

Abstract
Objective
To evaluate the efficacy and safety of 2 canakinumab monotherapy tapering regimens in order to maintain complete clinical remission in children with systemic juvenile idiopathic arthritis (JIA).
Methods
The study was designed as a 2‐part phase IIIb/IV open‐label, randomized trial. In the first part, patients received 4 mg/kg of canakinumab subcutaneously every 4 weeks and discontinued glucocorticoids and/or methotrexate as appropriate. Patients in whom clinical remission was achieved (inactive disease for at least 24 weeks) with canakinumab monotherapy were entered into the second part of the trial, in which they were randomized 1:1 into 1 of 2 treatment arms. In arm 1, the dose of canakinumab was reduced from 4 mg/kg to 2 mg/kg and then to 1 mg/kg, followed by discontinuation. In arm 2, the 4 mg/kg dose interval was prolonged from every 4 weeks, to every 8 weeks, and then to every 12 weeks, followed by discontinuation. In both arms, canakinumab exposure could be reduced provided systemic JIA remained in clinical remission for 24 weeks with each step. The primary objective was to assess whether >40% of randomized patients in either arm maintained clinical remission of systemic JIA for 24 weeks in the first part of the study.
Results
In part 1 of the study, 182 patients were enrolled, with 75 of those patients randomized before entering part 2 of the trial. Among the 75 randomized patients, clinical remission was maintained for 24 weeks in 27 (71%) of 38 patients in arm 1 (2 mg/kg every 4 weeks) and 31 (84%) of 37 patients in arm 2 (4 mg/kg every 8 weeks) (P ≤ 0.0001 for arm 1 versus arm 2 among those meeting the 40% threshold). Overall, 25 (33%) of 75 patients discontinued canakinumab, and clinical remission was maintained for at least 24 weeks in all 25 of these patients. No new safety signals were identified.
Conclusion
Reduction of canakinumab exposure may be feasible in patients who have achieved clinical remission of systemic JIA, but consistent interleukin‐1 inhibition appears necessary to maintain this response.
INTRODUCTION
Systemic juvenile idiopathic arthritis (JIA), the most severe category of JIA, is characterized by systemic inflammation and arthritis. Systemic inflammation is manifested through spiking quotidian fever, maculopapular rash, hepatosplenomegaly, lymphadenopathy, serositis, and elevated serum levels of inflammation markers such as C‐reactive protein (CRP), erythrocyte sedimentation rate, and ferritin (1, 2, 3, 4, 5). Systemic JIA is a systemic autoinflammatory disease, and the innate immune system plays a prominent role in its pathophysiology (6, 7).
Currently available therapeutic agents for systemic JIA include nonsteroidal antiinflammatory drugs (NSAIDs), glucocorticoids, and disease‐modifying antirheumatic drugs (DMARDs), including synthetic DMARDs and biologic DMARDs targeting interleukin‐1 (IL‐1) and IL‐6 (7, 8, 9, 10, 11, 12, 13, 14). Tumor necrosis factor (TNF) blockers can also be used, but their efficacy is generally lower in systemic JIA compared to other forms of JIA (15, 16). The goal of systemic JIA treatment is to achieve and maintain complete clinical remission in order to avoid complications and improve long‐term outcomes (17, 18). Therapeutic strategies aiming to minimize exposure to systemic glucocorticoids, and ideally, discontinue them, are of great importance to prevent severe and longstanding side effects associated with the long‐term use of these drugs, particularly growth inhibition and obesity (19, 20). In recent years, the paradigm of explicitly defining a treatment target and applying tight control and necessary adjustment of therapy to reach it has been incorporated into “treat‐to‐target” recommendations for several rheumatic diseases, including JIA (17, 18). Treat‐to‐target strategies may include the reduction of exposure to DMARDs, or even discontinuation, for patients with systemic JIA in clinical remission; however, very limited scientific evidence is currently available on the effects of tapering synthetic or biologic DMARDs in patients with systemic JIA.
IL‐1 plays a major role in the pathogenesis of systemic JIA (7), and blockade of IL‐1 can provide effective therapeutic control of disease activity (8, 9, 10, 11, 14, 21). Canakinumab, a human anti–IL‐1β monoclonal antibody, has shown sustained efficacy in treating systemic JIA patients and allowing the tapering of glucocorticoids (8, 10). In a pivotal phase III study of canakinumab in patients with active systemic JIA, the average glucocorticoid dose could be reduced from 0.34 mg/kg to 0.05 mg/kg in patients treated with canakinumab, with 42 (33%) of 128 patients discontinuing glucocorticoids (10). However, limited information is available on the effects of reducing exposure to, or even discontinuation of, canakinumab in patients with systemic JIA in clinical remission. In a long‐term extension study, tapering of canakinumab to a reduced dose of 2 mg/kg every 4 weeks was attempted, with 44 (25%) of 177 patients receiving at least 3 reduced doses over a median follow‐up of 25 months (8). The dose reduction in these 44 patients was maintained until study end in 26 (59%) of the patients. Therefore, in this study, we evaluated the efficacy and safety of 2 canakinumab monotherapy tapering schemes in patients with systemic JIA in clinical remission.
PATIENTS AND METHODS
Study design
The present study was designed as a randomized, open‐label, 2‐part clinical trial. As shown in Figure 1, the trial included 2 cohorts of patients with systemic JIA. Cohort 1 comprised patients with systemic JIA who had inactive disease at their last visit. These patients were included in a previous long‐term extension trial of canakinumab preceded by other pivotal studies (8, 10) (see Supplementary Figure 1 for a summary of the flow of patients in the clinical trials of the canakinumab development program, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41488/abstract). Inactive disease was considered achieved when the following criteria were met (22): no joints with active arthritis; no fever due to systemic JIA; no rash, serositis, splenomegaly, hepatomegaly, or generalized lymphadenopathy attributable to systemic JIA; normal CRP serum levels (or, if elevated, not attributable to systemic JIA); and a physician global assessment of disease activity score of ≤10 mm (on a 100‐mm visual analog scale [VAS]), indicating inactive disease. Cohort 2 comprised newly recruited patients with systemic JIA who had never been treated with canakinumab and who had active disease at the time of enrollment (see the Supplementary Appendix for inclusion and exclusion criteria, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41488/abstract).
Figure 1.
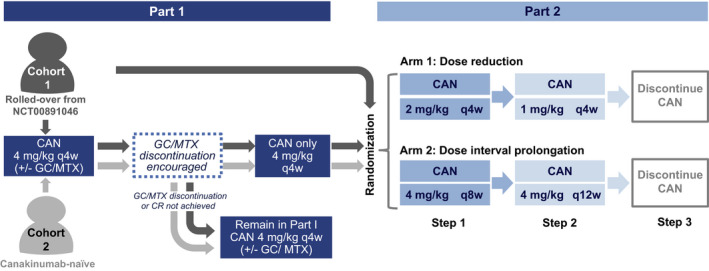
Flow chart of the study design. Cohort 1 comprised patients with systemic juvenile idiopathic arthritis in complete clinical remission (CR) at the last visit of a previous long‐term extension trial of canakinumab (CAN) (ClinicalTrials.gov identifier: NCT00891046) (8). Patients in cohort 1 who were already receiving canakinumab monotherapy could enter directly into part 2 of the study. Cohort 2 comprised newly recruited patients with active disease at baseline. Patients in whom clinical remission was achieved with canakinumab monotherapy for at least 4 weeks in part 1 entered part 2 of the study (as long as the randomization period was still open). Patients in whom these criteria were not met, or in whom criteria were met after randomization was closed, remained in part 1 until study end. Randomization was only to achieve balance between the treatment arms as no comparison between the cohorts was planned. GC = glucocorticoid; MTX = methotrexate; q4w = every 4 weeks.
In part 1 of the trial, all patients received 4 mg/kg of open‐label canakinumab subcutaneously every 4 weeks. Patients taking systemic glucocorticoids and/or methotrexate (MTX) at the time of study entry were encouraged, at the discretion of the investigator, to taper and discontinue these medications according to protocol‐specified guidelines (Supplementary Appendix, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41488/abstract) at any time after study entry (cohort 1) or to discontinue these medications 8 weeks after study initiation (cohort 2). Patients in whom clinical remission was achieved (which was classified as having maintained inactive disease for ≥24 weeks) and who had received canakinumab monotherapy for ≥4 weeks were considered eligible for randomization in part 2 of the study. A sample size of 76 patients was estimated to be sufficient for the evaluation of the primary objective (sample size calculations are available in the Supplementary Appendix), and therefore randomization was stopped once this predefined target was reached. Patients who met eligibility criteria after this time point did not enter the second part of the study and remained in part 1 until study end. Patients who were eligible to enroll in part 2 were randomized 1:1 into 1 of 2 treatment arms. Randomization was performed only to achieve balance between the treatment arms, and therefore it was not necessary to compare the number of patients deemed eligible before and after this time point.
In both treatment arms, open‐label canakinumab exposure was progressively reduced in 3 consecutive steps (24 weeks each) as long as clinical remission was maintained. In the dose reduction arm, the tapering schedule was as follows: reduction from 4 mg/kg of canakinumab every 4 weeks during the first part of the study to 2 mg/kg every 4 weeks (step 1), reduction from 2 mg/kg to 1 mg/kg of canakinumab every 4 weeks (step 2), and then finally discontinuation (step 3). In the dose interval prolongation arm, prolongation of the dose intervals in canakinumab therapy was as follows: prolongation of the 4 mg/kg dose of canakinumab during the first part of the study from every 4 weeks to every 8 weeks (step 1), then to every 12 weeks (step 2), and then finally discontinuation (step 3). At every step, patients were considered as having “regimen failure” and were required to return to a regimen of 4 mg/kg of canakinumab every 4 weeks if they experienced any of the following: a systemic JIA flare according to the criteria for disease flare in pediatric JIA (10, 23), need for up‐titration of medication, or need for treatment with systemic glucocorticoids or MTX. In patients with regimen failure, we required that achievement of clinical remission in these individuals be demonstrated again with 4 mg/kg of canakinumab monotherapy every 4 weeks. If clinical remission was maintained in these patients, they were then permitted to attempt tapering again. Finally, patients in whom clinical remission was not maintained and did not experience regimen failure remained in the reduced‐dose regimen. If clinical remission was achieved in these patients again, they were allowed to progress to the next tapering step and further reduce or discontinue canakinumab.
The present study was conducted from November 2014 to September 2017 in 45 centers from 16 countries following the ethics principles of the Declaration of Helsinki and the Good Clinical Practice guidelines and was approved by an independent ethics committee for each participating country. Participating centers were members of the Paediatric Rheumatology International Trials Organisation (PRINTO) and the Pediatric Rheumatology Collaborative Study Group (PRCSG) (see Appendix A for the full list of study investigatores) (24, 25). All patients, parents, or legal guardians of patients provided written informed consent.
Study objectives
The primary objective of the study was to assess whether, on initiation of these regimens in part 2 of the study in patients with systemic JIA in clinical remission who had been receiving 4 mg/kg of canakinumab monotherapy, at least 40% of patients remained in clinical remission for 24 consecutive weeks after randomization to either the dose reduction arm (dose reduced to 2 mg/kg every 4 weeks) or the dose interval prolongation arm (4 mg/kg dose interval prolonged to every 8 weeks). This 40% threshold was chosen after consultation with a panel of experts from the PRINTO and PRCSG networks and was estimated to be meaningful for clinical practice. The 24‐week time point was determined taking into account that 8‒12 weeks are required to reach steady‐state serum concentrations of canakinumab (26), and 12 additional weeks were estimated to be an appropriate observation period to assess the effect of the drug thereafter. The secondary objective of the study was to assess the long‐term safety and tolerability of canakinumab throughout the trial. Additional metrics included the proportion of patients treated with canakinumab who successfully discontinued MTX and glucocorticoids (in part 1 of the study), the efficacy of canakinumab over time as measured according to the American College of Rheumatology (ACR) preliminary definition of improvement in juvenile arthritis (27), and determination of the extent of disease activity according to the Juvenile Arthritis Disease Activity Score (JADAS) (28, 29, 30).
Outcome measures
In the present study, the following ACR core set measures adapted for JIA (27) were used to assess active disease: the physician global assessment of disease activity on a 0‒100‐mm VAS, patient/parent assessment of patient overall well‐being, number of joints with active arthritis according to the ACR definition, number of joints with limitation of motion, CRP values (normal range 0‒10 mg/liter), the cross‐culturally adapted and validated version of the Childhood Health Assessment Questionnaire (C‐HAQ) score (31), and fever (defined as having a body temperature of >38°C) in the preceding week due to systemic JIA. Inactive disease status was assessed every 4 weeks and at any point at which the investigators suspected worsening of systemic JIA symptoms.
To assess the extent of disease activity, the JADAS score in 27 joints using the CRP level (JADAS27‐CRP) was calculated as previously described (28, 29, 30, 32, 33). Clinical responses were measured according to achievement of 30%, 70%, and 90% levels of improvement in the ACR core set measures adapted for JIA (ACR Pediatric 30 [ACR Pedi 30], ACR Pedi 70, and ACR Pedi 90, respectively), as previously described (27, 34), along with the absence of fever.
The safety of canakinumab was assessed in terms of adverse events (AEs) and serious AEs (SAEs) classified according to version 20.1 of the Medical Dictionary for Regulatory Activities. All potential cases of macrophage activation syndrome (MAS), infections classified as SAEs, and malignancies were reviewed and adjudicated by independent adjudication committees that were established for each of these groups of AEs for the entire canakinumab development program (35). The clinical and laboratory assessments included the regular monitoring of hematology, blood chemistry, and urinalysis results as well as regular assessments of vital signs, physical condition, and body weight. Immunogenicity assessments were performed at baseline and every 24 weeks by testing for antidrug antibody concentrations in the serum over time, using a validated electrochemiluminescence immunoassay (36).
Statistical analysis
The study results were reported following the recommendations of the Consolidated Standards of Reporting Trials (CONSORT) statement (37) and the intent‐to‐treat (ITT) principle for parts 1 and 2 of the study, with imputation of missing data for nonresponders in analyses of the primary variables (as described in the Supplementary Appendix, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41488/abstract). The full analysis set and the safety set in part 1 were equivalent and consisted of all patients in cohort 1 or cohort 2 who received ≥1 dose of the study drug. The full analysis set in part 2 consisted of all patients who were randomized to undergo either dose reduction or dose interval prolongation of the study drug and who received ≥1 dose of canakinumab, and the safety set consisted of all patients who received ≥1 dose of the study drug.
Statistical hypotheses for the primary end point were tested by exact binomial test at the 2.5% level of significance. As randomization in part 2 was performed solely for the purpose of avoiding selection bias, no inferential between‐treatment comparisons were done. For safety evaluation, exposure‐adjusted rates of AEs per 100 patient‐days were summarized in part 1 of the study by cohorts and in part 2 by tapering regimen arm overall and for each tapering step within the regimen. Additional details on statistical methods and sample size calculation are provided in the Supplementary Appendix, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41488/abstract.
RESULTS
Study patients
The distribution of patients assessed in this study is depicted in Figure 2. Additionally, the flow of patients through clinical trials evaluating canakinumab is described in Supplementary Figure 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41488/abstract. Overall, 182 patients were enrolled in the present study. Cohort 1 included 84 (46.2%) of 182 patients, of whom 68 entered the first part of the study. The remaining 16 patients were directly eligible to enroll into the second part of the study, as clinical remission of systemic JIA was achieved in these patients with a canakinumab regimen that did not include glucocorticoids or MTX, and 15 of these patients were subsequently included in the second part of the study. Cohort 2 included 98 (53.8%) of 182 patients.
Figure 2.
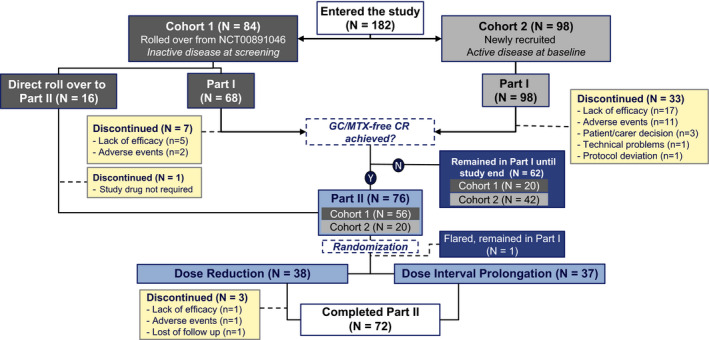
Flow chart of the study population. In total, 182 individuals entered the study, with 44 discontinuing the study and 138 completing it. Sixty‐three patients remained in part 1 until study end, and 72 patients completed part 2. After reaching the predefined population of 76 patients for part 2 (patients in whom clinical remission [CR] with canakinumab monotherapy was achieved without glucocorticoids [GCs] or methotrexate [MTX]), randomization enrollment was closed, and patients in whom clinical remission was achieved with canakinumab monotherapy after this time remained in part 1 of the study. Among the 62 patients who were not randomized and remained in part 1 until study end, protocol deviations were reported in 4 patients who were not randomized but progressed to the canakinumab tapering treatment arm, with 1 patient enrolled directly from the long‐term extension trial of canakinumab (ClinicalTrials.gov identifier: NCT00891046) (8) and receiving 1 dose according to the dose reduction scheme before discontinuing the study and 3 additional patients in part 1 progressing to the dose interval prolongation treatment arm. These patients were not included in the efficacy analyses of part 2. Y = yes; N = no.
In the first part of the study, 166 patients were treated with 4 mg/kg of canakinumab every 4 weeks, with tapering and discontinuation of glucocorticoids and MTX allowed and encouraged, as described in the Patients and Methods. Of these individuals, 40 patients (7 of 68 in cohort 1 [10.3%] and 33 of 98 in cohort 2 [33.7%]) discontinued the study, and 61 patients entered part 2. The remaining patients did not meet the eligibility criteria to enter the second part of the study before enrollment for randomization was closed, and therefore remained in part 1 until study end. The mean observation time in part 1 was 466 days in cohort 1 and 434 days in cohort 2.
In the second part of the study, 76 patients were randomized to receive canakinumab at either a reduced dose (n = 38) or a prolonged dose interval (n = 38). One patient randomized to the dose interval prolongation arm experienced a flare just after randomization, and therefore never initiated canakinumab tapering and moved back into part 1 of the study; this individual was not included in the analyses of the results derived from the second part of the study. The mean observation time in part 2 was 597 days for the dose reduction arm and 595 days for the interval‐prolongation arm.
Demographics and baseline characteristics
Baseline demographic details and characteristics of patients enrolled in parts 1 and 2 of the study are summarized in Table 1. In part 1, there were more young children (age <6 years) in cohort 2 (17 of 98, or 17%) compared to cohort 1 (6 of 68, or 8.8%). The median values for clinical parameters and disease activity scores at baseline were high among patients in cohort 2, including CRP level, number of joints with active disease, number of joints with limited range of motion, physician global assessment of disease activity scores, parent/patient global assessment of overall well‐being, and C‐HAQ scores. As expected, those parameters were much lower in cohort 1. In the second part of the study, baseline demographic characteristics were mostly comparable for the 2 treatment arms. At baseline, the mean time of prior exposure to canakinumab in cohort 1 patients was 3.1 years. Of note, only 8 of 84 patients in cohort 1 had previously attempted a dose reduction to 2 mg/kg of canakinumab every 4 weeks in the long‐term extension trial of canakinumab (8).
Table 1.
Patient demographic and baseline clinical characteristics*
| Part 1 | Part 2 | ||||
|---|---|---|---|---|---|
|
Cohort 1 (n = 68) |
Cohort 2 (n = 98) |
Total (n = 166) |
Dose reduction arm (n = 38) |
Dose interval prolongation arm (n = 37) |
|
| Age, years | 12 (8–15) | 8 (5–12) | 9 (6–14) | 10.5 (7–15) | 11 (9–14) |
| Female sex, no. (%) | 34 (50) | 56 (57.1) | 90 (54.2) | 19 (50) | 20 (54.1) |
| Weight, kg | 39.0 (28.4–54.3) | 26.5 (19.4–41.8) | 31.8 (22–48.4) | 35.1 (23.4–55.2) | 41.3 (29–54.2) |
| Parent or patient global assessment of overall well‐being, 0–100‐mm VAS | 2 (0–8) | 42 (17–65) | 18.5 (2–52) | 5 (0–46) | 1 (0–10) |
| Physician global assessment of disease activity, 0–100‐mm VAS | 0 (0–2) | 51 (38–63) | 32 (0–54) | 0 (0–11) | 0 (0–26) |
| C‐HAQ score | 0 (0–0.4) | 1.3 (0.5–1.8) | 0.7 (0–1.5) | 0 (0–1) | 0.1 (0–0.6) |
| Number of joints with active arthritis | 0 (0–0) | 3 (2–8) | 1 (0–5) | 0 (0–0) | 0 (0–0) |
| Standardized CRP level, mg/liter† | 5.6 (0.6–9.7) | 88.8 (24.3–255.7) | 18.5 (4–114.2) | 5.9 (0.5–10) | 5.5 (2–10) |
| Systemic glucocorticoid use at baseline, no. (%) | 23 (33.8) | 50 (51) | 73 (44) | 7 (18.4) | 10 (27) |
| Methotrexate use at baseline, no. (%) | 31 (45.6) | 32 (32.7) | 63 (38) | 11 (28.9) | 4 (10.8) |
| Prior use of anakinra, no. (%) | 22 (32.4) | 15 (15.3) | 37 (22.3) | 16 (42.1) | 11 (29.7) |
| Prior use of tocilizumab, no. (%) | 15 (22.1) | 20 (20.4) | 35 (21.1) | 9 (23.7) | 5 (13.5) |
| Prior use of any biologic agents, no. (%)‡ | 41 (60.3) | 36 (36.7) | 77 (46.4) | 23 (60.5) | 15 (40.5) |
Except where indicated otherwise, values are the median (interquartile range). VAS = visual analog scale; C‐HAQ = Childhood Health Assessment Questionnaire.
C‐reactive protein (CRP) analyses were performed by local laboratories and standardized for comparison across sites and patients.
Includes anakinra, tocilizumab, abatacept, etanercept, and adalimumab.
Efficacy
Tapering of glucocorticoids and MTX and achievement of clinical remission in part 1 of the study
In cohort 1, discontinuation of glucocorticoids during the first part of the study was achieved in 9 (39%) of 23 patients who had received them at baseline, and MTX was discontinued in 13 (42%) of 31 patients. During the first 24 weeks of part 1, inactive disease was maintained in a relatively constant proportion of the patients in cohort 1 who were receiving canakinumab monotherapy (Figure 3A). In total, clinical remission was achieved in 46 of 68 patients who received canakinumab monotherapy in part 1 of the study and in 41 patients who entered part 2 of the study. Calculated median JADAS27–CRP scores remained low during the first 24 weeks of the study (Figure 3B). Flares of systemic JIA were experienced by 14 (20%) of 68 patients.
Figure 3.
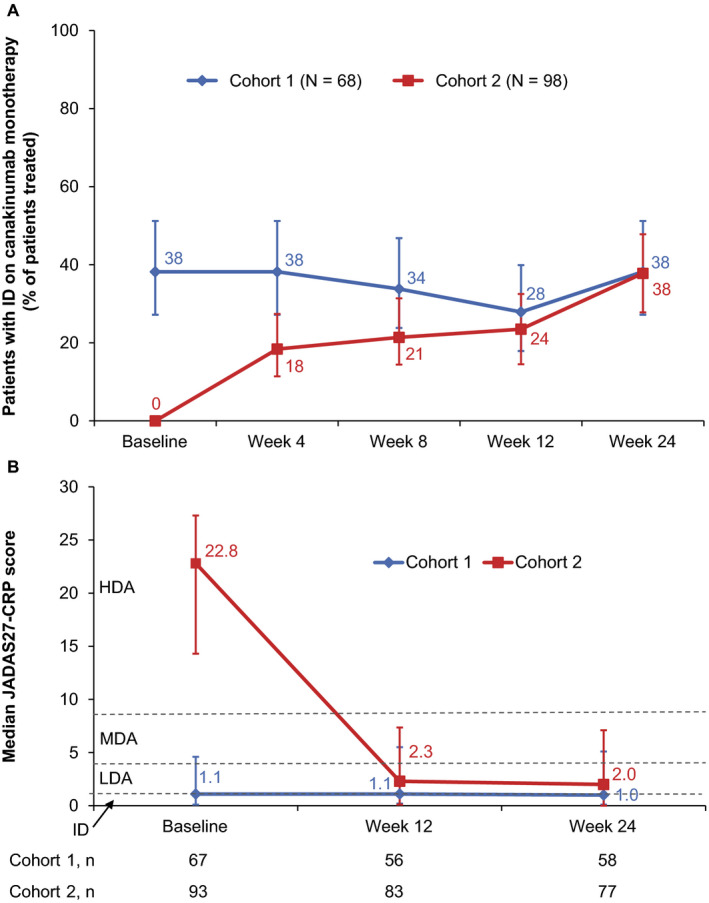
Efficacy of canakinumab in part 1 of the study. A, Percentage of systemic juvenile idiopathic arthritis (JIA) patients with inactive disease (ID) following canakinumab monotherapy. Intent‐to‐treat (ITT) analysis of the full analysis set is shown, with imputation of missing data for nonresponders in the initial 24 weeks of part 1. For each time point, the percentage of patients with inactive disease who received canakinumab monotherapy (with treatment maintained until the end of part 1) is presented. Percentages were calculated from the total ITT population, which included patients who had received glucocorticoids and/or methotrexate. Values indicate percentages, and error bars show the 95% confidence intervals. Of note, no adjustment for multiple comparisons has been performed, and line graphs used are only for descriptive purposes. B, Median Juvenile Arthritis Disease Activity Scores in 27 joints using the C‐reactive protein level (JADAS27‐CRP) in cohorts 1 and 2 (n = 68 and n = 98, respectively). Numbers under the line graph represent the actual number of patients evaluated at each time point. Values indicate median scores, and error bars show the interval between the first and third quartiles. Horizontal lines represent the JADAS cutoff values for high disease activity (HDA) (>8.5), moderate disease activity (MDA) (3.9–8.5), low disease activity (LDA) (1.1–3.8), and inactive disease (≤1). Of note, these cutoff values have been defined for polyarticular JIA and have not been evaluated specifically in systemic JIA.
In cohort 2, of the 50 patients who received glucocorticoids at baseline, glucocorticoids were discontinued in 17 patients (34%) by the end of part 1, whereas MTX was discontinued in 19 (59%) of 32 patients who had received it at baseline. The proportion of patients in cohort 2 in whom inactive disease was able to be maintained with canakinumab monotherapy increased progressively during the first 24 weeks (Figure 3A). The median time from the first dose of canakinumab to inactive disease achievement was 8 weeks (data not shown). In total, clinical remission was achieved in 49 of 98 patients from cohort 2 who received canakinumab monotherapy in part 1, and among these 49 patients, 20 entered the second part of the study.
A clear decrease was observed in the median JADAS27‐CRP scores from baseline to week 4, decreasing from a median score of 22.8 to 2.6, which was maintained until week 24 of the study (Figure 3B). The efficacy of canakinumab was also reflected by ACR Pedi 30, ACR Pedi 70, and ACR Pedi 90 responses observed in patients from cohort 2, with >60% of the patients presenting with ACR Pedi 90 responses at week 24 (Supplementary Figure 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41488/abstract). Flares of systemic JIA were experienced by 34 (35%) of 98 patients in cohort 2 during part 1.
Tapering of canakinumab in part 2 of the study
The proportion of patients in whom clinical remission was achieved for 24 weeks was assessed as a percentage with 97.5% confidence intervals (97.5% CIs) for patients in each group. At the first tapering event in step 1 of part 2 of the study, the proportion of patients in whom clinical remission was achieved for 24 weeks with a reduced dose of canakinumab of 2 mg/kg every 4 weeks was 27 of 38 (71% of patients [97.5% CI 52–86%]), and the proportion of patients in whom clinical remission was achieved with the prolonged dose interval regimen of 4 mg/kg every 8 weeks was 31 of 37 patients (84% of patients [97.5% CI 66–95%]) (Figure 4). These values significantly exceeded the predefined threshold of 40% (P ≤ 0.0001), indicating that the primary end point was met.
Figure 4.
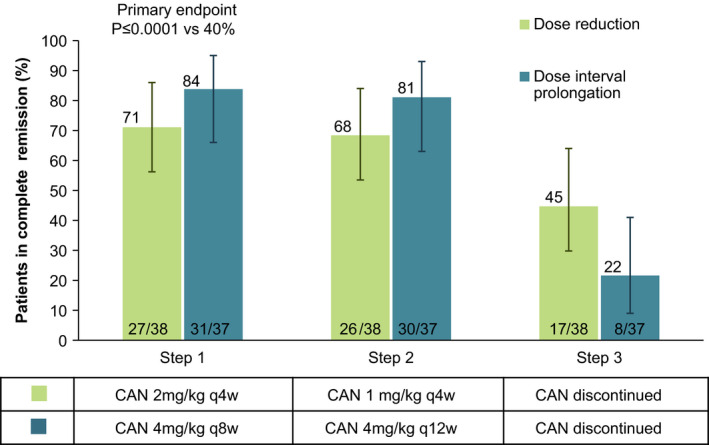
Proportion of patients in whom complete clinical remission (CR) of systemic juvenile idiopathic arthritis was maintained in part 2 of the study. Intent‐to‐treat (ITT) analysis of the full analysis set with imputation of missing data for nonresponders is shown. Values over bars indicate percentages, values within bars are the number of patients/total number assessed, and error bars show the 97.5% confidence intervals. Only patients in whom clinical remission was maintained for 24 weeks on the first attempt of dose reduction or dose interval prolongation of canakinumab (CAN) were categorized as responders. The proportion of patients in whom clinical remission was maintained in step 1 was significantly higher than the predefined 40% threshold (primary end point) for both treatment arms. No adjustment for multiple comparisons was performed for the remaining data shown, and therefore their presentation is only for descriptive purposes. q4w = every 4 weeks.
Analyses using an exact logistic regression model showed that prior exposure to canakinumab (which occurred in cohort 1 but not in cohort 2) did not impact the probability of being able to maintain clinical remission in the patients at step 1 (data not shown). When including patients in whom clinical remission was not maintained after the initial attempt and who underwent a second attempt, the proportions of patients in whom clinical remission was maintained in step 1 were slightly higher, with 29 (76%) of 38 patients in the dose reduction arm and 33 (89%) of 37 patients in the dose interval prolongation arm maintaining clinical remission. Of note, 7 patients (4 in the dose reduction arm and 3 in the dose interval prolongation arm) did not meet the criteria for inactive disease at all visits during the 24‐week period in step 1, and were therefore not considered to be responders at the primary end point, but also did not meet the criteria for regimen failure (as defined in the Patients and Methods), and therefore investigators chose to keep those patients in the tapering regimens. A supportive analysis including these patients as responders showed higher success rates for both arms, with 31 (82%) of 38 patients in the dose reduction arm and 34 (92%) of 37 patients in the dose prolongation arm maintaining these regimens for at least 24 weeks during their first attempt in step 1.
In Figure 4, proportions of randomized patients in each arm in whom canakinumab exposure was further decreased at their first attempt in step 2 and who subsequently discontinued canakinumab at their first attempt in step 3 with maintenance of clinical remission for 24 weeks are displayed. Overall, 25 (33%) of 75 patients who were randomized in part 2 discontinued canakinumab and systemic JIA remained in clinical remission in step 3.
The proportion of patients with regimen failure in the dose reduction arm was 7 (18%) of 38, 3 (10%) of 28, and 2 (8.0%) of 25 for steps 1, 2, and 3, respectively. In the dose prolongation arm, these proportions were 1 (2.7%) of 37, 2 (6.1%) of 33, and 4 (15%) of 27. In all patients except 1 (in the dose reduction arm), systemic JIA returned to a state of inactive disease when canakinumab was up‐titrated to 4 mg/kg every 4 weeks, according to the study protocol. Overall, 14 (19%) of 75 patients experienced disease flares in part 2, with similar proportions experiencing flares in the 2 treatment arms. The mean number of flares per patient was 0.1 in steps 1, 2, and 3 for the dose reduction arm and 0, 0, and 0.2 for steps 1, 2, and 3 in the dose prolongation arm.
Safety
Incidence of AEs
The mean duration of canakinumab exposure among patients in cohorts 1 and 2 enrolled in part 1 of the study was 466 days and 434 days, respectively. The incidence rate of AEs following adjustment for exposure was higher in cohort 2 (2.45 per 100 patient‐days) than in cohort 1 (1.64 per 100 patient‐days), with the most commonly reported AEs being pyrexia, nasopharyngitis, headache, arthralgia, and diarrhea (Table 2).
Table 2.
Incidence of AEs in parts 1 and 2 of the study following adjustment for canakinumab exposure*
| Part 1 | Part 2 | ||||
|---|---|---|---|---|---|
| Cohort 1 (n = 68) | Cohort 2(n = 98) | Total (n = 166) | Dose reduction arm (n = 38) | Dose interval prolongation arm (n = 37) | |
| Total AEs | 1.64 (521) | 2.45 (1,043) | 2.11 (1,564) | 1.59 (361) | 1.32 (291) |
| Common AEs† | |||||
| Pyrexia | 0.04 (13) | 0.15 (63) | 0.10 (76) | 0.07 (17) | 0.06 (13) |
| Nasopharyngitis | 0.10 (32) | 0.09 (38) | 0.09 (70) | 0.07 (16) | 0.08 (18) |
| Headache | 0.07 (21) | 0.09 (37) | 0.08 (58) | 0.04 (10) | 0.07 (16) |
| Arthralgia | 0.04 (14) | 0.08 (36) | 0.07 (50) | 0.07 (15) | 0.05 (11) |
| Diarrhea | 0.01 (3) | 0.09 (39) | 0.06 (42) | 0.03 (7) | 0.01 (2) |
| Cough | 0.03 (10) | 0.07 (29) | 0.05 (39) | 0.05 (11) | 0.01 (3) |
| Rash | 0.02 (5) | 0.06 (25) | 0.04 (30) | 0.02 (4) | 0.03 (7) |
| Upper respiratory tract infection | 0.07 (22) | 0.04 (18) | 0.05 (40) | 0.07 (16) | 0.03 (6) |
| Viral infection | 0.01 (4) | 0.02 (8) | 0.02 (12) | 0.05 (12) | 0.01 (3) |
| Serious AEs | 0.05 (15) | 0.08 (34) | 0.07 (49 | 0.02 (4) | 0.01 (2) |
| Study drug discontinuation due to AEs, no. | 3 | 12 | 15 | 1 | 0 |
Except where indicated otherwise, values are the number of adverse events (AEs) per 100 patient‐days in the safety set (total number of AEs).
Common AEs were defined as those with incidence rates of >0.05 per 100 patient‐days in either cohort or treatment arm.
The mean duration of canakinumab exposure for patients randomized for enrollment in part 2 of the study was 489 days for the dose reduction arm and 495 days for the dose prolongation arm. Incidence rates for AEs following adjustment for exposure were 1.59 per 100 patient‐days in the dose reduction arm and 1.32 per 100 patient‐days in the dose interval prolongation arm (Table 2), with the more commonly reported AEs being pyrexia, nasopharyngitis, upper respiratory tract infection, arthralgia, and headache. Incidence of AEs for each tapering regimen overall as well as for each tapering step is shown in Supplementary Table 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41488/abstract. There was no apparent correlation between incidence of AEs and the tapering regimens or tapering steps used in the present study.
Serious AEs and AEs leading to drug discontinuation
Overall, 33 patients (19.9%) in part 1 experienced at least 1 SAE. Incidence rates of SAEs following adjustment for exposure were 0.05 per 100 patient‐days in cohort 1 and 0.08 per 100 patient‐days in cohort 2 (Table 2). In part 2, SAEs were reported in 4 patients (10.5%) in the dose reduction arm and 1 patient (2.7%) in the dose interval prolongation arm, with exposure‐adjusted incidence rates of 0.02 and 0.01 per 100 patient‐days, respectively (Table 2). Most SAEs represented isolated events, with infections and infestations being the most commonly reported system organ class affected (Supplementary Table 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41488/abstract). Most SAEs classified as infections or infestations occurred in part 1 of the study, and they were more frequently reported in cohort 2 (9 events, or 0.02 per 100 patient‐days) than in cohort 1 (4 events, or 0.01 per 100 patient‐days) (Supplementary Table 2). No SAEs of infections were indicated as being opportunistic infections, as adjudicated by an independent committee. Two SAEs related to lung disease were reported, including a mild case of interstitial lung disease and a severe case of alveolar proteinosis that led to study discontinuation for that individual. No deaths were reported during the study.
In part 1, study treatment discontinuation due to AEs occurred in a higher proportion of patients in cohort 2 (12.2%) compared to cohort 1 (4.4%) (Table 2). The most common reasons for discontinuation were disease‐related, namely MAS and AEs related to systemic JIA worsening or disease flares. Only 1 patient in part 1 (in the dose reduction arm) experienced an AE (blepharitis) that led to study discontinuation.
Macrophage activation syndrome
In part 1, 5 events were assessed as probable MAS by an independent MAS adjudication committee. These included 2 SAEs of MAS reported in 2 patients in cohort 2 and 3 patients (1 in cohort 1 and 2 in cohort 2) with laboratory abnormalities consistent with MAS (ferritin level of ≥500 μg/liter and elevated levels of liver transaminases). In part 2, 1 SAE of MAS in a patient in the dose reduction arm was adjudicated as probable MAS.
Immunogenicity
No antidrug antibodies were detected during the study, except in 1 patient in cohort 1 in whom non‐neutralizing antidrug antibodies were detected at an unplanned visit (82 days after last dose of the study drug) and in whom no detectable antibodies were observed at all other visits.
DISCUSSION
In this study, a treat‐to‐target strategy was used in patients with systemic JIA who were treated with canakinumab. Achievement of clinical remission was chosen as the treatment target, and therapeutic interventions were adjusted to maintain it. Results from this study indicate that systemic JIA patients in whom clinical remission has been maintained with the recommended canakinumab monotherapy regimen of 4 mg/kg every 4 weeks could reduce their exposure to canakinumab either by dose reduction or dose interval prolongation and still be able to maintain clinical remission. No substantial differences between the 2 tapering strategies were apparent, although the study was not designed or powered to compare between the reduced dose and the increased dosing interval therapies, and as such, meaningful comparisons cannot be made.
Clinical remission could be maintained in most patients who were randomized in part 2 of this trial to receive either 2 mg/kg of canakinumab monotherapy every 4 weeks or 4 mg/kg every 8 weeks. Moreover, in almost all patients in whom clinical remission was maintained with 2 mg/kg of canakinumab every 4 weeks, the dose could be further reduced to 1 mg/kg every 4 weeks in step 2. Similarly, in almost all patients in whom clinical remission was maintained with 4 mg/kg of canakinumab every 8 weeks, the dose interval could be further increased to 12 weeks. These results suggest that the progressive tapering methods used in this trial are appropriate to reduce canakinumab exposure in this patient population, with a low incidence of regimen failure, which is defined as occurrence of systemic JIA flares or requirement of canakinumab up‐titration or treatment with systemic glucocorticoids or MTX. Nevertheless, only one‐third of the patients could discontinue canakinumab (step 3 in this study) with maintained clinical remission for at least 24 weeks, indicating that a certain level of IL‐1 inhibition may be important to maintain clinical remission in the majority of these patients.
The AEs recorded in parts 1 and 2 of this trial were consistent with the known safety profile of canakinumab. Overall, there was no apparent association between progressively tapering canakinumab (either by dose reduction or dose interval prolongation) and the incidence of any class of AEs (including events of MAS) or systemic JIA flares.
Very limited clinical evidence is currently available to support appropriate dose reduction or tapering of DMARDs in patients with systemic JIA or other JIA subtypes. Foell and colleagues compared relapse rates in patients with several types of JIA in clinical remission who discontinued MTX after 6 or 12 months (38). They found no differences between the 2 groups, with ~50% of the patients experiencing a flare within 24 months following study inclusion. Lovell et al reported that discontinuation of anti‐TNF treatment in JIA patients resulted in disease flare in 37% of patients by 8 months (39). Results from a previous prospective observational single‐center cohort study have demonstrated the efficacy of first‐line treatment with IL‐1 receptor antagonist anakinra administered daily as monotherapy in 42 patients with newly diagnosed systemic JIA (14). In that study, anakinra was discontinued in patients with an ACR Pedi 90 response for 3 consecutive months (14, 34). Results showed that 22 patients had discontinued anakinra and presented with inactive disease after 1 year of follow‐up, 25 patients after 3 years, and 18 patients after 5 years.
Part 1 of this trial investigated the efficacy of administering 4 mg/kg of canakinumab every 4 weeks in patients with systemic JIA, to assess whether clinical remission could be achieved without adjunct therapy with glucocorticoids and/or MTX. In treating patients with active disease (cohort 2), efficacy with canakinumab was consistent with the results from previous pivotal phase III trials (10), and clinical remission could be achieved in 50% of the patients receiving canakinumab monotherapy. Also consistent with reported data from the previous pivotal phase III and long‐term extension studies was that the discontinuation of glucocorticoids was successful in one‐third of the patients who received them at baseline (8, 10). These results underline the difficulty in achieving discontinuation of glucocorticoids in patients with systemic JIA. In addition, this study was the first to evaluate the effectiveness of canakinumab in enabling discontinuation of MTX, with nearly 50% of the patients achieving this outcome.
Key limitations of the present study include the open‐label nature of canakinumab treatment, lack of a comparator arm in part 2 that included sustained canakinumab monotherapy without tapering, lack of sufficient power to compare between reduced dose and reduced frequency tapering regimens, and the decision to consider the maintenance of 40% of patients in whom clinical remission was achieved as meaningful in clinical practice.
In summary, we observed achievement of clinical remission with canakinumab monotherapy in >50% of the patients in part 1 of the study and maintenance of clinical remission with reduced‐exposure canakinumab monotherapy in a high proportion of patients in part 2. We believe that these results are relevant for clinical practice, particularly for designing personalized tapering strategies that can allow an adequate control of disease while minimizing the side effects of certain medications, notably glucocorticoids (17). Our results also show that while clinical remission was maintained in the majority of systemic JIA patients during canakinumab tapering, only a minority of patients could eventually discontinue canakinumab and maintain clinical remission of systemic JIA. Therefore, a certain level of sustained inhibition of the IL‐1 pathway seems important for the maintenance of clinical remission in most patients with systemic JIA.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Quartier had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Quartier, Alexeeva, Constantin, Palmblad, Wouters, Brunner, Martini, Wei, Slade, Ruperto, Abrams.
Acquisition of data
Quartier, Constantin, Chasnyk, Wulffraat, Palmblad, Marzan, Schneider, Horneff, Anton, Wei, Slade, Ruperto, Abrams.
Analysis and interpretation of data
Quartier, Palmblad, Brunner, Horneff, Wei, Slade, Ruperto, Abrams.
ROLE OF THE STUDY SPONSOR
Novartis had no role in the writing of the manuscript or the decision to submit the manuscript for publication. Publication of this article was not contingent upon approval by Novartis. Authors Wei, Slade, and Abrams are employees of Novartis. We thank Rajeeb Ghosh, of Novartis Healthcare, and Marco Migliaccio for medical writing assistance, which was funded by Novartis Pharma AG, Basel, Switzerland.
Supporting information
Supplementary Appendix
Supplementary Figure S1
Supplementary Figure S2
Supplementary Tables S1 and S2
Supplementary Figure Legends S1 and S2
APPENDIX A. THE PRINTO and PRCSG INVESTIGATORS
The PRINTO and PRCSG Investigators are as follows: W. Emminger, A. Ulbrich, S. Fodor (Vienna, Austria), C. Wouters, L. Desomer (Leuven, Belgium), B. Lauwerys, B. Brichard, C. Boulanger, G. Levy, L. Goffin, P.‐Q. Le (Brussels, Belgium), M. Bandeira, C. Feitosa Pelajo (Curitiba, Brazil), S. Knupp Feitosa, C. Costa, M. Felix Rodrigues, C. Almeida da Silva, L. Mattei de, K. Kozu (Rio de Janeiro, Brazil), Ronald Laxer (Toronto, Canada), K. Houghton, L. Tucker, K. Morishita (Vancouver, Canada), A. Mogenet, R. Mouy, B. Bader Meunier, C. Meyzer, M. Semeraro, O. Ben‐Brahim (Paris, France), I. Kone‐Paut, C. Galeotti, L. Rossi, P. Dusser, B. Cherquaoui (Le Kremlin Bicetre, France), A. Belot, A. Duquesne, F. Caroline, L. Audrey, M. Desjonqueres (Bron Cedex, France), I. Foeldvari, A. Kienast, B. Willig, E. Weissbarth‐Riedel, A. Froehlich (Hamburg, Germany), D. Barthel, J. Peitz, S. Wintrich, T. Geikowski, A. Schulz (St. Augustin, Germany), M. Hufnagel, M. Hirdes, R. Kubicki, J. Kirschner, A. Janda, A. Jacob, C. Emerich (Freiburg, Germany), A. Raab, G. Ngoumou, K. Minden, M. Lieber, S.‐L. von Stuckrad, R. Trauzeddel, D. Haselbusch, H. Kolbeck (Berlin, Germany), J. Kuemmerle Deschner, S. Hansmann, T. Schleich, I. Magunia, J. Riethmuller, N. Anders (Tübingen, Germany), H. Lehmann, J. de Laffolie (Giessen, Germany), T. Lutz, J. Grulich‐Henn, J. Pfeil, A. Helling‐Bakki (Heidelberg, Germany), A. Ponyi, D. Garan, I. Orban, K. Sevcic (Budapest, Hungary), Y. Butbul, R. Brik, M. Helo (Haifa, Israel), P. Hashkes, O. Toker, R. Haviv (Jerusalem, Israel), Y. Uziel, R. Haviv, V. Moshe, M. Rothschild (Kfar‐Saba, Israel), L. Harel, G. Amarilyo, R. Tal, M. Said (Petach‐Tikva, Israel), I. Tirosh, S. Spielman, M. Gerstein (Ramat Gan, Israel), A. Ravelli, B. Schiappapietra, G. Varnier, M. Finetti, M. Marasini, R. Caorsi, S. Rosina, S. Federici (Genova, Italy), I. Pontikaki, P. Meroni, V. Gerloni, N. Ughi, T. Ubiali (Milano, Italy), M. Alessio, R. Della Casa (Nepoli, Italy), S. Vastert, J. Swart, A. van Royen‐Kerhof, E. Schatorje, G. Van Iperen‐Schutte (Utrecht, Netherlands), L. Rutkowska‐Sak, I. Szczygielska, M. Kwiatkowska, M. Marusak‐Banacka, P. Gietka, (Warsaw, Poland), K. Isaeva, R. Denisova (Moscow, Russia), L. Snegireva, M. Dubko, M. Kostik, N. Buchinskaia, O. Kalashnikova, S. Avrusin, V. Masalova (Saint‐Petersburg, Russia), E. Nunez Cuadros, G. Diez, R. Galindo Zavala (Málaga, Spain), R. Bou Torrent, E. Iglesias, J. Calzada, V. Bittermann (Barcelona, Spain), A. Boteanu, M. L. Gamir, D. Clemente Garulo, J. C. Lopez Robledillo, R. Merino, R. Alcobendas, A. Remesal, S. Murias (Madrid, Spain), I. Calvo, B. Lopez, I. Gonzalez, L. Fernandez (Valencia, Spain), Bo Magnusson (Stockholm, Sweden), O. Kasapcopur, K. Barut, A. Adrovic, S. Sahin, M. Erguven, R. Gozdenur Savci (Istanbul, Turkey), S. Ozen, S. Demir, Y. Bilginer, Z. S. Avci, E. D. Batu (Ankara, Turkey), Andreas Reiff, Anusha Ramanatham, Diana Brown, Bracha Shaham, Shirley Parks, Michal Cidon (Los Angeles, California, US), G. Higgins, C. Spencer, J. Rossette, K. Jones, S. Bout Tabaku, S. Farley, S. Akoghlanian (Columbus, Ohio, US).
ClinicalTrials.gov identifier: NCT02296424.
Supported by Novartis Pharma AG.
Dr. Quartier has received consulting fees, speaking fees, and/or honoraria from Novartis (less than $10,000). Dr. Brunner has received consulting fees, speaking fees, and/or honoraria from GlaxoSmithKline (less than $10,000) and from Roche and Novartis (more than $10,000 each). Dr. Schneider has received consulting fees from Novartis, NovImmune, Sobi, and Roche (less than $10,000 each). Dr. Horneff has received consulting fees, speaking fees, and/or honoraria from Eli Lilly, GSK, Novartis, Pfizer, and Sobi (less than $10,000 each). Dr. Martini has received consulting fees, speaking fees, and or honoraria from Eli Lilly, EMD Serono, Janssen, Novartis, Pfizer, and AbbVie (less than $10,000 each). Dr. Anton has received consulting fees, speaking fees, and/or honoraria from Novartis, Sobi, Roche, Sanofi, AbbVie, Pfizer, and NovImmune (less than $10,000 each). Dr. Wei owns stock or stock options in Novartis. Dr. Ruperto has received consulting fees from Ablynx, AbbVie, AstraZeneca‐Medimmune, Biogen, Boehringer, Bristol Myers Squibb, Eli Lilly, EMD Serono, GlaxoSmithKline, Hoffmann‐La Roche, Janssen, Merck, Novartis, Pfizer, R‐Pharma, Sanofi, Servier, Sinergie, Sobi, and Takeda (less than $10,000 each). No other disclosures relevant to this article were reported.
Contributor Information
Pierre Quartier, Email: pierre.quartier@aphp.fr.
the Paediatric Rheumatology International Trials Organisation and the Pediatric Rheumatology Collaborative Study Group:
W. Emminger, A. Ulbrich, S. Fodor, C. Wouters, L. Desomer, B. Lauwerys, B. Brichard, C. Boulanger, G. Levy, L. Goffin, P.‐Q. Le, M. Bandeira, C. Feitosa Pelajo, S. Knupp Feitosa, C. Costa, M. Felix Rodrigues, C. Almeida da Silva, L. Mattei de, K. Kozu, Ronald Laxer, K. Houghton, L. Tucker, K. Morishita, A. Mogenet, R. Mouy, B. Bader Meunier, C. Meyzer, M. Semeraro, O. Ben‐Brahim, I. Kone‐Paut, C. Galeotti, L. Rossi, P. Dusser, B. Cherquaoui, A. Belot, A. Duquesne, F. Caroline, L. Audrey, M. Desjonqueres, I. Foeldvari, A. Kienast, B. Willig, E. Weissbarth‐Riedel, A. Froehlich, D. Barthel, J. Peitz, S. Wintrich, T. Geikowski, A. Schulz, M. Hufnagel, M. Hirdes, R. Kubicki, J. Kirschner, A. Janda, A. Jacob, C. Emerich, A. Raab, G. Ngoumou, K. Minden, M. Lieber, S.‐L. von Stuckrad, R. Trauzeddel, D. Haselbusch, H. Kolbeck, J. Kuemmerle Deschner, S. Hansmann, T. Schleich, I. Magunia, J. Riethmuller, N. Anders, H. Lehmann, J. de Laffolie, T. Lutz, J. Grulich‐Henn, J. Pfeil, A. Helling‐Bakki, A. Ponyi, D. Garan, I. Orban, K. Sevcic, Y. Butbul, R. Brik, M. Helo, P. Hashkes, O. Toker, R. Haviv, Y. Uziel, R. Haviv, V. Moshe, M. Rothschild, L. Harel, G. Amarilyo, R. Tal, M. Said, I. Tirosh, S. Spielman, M. Gerstein, A. Ravelli, B. Schiappapietra, G. Varnier, M. Finetti, M. Marasini, R. Caorsi, S. Rosina, S. Federici, I. Pontikaki, P. Meroni, V. Gerloni, N. Ughi, T. Ubiali, M. Alessio, R. Della Casa, S. Vastert, J. Swart, A. van Royen‐Kerhof, E. Schatorje, G. Van Iperen‐Schutte, L. Rutkowska‐Sak, I. Szczygielska, M. Kwiatkowska, M. Marusak‐Banacka, P. Gietka, K. Isaeva, R. Denisova, L. Snegireva, M. Dubko, M. Kostik, N. Buchinskaia, O. Kalashnikova, S. Avrusin, V. Masalova, E. Nunez Cuadros, G. Diez, R. Galindo Zavala, R. Bou Torrent, E. Iglesias, J. Calzada, V. Bittermann, A. Boteanu, M. L. Gamir, D. Clemente Garulo, J. C. Lopez Robledillo, R. Merino, R. Alcobendas, A. Remesal, S. Murias, I. Calvo, B. Lopez, I. Gonzalez, L. Fernandez, Bo Magnusson, O. Kasapcopur, K. Barut, A. Adrovic, S. Sahin, M. Erguven, R. Gozdenur Savci, S. Ozen, S. Demir, Y. Bilginer, Z. S. Avci, E. D. Batu, Andreas Reiff, Anusha Ramanatham, Diana Brown, Bracha Shaham, Shirley Parks, Michal Cidon, G. Higgins, C. Spencer, J. Rossette, K. Jones, S. Bout Tabaku, S. Farley, and S. Akoghlanian
References
- 1. Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 2004;31:390–2. [PubMed] [Google Scholar]
- 2. Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet 2011;377:2138–49. [DOI] [PubMed] [Google Scholar]
- 3. Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet 2007;369:767–78. [DOI] [PubMed] [Google Scholar]
- 4. Martini A, Ravelli A, di Fuccia G, Rosti V, Cazzola M, Barosi G. Intravenous iron therapy for severe anaemia in systemic‐onset juvenile chronic arthritis. Lancet 1994;344:1052–4. [DOI] [PubMed] [Google Scholar]
- 5. Consolaro A, Giancane G, Alongi A, van Dijkhuizen EH, Aggarwal A, al‐Mayouf SM, et al. Phenotypic variability and disparities in treatment and outcomes of childhood arthritis throughout the world: an observational cohort study. Lancet Child Adolesc Heal 2019;3:255–63. [DOI] [PubMed] [Google Scholar]
- 6. Vastert SJ, Kuis W, Grom AA. Systemic JIA: new developments in the understanding of the pathophysiology and therapy. Best Pract Res Clin Rheumatol 2009;23:655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin‐1 (IL‐1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL‐1 blockade. J Exp Med 2005;201:1479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat NM, Horneff G, et al. Canakinumab in patients with systemic juvenile idiopathic arthritis and active systemic features: results from the 5‐year long‐term extension of the phase III pivotal trials. Ann Rheum Dis 2018;77:1710–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ruperto N, Quartier P, Wulffraat N, Woo P, Ravelli A, Mouy R, et al. A phase II, multicenter, open‐label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum 2012;64:557–67. [DOI] [PubMed] [Google Scholar]
- 10. Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2396–406. [DOI] [PubMed] [Google Scholar]
- 11. Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, et al. A multicentre, randomised, double‐blind, placebo‐controlled trial with the interleukin‐1 receptor antagonist anakinra in patients with systemic‐onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis 2011;70:747–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Benedetti F, Brunner HI, Ruperto N, Kenwright A, Wright S, Calvo I, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2385–95. [DOI] [PubMed] [Google Scholar]
- 13. Ruperto N, Martini A. Current and future perspectives in the management of juvenile idiopathic arthritis. Lancet Child Adolesc Heal 2018;2:360–70. [DOI] [PubMed] [Google Scholar]
- 14. Ter Haar NM, van Dijkhuizen EH, Swart JF, van Royen‐Kerkhof A, el Idrissi A, Leek AP, et al. Treatment to target using recombinant interleukin‐1 receptor antagonist as first‐line monotherapy in new‐onset systemic juvenile idiopathic arthritis: results from a five‐year follow‐up study. Arthritis Rheumatol 2019;71:1163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Quartier P, Taupin P, Bourdeaut F, Lemelle I, Pillet P, Bost M, et al. Efficacy of etanercept for the treatment of juvenile idiopathic arthritis according to the onset type. Arthritis Rheum 2003;48:1093–101. [DOI] [PubMed] [Google Scholar]
- 16. Horneff G, Schmeling H, Biedermann T, Foeldvari I, Ganser G, Girschick HJ, et al. The German etanercept registry for treatment of juvenile idiopathic arthritis. Ann Rheum Dis 2004;63:1638–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ravelli A, Consolaro A, Horneff G, Laxer RM, Lovell DJ, Wulffraat NM, et al. Treating juvenile idiopathic arthritis to target: recommendations of an international task force. Ann Rheum Dis 2018;77:819–28. [DOI] [PubMed] [Google Scholar]
- 18. Magnani A, Pistorio A, Magni‐Manzoni S, Falcone A, Lombardini G, Bandeira M, et al. Achievement of a state of inactive disease at least once in the first 5 years predicts better outcome of patients with polyarticular juvenile idiopathic arthritis. J Rheumatol 2009;36:628–34. [DOI] [PubMed] [Google Scholar]
- 19. Vannucci G, Cantarini L, Giani T, Marrani E, Moretti D, Pagnini I, et al. Glucocorticoids in the management of systemic juvenile idiopathic arthritis. Pediatr Drugs 2013;15:343–9. [DOI] [PubMed] [Google Scholar]
- 20. Guzman J, Kerr T, Ward LM, Ma J, Oen K, Rosenberg AM, et al. Growth and weight gain in children with juvenile idiopathic arthritis: results from the ReACCh‐Out cohort. Pediatr Rheumatol 2017;15:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lovell DJ, Giannini EH, Reiff AO, Kimura Y, Li S, Hashkes PJ, et al. Long‐term safety and efficacy of rilonacept in patients with systemic juvenile idiopathic arthritis. Arthritis Rheum 2013;65:2486–96. [DOI] [PubMed] [Google Scholar]
- 22. Wallace CA, Ruperto N, Giannini E, on behalf of the Childhood Arthritis and Rheumatology Research Alliance, Pediatric Rheumatology International Trials Organization, and the Pediatric Rheumatology Collaborative Study Group . Preliminary criteria for clinical remission for select categories of juvenile idiopathic arthritis. J Rheumatol 2004;31:2290–4. [PubMed] [Google Scholar]
- 23. Brunner HI, Lovell DJ, Finck BK, Giannini EH. Preliminary definition of disease flare in juvenile rheumatoid arthritis. J Rheumatol 2002;29:1058–64. [PubMed] [Google Scholar]
- 24. Brunner HI, Rider LG, Kingsbury DJ, Co D, Schneider R, Goldmuntz E, et al. Pediatric Rheumatology Collaborative Study Group: over four decades of pivotal clinical drug research in pediatric rheumatology [review]. Pediatr Rheumatol Online J 2018;16:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ruperto N, Martini A. Networking in paediatrics: the example of the Paediatric Rheumatology International Trials Organisation (PRINTO). Arch Dis Child 2011;96:596–601. [DOI] [PubMed] [Google Scholar]
- 26. Sun H, Van LM, Floch D, Jiang X, Klein UR, Abrams K, et al. Pharmacokinetics and pharmacodynamics of canakinumab in patients with systemic juvenile idiopathic arthritis. J Clin Pharmacol 2016;56:1516–27. [DOI] [PubMed] [Google Scholar]
- 27. Giannini EH, Ruperto N, Ravelli A, Lovell DJ, Felson DT, Martini A. Preliminary definition of improvement in juvenile arthritis. Arthritis Rheum 1997;40:1202–9. [DOI] [PubMed] [Google Scholar]
- 28. Consolaro A, Bracciolini G, Ruperto N, Pistorio A, Magni‐Manzoni S, Malattia C, et al. Remission, minimal disease activity, and acceptable symptom state in juvenile idiopathic arthritis: defining criteria based on the juvenile arthritis disease activity score. Arthritis Rheum 2012;64:2366–74. [DOI] [PubMed] [Google Scholar]
- 29. Consolaro A, Ruperto N, Bracciolini G, Frisina A, Gallo MC, Pistorio A, et al. Defining criteria for high disease activity in juvenile idiopathic arthritis based on the Juvenile Arthritis Disease Activity Score. Ann Rheum Dis 2014;73:1380–3. [DOI] [PubMed] [Google Scholar]
- 30. Consolaro A, Ruperto N, Bazso A, Pistorio A, Magni‐Manzoni S, Filocamo G, et al. Development and validation of a composite disease activity score for juvenile idiopathic arthritis. Arthritis Care Res 2009;61:658–66. [DOI] [PubMed] [Google Scholar]
- 31. Ruperto N, Ravelli A, Pistorio A, Malattia C, Cavuto S, Gado‐West L, et al. Cross‐cultural adaptation and psychometric evaluation of the Childhood Health Assessment Questionnaire (CHAQ) and the Child Health Questionnaire (CHQ) in 32 countries: review of the general methodology. Clin Exp Rheumatol 2001;19:S1–9. [PubMed] [Google Scholar]
- 32. Nordal EB, Zak M, Aalto K, Berntson L, Fasth A, Herlin T, et al. Validity and predictive ability of the juvenile arthritis disease activity score based on CRP versus ESR in a Nordic population‐based setting. Ann Rheum Dis 2012;71:1122–7. [DOI] [PubMed] [Google Scholar]
- 33. McErlane F, Beresford MW, Baildam EM, Chieng SEA, Davidson JE, Foster HE, et al. Validity of a three‐variable Juvenile Arthritis Disease Activity Score in children with new‐onset juvenile idiopathic arthritis. Ann Rheum Dis 2013;72:1983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vastert SJ, de Jager W, Noordman BJ, Holzinger D, Kuis W, Prakken BJ, et al. Effectiveness of first‐line treatment with recombinant interleukin‐1 receptor antagonist in steroid‐naive patients with new‐onset systemic juvenile idiopathic arthritis: results of a prospective cohort study. Arthritis Rheumatol 2014;66:1034–43. [DOI] [PubMed] [Google Scholar]
- 35. Grom AA, Ilowite NT, Pascual V, Brunner HI, Martini A, Lovell D, et al. Rate and clinical presentation of macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis treated with canakinumab. Arthritis Rheumatol 2016;68:218–28. [DOI] [PubMed] [Google Scholar]
- 36. Committee for Medicinal Products for Human Use . Guideline on immunogenicity assessment of therapeutic proteins. URL: https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐immunogenicity‐assessment‐therapeutic‐proteins‐revision‐1_en.pdf. European Medicines Agency. May 2017. [Google Scholar]
- 37. Schulz KF, Altman DG, Moher D, on behalf of the CONSORT Group . CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMC Med 2010;8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Foell D, Wulffraat N, Wedderburn LR, Wittkowski H, Frosch M, Gerss J, et al. Methotrexate withdrawal at 6 vs 12 months in juvenile idiopathic arthritis in remission: a randomized clinical trial. JAMA 2010;303:1266–73. [DOI] [PubMed] [Google Scholar]
- 39. Lovell DJ, Johnson AL, Huang B, Gottlieb BS, Morris PW, Kimura Y, et al. Risk, timing, and predictors of disease flare after discontinuation of anti–tumor necrosis factor therapy in children with polyarticular forms of juvenile idiopathic arthritis with clinically inactive disease. Arthritis Rheumatol 2018;70:1508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Appendix
Supplementary Figure S1
Supplementary Figure S2
Supplementary Tables S1 and S2
Supplementary Figure Legends S1 and S2