

Abstract
The softness of nanohydrogels results in unique properties and recently attracted tremendous interest due to the multi‐functionalization of interfaces. Herein, we study extremely soft temperature‐sensitive ultra‐low cross‐linked (ULC) nanogels adsorbed to the solid/water interface by atomic force microscopy (AFM). The ultra‐soft nanogels seem to disappear in classical imaging modes since a sharp tip fully penetrates these porous networks with very low forces in the range of steric interactions (ca. 100 pN). However, the detailed evaluation of Force Volume mode measurements allows us to resolve their overall shape and at the same time their internal structure in all three dimensions. The nanogels exhibit an extraordinary disk‐like and entirely homogeneous but extremely soft structure—even softer than polymer brushes. Moreover, the temperature‐sensitive nanogels can be switched on demand between the ultra‐soft and a very stiff state.
Keywords: atomic force microscopy, soft matter, stiffness tomography, surface analysis, temperature-sensitive nanogels
Extremely soft temperature‐sensitive nanogels adsorbed to the solid/water interface were studied by atomic force microscopy. The ultra‐soft nanogels disappeared in the classical 2D height image. By detailed analysis of Force Volume data the extraordinary disk‐like and entirely homogeneous but extremely soft structure of the nanogels was resolved.
Introduction
Soft hydrogels, hydrogel layers and coatings of soft nanohydrogels play a major role in various fields as, e.g., tissue engineering, [1] cell adhesion, [2] antifouling,[ 3 , 4 , 5 ] sensors,[ 6 , 7 ] water purification, [8] self‐repairing and superolephobic materials, [9] or fertilizer. [10] Exemplary, soft water‐swollen hydrogels are widely used for 3D cell culture[ 11 , 12 ] as they mimic the mechanics of the extracellular matrix and lead stem cells to differentiate into distinct cell lines.[ 13 , 14 ] The hydrogels can further guide the cell growth direction [15] and nanohydrogel coatings are highly suitable for harvesting of cell‐sheets. [2] Besides their multi‐applicability in the biomedical field, nanogel coatings serve also as sensors in optics [6] or for pesticides, [7] and filter wastewater due to their porosity. [8] Moreover, nanogels coated on leaves increase the plant health as they permanently transport nutrients. [10]
In all these cases, the internal structure and related to that, the mechanical properties of the hydrogel network are relevant. However, determining the internal structure of soft hydrogel networks on length scales below 100 nm is extremely challenging.
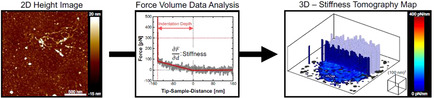
Generally, the structure can be probed by two distinct approaches: reciprocal‐ and real‐space techniques. Reciprocal‐space techniques as, e.g., ellipsometry [16] or grazing‐incidence small‐angle scattering [17] provide information like film thickness, surface roughness, or internal density distribution profiles, however, averaged over a rather large sample area and, thus, precluding information on local heterogeneities. Direct, real‐space imaging is achieved, e.g., by cryogenic electron microscopy, [18] which faces poor contrast in hydrogel samples. Super‐resolution fluorescence microscopy,[ 19 , 20 ] can be used to visualize heterogeneities on lengths scales <100 nm but the resolution in the z‐direction is still limited and requires suitable labeling. Atomic force microscopy (AFM) is a powerful tool beyond imaging. AFM relies on the sensing of forces and, thus, allows to investigate mechanical properties, that are related to the internal structure of the material.[ 21 , 22 , 23 , 24 ] Monitoring the interaction forces while scanning the sample results not only in a surface (two‐dimensional) characterization.[ 25 , 26 ] Force Volume measurements allow determining the stiffness as a function of the three‐dimensional position. [27] The so‐called stiffness tomography was used to resolve the internal structure of soft objects such as cells, [28] bacteria, [29] or hydrogels. [30]
Recent progress in synthesis enabled preparation nanogels with internal compartments and various shapes.[ 31 , 32 , 33 , 34 , 35 ] Furthermore, is was observed that the extreme softness of ultra‐low cross‐linked (ULC) nanogels leads to unique properties. [36] ULC nanogels exhibit high swellability and flexibility.[ 37 , 38 ] Their extreme softness results in even more complex phase behavior when compared to conventionally cross‐linked nanogels and pronounced polymer‐hard colloid duality.[ 39 , 40 ] On one hand, the ULC nanogels show a three‐dimensional behavior comparable to colloids but interacting with soft potentials while at the liquid/liquid interface they resemble more flexible polymers. [40] On the other hand, at the solid/liquid interface both colloidal and polymeric behavior can be observed dependently on the adsorption process. [41] It is their extreme softness that makes them appealing for advanced applications as synthetic platelet particles, [42] and the enhancement of the invasion of cells, [43] or of the catalytic activity of compartments. [44]
In this study, we reveal how to gain structural information with AFM beyond the surface for temperature‐sensitive, ultra‐soft nanogels. Force Volume mode measurements were performed for two kinds of N‐isopropylacrylamide (NIPAM) based nanogels: (i) conventional, with a more cross‐linked core and a fuzzy shell, and (ii) ultra‐low cross‐linked nanogels. A sharp AFM tip is used to penetrate the porous nanogel network such that the local contact resistance is monitored. The ULC nanogels are so soft that even the minimal force of 100 pN leads to penetration through the entire nanogel, which greatly impedes imaging. We demonstrate that detailed analysis of the Force Volume measurements allows to receive on the one hand topographic maps and other hand provide additional information on the three‐dimensional internal structure of the ultra‐soft nanogels. This information on such soft objects is hardly or not obtainable by any other technique.
Results and Discussion
Nanogels based on N‐isopropylacrylamide (NIPAM) as the main monomer show a fast response to temperature. Their volume phase transition temperature (VPTT) is at 32 °C in water. [45] Below the VPTT, the nanogels are swollen but collapse upon heating above the VPTT, i.e., they expel water and reduce in size.[ 46 , 47 ] In this study, we compare two types of poly(N‐isopropylacrylamide) (PNIPAM) nanogels exhibiting a different amount of incorporated cross‐linker: (i) ultra‐low cross‐linked (ULC, synthesized without cross‐linker), [36] and (ii) nanogels containing 5 mol % of the cross‐linking agent N,N′‐methylenebisacrylamide (BIS), hereinafter denoted as conventional nanogels.
The bulk solution properties of these ULC and conventional nanogels were investigated in detail by various scattering techniques (DLS, SLS, and SANS) previously [41] and detailed structural information are reported in the Supporting Information (Figure S2). In the collapsed state (T>VPTT), both nanogels have a similar hydrodynamic radius. The nanogels swell upon cooling below the VPTT, however, to a very different extent: While the volume of the conventional nanogels increases by a factor of 5.7, the ULC nanogels swell by the factor 35.
To reveal the behavior of the nanogels when being adsorbed to a solid support, atomic force microscopy (AFM) is employed to study the morphology at the solid/liquid interface. However, the imaging of soft structures is rather challenging. Especially the applied force during imaging needs to be considered with care, since such soft objects can be easily deformed by the mechanical probe [48] —as previously observed for core–shell and hollow microgels. [49]
In this manner, the very soft ULC nanogels were adsorbed in situ at T=27 °C onto a PAH‐coated glass substrate and investigated at various temperatures at the solid/water interface. Topographic images were taken in the Peak Force tapping mode applying a minimum force to obtain a stable image (SP=300/500 pN, Figure 1). The same area is imaged at all temperatures recognizable by the contamination of the interface with a fiber (Figure 1 F, marked in black).
Figure 1.
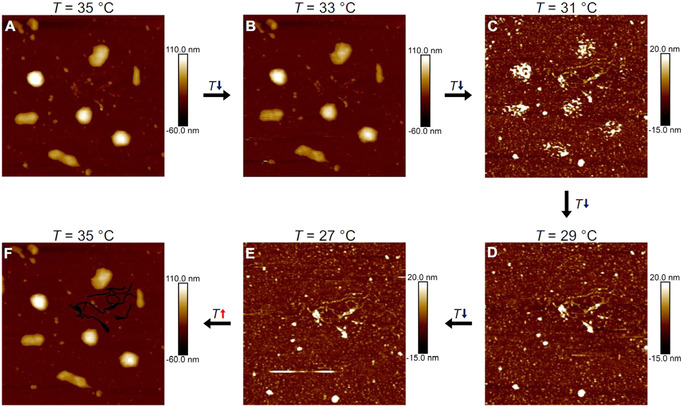
AFM height images (2.5×2.5 μm2) of ULC nanogels in situ adsorbed on a PAH‐coated glass substrate in the hydrated state at various temperatures (above and below the VPTT). The same position is imaged at the different temperatures as identified by the dirt contamination (F, marked in black). The images were obtained by applying a force of SP=500 pN resp. 300 pN (in collapsed resp. swollen state) in Peak Force tapping mode. The temperature was varied in 2 °C‐steps (A–E) starting at T=35 °C (A), decreasing until a temperature of T=27 °C is reached (E) and afterwards heating again to T=35 °C (F).
Figure 1 A shows the height image of the ULC nanogels in the collapsed state at T=35 °C. Two distinct structures at the interface can be identified: broken and intact nanogels. Some nanogels break apart due to the adsorption‐induced deformation of the nanogels, highlighting again the extreme softness of the ULC nanogels. [41] Those who stay intact, exhibit half‐spherical shapes. Lowering the temperature to T=33 °C (Figure 1 B) does not affect the nanogels’ morphology. However, a strong change can be observed when further decreasing the temperature below the VPTT. The lateral dimensions slightly increase, and it is accompanied by a transition of the shape from the smooth hemispherical shape into inhomogeneous and rough objects at T=31 °C (Figure 1 C). Significantly below the VPTT (Figures 1 D, E), the nanogels are not visible anymore and only the substrate can be imaged at the nanogels’ positions. However, heating above the VPTT lets the nanogels appear again with the same structure as before the cooling (Figure 1 F). This indicates that the nanogels do not desorb, but that they get “invisible” below the VPTT.
In contrast, conventional nanogels are still observable in the swollen state (Figure S3B). This is in agreement with the Peak Force tapping mode measurements of core–shell and hollow microgels. [49] Incomprehensively, the ULC nanogels are not mappable below the VPTT, even though the applied force was kept to a minimum. Therefore, force‐spectroscopic measurements were performed across the entire sample to disclose this special case. Force Volume mode images were taken (at the identical position as in Figure 1) at T=27 °C (Figure 2).
Figure 2.
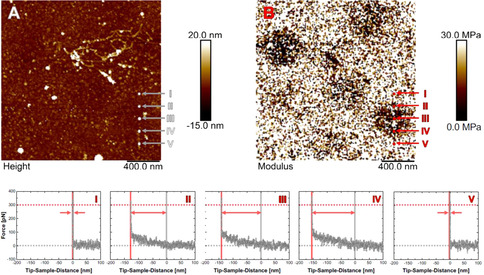
AFM height (A) and the corresponding modulus (B) image of ULC nanogels in situ adsorbed onto a PAH‐coated glass substrate in water at T=27 °C at the same position as in Figure 1 E. Images were obtained from Force Volume measurements. I‐V: Force‐distance curves at the five positions as indicated in the images (A and B). The curves were first‐order baseline corrected and shifted by the contact point. The applied force for imaging of F=300 pN (force threshold) is marked as well as the contact point and the tip‐sample‐distance, at which the force threshold is reached. The difference in distances is visualized by an arrow and corresponds to the indentation depth into the nanogel, by which the visualized height has to be corrected.
The Force Volume mode measurements enable to obtain force‐distance curves on the whole 2.0×2.0 μm2 scan with a resolution of 192×192 pixels. Even though no nanogels are detectable in the height image (Figure 2 A), areas of dark contrast are visible in the corresponding E‐modulus image (Figure 2 B). These dark areas, therefore, allow the localization of the swollen ULC nanogels. For one of the intact ULC nanogels, a series of force‐distance curves across the nanogel is shown as example (Figures 2 I–V). The spots, where the five curves were taken, are marked in the height and modulus image.
Curves I and V were obtained outside the dark contrast in the modulus image. For positive tip‐sample‐distances the force is F=0 pN with a noise level of ca. 40 pN. After the contact point, i.e., for “negative” tip‐sample‐distances, the force increases steeply. This steep increase results from the contact of the tip with the solid substrate and is the reason for the light contrast in the modulus image (high modulus value). In contrast, curves II–IV are measured at positions with a dark contrast. At those positions, the part of the curve, where tip and sample are in contact, can be divided into two regions: (i) a shallow, but monotonic increase directly after the contact point with expansion over ca. 150 nm, and (ii) a rapid increase as observed on the bare substrate. The force signal does not exceed F≈100 pN before the second region is reached.
For imaging, the minimum force of F=300 pN was applied to obtain a stable image. The shallow increase of the force in the curves II‐IV shows that the AFM senses something different than the bare substrate before the threshold force value is reached. At F=300 pN the force increases steeply, i.e., the tip already probes the underlying solid substrate. The real contact point between the tip and the sample, however, is before the first slight increase of the force, at F=0 pN. The height information has to be corrected to the real point of contact to obtain a height map and, thus, resolve the entire sample. The difference in traveling‐distance of the tip between the contact point (F=0 pN) and the distance at which the force threshold is reached is the height information that is missed during imaging (visualized by arrows in Figures 2 I–V). This value corresponds to the indentation depth resulting from the application of the imaging force. On the bare glass substrate the indentation depth is zero, but towards the center of the nanogel (Curve III, as extracted from the modulus image) it is increasing up to ca. 150 nm. Using these depths of indentation, the height information has to be corrected in every pixel to obtain the real morphology of the swollen ULC nanogels.
Figure 3 A shows the height image of the ULC nanogels at T=27 °C after this correction. The ULC nanogels are now clearly observable even in the swollen state. Three intact ULC nanogels at the same positions as in the collapsed state are distinguishable from the flat background. The nanogels exhibit a maximum height of ca. 150 nm, which surprisingly is constant of the entire lateral dimensions, i.e., the nanogels take a “disk‐like” shape (Figure 3 B).
Figure 3.
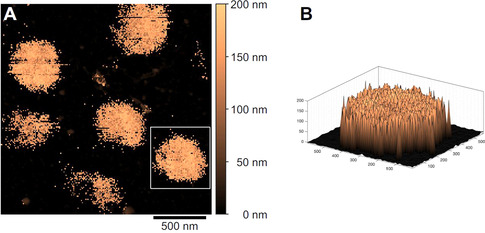
Corrected height image of ULC nanogels in situ adsorbed onto a PAH‐coated glass substrate in water at T=27 °C (A). The height information is corrected by the indentation depth in every pixel due to the application of a force of F=300 pN for imaging via the Force Volume mode. A three‐dimensional height map (x, y, and z in nanometer) of a single nanogel is given (B, the white rectangle marks the corresponding section in the 2D image).
Force Volume mode measurements can be used to extract information beyond the topography of the nanogels since the force‐distance curves allow the determination of the mechanical properties inside the nanogels.[ 4 , 22 , 50 ] As shown previously, [49] a sharp tip mainly penetrates the porous nanogel network instead of compressing it. During the vertical movement of the probe towards the solid substrate, a local resistance is monitored by the probe to enter the nanogel network. This local resistance corresponds to the slope of the force‐distance curve and is a contact stiffness, i.e., a measure of the network density in vertical direction. [49] Representative force‐distance curves obtained in the center of the ULC and conventional nanogels below (T=27 °C) and above (T=35 °C) the VPTT are shown in Figure 4.
Figure 4.
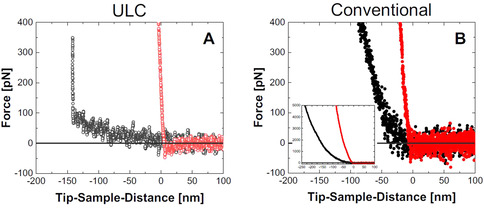
Force‐distance curves for the vertical approach of the AFM tip toward the solid substrate at the center of a ULC (A, open circles) and conventional nanogel (B, filled circles) at the solid/liquid interface at T=27 °C (gray/black) and T=35 °C (light red/red). Inset in B: Zoom‐out graph to visualize the full force scale investigated for the conventional nanogels.
In the collapsed state, the nanogels are rather stiff objects and the tip cannot penetrate the collapsed ULC and conventional nanogels as shown by the steep force‐distance curves in Figure 4 A (light red open circles) and B (red filled circles). Very different behavior is observed in the swollen state.
As discussed above, for the ULC nanogels in the swollen state (Figure 4 A, gray open circles) two regions of the force‐distance curve in the contact regime can be identified: (i) a shallow, and (ii) a steep increase in force with increasing indentation. Obviously, the sharp tip penetrates the entire ULC nanogel even with a minimal force of less than 100 pN and then reaches the underlying solid support. It means, that information about the internal network density can be obtained for the entire ULC nanogel. Despite the low signal‐to‐noise ratio, one can recognize the very small slope of the curve. The slope is nearly constant, i.e., the nanogel network is extremely soft without any gradient perpendicular to the substrate (see also the extracted contact stiffness vs. indentation depth curve in Figure S5C).
The force‐distance curve of the conventional nanogels in the swollen state (Figure 4 B, black filled circles) is very different as compared to the ULC nanogels. Firstly, the slope of the curve increases with indentation (see also the contact stiffness vs. indentation depth curve, Figure S5D) which indicates a heterogeneous internal structure perpendicular to the substrate. Secondly, even with a force of F=5.0 nN and, therefore, a significant indentation depth of Δd≈230 nm (see inset in Figure 4 B), the underlying substrate is not reached, i.e., the nanogel cannot be penetrated entirely which is distinctly different from the fully penetrable ultra‐soft ULC nanogels.
Until now we have focused on the vertical variation in contact stiffness at one position, namely the nanogel center. However, obtaining information in all three dimensions is of outstanding interest as the structure of adsorbed nanogels might be inhomogeneous in the horizontal direction parallel to the substrate.[ 4 , 24 , 50 , 51 , 52 ] Therefore, we measured force‐distance curves in a 192×192 grid across a 2.0×2.0 μm2 scan and extracted the contact stiffness across the volume of the entire nanogel. The huge amount of information gathered from multiple force‐spectroscopy measurements can be easily grasped by the visualization in a map comparable to the so‐called stiffness tomography[ 27 , 28 , 30 ] (for details regarding the processing of the Force Volume data refer to Section “Force Volume data analysis” of the Experimental Section).
Figure 5 shows cut‐offs of the extracted contact stiffness maps as obtained for the ULC (A) and conventional nanogels (B) in the swollen state. The corresponding maps in the collapsed state can be found in the Supporting Information (Figure S6).
Figure 5.
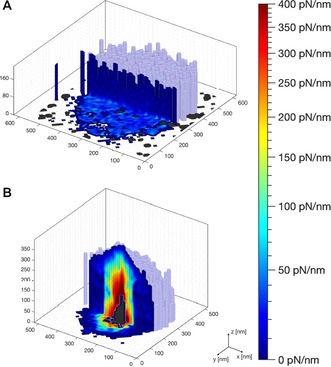
Cut‐offs of the contact stiffness maps of individual ULC (A) and conventional (B) nanogels at the solid/liquid interface at T=27 °C. The x, y, and z‐axes describe the topographic information (light blue surface extracted for contact stiffness equals 0 pN nm−1), while the color code within the cut‐off planes signifies the local contact stiffness value. The dark gray color reflects the area which cannot be accessed by the AFM tip.
Both, topography and internal structure, are visualized in that one map. While x, y, and z map the topographic information (light blue surface extracted for contact stiffness equals zero), the additional information—beyond the sample surface—is captured by a color code within the cut‐off plane (Figure 5). The color indicates the contact stiffness: Regions of low contact stiffness are colored in blue, whereas regions of higher stiffness are marked in red.
The conventional nanogel reveals a strongly heterogeneous internal structure (Figure 5 B). The color within the hemiellipsoidally shaped nanogel changes in lateral as well as in vertical directions. The contact stiffness increases from ca. 10 pN nm−1 at the periphery up to 400 pN nm−1 towards the nanogel center. The very center of the conventional nanogel in proximity to the surface cannot be penetrated by the sharp tip (dark gray area). The increase in stiffness towards the nanogel center agrees with what is known about the structure in bulk solution: the faster incorporation of the cross‐linker BIS in comparison to the monomer during the nanogel co‐polymerization reaction [46] leads to a heterogeneous structure with a polymer density that is high in the center and fades out towards the periphery and finally into dangling polymer chains (see Figure S2D).
In contrast, the swollen ULC nanogel reveals a completely homogeneous contact stiffness map (Figure 5 A). The contact stiffness inside the ULC nanogels is ca. 10 pN nm−1 and, thus, ca. 40 times smaller as compared to the conventional nanogels. Furthermore, this ultra‐low value is found within the entire nanogel, i.e., in both vertical and horizontal directions. In bulk solution, the ULC nanogels have low polymer density, which is homogeneous of the entire volume (see Figure S2E). In contrast to the conventional nanogels, ULC nanogels are synthesized in a homo‐polymerization of NIPAM, i.e., in the absence of the cross‐linking agent BIS. During this homo‐polymerization, a chemically cross‐linked network forms due to self‐cross‐linking of the PNIPAM polymer chains resulting from hydrogen atom abstraction reactions (preferably at the tertiary carbon atom of the isopropyl group). [53] Throughout the polymer chains the monomer repeating units are identical and, thus, the self‐cross‐linking can happen at every position with the same reactivity. This leads to a considerably more homogeneous distribution of cross‐links and, therefore, network density. Figure 5 A demonstrates that the internal structure of the ULC nanogels remains homogeneous also at the solid interface despite the fact that the nanogel is strongly deformed into a disk‐like shape. Remarkably, upon an increase in temperature, their spherical character is recapped since they reversibly collapse into stiff hemispheres (see Figure S6A and the 3D height map, Figure S4A). Previous studies of ULC nanogels at the solid/liquid interface by AFM are rare, and the temperature‐induced changes have not been investigated before. Densely packed monolayers of ULC nanogels were imaged,[ 37 , 54 ] however, it has been shown, that the interfacial properties of soft ULC nanogels are strongly affected by the two‐dimensional packing density. [40] Therefore, properties of individual microgels are hardly accessible from densely packed nanogel layers.
The very low stiffness that we find for the ULC nanogel at the surface as well as in the interior is the same as for the outermost region of the conventional nanogel. In other words, the ULC nanogels are entirely extremely soft—as soft as the dangling chains of the fuzzy shell of their regularly cross‐linked counterpart.
Interestingly, the force insertion profile of the ULC nanogels is also clearly different from the ones observed for polymer brushes—systems without any cross‐links. An exponential increase in force is typically observed for sharp tips penetrating a polymer brush, regardless of the density of grafting.[ 55 , 56 , 57 , 58 , 59 ] At high grafting densities, the linear chains are stretched into the typical brush structure, and the polymer segment density is expected to be much higher in comparison to the ULC nanogels, i.e., the response is more comparable to the conventional nanogels. At low grafting densities, where conformational freedom is given and so‐called “mushrooms” are formed, the resistance to penetration is still increasing exponentially,[ 58 , 60 ] being in clear contrast to the ultra‐soft ULC nanogels in the swollen state studied here. It suggests, that the loosely cross‐linked polymer network of the ULC nanogels is even less dense than individually grafted polymer chains in mushroom conformation. However, it has to be mentioned that the brushes were probed with less sharp tips (R probe≥10 nm) than the ones used for this study.
Conclusion
We introduced a method to resolve the internal structure of ultra‐soft materials, namely ultra‐low cross‐linked (ULC) nanogels, via atomic force microscopy (AFM). Imaging soft, heterogeneous structures such as supported lipid bilayers, hydrogels or cells is challenging since it is always connected to the application of forces which result in deformation. For the extremely soft ULC nanogels, the edge of the resolution limit is reached: Minimal forces corresponding to steric interactions lead the sharp tip to penetrate the entire polymeric network so that the nanogels disappear in classical height images. However, during the penetration a local contact resistance is monitored and correlates closely to the density profiles of the nanogels in bulk solution, i.e., the internal polymer density distribution orthogonal to the interface is resolved. Force Volume mode measurements result in three‐dimensional maps of the network density referred to as stiffness tomography: Topography and internal network density can be extracted simultaneously.
Here, we resolved the entirely homogeneous, ultra‐soft, and extraordinary disk‐like structure of ULC nanogels at the solid/water interface. In contrast, the hemiellipsoidally shaped conventional nanogels reveal a strongly heterogeneous and stiffer interior. The ULC nanogels are even more penetrable than polymer brushes, highlighting their extremely soft character. In addition, the nanogels are temperature‐sensitive and can be vigorously switched in stiffness by slight changes in temperature.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge helpful discussions with Prof. Laura De Laporte. The authors acknowledge the Deutsche Forschungsgemeinschaft (DFG) for financial support within the Sonderforschungsbereich SFB 985 “Functional Microgels and Microgel Systems” (Projects A3 and B8). Open access funding enabled and organized by Projekt DEAL.
M. F. Schulte, S. Bochenek, M. Brugnoni, A. Scotti, A. Mourran, W. Richtering, Angew. Chem. Int. Ed. 2021, 60, 2280.
References
- 1. Rose J. C., De Laporte L., Adv. Healthcare Mater. 2018, 7, 1701067. [Google Scholar]
- 2. Kim H., Witt H., Oswald T. A., Tarantola M., ACS Appl. Mater. Interfaces 2020, 12, 33516–33529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. South A. B., Whitmire R. E., Garcia A. J., Lyon L. A., ACS Appl. Mater. Interfaces 2009, 1, 2747–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schmidt S., Zeiser M., Hellweg T., Duschl C., Fery A., Möhwald H., Adv. Funct. Mater. 2010, 20, 3235–3243. [Google Scholar]
- 5. Keskin D., Mergel O., van der Mei H. C., Busscher H. J., van Rijn P., Biomacromolecules 2019, 20, 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Islam M. R., Ahiabu A., Li X., Serpe M. J., Sensors 2014, 14, 8984–8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sigolaeva L. V., Gladyr S. Y., Mergel O., Gelissen A. P. H., Noyong M., Simon U., Pergushov D. V., Kurochkin I. N., Plamper F. A., Richtering W., Anal. Chem. 2017, 89, 6091–6098. [DOI] [PubMed] [Google Scholar]
- 8. Tripathi B. P., Dubey N. C., Stamm M., ACS Appl. Mater. Interfaces 2014, 6, 17702–17712. [DOI] [PubMed] [Google Scholar]
- 9. Chen K., Zhou S., Wu L., ACS Nano 2016, 10, 1386–1394. [DOI] [PubMed] [Google Scholar]
- 10. Meurer R. A., Kemper S., Knopp S., Eichert T., Jakob F., Goldbach H. E., Schwaneberg U., Pich A., Angew. Chem. Int. Ed. 2017, 56, 7380–7386; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7486–7492. [Google Scholar]
- 11. Tibbitt M. W., Anseth K. S., Biotechnol. Bioeng. 2009, 103, 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Caliari S. R., Burdick J. A., Nat. Methods 2016, 13, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Discher D. E., Janmey P., Wang Y.-L., Science 2005, 310, 1139–1143. [DOI] [PubMed] [Google Scholar]
- 14. Wen J. H., Vincent L. G., Fuhrmann A., Choi Y. S., Hribar K. C., Taylor-Weiner H., Chen S., Engler A. J., Nat. Mater. 2014, 13, 979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rose J. C., Camara-Torres M., Rahimi K., Kohler J., Moller M., De Laporte L., Nano Lett. 2017, 17, 3782–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maestro A., Jones D., Sanchez de Rojas Candela C., Guzman E., Duits M. H. G., Cicuta P., Langmuir 2018, 34, 7067–7076. [DOI] [PubMed] [Google Scholar]
- 17. Kyrey T., Ganeva M., Gawlitza K., Witte J., von Klitzing R., Soltwedel O., Di Z., Wellert S., Holderer O., Phys. B 2018, 551, 172–178. [Google Scholar]
- 18. Gelissen A. P. H., Oppermann A., Caumanns T., Hebbeker P., Turnhoff S. K., Tiwari R., Eisold S., Simon U., Lu Y., Mayer J., Richtering W., Walther A., Wöll D., Nano Lett. 2016, 16, 7295–7301. [DOI] [PubMed] [Google Scholar]
- 19. Siemes E., Nevskyi O., Sysoiev D., Turnhoff S. K., Oppermann A., Huhn T., Richtering W., Wöll D., Angew. Chem. Int. Ed. 2018, 57, 12280–12284; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12460–12464. [Google Scholar]
- 20. Karanastasis A. A., Zhang Y., Kenath G. S., Lessard M. D., Bewersdorf J., Ullal C. K., Mater. Horiz. 2018, 5, 1130–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Puntheeranurak T., Neundlinger I., Kinne R. K., Hinterdorfer P., Nat. Protoc. 2011, 6, 1443–1452. [DOI] [PubMed] [Google Scholar]
- 22. Chyasnavichyus M., Young S. L., Tsukruk V. V., Polymer 2014, 55, 6091–6101. [Google Scholar]
- 23. Kadem L. F., Suana K. G., Holz M., Wang W., Westerhaus H., Herges R., Selhuber-Unkel C., Angew. Chem. Int. Ed. 2017, 56, 225–229; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 231–235. [Google Scholar]
- 24. Nishizawa Y., Matsui S., Urayama K., Kureha T., Shibayama M., Uchihashi T., Suzuki D., Angew. Chem. Int. Ed. 2019, 58, 8809–8813; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8901–8905. [Google Scholar]
- 25. Spitzner E.-C., Riesch C., Magerle R., ACS Nano 2011, 5, 315–320. [DOI] [PubMed] [Google Scholar]
- 26. Ebeling D., Eslami B., De Jesus Solares S., ACS Nano 2013, 7, 10387–10396. [DOI] [PubMed] [Google Scholar]
- 27. Roduit C., Sekatski S., Dietler G., Catsicas S., Lafont F., Kasas S., Biophys. J. 2009, 97, 674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Roduit C., Saha B., Alonso-Sarduy L., Volterra A., Dietler G., Kasas S., Nat. Methods 2012, 9, 774–775. [DOI] [PubMed] [Google Scholar]
- 29. Longo G., Rio L. M., Roduit C., Trampuz A., Bizzini A., Dietler G., Kasas S., J. Mol. Recognit. 2012, 25, 278–284. [DOI] [PubMed] [Google Scholar]
- 30. Galluzzi M., Tang G., Biswas C. S., Zhao J., Chen S., Stadler F., Nat. Commun. 2018, 9, 3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li F., Wang C., Guo W., Adv. Funct. Mater. 2018, 28, 1705876. [Google Scholar]
- 32. Nickel A. C., Scotti A., Houston J. E., Ito T., Crassous J., Pedersen J. S., Richtering W., Nano Lett. 2019, 19, 8161–8170. [DOI] [PubMed] [Google Scholar]
- 33. Krüger A. J. D., Bakirman O., Guerzoni L. P. B., Jans A., Gehlen D. B., Rommel D., Haraszti T., Kuehne A. J. C., De Laporte L., Adv. Mater. 2019, 31, 1903668. [DOI] [PubMed] [Google Scholar]
- 34. Xu W., Rudov A., Oppermann A., Wypysek S., Kather M., Schroeder R., Richtering W., Potemkin I., Woll D., Pich A., Angew. Chem. Int. Ed. 2020, 59, 1248–1255; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1264–1271. [Google Scholar]
- 35. Schmitt J., Hartwig C., Crassous J. J., Mihut A. M., Schurtenberger P., Alfredsson V., RSC Adv. 2020, 10, 25393–25401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gao J., Frisken B. J., Langmuir 2003, 19, 5212–5216. [Google Scholar]
- 37. Bachman H., Brown A. C., Clarke K. C., Dhada K. S., Douglas A., Hansen C. E., Herman E., Hyatt J. S., Kodlekere P., Meng Z., Saxena S., Spears J., Welsch N., Lyon L. A., Soft Matter 2015, 11, 2018–2028. [DOI] [PubMed] [Google Scholar]
- 38. Hu X., Lyon L. A., ACS Macro Lett. 2015, 4, 302–307. [DOI] [PubMed] [Google Scholar]
- 39. Conley G. M., Zhang C., Aebischer P., Harden J. L., Scheffold F., Nat. Commun. 2019, 10, 2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scotti A., Bochenek S., Brugnoni M., Fernandez-Rodriguez M. A., Schulte M. F., Houston J. E., Gelissen A. P. H., Potemkin I. I., Isa L., Richtering W., Nat. Commun. 2019, 10, 1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schulte M. F., Scotti A., Brugnoni M., Bochenek S., Mourran A., Richtering W., Langmuir 2019, 35, 14769–14781. [DOI] [PubMed] [Google Scholar]
- 42. Brown A. C., Stabenfeldt S. E., Ahn B., Hannan R. T., Dhada K. S., Herman E. S., Stefanelli V., Guzzetta N., Alexeev A., Lam W. A., Lyon L. A., Barker T. H., Nat. Mater. 2014, 13, 1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Douglas A. M., Fragkopoulos A. A., Gaines M. K., Lyon L. A., Fernandez-Nieves A., Barker T. H., Proc. Natl. Acad. Sci. USA 2017, 114, 885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang J., Liu Y., Li X., Luo Y., Zheng L., Hu J., Chen G., Chen H., Macromol. Rapid Commun. 2020, 41, 2000135. [DOI] [PubMed] [Google Scholar]
- 45. Pelton R., Adv. Colloid Interface Sci. 2000, 85, 1–33. [DOI] [PubMed] [Google Scholar]
- 46. Stieger M., Richtering W., Pedersen J. S., Lindner P., J. Chem. Phys. 2004, 120, 6197–6206. [DOI] [PubMed] [Google Scholar]
- 47. Keidel R., Ghavami A., Lugo D. M., Lotze G., Virtanen O., Beumers P., Pedersen J. S., Bardow A., Winkler R. G., Richtering W., Sci. Adv. 2018, 4, eaao7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aufderhorst-Roberts A., Baker D., Foster R. J., Cayre O., Mattsson J., Connell S. D., Nanoscale 2018, 10, 16050–16061. [DOI] [PubMed] [Google Scholar]
- 49. Schulte M. F., Scotti A., Gelissen A. P. H., Richtering W., Mourran A., Langmuir 2018, 34, 4150–4158. [DOI] [PubMed] [Google Scholar]
- 50. Burmistrova A., Richter M., Üzüm C., Klitzing R. v., Colloid Polym. Sci. 2011, 289, 613–624. [Google Scholar]
- 51. Camerin F., Fernandez-Rodriguez M. A., Rovigatti L., Antonopoulou M.-N., Gnan N., Ninarello A., Isa L., Zaccarelli E., ACS Nano 2019, 13, 4548–4559. [DOI] [PubMed] [Google Scholar]
- 52. Deshmukh O. S., Maestro A., Duits M. H. G., van den Ende D., Stuart M. C., Mugele F., Soft Matter 2014, 10, 7045–7050. [DOI] [PubMed] [Google Scholar]
- 53. Brugnoni M., Nickel A. C., Kröger L. C., Scotti A., Pich A., Leonhard K., Richtering W., Polym. Chem. 2019, 10, 2397–2405. [Google Scholar]
- 54. Welsch N., Lyon L. A., PLoS One 2017, 12, e0181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. O'Shea S. J., Welland M. E., Rayment T., Langmuir 1993, 9, 1826–1835. [Google Scholar]
- 56. Kelley T. W., Schorr P. A., Johnson K. D., Tirrell M., Frisbie C. D., Macromolecules 1998, 31, 4297–4300. [Google Scholar]
- 57. Butt H.-J., Kappl M., Mueller H., Raiteri R., Meyer W., Rühe J., Langmuir 1999, 15, 2559–2565. [Google Scholar]
- 58. Nnebe I. M., Schneider J. W., Macromolecules 2006, 39, 3616–3621. [Google Scholar]
- 59. Mendez S., Andrzejewski B. P., Canavan H. E., Keller D. J., McCoy J. D., Lopez G. P., Curro J. G., Langmuir 2009, 25, 10624–10632. [DOI] [PubMed] [Google Scholar]
- 60. Israelachvili J. N., Intramolecular and Surface Forces , 3rd ed., Elsevier Inc., Waltham, San Diego, Oxford, Amsterdam, 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary