Biol. Chem. 2007, 282, 24131–24145. [DOI] [PubMed] [Google Scholar]
- 25. Edwards G., Moreno-Gonzalez I., Soto C., Biochem. Biophys. Res. Commun. 2017, 483, 1137–1142. [DOI] [PubMed] [Google Scholar]
- 26. Ramos-Cejudo J., Wisniewski T., Marmar C., Zetterberg H., Blennow K., de Leon M. J., Fossati S., EBioMedicine 2018, 28, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boyce M., Bryant K. F., Jousse C., Long K., Harding H. P., Scheuner D., Kaufman R. J., Ma D. W., Coen D. M., Ron D., Yuan J. Y., Science 2005, 307, 935–939. [DOI] [PubMed] [Google Scholar]
- 28. Das I., Krzyzosiak A., Schneider K., Wrabetz L., D'Antonio M., Barry N., Sigurdardottir A., Bertolotti A., Science 2015, 348, 239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Colla E., Coune P., Liu Y., Pletnikova O., Troncoso J. C., Iwatsubo T., Schneider B. L., Lee M. K., J. Neurosci. 2012, 32, 3306–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rubovitch V., Barak S., Rachmany L., Goldstein R. B., Zilberstein Y., Pick C. G., Neuromol. Med. 2015, 17, 58–70. [DOI] [PubMed] [Google Scholar]
- 31. Sidrauski C., Acosta-Alvear D., Khoutorsky A., Vedantham P., Hearn B. R., Li H., Gamache K., Gallagher C. M., Ang K. K. H., Wilson C., et al., eLife 2013, 2, e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.
- 32a. Blackwood E. A., Azizi K., Thuerauf D. J., Paxman R. J., Plate L., Kelly J. W., Wiseman R. L., Glembotski C. C., Nat. Commun. 2019, 10, 187; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32b. Plate L., Cooley C. B., Chen J. J., Paxman R. J., Gallagher C. M., Madoux F., Genereux J. C., Dobbs W., Garza D., Spicer T. P., et al., eLife 2016, 5, e15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary
Supplementary