

Abstract
Aim
To compare the pharmacokinetic exposure of SAR341402 Mix 70/30 (SARAsp‐Mix) with US‐ and European (EU)‐approved versions of insulin aspart Mix 70/30 (NovoLog Mix 70/30 [NN‐Mix‐US]/NovoMix 30 [NN‐Mix‐EU]) and SAR341402 insulin aspart solution (SAR‐Asp) in subjects with type 1 diabetes.
Materials and Methods
This was a randomized, double‐blind, crossover trial in two cohorts. Fifty‐two subjects received a single subcutaneous 0.3 U/kg dose of each treatment and underwent a euglycaemic clamp procedure lasting for a maximum of 24 hours after dosing. In cohort 1, subjects (N = 36) were exposed once each to SARAsp‐Mix, NN‐Mix‐US and NN‐Mix‐EU. In cohort 2, subjects (N = 16) were exposed once each to SARAsp‐Mix and SAR‐Asp.
Results
Of the 52 subjects randomized, 48 completed all treatment periods. In cohort 1, the extent of exposure (total and maximum concentration) was similar among the three treatments, with the 90% confidence intervals for pairwise treatment ratios meeting the predefined acceptance range (0.80 to 1.25). In cohort 2, statistically significant differences (P < .001) in early (0‐4 hours) and intermediate (4‐12 hours) exposure to SARAsp‐Mix compared with SAR‐Asp were observed, all exceeding a 20% difference. Pharmacodynamic results were in support of the pharmacokinetic findings for both cohorts. All treatments were well tolerated and there were no relevant differences in safety variables among treatments.
Conclusions
SARAsp‐Mix showed similar pharmacokinetic exposure to commercially available insulin aspart Mix 70/30 formulations, and a distinct exposure profile compared with SAR‐Asp.
Keywords: biosimilar, insulin aspart mix, pharmacodynamics, pharmacokinetics, phase I study, premix, type 1 diabetes
1. INTRODUCTION
SAR341402 (SAR‐Asp; Sanofi, Paris, France) is a biosimilar insulin aspart product, a human insulin analogue of recombinant DNA origin. 1 Insulin aspart is the active ingredient of a rapid‐acting insulin product currently marketed as NovoLog in the United States 2 and as NovoRapid in Europe (EU), 3 and of a premixed rapid‐ and intermediate‐acting insulin product currently marketed as NovoLog Mix 70/30 4 in the United States and as NovoMix 30 5 in Europe for the treatment of diabetes. SAR‐Asp has the same amino acid sequence and structure as insulin aspart in the reference medicinal product, NovoLog/NovoRapid (NN‐Asp; Novo Nordisk, Bagsværd, Denmark) and is formulated at a concentration of 100 U/mL.
In accordance with relevant US and EU guidelines, 6 , 7 , 8 , 9 a stepwise approach has been utilized to show that SAR‐Asp is similar to NN‐Asp in physicochemical analyses, non‐clinical and clinical studies. Similar pharmacokinetic exposure and pharmacodynamic activity were shown for SAR‐Asp versus both US‐approved (NovoLog) and EU‐approved (NovoRapid) NN‐Asp, as well as between US‐approved and EU‐approved NN‐Asp in a study in subjects with type 1 diabetes (T1D) using the euglycaemic clamp technique. 10 In a separate study, SAR‐Asp also showed similar exposure and activity compared with Japan‐approved NN‐Asp (NovoRapid) in healthy subjects. 11 In addition, similar efficacy, safety and immunogenicity of SAR‐Asp and NN‐Asp were reported in a multinational, open‐label, randomized phase 3 study in participants with T1D and type 2 diabetes (T2D) using insulin glargine 100 U/mL (Lantus) as the basal insulin. 12 , 13 SAR‐Asp and NN‐Asp were also well tolerated and had similar infusion set occlusions over a 4‐week period in insulin pump users with T1D. 14
In addition to the rapid‐acting solution, SAR‐Asp is in development as a premixed formulation with 70% protamine‐crystallized insulin aspart suspension and 30% insulin aspart solution (SARAsp‐Mix; Sanofi, Paris, France) that provides coverage of prandial and basal insulin in a single injection. The originator premixed insulin aspart product, US‐approved NovoLog Mix 70/30 (NN‐Mix‐US) and EU‐approved NovoMix 30 (NN‐Mix‐EU), has been marketed for use in adults and children (EU only) with T1D and T2D in many countries for ∼20 years, with a well characterized pharmacological, efficacy and safety profile. 15 , 16
Intensive insulin treatment with multiple daily injection (MDI) therapy of basal and mealtime insulin is recommended for people with T1D and for those individuals with T2D who require more intensive glycaemic control. 17 For people with T2D who require intensification, the initial addition of basal insulin to oral medication regimens is well established and shows less hypoglycaemia and weight gain than combinations using premixed insulin formulations or prandial insulin. 18 People unable to maintain glycaemic targets on basal insulin may then require basal‐bolus treatment. Although a basal‐bolus insulin regimen consisting of MDI therapy of prandial rapid‐acting insulin in combination with a long‐acting basal insulin most closely mimics normal insulin secretion, 19 the complexity and intensity of this regimen can be intrusive for some patients and lead to poor compliance. For this reason, people who are unable to cope with the demands of an intensive basal‐bolus regimen may choose a simpler biphasic premixed regimen. Premixed insulins remain widely used in different regions of the world, 20 , 21 with efficacy and safety outcomes similar to those of basal or basal‐bolus insulin. 22 , 23 These insulins offer an attractive option of delivering both rapid and longer‐acting insulin in a single convenient injection, and theoretically address fasting, nocturnal and prandial aspects of glucose management. 24 Although premixed insulins are available in various basal/bolus ratios, the 70/30 ratio of intermediate‐acting and rapid‐acting solution remains the most common one used in clinical practice. 25 The use of biosimilar premixed insulin formulations has the potential to reduce drug treatment costs as they are priced lower than the originator products, thereby facilitating greater access to insulin treatment. 26
The clinical development programme of SARAsp‐Mix was designed to show similarity in the exposure of SARAsp‐Mix versus the reference therapy (NN‐Mix‐US and NN‐Mix‐EU), and distinctiveness in early and intermediate exposure of SARAsp‐Mix compared with the individual insulin component SAR‐Asp (rapid‐acting solution) before clinical evaluation in a phase 3 study. 27 The pharmacodynamic activity should support the pharmacokinetic findings. In this study, we report the results of a euglycaemic clamp study in subjects with T1D to achieve these objectives.
2. MATERIALS AND METHODS
This was a single‐centre, randomized, double‐blind, controlled, single‐dose, four‐treatment, euglycaemic clamp crossover study in two cohorts (ClinicalTrials.gov identifier: NCT03916601) undertaken during 2017‐2018. Cohort 1 had three treatments, three periods and three sequences; cohort 2 had two treatments, two periods and two sequences. Subjects were exposed to each treatment once only. The study protocol was approved by an independent Ethics Committee and was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all subjects before study entry.
2.1. Subjects
Participants were enrolled at the Profil Institute (Neuss, Germany) and included male and female subjects aged 18‐64 years (both inclusive) with T1D (duration >1 year) but otherwise healthy, with a body mass index of 18 to 30 kg/m2, a fasting negative serum C‐peptide level of less than 0.3 nmol/L, an HbA1c level of 75 mmol/mol or lower (≤9%), an anti‐insulin antibody titre of 30.0 kU/L or less based on local laboratory assessment, a total insulin dose of less than 1.0 U/kg/day, a total basal insulin dose of 0.4 U/kg/day or less, and a stable insulin regimen for at least 2 months prior to the study.
2.2. Study design and treatments
An overview of the study design is outlined in Figure S1. In both cohorts, a screening period of up to 28 days was followed by three (cohort 1) or two (cohort 2) inpatient treatment periods (each of 2 days' duration), each separated by a 5‐ to 18‐day washout period (preferentially 7 days) and a follow‐up visit at least 5 days after the last dose.
Subjects were randomly assigned using a computer‐generated list (one each for cohort 1 and cohort 2) to receive one of three possible treatment sequences (cohort 1) or one of two treatment sequences (cohort 2) of single 0.3 U/kg doses of SARAsp‐Mix (test product), NN‐Mix‐US (100 U/mL), NN‐Mix‐EU (100 U/mL) or SAR‐Asp. SARAsp‐Mix and SAR‐Asp were manufactured by Sanofi (Frankfurt, Germany) and provided as a suspension (SARAsp‐Mix) and solution (SAR‐Asp) for injection at a concentration of 100 U/mL. NN‐Mix‐US and NN‐Mix‐EU were provided as commercial formulations. To maintain double‐blinding and consistency of dosing methodology, study treatments were withdrawn by an unblinded investigator from cartridges with insulin syringes, with SARAsp‐Mix, NN‐Mix‐US and NN‐Mix‐EU mixed according to prescribing information to ensure a complete resuspension 4 , 5 ; this individual was not otherwise involved in the study.
Before each dosing visit, subjects underwent a washout of their usual insulin therapy, with the last dose of basal insulin received at least 24 hours before dosing for NPH insulin or other intermediate‐acting insulins, 48 hours before for first‐generation basal insulins (Lantus, insulin detemir), and 72 hours for second‐generation basal insulins (insulin degludec, insulin glargine 300 U/mL). During this period, subjects using long‐acting insulin products were switched to intermediate insulin products, and those using intermediate products to short‐acting products at least 24 hours prior to the clamp. All were switched to a short‐acting insulin by 24 hours before dosing. Subjects were required to abstain from injection or infusion of short‐acting insulin products (other than insulin aspart) for a minimum of 8 hours before dosing (see Figure S2 for further details).
2.3. Pharmacokinetic evaluation
Venous blood samples were collected before and then at frequent times after dosing in each treatment period, that is, every 10 minutes for the first 2 hours, every 30 minutes from 2 to 5 hours, every 60 minutes from 5 to 10 hours, every 120 minutes from 10 to 20 hours, and then at 24 hours. All samples were centrifuged within 20 minutes of collection. Plasma was transferred to separate tubes, frozen immediately and stored at −60 to −80°C until analysis.
Plasma concentrations of insulin aspart following administration of SARAsp‐Mix and NN‐Mix‐US/NN‐Mix‐EU were analysed using a validated liquid chromatography–tandem mass spectrometry assay at Syneos Health (Québec, Canada). The measurement range of the assay was 100‐8000 pg/mL with 100 pg/mL defined as the lower limit of quantification. Cross‐validations of the assay with respective marketed insulin aspart products available in the United States and EU were also performed, with the assay shown to be precise, accurate, sensitive and selective over the validated range. For subjects receiving a rescue insulin during the clamp procedure (after dosing of study drugs), plasma concentrations of insulin aspart following administration of SARAsp‐Mix and NN‐Mix‐US/NN‐Mix‐EU were analysed up to the start time of the rescue insulin administration.
2.4. Pharmacodynamic evaluation using euglycaemic clamp
The pharmacodynamic effect of the insulin aspart products was evaluated using the euglycaemic clamp technique, as described previously. 10 During the euglycaemic clamp, the blood glucose (BG) concentration and the glucose infusion rate (GIR), representing the amount of external glucose needed to keep a subject's BG concentration at its target level, were continuously measured and recorded by the clamp device (ClampArt, Profil, Neuss, Germany). The amount of glucose required (area under the body weight‐standardized GIR time curve [GIR‐AUC]) is a measure of the metabolic activity of the investigated insulin (decrease in endogenous glucose production and variable peripheral glucose uptake). The clamp device determined BG levels at 1‐minute intervals and adjusted the GIR in response to changes in BG using a predefined algorithm to keep a euglycaemic BG level of 100 mg/dL (5.5 mmol/L). During the clamp, arterialized venous BG concentration, 28 which reflects the supply for total glucose utilization of all tissues, as well as GIRs, was continuously monitored. In addition, blood samples (∼0.1 mL) were collected at 30‐minute intervals for concurrent clamp device adjustment as part of the calibration procedure to maintain the glycaemic clamp.
The clamp procedure was performed under fasting conditions following an overnight fasting period. Once connected to the clamp device, a variable basal intravenous infusion of insulin glulisine (0.3 U/mL) or 20% glucose solution was initiated to achieve a BG target level of 100 mg/dL (5.5 mmol/L). After BG levels were stabilized for at least 1 hour without any glucose infusion, the insulin aspart products were administered. The insulin glulisine infusion was discontinued at least 20 minutes before dosing. Based on the duration of action of insulin premixes after subcutaneous injection, a maximum clamp duration of 24 hours was considered sufficient to adequately monitor subjects and account for individual variations in insulin elimination and the duration of pharmacodynamic activity. The clamp was prematurely terminated if BG consistently exceeded 200 mg/dL (11.1 mmol/L) with no glucose infusion for the last 30 minutes. In the case of early clamp termination, subjects received insulin glulisine as rescue insulin.
2.5. Safety evaluation
The safety and tolerability of SARAsp‐Mix was assessed by 12‐lead ECG, vital signs, routine laboratory variables, physical examination, reporting of adverse events (AEs) and injection site reactions, and any episodes of hypoglycaemia. AEs were coded using Medical Dictionary for Regulatory Activities version 20.1.
2.6. Pharmacokinetic and pharmacodynamic variables
Pharmacokinetic variable estimates for insulin aspart after administration of SARAsp‐Mix, SAR‐Asp solution and NN‐Mix‐US/NN‐Mix‐EU were calculated using standard non‐compartmental methods with Phoenix WinNonlin version 6.4 (Certara USA, Inc., Princeton, NJ, USA). Area under the plasma insulin aspart concentration‐time curve was calculated using the log‐linear trapezoidal rule from time zero (predose) up to the time of the last quantifiable concentration (INS‐AUClast) and extrapolated to infinity (INS‐AUCinf). For cohort 1, the primary pharmacokinetic variables were the maximum observed plasma insulin aspart concentration (INS‐Cmax) and INS‐AUClast. Secondary pharmacokinetic endpoints in cohort 1 included area under the insulin concentration time curve from 0 to 4 hours postadministration (INS‐AUC0‐4h) and from 4 to 24 hours postadministration (INS‐AUC4‐24h). For cohort 2, the primary pharmacokinetic variables were INS‐AUC0‐4h, from 4 to 12 hours postadministration (INS‐AUC4‐12h) and INS‐Cmax.
Secondary pharmacodynamic variables were recorded during the glucose clamp procedure, where the GIR over time was used as a measure of insulin effect during each clamp period. The calculated variables included the GIR‐AUC from 0 to 24 hours (GIR‐AUC0‐24h) and the maximum body weight‐standardized GIR (GIRmax) for cohort 1; and the GIR‐AUC from 0 to 4 hours (GIR‐AUC0‐4h), GIR‐AUC4‐12h and GIRmax for cohort 2. The maximum of the raw (unsmoothed) body weight‐standardized GIR was subject to noise in the GIR adjustment. Thus, the derivation of GIRmax was based on smoothing technique (locally weighted regression in smoothing scatter [LOESS] plots; SAS, PROC LOESS, factor 0.15) for the raw body weight‐standardized GIR data and BG levels. Because of the expected morphology of the GIR profiles, a smoothing factor of 15% was used.
2.7. Sample size and statistical analyses
The primary aim of the cohort 1 analysis was to show similarity (equivalence) in exposure of SARAsp‐Mix to NN‐Mix‐US and NN‐Mix‐EU. To achieve this, a total of 30 evaluable subjects was required, assuming a true within‐subject standard deviation (SD) of 0.20 or lower for natural log‐transformed INS‐Cmax and INS‐AUClast, for a true treatment ratio of between 0.95 and 1.05. Sample sizes were planned to provide at least 95% power to show equivalence with 5% type 1 error for the pharmacokinetic variables using the 0.80 to 1.25 acceptance range, in agreement with regulatory guidance. 6 To allow for drop‐outs, cohort 1 planned to recruit 36 subjects.
The primary aim of the cohort 2 analysis was to show distinctiveness in exposure of SARAsp‐Mix and SAR‐Asp. Regulatory guidance recommends confirmation that the premixed insulin product (i.e. SARAsp‐Mix) has a distinct pharmacokinetic and pharmacodynamic profile compared with its individual insulin component (i.e. SAR‐Asp), defined in this study as a target difference of at least 20% as per guidance. 7 To show a significant difference in exposure between SAR‐Mix and SAR‐Asp, a total of 12 evaluable subjects was required, assuming a true within‐subject SD of 0.20 or lower for log‐transformed INS‐Cmax, INS‐AUC0‐4h and INS‐AUC4‐12h, for a true treatment ratio of 1.43 or higher. Sample sizes were planned to provide at least 95% power to show a significant difference with 5% type 1 error (two‐sided) for the pharmacokinetic variables using a 0.7 or lower and 1.43 or higher treatment ratio range. To allow for drop‐outs, cohort 2 planned to recruit 16 subjects.
In cohort 1, the log‐transformed INS‐Cmax and INS‐AUClast were evaluated with a linear mixed effects model including subject within sequence as a random effect with period, sequence and treatment as fixed effects. For each variable, the estimated difference in treatment means along with the 90% confidence limits was back‐transformed to estimate the treatment ratio of geometric means and the confidence limits. Similarity for the primary pharmacokinetic variables INS‐Cmax and INS‐AUClast was concluded if the 90% confidence intervals (CIs) of the treatment ratios of the geometric means were entirely within the 0.80 to 1.25 equivalence interval, in keeping with regulatory guidance for showing bioequivalence. 6 In cohort 2, log‐transformed INS‐Cmax, INS‐AUC0‐4h and INS‐AUC4‐12h were analysed using a similar statistical model to that used for cohort 1. The ratio was tested for significant deviation from the value 1.0 (two‐sided, alpha = .05), and a P‐value was derived. Correspondingly, the 95% CIs of the treatment ratios of the geometric means were presented, consistent with regulatory guidance for showing differences between two treatments. 7
Pharmacodynamic variables were summarized by treatment per cohort using descriptive statistics. Pharmacodynamic analyses were secondary and were performed without the aim of formal inferential statistics. In each cohort, similar analyses to the pharmacokinetic variables were performed. Statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).
Clamp quality was assessed by determining the time span of euglycaemia, defined as the time from dosing to the last value of the smoothed BG concentration curve at or below 105 mg/dL (5.8 mmol/L); the duration of euglycaemia; the individual mean of the BG concentrations; the precision, defined as the individual coefficient of variation (CV, %) of BG device measurements during euglycaemia; and the control deviation, defined as the mean absolute difference of individual mean BG measurements from the clamp target level, as described previously. 10
3. RESULTS
Fifty‐two subjects were randomized and treated, with 48 completing the study. In cohort 1, 36 subjects were randomized, and 34 subjects completed all three study treatment periods. In cohort 2, 16 subjects were randomized, and 14 subjects completed both treatment periods. Baseline characteristics of the randomized subjects are given in Table 1. All participants were Caucasian, 43 were male and nine were female, with an overall mean age of 44.2 years. Participant disposition and details of the four subjects who withdrew from the study are presented in Table S1. Per treatment, 35 profiles were evaluable for cohort 1 and 15 profiles were evaluable for cohort 2.
TABLE 1.
Baseline characteristics of the study population (safety population)
| Cohort 1 (N = 36) | Cohort 2 (N = 16) | |
|---|---|---|
| Male, n (%) | 32 (88.9) | 11 (68.8) |
| Age (years) | 45.7 ± 10.3 [27‐62] | 40.8 ± 12.7 [20‐60] |
| White race, n (%) | 36 (100.0) | 16 (100.0) |
| Body weight | 83.0 ± 8.5 [68.4‐96.9] | 77.8 ± 9.1 [58.3‐96.4] |
| Body mass index (kg/m2) | 26.0 ± 2.3 [22.3‐29.8] | 25.3 ± 2.4 [21.7‐28.6] |
| Duration of diabetes, years | 27.1 ± 10.6 [8‐54] | 19.2 ± 10.8 [2‐45] |
| HbA1c, % | 7.30 ± 0.86 [5.5‐8.9] | 7.20 ± 0.62 [5.9‐8.3] |
Note: All average data are mean ± standard deviation [range].
3.1. Pharmacokinetics
In cohort 1, the pharmacokinetic profiles of the three insulin aspart mix products were virtually superimposable, as shown when plotting the mean insulin aspart concentration versus time for SARAsp‐Mix, NN‐Mix‐US and NN‐Mix‐EU (Figure 1A). The 90% CIs of the treatment ratios for primary pharmacokinetic variables INS‐Cmax and INS‐AUClast were entirely within the predefined acceptance interval of 0.80 to 1.25 (Table 2), confirming equivalent exposure of SARAsp‐Mix compared with NN‐Mix‐US and NN‐Mix‐EU. Descriptive statistics per treatment for pharmacokinetic variables are shown in Table S2. The pharmacokinetic variables show low to moderate between‐subject variability, as shown by CVs between 32% and 37%. The 90% CIs of the treatment ratios for secondary pharmacokinetic variables INS‐AUC0‐4h and INS‐AUC4‐24h were also entirely contained within the 0.80 to 1.25 acceptance range (Table S3).
FIGURE 1.
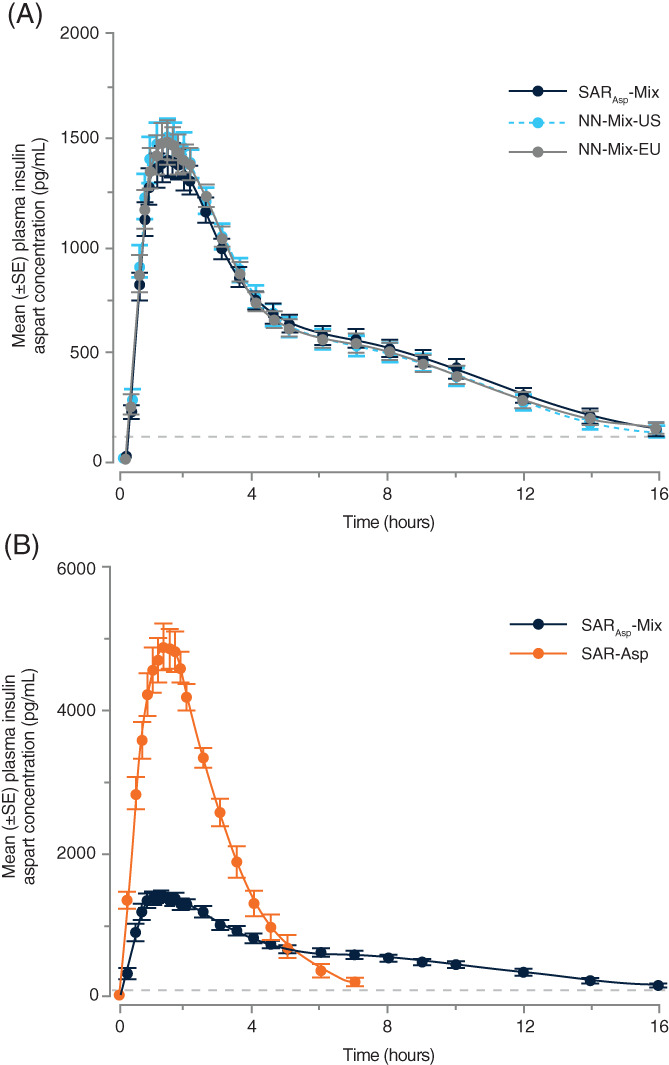
Pharmacokinetic profiles for SARAsp‐Mix, NN‐Mix‐US, NN‐Mix‐EU and SAR‐Asp versus time. Mean insulin aspart plasma concentrations in (A) cohort 1 and (B) cohort 2. The horizontal dotted line represents the lower limit of quantification (100 pg/mL)
TABLE 2.
Primary pharmacokinetic variables in cohorts 1 and 2
| Group | Variable | Treatment ratio | Point estimate (CI) |
|---|---|---|---|
| Cohort 1 | (90% CI) a | ||
| INS‐Cmax | SARAsp‐Mix vs. NN‐Mix‐US | 0.91 (0.84 to 0.99) | |
| SARAsp‐Mix vs. NN‐Mix‐EU | 0.92 (0.85 to 1.00) | ||
| INS‐AUClast | SARAsp‐Mix vs. NN‐Mix‐US | 1.00 (0.92 to 1.08) | |
| SARAsp‐Mix vs. NN‐Mix‐EU | 0.99 (0.91 to 1.07) | ||
| Cohort 2 | (95% CI) b | ||
| INS‐Cmax | SARAsp‐Mix vs. SAR‐Asp | 0.29 (0.25 to 0.33)* | |
| INS‐AUC0‐4h | SARAsp‐Mix vs. SAR‐Asp | 0.33 (0.29 to 0.36)** | |
| INS‐AUC4‐12h | SARAsp‐Mix vs. SAR‐Asp | 2.89 (1.71 to 4.89)*** |
Abbreviations: CI, confidence interval; INS‐AUClast, area under the drug plasma concentration‐time curve from time 0 to the time of the last quantifiable data point; INS‐Cmax, maximum insulin aspart concentration in plasma.
Note: P‐values for the effect of treatment on primary pharmacokinetic variables: *P < .0001, **P < .0001, ***P < .0008.
90% CI for the pairwise treatment ratios in keeping with regulatory guidance for showing bioequivalence between treatments. 6
95% CI for the pairwise treatment ratios in keeping with regulatory guidance for confirming a difference between treatments. 7
In cohort 2, the pharmacokinetic profile of SARAsp‐Mix was generally flatter than that of the rapid‐acting solution SAR‐Asp when plotting the mean insulin aspart concentration versus time (Figure 1B). The 95% CIs for the ratios of the geometric means (SARAsp‐Mix vs. SAR‐Asp) for the primary pharmacokinetic variables (INS‐Cmax, INS‐AUC0‐4h and INS‐AUC4‐12h) exclude the value 1.0, thereby showing statistically significant differences in early and intermediate pharmacokinetic variables between SARAsp‐Mix and SAR‐Asp (Table 2). The P‐values for treatment effect were all below .001. Point estimates indicate a clear distinction and were greater than 20% between the two products. Descriptive statistics per treatment for the primary pharmacokinetic variables (INS‐Cmax, INS‐AUC0‐4h and INS‐AUC4‐12h) are shown in Table S4. The pharmacokinetic variables show low to moderate between‐subject variability, as shown by CVs between 12% and 37%.
3.2. Pharmacodynamics
In cohort 1, the pharmacodynamic profiles of the three insulin aspart mix products were similar, as shown in plots of the mean smoothed standardized GIR profiles (Figure S3A) and the median of percentage of cumulative glucose infusion based on GIR‐AUC0‐24h versus time (Figure S3C). The results were consistent and supportive of the pharmacokinetic findings. The onset and duration of action were similar, with SARAsp‐Mix reaching a slightly lower mean peak value compared with the other two treatments. Descriptive statistics per treatment for secondary pharmacodynamic variables are shown in Table S2. The extent of the glucose‐lowering effect, as indicated by GIR‐AUC0‐24h and GIRmax, was similar among the three treatments (Table S5). For GIR‐AUC0‐24h, the 90% CIs for the pairwise treatment ratio were within the classical bioequivalence range of 0.80 to 1.25 used for pharmacokinetic variables, whereas for GIRmax, the lower boundaries of the 90% CIs were slightly below 0.80. Between‐subject variability for GIR‐AUC0‐24h and GIRmax was moderate, as shown by CVs per treatment between 33% and 50% (Table S2).
In cohort 2, the pharmacodynamic profiles of the two insulin treatments showed a clear difference in activity (Figure S3B and D), with SARAsp‐Mix having a flatter profile with a much slower onset and a longer duration of action compared with SAR‐Asp rapid‐acting solution. Descriptive statistics per treatment for pharmacodynamic variables are shown in Table S4. The differences in the mean GIR‐AUC and mean GIRmax values for SARAsp‐Mix and SAR‐Asp reflect the differences in the formulations. Point estimates for the GIRmax, GIR‐AUC0‐4h and GIR‐AUC4‐12h ratios comparing SARAsp‐Mix and SAR‐Asp, and the corresponding 95% CIs, are shown in Table S5. The 95% CIs for the ratios of the geometric means (SARAsp‐Mix and SAR‐Asp) exclude the value 1.0, thus showing statistically significant differences in early and intermediate pharmacodynamic variables between SAR‐Asp and SARAsp‐Mix. P‐values in the model for treatment effect were below .001 for GIRmax and GIR‐AUC0‐4h. Point estimates indicated a clear distinction among the two formulations, exceeding the recommended target difference of 20%, with a flatter profile for SARAsp‐Mix.
3.3. Clamp quality
Clamp quality was high and comparable among treatments (Table S6). The precision (i.e. the CV% of the ClampArt BG measurements) showed median values of 4.0%‐4.5% for the three treatments in cohort 1 and 3.9%‐5.1% for the two treatments in cohort 2. Similarly, the mean difference between the ClampArt BG measurements and the target BG value (control deviation) was generally small, being 0.84‐1.05 mg/dL in cohort 1 and 0.63‐0.73 mg/dL in cohort 2. As expected for cohort 2, the duration of euglycaemia during the clamp procedure was longer for SARAsp‐Mix than for the rapid‐acting solution.
3.4. Safety and tolerability
All treatments in cohorts 1 and 2 were well tolerated and there were no relevant differences in safety variables among treatments. There were no serious AEs or AEs of special interest reported during the study, and no severe/serious hypoglycaemic events were observed in either cohort. In both cohorts, the majority of treatment‐emergent AEs (TEAEs) were of mild intensity, none were of severe intensity and none were considered to be related to study treatment by the investigator. One patient in cohort 2 had a fall (reported as a TEAE), which led to a TEAE of contusion (not considered to be related to study treatment) in treatment period 1; the events led to discontinuation of the patient and this patient only received one of the treatments (SAR‐Asp) during the study. There were no clinically relevant changes in any of the standard clinical laboratory and haematology variables during the treatment period in either cohort (data not shown).
4. DISCUSSION
The key finding of the current study, which is the first to investigate the pharmacokinetic and pharmacodynamic properties of a biosimilar insulin aspart premix, was that in individuals with T1D, SARAsp‐Mix showed a similarity in exposure compared with both NN‐Mix‐US and NN‐Mix‐EU, with 90% CIs of the treatment ratios meeting the predefined acceptance range (0.80 to 1.25). In addition, SARAsp‐Mix showed statistically significant differences (P < .001) in early and intermediate exposure compared with the rapid‐acting solution SAR‐Asp, each showing a greater than 20% recommended difference between treatments. 7 The pharmacodynamic results, a secondary outcome of the study, were in support of the pharmacokinetic findings for both cohorts. All treatments were well tolerated and there were no relevant differences in safety variables among treatments.
The single‐dose, crossover design and the euglycaemic glucose clamp used in this study are consistent with regulatory guidance for the development of biosimilar insulin products. 6 Crossover studies are mandatory to show similarity in exposure and activity as they allow each subject to receive all treatments, so that a comparison among the treatments can be made on the same subject. In addition, evaluation of insulin premix products requires differentiation from their rapid‐acting products by concluding that key pharmacokinetic and pharmacodynamic variables are outside the usual 80% to 125% acceptance limits for concluding equivalence. To achieve these aims, a crossover study comparing four different treatments (SARAsp‐Mix, NN‐Mix‐US, NN‐Mix‐EU and SAR‐Asp) was required. 7 To reduce the burden on study participants undergoing repeated 24‐hour euglycaemic clamp procedures, two separate cohorts were used in place of a four‐way crossover study, thereby simultaneously testing corresponding products for similarity and differentiating the rapid‐acting and premixed products. Cohort 1 was a Latin square design with three treatments, three periods and three sequences, and cohort 2 was a two‐treatment, two‐period, two‐sequence crossover study. Subjects were exposed to each treatment once only.
The endpoints used in the study also followed regulatory requirements for assessment of biosimilar premix insulin preparations containing the same active ingredient. 6 The study involved evaluation of a premix preparation (SARAsp‐Mix) that contains the same active ingredient as the rapid‐acting solution (SAR‐Asp). In view of the similar pharmacokinetic and pharmacodynamic profiles shown for the soluble insulin preparation SAR‐Asp and its reference medicinal product (NN‐Asp), 10 the pharmacodynamic data for SARAsp‐Mix, although not needed, are presented in keeping with regulatory guidelines that recommend reporting of all data collected during the conduct of these studies. Pharmacodynamic analyses were therefore secondary, and statistical analyses of pharmacodynamic variables were prespecified as non‐confirmative. The clamp quality, assessed by the individual CV% of BG over the clamp duration (from 0 to the end of euglycaemia), was reliably maintained within reasonable variability, which was indicative of successful performance of the euglycaemic clamp technology.
The strengths of the current study include its crossover design using single subcutaneous doses that enabled the subjects to act as their own control, along with enrolment of subjects with T1D that allowed comparison of exogenous insulins with respect to glucose‐lowering effect without interference from endogenous insulin. 29 Investigator‐related bias was avoided by the automated glucose clamp procedure and the use of a double‐blind design. Because of the duration of action of insulin premixes after subcutaneous injection, a maximum clamp duration of 24 hours was considered sufficient to adequately monitor subjects and account for individual variations in insulin pharmacokinetic and pharmacodynamic profiles. A minimum washout period of 5 to 18 days (preferred 7 days) between dosing occasions was considered acceptable to enable the subjects to re‐establish (as applicable) their usual individual insulin regimens. Similarly, the insulin dose of 0.3 U/kg selected for this study is well characterized to provide euglycaemia in subjects with T1D and has been readily investigated in other similarly designed clamp studies. 6
In summary, SARAsp‐Mix showed similar pharmacokinetic exposure and glucodynamic activity to commercially available biphasic insulin aspart formulations, supporting further clinical evaluation of SARAsp‐Mix as a biosimilar product.
CONFLICT OF INTEREST
C.K. is an employee and co‐owner of Profil, which has received research funds from Adocia, Biocon, Boehringer Ingelheim Pharmaceuticals, Dance Biopharm, Eli Lilly and Company, Gan & Lee Pharmaceuticals, MedImmune, Mylan, Nordic Bioscience, Nestlé, Novo Nordisk, Poxel SA, Sanofi‐Aventis, Wockhardt, Xeris Pharmaceuticals and Zealand Pharma. L.N. is an employee of Profil. W.S., L.T. and B.M. are employees of Sanofi. I.N. is a former employee of Sanofi and is a current consultant to Sanofi.
AUTHOR CONTRIBUTIONS
All the authors participated in the drafting, critical revision and approval of the final version of the manuscript. C.K., L.N., W.S., L.T. and I.N. were involved in the study design, and C.K. and L.N. were investigators in the study. C.K., L.N., W.S., L.T. and I.N. were involved in data acquisition, and L.T. conducted the statistical analyses. All the authors were involved in interpretation of the study results.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/dom.14260.
Supporting information
Appendix S1: Supporting information
ACKNOWLEDGEMENTS
This study was funded by Sanofi. The funder participated in trial design, data collection and analysis. We thank Irma Slomp, Baerbel Rotthaeuser, Christine Einig and Nadine Almeras, all employed by Sanofi, for their assistance during the conduct and analysis of this study. Editorial support was provided by Oberon Ltd (London, UK), funded by Sanofi.
Kapitza C, Nosek L, Schmider W, Teichert L, Mukherjee B, Nowotny I. A single‐dose euglycaemic clamp study in two cohorts to compare the exposure of SAR341402 (insulin aspart) Mix 70/30 with US‐ and European‐approved versions of insulin aspart Mix 70/30 and SAR341402 rapid‐acting solution in subjects with type 1 diabetes. Diabetes Obes Metab. 2021;23:674–681. 10.1111/dom.14260
Funding information Sanofi
DATA AVAILABILITY STATEMENT
Qualified researchers may request access to participant level data and related study documents including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan, and dataset specifications. Participant level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi's data sharing criteria, eligible studies, and process for requesting access can be found at: https://www.clinicalstudydatarequest.com/
REFERENCES
- 1. European Medicines Agency . Insulin aspart Sanofi. Summary of product characteristics, 2020. https://www.ema.europa.eu/en/documents/product-information/insulin-aspart-sanofi-epar-product-information_en.pdf. Accessed July 22, 2020.
- 2. European Medicines Agency . NovoRapid. Summary of product characteristics, 2019. https://www.ema.europa.eu/en/documents/product‐information/novorapid‐epar‐product‐information_en.pdf. Accessed July 22, 2020.
- 3. Novo Nordisk . NovoLog, insulin aspart injection 100 units/mL, NovoNordisk, 2019. https://www.novo-pi.com/novolog.pdf. Accessed July 22, 2020.
- 4. Novo Nordisk . NovoLog Mix 70/30 insulin aspart protamine and insulin aspart injectable suspesion 100 units/mL, NovoNordisk, 2019. https://www.novo-pi.com/novologmix7030.pdf. Accessed July 22, 2020.
- 5. European Medicines Agency . NovoMix 30 (30% insulin aspart and 70% insulin aspart protamine). Summary of product characteristics, updated August 16, 2019. https://www.ema.europa.eu/en/documents/product-information/novomix-epar-product-information_en.pdf. Accessed July 22, 2020.
- 6. European Medicines Agency . Guideline on non‐clinical and clinical development of similar biological medicinal products containing recombinant human insulin and insulin analogues, February 26, 2015. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-non-clinical-clinical-development-similar-biological-medicinal-products-containing_en-0.pdf. Accessed July 22, 2020.
- 7. US Food and Drug Administration Center for Drug Evaluation and Research (CDER) . Guidance for industry. Diabetes Mellitus: Developing Drugs and Therapeutic Biologics for Treatment and Prevention. February 2008:1–30. https://wayback.archive-it.org/7993/20191211232220/https://www.fda.gov/media/71289/download. Accessed July 22, 2020.
- 8. US Food and Drug Administration, Center for Drug Evaluation and Research (CDER) . Guidance for Industry: Questions and Answers on Biosimilar Development and the BPCI Act (Revision 1), December 2018:1–19. https://www.fda.gov/media/119258/download. Accessed July 22, 2020.
- 9. US Food and Drug Administration, Center for Drug Evaluation and Research (CDER) . Guidance for Industry. New and Revised Draft Q&As on Biosimilar Development and the BPCI Act (Revision 2) (Draft Guidance), December 2018:1–14. https://www.fda.gov/media/119278/download. Accessed July 22, 2020.
- 10. Kapitza C, Nosek L, Schmider W, Teichert L, Nowotny I. Single‐dose euglycemic clamp study demonstrating pharmacokinetic and pharmacodynamic similarity between SAR341402 insulin aspart and US‐ and EU‐approved versions of insulin aspart in subjects with type 1 diabetes. Diabetes Technol Ther. 2020;22:278‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. ClinicalTrials.jp . A randomized, double‐blind, single‐dose, 2‐treatment, 2‐period, 2‐sequence crossover study to compare exposure and activity of SAR341402 to NovoRapid using the euglycemic clamp technique, in healthy Japanese male subjects. JapicCTI‐No. JapicCTI‐205379. https://www.clinicaltrials.jp/cti-user/trial/ShowDirect.jsp?japicId=JapicCTI-205379. Accessed July 22, 2020.
- 12. Garg SK, Wernicke‐Panten K, Wardecki M, et al. Efficacy and safety of insulin aspart biosimilar SAR341402 versus originator insulin aspart in people with diabetes treated for 26 weeks with multiple daily injections in combination with insulin glargine: a randomized open‐label trial (GEMELLI 1). Diabetes Technol Ther. 2020;22:85‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garg SK, Wernicke‐Panten K, Wardecki M, et al. Safety, immunogenicity, and glycemic control of insulin aspart biosimilar SAR341402 versus originator insulin aspart in people with diabetes also using insulin glargine: 12‐month results from the GEMELLI 1 trial. Diabetes Technol Ther. 2020;22:516‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thrasher J, Polsky S, Hovsepian L, et al. Safety and tolerability of insulin aspart biosimilar SAR341402 versus originator insulin aspart (novolog) when used in insulin pumps in adults with type 1 diabetes: a randomized, open‐label clinical trial. Diabetes Technol Ther. 2020;22:666‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liebl A, Prusty V, Valensi P, Yang W, Strojek K, Linjawi S. Ten years of experience with biphasic insulin aspart 30: from drug development to the latest clinical findings. Drugs. 2012;72:1495‐1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liebl A, Mohan V, Yang W, Strojek K, Linjawi S. 15 years of experience with biphasic insulin aspart 30 in type 2 diabetes. Drugs R D. 2018;18:27‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. American Diabetes Association: 9 . Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes‐2020. Diabetes Care. 2020;43(suppl 1):S98‐S110. [DOI] [PubMed] [Google Scholar]
- 18. Davies MJ, D'Alessio DA, Fradkin J, et al. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2018;41:2669‐2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anyanwagu U, Mamza J, Gordon J, Donnelly R, Idris I. Premixed vs basal‐bolus insulin regimen in type 2 diabetes: comparison of clinical outcomes from randomized controlled trials and real‐world data. Diabet Med. 2017;34:1728‐1736. [DOI] [PubMed] [Google Scholar]
- 20. Jia W, Xiao X, Ji Q, et al. Comparison of thrice‐daily premixed insulin (insulin lispro premix) with basal‐bolus (insulin glargine once‐daily plus thrice‐daily prandial insulin lispro) therapy in east Asian patients with type 2 diabetes insufficiently controlled with twice‐daily premixed insulin: an open‐label, randomised, controlled trial. Lancet Diabetes Endocrinol. 2015;3:254‐262. [DOI] [PubMed] [Google Scholar]
- 21. Ray KK, Kendall DM, Zhao Z, et al. A multinational observational study assessing insulin use: understanding the determinants associated with progression of therapy. Diabetes Obes Metab. 2019;21:1101‐1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giugliano D, Chiodini P, Maiorino MI, Bellastella G, Esposito K. Intensification of insulin therapy with basal‐bolus or premixed insulin regimens in type 2 diabetes: a systematic review and meta‐analysis of randomized controlled trials. Endocrine. 2016;51:417‐428. [DOI] [PubMed] [Google Scholar]
- 23. Kalra S, Czupryniak L, Kilov G, et al. Expert opinion: patient selection for premixed insulin formulations in diabetes care. Diabetes Ther. 2018;9:2185‐2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rizvi AA. Treatment of type 2 diabetes with biphasic insulin analogues. Eur Med J Diabetes. 2016;4:74‐83. [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar A, Awata T, Bain SC, et al. Clinical use of the co‐formulation of insulin degludec and insulin aspart. Int J Clin Pract. 2016;70:657‐667. [DOI] [PubMed] [Google Scholar]
- 26. Fralick M, Kesselheim AS. The U.S. insulin crisis — rationing a lifesaving medication discovered in the 1920s. New Engl J Med. 2019;381:1793‐1795. [DOI] [PubMed] [Google Scholar]
- 27. EU Clinical Trials Register . A 26‐week, randomized, open‐label, parallel‐group comparison of SAR341402 Mix 70/30 to NovoMix®30 in adult patients with diabetes mellitus using pre‐mix insulin analogs. EudraCT Number: 2017‐000092‐84. https://www.clinicaltrialsregister.eu/ctr-search/trial/2017-000092-84/PL. Accessed July 22, 2020.
- 28. Heise T, Zijlstra E, Nosek L, Heckermann S, Plum‐Mörschel L, Forst T. Euglycaemic glucose clamp: what it can and cannot do, and how to do it. Diabetes Obes Metab. 2016;18:962‐972. [DOI] [PubMed] [Google Scholar]
- 29. Swinnen SG, Holleman F, DeVries JH. The interpretation of glucose clamp studies of long‐acting insulin analogues: from physiology to marketing and back. Diabetologia. 2008;51:1790‐1795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information
Data Availability Statement
Qualified researchers may request access to participant level data and related study documents including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan, and dataset specifications. Participant level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi's data sharing criteria, eligible studies, and process for requesting access can be found at: https://www.clinicalstudydatarequest.com/