

Abstract
A six‐step synthesis towards a tribenzotriquinacene (TBTQ) bearing three quinoxalinophenanthrophenazine (QPP) units is presented. The optoelectronic properties are investigated and the effect of the three‐dimensional arrangement of the individual QPP planes is examined using optical spectroscopy, electrochemical analysis and quantum‐chemical calculations.
Keywords: electronic communication, heterocycles, pyrenes, quinoxalinophenanthrophenazine, tribenzotriquinacene
A six‐step synthesis towards a tribenzotriquinacene (TBTQ) bearing three quinoxalinophenanthrophenazine (QPP) units is presented. The optoelectronic properties are investigated and the effect of the three‐dimensional arrangement of the individual QPP planes is examined using optical spectroscopy, electrochemical analysis and quantum‐chemical calculations.
Introduction
Tribenzotriquinacene (TBTQ) [1] offers a conformational rigid bowl‐shaped framework of high symmetry (C 3v). Its unique orthogonal geometry, in which the three indane moieties are aligned along Cartesian axes, makes TBTQ an interesting building block for the synthesis of three‐dimensional structures, such as curved nonplanar polycyclic hydrocarbons. [2] In the field of supramolecular chemistry, TBTQ has been used for the construction of three‐dimensional assemblies, like for example an octameric hydrogen‐bonded capsule, [3] organic nanocubes, [4] metallosquares [5] or as hosts for complexation of quaternary ammonium cations [6] or C60 and C70 fullerenes. [7] It can also be used to arrange several π systems within the same molecule close in space but separated by an sp3 carbon atom. Examples are a linearly extended TBTQ derivative with three tetracene units [8] or perylene bisimide substituted TBTQ derivatives. [9] Whereas in triptycene based compounds the π planes are perpendicular to a common plane and are oriented mutually at 120°, [10] TBTQ allows a 90° arrangement, which is otherwise difficult to achieve, [2a] except for spiro‐bridged compounds. [11] In the field of organic electronics, molecules with more than one chromophore, which can interact with each other either “through‐bond” or “through‐space”,[ 11a , 12 ] open up a wide area of interesting features, ranging from exciton effects, [13] energy [14] and electron transfer processes [15] or intramolecular singlet fission.[ 11a , 12a , 16 ] Especially for the latter one, an orthogonal orientation has been proved beneficial.[ 11a , 17 ] Apart from this the integration of 2D building blocks into a 3D structure is also an effective method to tune the morphology of thin‐films of pentacenes and tetraazapentacenes [18] or to reduce aggregation and therefore to enhance solubility. This has been demonstrated by incorporating pyrene‐fused pyrazaacenes (PFPs) into a 3D triptycene‐based structure. By this approach, soluble PFPs with up to 22 fused rings could be synthesized. Its diameter is approximately 11 nm, which is up to date the largest soluble N‐heteroacene derivative. [19] PFPs combine high stability with desirable electronic properties, [20] that can further be tuned by appropriate substituents, [21] making them versatile target compounds for materials used for a wide spectrum of applications, including organic field‐effect transistors (OFETs),[ 21g , 22 ] organic light emitting diodes (OLEDs) [23] or photovoltaic cells.[ 22e , 23c , 23d , 23g , 24 ]
While there are examples for triptycene‐based PFPs,[ 19 , 25 ] planar, star shaped PFPs [26] or phenylene bridged PFP dimers, [27] no TBTQ based analogue has been realized so far.
Here, we present the synthesis of such a TBTQ‐tris(quinoxalinophenanthrophenazine (TBTQ‐TQPP) hybrid (Figure 1) bearing three QPP units in an orthogonal arrangement. The electronic communication between the individual QPP units were studied by cyclovoltammetric methods and compared with a structurally related triptycene based TQPP, as well as with smaller model compounds.
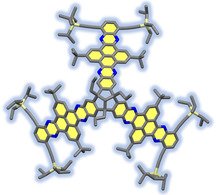
Figure 1.
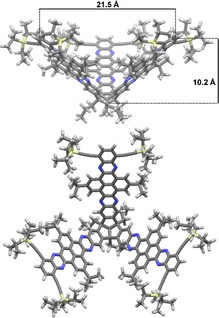
MM2‐optimized model of targeted TBTQ‐TQPP hybrid (9) top: side view; bottom: view into the concave cavity.
Results and Discussion
Synthesis and characterization
The six step synthesis started from C 1 symmetrical triamino‐TBTQ 1 (Scheme 1).[ 4 , 9 ] First, the amino groups were protected as acetamides, giving TBTQ 2 in quantitative yields, followed by a nitration with KNO3 in trifluoracetic acid to give TBTQ 3 in 71 % yield. Subsequently, the amide groups were cleaved under acidic conditions to TBTQ 4, which occurred in 50 % yield. By the reduction with tin(II) chloride in conc. hydrochloric acid hexaammonium hydrochloride salt TBTQ 5 was obtained and used without further purification for the next step. [28] The corresponding hexaamino TBTQ was generated in situ in the presence of potassium acetate, [28] and condensed with a slight excess (3.6 equiv.) of pyrene tetraone 6 [29] under acidic conditions to give TBTQ hexaone 7 in 63 % yield. The target compound 9 was finally synthesized by a second condensation with phenylene diamine 8 [30] and isolated as bright yellow powder in 51 % yield after recrystallization from ethanol/chloroform. The TIPS ethynyl groups are necessary to enhance solubility, as the unsubstituted TBTQ‐TQPP congener was insoluble in all tested solvents such as CHCl3, tetrachloroethane or ortho‐dichlorobenzene. In contrast, TBTQ 9 showed good solubility in most organic solvents, except for polar ones, such as MeOH or acetonitrile. All compounds were characterized by 1H and 13C NMR spectroscopy, IR spectroscopy, high resolution mass spectrometry and elemental analysis (see Supporting Information). Additionally, the structure of TBTQ 4 was confirmed by single‐crystal X‐ray diffraction (see the Supporting Information). We compared TBTQ‐TQPP 9 with its triptycene based analogue 10, which was already published in a different context. [25c] In both structures three QPP planes are connected through a rigid core (TBTQ vs. triptycene) and adopt a different geometrical arrangement (C 3v with orthogonal arrangement of the extended planes vs. D 3h with 120° orientation of the planes). By comparison between these two TQPPs the effect of the different spatial arrangement of the QPP units onto the optoelectronic properties should become clear. In order to gain a deeper insight into the structure‐property relationship of the orthogonal arrangement of three extended π planes, mono‐extended model compounds for both TQPP derivatives were synthesized as well. TBTQ‐QPP 16 was synthesized via a five‐step route starting from TBTQ amine 11 [31] (Scheme 2). Adopted from a procedure for the corresponding triptycene congener[ 21p , 32 ] TBTQ amine 11 was first converted to its acetamide by treatment with acetic anhydride. Subsequent addition of p‐toluene sulfonic acid and an equimolar amount of KNO3 to the reaction mixture gave nitro‐TBTQ acetamide 12 in 64 % yield. Hydrolysis of 12 under basic conditions (NaOH) gave nitro‐TBTQ amine 13 (77 %), which was then reduced with tin(II) chloride in conc. hydrochloric acid and used for the next step without further purification (78 % yield). The diammonium dihydrochloride salt 14 was then condensed with pyrene tetraone 6 as previously described. After column chromatography, dione 15 was obtained in 48 % yield. A second condensation with diamine 8 gave model QPP 16 in 72 % yield after column chromatography. Similarly, QPP 18 was synthesized from dione 17 [21q] in 82 % yield. All compounds were fully characterized (See the Supporting Information for analytical data and spectra).
Scheme 1.
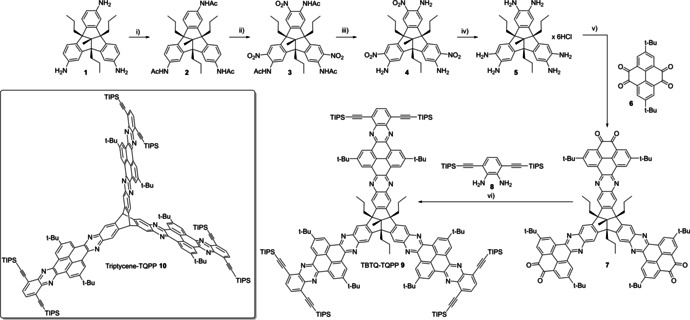
Synthesis of TBTQ‐TQPP 9. Conditions: i) Ac2O, 4 h, rt, 99 %; ii) KNO3, TFA, rt, 5 h, 71 %; iii) EtOH, conc. HCl, 85 °c, rt, 51 %; iv) SnCl2⋅2 H2O, conc. HCl, 90 °C, 18 h; inserted box bottom left: triptycene TQPP 10. [25c]
Scheme 2.
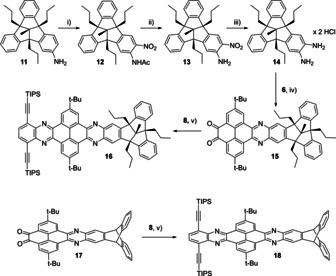
Synthesis of model QPPs 16 and 18. Conditions: i) 1. Ac2O, rt, 2.5 h; 2. KNO3, p‐toluene sulfonic acid, rt, 18 h, 64 %; ii) NaOH, EtOH/H2O, 80 °C, 5 h, 77 %; iii) SnCl2⋅2 H2O, HCl (37 %), CHCl3, 90 °C, 24 h, 78 %; iv) CHCl3/AcOH, 70 °C, 20 h, 48 %; v) CHCl3/AcOH, 70 °C, 20 h, 72 % (16), 82 % (18).
Single crystals suitable for X‐ray diffraction of QPP 18 could be grown by vapor diffusion of MeOH in solutions of 18 in either CHCl3 (solvate α), toluene (solvate β) or by slowly cooling a hot saturated solution in mesitylene (solvate γ). In the α and β solvate the QPP molecules form antiparallel π stacked dimers with d π = 3.38–3.52 Å (Figure 2 a, b), which is frequently found for triptycene end‐capped compounds.[ 21r , 21s , 33 ] Adjacent dimers arrange in an edge‐to‐face orientation in a herringbone‐type motif and interact via van der Waals forces (Figure 2 c). In the β solvate non π stacked QPP molecules alternate with π stacked dimers (Figure 2 d). In the γ structure, mesitylene is π stacked onto the QPP plane (Figure 2 e), forming one‐dimensional mixed stacked columns along the crystallographic c axis (Figure 2 f). Noteworthy, out of 24 structures of triptycene end‐capped QPPs,[ 21r , 21s ] this is the only structure, in which no antiparallel π dimers are found (For a detailed crystallographic discussion, see the Supporting Information).
Figure 2.
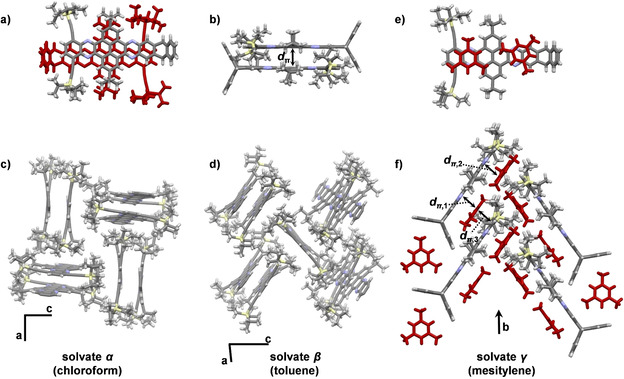
Single crystal X‐ray structures of QPP 18. a, b) π Stacked dimers found in crystals grown from CHCl3 and toluene (d π=3.38–3.52 Å). c) Solvate α obtained by vapor diffusion of MeOH into a CHCl3 solution, triptycene units omitted for clarity. d) Solvate β obtained by vapor diffusion of MeOH into a toluene solution, triptycene units and solvent molecules omitted for clarity. e) Face‐to‐face π stacking between QPP and mesitylene in solvate γ. f) Solvate γ obtained by thermal crystallization from mesitylene, (d π,1=3.46 Å, d π,2=3.41 Å, d π,3=3.39 Å).
Optoelectronic properties
TQPPs 9 and 10 and model QPPs 16 and 18 have been studied by UV/Vis and fluorescence spectroscopy in n‐hexane. Similar spectra for all four compounds were obtained. Vibronically well‐resolved absorption peaks for 9 are found at λ abs = 437, 428, 413, 404, 335 and 321 corresponding to π–π* transitions (Figure 3 a, top). For triptycene analogue 10 the relative intensities of the bathochromic transitions, especially the absorption at λ abs = 428 nm are increased compared to 9. For QPPs 16 and 18 dominant bands at 438, 421 and 398 nm for 16 and 439, 416 and 394 nm for 18 in the red‐shifted region are found (Figure 3 a). To get a better understanding of the differences found in the spectra of mono‐ and threefold extended compounds, TD‐DFT calculations (B3LYP/6–311G(d,p)) were carried out for TQPP 9 and QPP 16 (see Supporting Information). For QPP 16 the π–π* absorption bands are mainly attributed to HOMO−1–LUMO and HOMO−1–LUMO+1 transitions, whereas the HOMO–LUMO transition has only a weak oscillator strength. For TQPP 9 the situation is more complex and a combination of several transitions involving the HOMO/HOMO−1/−2/−3/−4/−5 and LUMO/LUMO+1/+2/+3 orbitals contribute to each absorption band, without a distinct HOMO–LUMO transition, as this is symmetry‐forbidden. The optical gap is nearly the same for all compounds (approx. 2.8 eV). As expected, the extinction coefficients are much larger for TQPP 9 than for QPPs 16 and 18. All four compounds have almost identical emission spectra (Figure 3 b). TQPPs 9 and 10 emit with a photoluminescent quantum yields (PLQY) of 3.5 % (9), respectively 5 % (10) [25c] with maxima at λ em=438 and 466 nm and two shoulders at 450 nm and 493 nm. For QPPs 16 and 18 the emission is similar, but bathochromically shifted by 2 nm with a PLQY of 10 % for 16 and 8 % for 18. The PLQY of TBTQ‐TQPP 9 increased to 11 % in chloroform and THF. The Stokes shifts are E Stokes=52 cm−1 for 9 and 18 and increase to 157 cm−1 for 10 and 207 cm−1 for 16.
Figure 3.
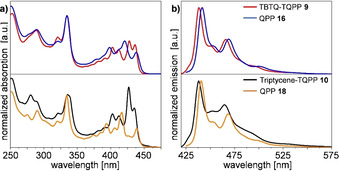
Absorption (a) and emission spectra (b) of TQPPs 9 and 10 and QPPs 16 and 18, measured in n‐hexane (1×10−6 mol L−1) at room temperature.
Absorption and emission spectra of TBTQ‐TQPP 9 have been also measured in different solvents (CHCl3, CH2Cl2, THF, toluene) and no pronounced differences are seen in the absorption spectra, however due to broader bands less peaks can be distinguished compared to the spectrum in n‐hexane (see the Supporting Information). In polar solvents such as MeOH and acetonitrile no spectra could be obtained due to the compounds insolubility. In the corresponding emission spectra the peak maximum shifts bathochromically from λ ex=438 nm for n‐hexane, to λ ex=446 nm for toluene over λ ex=455 for THF to λ ex=458 nm for CH3Cl and λ ex=464 nm for CH2Cl2 (see the Supporting Information).
All compounds were investigated by cyclic voltammetry (CV) and square‐wave voltammetry (SWV) in dichloromethane (Figure 4). The first reduction potential was almost identical for all four compounds (E red,1 = −1.62 to −1.65 V). From the first reduction potential at E red,1 = −1.62 V the electron affinity was estimated to be EA = −3.2 eV via the commonly used expression . [21a] For both model QPPs 16 and 18 three reduction potentials are found at E red = −1.65, −1.98 and −2.39 V and E red = −1.64, −2.02 and −2.40 V, respectively. While the CVs of the TQPPs are poorly resolved due to overlapping peaks, five distinct reduction potentials at E red = −1.62, −1.88, −2.02, −2.16 and −2.44 V can be distinguished in the SWV spectrum of TBTQ 9 and for triptycene 10 (−1.64, −1.90, −2.01, −2.20 and −2.44 V). These differences found in the voltammograms of both TQPP derivatives 9 and 10 compared to their corresponding monosubstituted model compounds 16 and 18 clearly illustrate that the three QPP units are electronically coupled in both compounds like in triptycene, [34] and is not in agreement with previous assumptions that no electronic coupling between individual chromophores occurs, which was based on photophysical investigations with three equal chromophores exclusively.[ 2a , 9 , 35 ]
Figure 4.
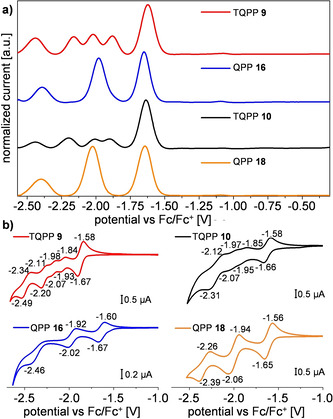
a) Normalized square wave voltammograms (CH2Cl2, nBu4NPF6 (0.1 m), amplitude: 20 mV, frequency: 25 Hz, scanning increment: 5 mV; baseline‐corrected) measured at room temperature with a Pt electrode and a Ag/Ag+ pseudo‐reference electrode and Fc/Fc+ as internal reference; b) cyclic voltammograms (scanning speed: 100 mV s−1) of TQPPs 9 and 10 and QPPs 16 and 18.
To gain a deeper insight into the electronic structure and how it is affected by the geometrical arrangement of the QPP blades, the frontier molecular orbitals of all four compounds were calculated by quantum chemical calculations (DFT‐B3LYP/6–311++G**) (Figures 5 and 6; Table 1). The band gaps between HOMO and LUMO levels are for all compounds in the same range (between 3.0 and 3.2 eV) and comparable to the ones estimated from the UV/Vis measurements (see above).
Figure 5.
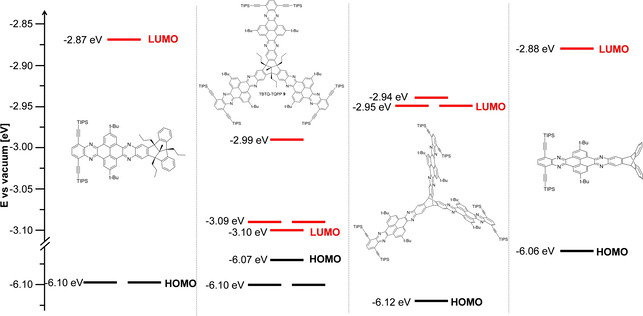
Molecular orbital levels versus vacuum of TBTQ QPP 16, TBTQ‐TQPP 9, triptycene TQPP 10, and triptycene QPP 18 calculated using DFT‐B3LYP:6–311++G**. Propyl and tert‐butyl groups were substituted by methyl groups and TIPS groups were omitted to reduce calculation time.
Figure 6.
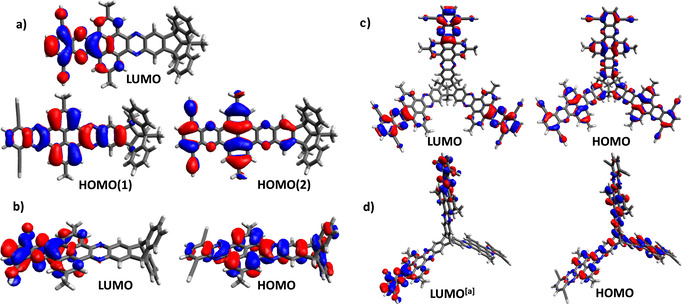
Comparison of Kohn–Sham molecular orbitals of all compounds calculated using DFT‐B3LYP:6–311++G**. a) QPP 16, b) QPP 18; c) TQPP 9, and d) TQPP 10. [a] Please note that only one of the symmetrically identical LUMOs of triptycene 10 is depicted. All MOs are found in the Supporting Information.
Table 1.
Optoelectronic and electrochemical properties of TQPPs 9 and 10 and QPPs 16 and 18.
|
Cmpd |
λ abs [a,b] [nm] (lg ϵ) |
λ onset [a] [nm] |
λ em [nm] (λ ex [nm])[a] |
[cm−1] |
Φ [%] |
[eV] |
[V] |
[mV] |
|
EA [g] [eV] |
[eV] |
[eV] |
[eV] |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
9 |
437 (5.25) |
444 |
438 (427) |
52 |
3.5 (n‐hexane) |
2.8 |
−1.62 (−1.62) |
90 |
3 |
−3.2 |
−6.07 |
−3.10 |
2.97 |
||||||||
|
|
|
|
|
|
11 (CHCl3) |
|
−1.88 (−1.88) |
90 |
1 |
|
|
|
|
||||||||
|
|
|
|
|
|
11 (THF) |
|
−2.02 (−2.02) |
90 |
1 |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
−2.16 (−2.16) |
90 |
1 |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
−2.44 (−2.42) |
150 |
1 |
|
|
|
|
||||||||
|
16 |
438 (4.34) |
447 |
442 (428) |
207 |
10 (n‐hexane) |
2.8 |
−1.65 (−1.64) |
70 |
3 |
−3.2 |
−6.10 |
−2.87 |
3.23 |
||||||||
|
|
|
|
|
|
|
|
−1.98 (−1.97) |
100 |
3 |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
−2.39 (n.a.) |
n.a. |
1 |
|
|
|
|
||||||||
|
10 |
435 |
443 |
438 (415) |
157 |
5 (n‐hexane)[i] |
2.8 |
−1.64 (−1.62) |
80 |
6 |
−3.2 |
−6.12 |
−2.95 |
3.17 |
||||||||
|
|
|
|
|
|
|
|
−1.90 (−1.90) |
100 |
1 |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
−2.01 (−2.02) |
100 |
1 |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
−2.20 (−2.21) |
190 |
1 |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
−2.44 (n.a.) |
n.a. |
1 |
|
|
|
|
||||||||
|
18 |
439 (4.58) |
446 |
440 (419) |
52 |
8 (n‐hexane) |
2.8 |
−1.64 (−1.62) |
90 |
2 |
−3.2 |
−6.06 |
−2.88 |
3.18 |
||||||||
|
|
|
|
|
|
19 (CHCl3) |
|
−2.02 (−2.00) |
120 |
2 |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
−2.40 (−2.32) |
130 |
1 |
[a] Measured in n‐hexane at rt. [b] Absorption maximum at the longest wavelength. [c] Estimated from onset; E g(opt)= . [d] Reduction potentials measured by cyclic voltammetry (CV) (Scan speed: 50 mV s−1) or square‐wave voltammetry (SQV) with a Pt electrode and nBu4NPF6 as electrolyte; ferrocene/ferrocenium (Fc/Fc+) was used as internal reference. [e] Peak‐to‐peak separation (CV); . [f] Relative peak integral (SQV); the integral of the peak at −2.42 V was normalized to 1. [g] Electron affinity estimated from . [h] Obtained from quantum‐chemical calculations using DFT‐B3LYP/6–311++G**. [i] Taken from ref. [25c].
TBTQ QPP 16 has two degenerated HOMOs (E HOMO = −6.10 eV) which are comparable in energy to the HOMO and the two HOMO−1’s of the three‐blades TBTQ‐TQPP 9 (E HOMO = −6.07 eV, E HOMO−1 = −6.07 eV). The twofold degeneration is typical for molecules of C 3 symmetry.[ 8 , 26 , 34c , 36 ] The atom orbital coefficients of these orbitals of 16 show no contributions delocalized at the TBTQ core. This is in contrast to triptycene based model compound 18. Here the orbital coefficients of the HOMO (E HOMO = −6.06 eV) are distributed over the whole molecule including the triptycene moiety, which is also found for the HOMO of triptycene TQPP 10 (E HOMO = −6.06 eV). This is in agreement with a recently demonstrated example of homoconjugation.[ 34c , 34d ] The LUMOs and LUMO+1s of both three‐bladed QPPs 9 and 10 show a distribution of orbital coefficients mainly found in the periphery of the molecules, thus suggesting that the first redox potential found by CV and SWV for both compounds are according to three electrons, creating three negative charges located at the periphery of the arms. Every additional electron is repulsed by Coulomb interactions even stronger, which probably is the reason for the threefold splitting of the “second” reduction waves.
The calculated electronic band gaps of all four compounds are larger than the optical gaps and lie within E g = 2.97 eV (TBTQ‐TQPP 9) and E g = 3.23 eV (QPP 16). It has to be mentioned that this deviation of a few tenth of an eV is not unusual due to the exciton binding energy stabilized by surrounding solvent molecules of the excited state, which is absent in DFT calculations performed for vacuum. [37]
Conclusions
Tribenzotriqinacenes as well as triptycenes with one or three QPP blades were synthesized and compared by absorption and emission spectroscopy, cyclic voltammetry and quantum‐chemical calculations. For both triptycene as well as the TBTQ based QPP hints for an electronic communication of the three blades via homoconjugation was observed during electrochemical reduction processes, which is contrary to previous findings for TBTQ derivatives. Further and more detailed photophysical studies of both the triptycene as well as the TBTQ QPPs as potential molecules for singlet fission will be studied in due course.
Experimental Section
Crystallographic data: Deposition numbers 2005888, 2008016, 2008017, and 2008018 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors are grateful to “Deutsche Forschungsgemeinschaft” supporting this project (within the collaborative research center SFB1249 “N‐heteropolycyclic compounds as functional materials” (TP‐A04). We like to thank Philippe Wagner for providing synthetic precursors. Open access funding enabled and organized by Projekt DEAL.
L. Ueberricke, B. Punja Benke, T. Kirschbaum, S. Hahn, F. Rominger, U. H. F. Bunz, M. Mastalerz, Chem. Eur. J. 2021, 27, 2043.
References
- 1.
- 1a. Kuck D., Angew. Chem. Int. Ed. Engl. 1984, 23, 508–509; [Google Scholar]; Angew. Chem. 1984, 96, 515–516; [Google Scholar]
- 1b. Kuck D., Lindenthal T., Schuster A., Chem. Ber. 1992, 125, 1449–1460. [Google Scholar]
- 2.
- 2a. Dietmar K., Pure Appl. Chem. 2006, 78, 749–775; [Google Scholar]
- 2b. He L., Ng C.-F., Li Y., Liu Z., Kuck D., Chow H.-F., Angew. Chem. Int. Ed. 2018, 57, 13635–13639; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13823–13827. [Google Scholar]
- 3. Beaudoin D., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2016, 55, 15599–15603; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15828–15832. [Google Scholar]
- 4. Strübe J., Neumann B., Stammler H.-G., Kuck D., Chem. Eur. J. 2009, 15, 2256–2260. [DOI] [PubMed] [Google Scholar]
- 5. Xu W.-R., Xia G.-J., Chow H.-F., Cao X.-P., Kuck D., Chem. Eur. J. 2015, 21, 12011–12017. [DOI] [PubMed] [Google Scholar]
- 6. Ng C.-F., Chow H.-F., Kuck D., Mak T. C. W., Cryst. Growth Des. 2017, 17, 2822–2827. [Google Scholar]
- 7.
- 7a. Georghiou P. E., Dawe L. N., Tran H.-A., Strübe J., Neumann B., Stammler H.-G., Kuck D., J. Org. Chem. 2008, 73, 9040–9047; [DOI] [PubMed] [Google Scholar]
- 7b. Wang T., Li Z.-Y., Xie A.-L., Yao X.-J., Cao X.-P., Kuck D., J. Org. Chem. 2011, 76, 3231–3238. [DOI] [PubMed] [Google Scholar]
- 8. Mughal E. U., Eberhard J., Kuck D., Chem. Eur. J. 2013, 19, 16029–16035. [DOI] [PubMed] [Google Scholar]
- 9. Langhals H., Rauscher M., Strübe J., Kuck D., J. Org. Chem. 2008, 73, 1113–1116. [DOI] [PubMed] [Google Scholar]
- 10. Long T. M., Swager T. M., Adv. Mater. 2001, 13, 601–604. [Google Scholar]
- 11.
- 11a. Kumarasamy E., Sanders S. N., Tayebjee M. J. Y., Asadpoordarvish A., Hele T. J. H., Fuemmeler E. G., Pun A. B., Yablon L. M., Low J. Z., Paley D. W., Dean J. C., Choi B., Scholes G. D., Steigerwald M. L., Ananth N., McCamey D. R., Sfeir M. Y., Campos L. M., J. Am. Chem. Soc. 2017, 139, 12488–12494; [DOI] [PubMed] [Google Scholar]
- 11b. Ahrens L., Butscher J., Brosius V., Rominger F., Freudenberg J., Vaynzof Y., Bunz U. H. F., Chem. Eur. J. 2020, 26, 412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Zirzlmeier J., Lehnherr D., Coto P. B., Chernick E. T., Casillas R., Basel B. S., Thoss M., Tykwinski R. R., Guldi D. M., Proc. Natl. Acad. Sci. USA 2015, 112, 5325; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Alagna N., Pérez Lustres J. L., Wollscheid N., Luo Q., Han J., Dreuw A., Geyer F. L., Brosius V., Bunz U. H. F., Buckup T., Motzkus M., J. Phys. Chem. B 2019, 123, 10780–10793. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Kuhn W., Trans. Faraday Soc. 1930, 26, 293–308; [Google Scholar]
- 13b. Davydov A. S., Kasha M., M. Oppenheimer, Jr. , Theory of Molecular Excitons, McGraw–Hill, 1962; [Google Scholar]
- 13c. Kasha M., Rawls H. R., El-Bayoumi M. A., Pure Appl. Chem. 1965, 11, 371–392. [Google Scholar]
- 14. Forster T., Fluoreszenz organischer Verbindungen, Vandenhoeck und Ruprecht, Göttingen, 1951. [Google Scholar]
- 15. Mattay J., Photoinduced Electron Transfer I, 1st ed., Springer, Heidelberg, 1990. [Google Scholar]
- 16. Low J. Z., Sanders S. N., Campos L. M., Chem. Mater. 2015, 27, 5453–5463. [Google Scholar]
- 17. Lukman S., Musser A. J., Chen K., Athanasopoulos S., Yong C. K., Zeng Z., Ye Q., Chi C., Hodgkiss J. M., Wu J., Friend R. H., Greenham N. C., Adv. Funct. Mater. 2015, 25, 5452–5461. [Google Scholar]
- 18.
- 18a. Geyer F. L., Schmid S., Brosius V., Bojanowski N. M., Bollmann G., Brödner K., Bunz U. H. F., Chem. Commun. 2016, 52, 5702–5705; [DOI] [PubMed] [Google Scholar]
- 18b. Geyer F. L., Koser S., Bojanowski M. N., Ullrich F., Brosius V., Hahn S., Brödner K., Mankel E., Marszalek T., Pisula W., Hinkel F., Bunz U. H. F., Chem. Commun. 2018, 54, 1045–1048. [DOI] [PubMed] [Google Scholar]
- 19. Hu B.-L., An C., Wagner M., Ivanova G., Ivanova A., Baumgarten M., J. Am. Chem. Soc. 2019, 141, 5130–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Mateo-Alonso A., Chem. Soc. Rev. 2014, 43, 6311–6324; [DOI] [PubMed] [Google Scholar]
- 20b. Mateo-Alonso A., Kulisic N., Valenti G., Marcaccio M., Paolucci F., Prato M., Chem. Asian J. 2010, 5, 482–485. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Kulisic N., More S., Mateo-Alonso A., Chem. Commun. 2011, 47, 514–516; [DOI] [PubMed] [Google Scholar]
- 21b. More S., Bhosale R., Choudhary S., Mateo-Alonso A., Org. Lett. 2012, 14, 4170–4173; [DOI] [PubMed] [Google Scholar]
- 21c. More S., Bhosale R., Mateo-Alonso A., Chem. Eur. J. 2014, 20, 10626–10631; [DOI] [PubMed] [Google Scholar]
- 21d. García R., Melle-Franco M., Mateo-Alonso A., Chem. Commun. 2015, 51, 8037–8040; [DOI] [PubMed] [Google Scholar]
- 21e. Cortizo-Lacalle D., Pertegás A., Melle-Franco M., Bolink H. J., Mateo-Alonso A., Org. Chem. Front. 2017, 4, 876–881; [Google Scholar]
- 21f. Min Y., Dou C., Liu D., Dong H., Liu J., J. Am. Chem. Soc. 2019, 141, 17015–17021; [DOI] [PubMed] [Google Scholar]
- 21g. Marco A. B., Cortizo-Lacalle D., Gozalvez C., Olano M., Atxabal A., Sun X., Melle-Franco M., Hueso L. E., Mateo-Alonso A., Chem. Commun. 2015, 51, 10754–10757; [DOI] [PubMed] [Google Scholar]
- 21h. Antonicelli G., Gozalvez C., Atxabal A., Melle-Franco M., Hueso L. E., Mateo-Alonso A., Org. Lett. 2016, 18, 4694–4697; [DOI] [PubMed] [Google Scholar]
- 21i. Mateo-Alonso A., Eur. J. Org. Chem. 2017, 7006–7011; [Google Scholar]
- 21j. Cortizo-Lacalle D., Gozalvez C., Melle-Franco M., Mateo-Alonso A., Nanoscale 2018, 10, 11297–11301; [DOI] [PubMed] [Google Scholar]
- 21k. Cortizo-Lacalle D., Mora-Fuentes J. P., Strutyński K., Saeki A., Melle-Franco M., Mateo-Alonso A., Angew. Chem. Int. Ed. 2018, 57, 703–708; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 711–716; [Google Scholar]
- 21l. Li G., Abiyasa A. P., Gao J., Divayana Y., Chen W., Zhao Y., Sun X. W., Zhang Q., Asian J. Org. Chem. 2012, 1, 346–351; [Google Scholar]
- 21m. Gu P.-Y., Wang Z., Liu G., Yao H., Wang Z., Li Y., Zhu J., Li S., Zhang Q., Chem. Mater. 2017, 29, 4172–4175; [Google Scholar]
- 21n. Wang Z., Gu P., Liu G., Yao H., Wu Y., Li Y., Rakesh G., Zhu J., Fu H., Zhang Q., Chem. Commun. 2017, 53, 7772–7775; [DOI] [PubMed] [Google Scholar]
- 21o. Hu B.-L., Zhang K., An C., Schollmeyer D., Pisula W., Baumgarten M., Angew. Chem. Int. Ed. 2018, 57, 12375–12379; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12555–12559; [Google Scholar]
- 21p. Kohl B., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2015, 54, 6051–6056; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6149–6154; [Google Scholar]
- 21q. Kohl B., Bohnwagner M. V., Rominger F., Wadepohl H., Dreuw A., Mastalerz M., Chem. Eur. J. 2016, 22, 646–655; [DOI] [PubMed] [Google Scholar]
- 21r. Ueberricke L., Holub D., Kranz J., Rominger F., Elstner M., Mastalerz M., Chem. Eur. J. 2019, 25, 11121–11134; [DOI] [PubMed] [Google Scholar]
- 21s. Ueberricke L., Wieland S., Rominger F., Mastalerz M., Organic Materials 2019, 01, 050–062; [Google Scholar]
- 21t. Gao B., Wang M., Cheng Y., Wang L., Jing X., Wang F., J. Am. Chem. Soc. 2008, 130, 8297–8306; [DOI] [PubMed] [Google Scholar]
- 21u. Yang T., Liang B., Cheng Z., Li C., Lu G., Wang Y., J. Phys. Chem. C 2019, 123, 18585–18592; [Google Scholar]
- 21v. Ji J. D., Zhu L., Klug C. L., Smith M. D., Miao S., Synthesis 2015, 47, 871–874. [Google Scholar]
- 22.
- 22a. Newman C. R., Frisbie C. D., da Silva Filho D. A., Brédas J.-L., Ewbank P. C., Mann K. R., Chem. Mater. 2004, 16, 4436–4451; [Google Scholar]
- 22b. Zaumseil J., Sirringhaus H., Chem. Rev. 2007, 107, 1296–1323; [DOI] [PubMed] [Google Scholar]
- 22c. Wang C., Dong H., Hu W., Liu Y., Zhu D., Chem. Rev. 2012, 112, 2208–2267; [DOI] [PubMed] [Google Scholar]
- 22d. Wen Y., Liu Y., Adv. Mater. 2010, 22, 1331–1345; [DOI] [PubMed] [Google Scholar]
- 22e. Guo J., Xu Y., Jin S., Chen L., Kaji T., Honsho Y., Addicoat M. A., Kim J., Saeki A., Ihee H., Seki S., Irle S., Hiramoto M., Gao J., Jiang D., Nat. Commun. 2013, 4, 2736; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22f. Wex B., El-Ballouli A. a. O., Vanvooren A., Zschieschang U., Klauk H., Krause J. A., Cornil J., Kaafarani B. R., J. Mol. Struct. 2015, 1093, 144–149. [Google Scholar]
- 23.
- 23a. Xu R.-P., Li Y.-Q., Tang J.-X., J. Mater. Chem. C 2016, 4, 9116–9142; [Google Scholar]
- 23b. Salehi A., Fu X., Shin D.-H., So F., Adv. Funct. Mater. 2019, 29, 1808803; [Google Scholar]
- 23c. Hu J., Zhang D., Jin S., Cheng S. Z. D., Harris F. W., Chem. Mater. 2004, 16, 4912–4915; [Google Scholar]
- 23d. Luo M., Shadnia H., Qian G., Du X., Yu D., Ma D., Wright J. S., Wang Z. Y., Chem. Eur. J. 2009, 15, 8902–8908; [DOI] [PubMed] [Google Scholar]
- 23e. Wang M., Tong H., Cheng Y., Xie Z., Wang L., Jing X., Wang F., J. Polym. Sci. Part A 2010, 48, 1990–1999; [Google Scholar]
- 23f. Wang M., Li Y., Xie Z., Wang L., Sci. China: Chem. 2011, 54, 656–665; [Google Scholar]
- 23g. Li Q., Li J., Ren H., Gao Z., Liu D., Synth. Commun. 2011, 41, 3325–3333. [Google Scholar]
- 24.
- 24a. Zhan X., Facchetti A., Barlow S., Marks T. J., Ratner M. A., Wasielewski M. R., Marder S. R., Adv. Mater. 2011, 23, 268–284; [DOI] [PubMed] [Google Scholar]
- 24b. Cao W., Xue J., Energy Environ. Sci. 2014, 7, 2123–2144. [Google Scholar]
- 25.
- 25a. Kohl B., Rominger F., Mastalerz M., Org. Lett. 2014, 16, 704–707; [DOI] [PubMed] [Google Scholar]
- 25b. Kohl B., Rominger F., Mastalerz M., Chem. Eur. J. 2015, 21, 17308–17313; [DOI] [PubMed] [Google Scholar]
- 25c. Prantl E., Kohl B., Ryvlin D., Biegger P., Wadepohl H., Rominger F., Bunz U. H. F., Mastalerz M., Waldvogel S. R., ChemPlusChem 2019, 84, 1239–1244. [DOI] [PubMed] [Google Scholar]
- 26. Mora-Fuentes J. P., Riaño A., Cortizo-Lacalle D., Saeki A., Melle-Franco M., Mateo-Alonso A., Angew. Chem. Int. Ed. 2019, 58, 552–556; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 562–566. [Google Scholar]
- 27. Mora-Fuentes J. P., Papadopoulos I., Thiel D., Álvarez-Boto R., Cortizo-Lacalle D., Clark T., Melle-Franco M., Guldi D. M., Mateo-Alonso A., Angew. Chem. Int. Ed. 2020, 59, 1113–1117; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1129–1133. [Google Scholar]
- 28. Mastalerz M., Sieste S., Cenić M., Oppel I. M., J. Org. Chem. 2011, 76, 6389–6393. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Yamato T., Miyazawa A., Tashiro M., Chem. Ber. 1993, 126, 2505–2511; [Google Scholar]
- 29b. Hu J., Zhang D., Harris F. W., J. Org. Chem. 2005, 70, 707–708. [DOI] [PubMed] [Google Scholar]
- 30. Coombs B. A., Lindner B. D., Edkins R. M., Rominger F., Beeby A., Bunz U. H. F., New J. Chem. 2012, 36, 550–553. [Google Scholar]
- 31. Li Z.-M., Li Y.-W., Cao X.-P., Chow H.-F., Kuck D., J. Org. Chem. 2018, 83, 3433–3440. [DOI] [PubMed] [Google Scholar]
- 32. Chong J. H., MacLachlan M. J., Inorg. Chem. 2006, 45, 1442–1444. [DOI] [PubMed] [Google Scholar]
- 33. Ueberricke L., Schwarz J., Ghalami F., Matthiesen M., Rominger F., Elbert S. M., Zaumseil J., Elstner M., Mastalerz M., Chem. Eur. J. 2020, 26, 12596–12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.
- 34a. Doerner T., Gleiter R., Robbins T. A., Chayangkoon P., Lightner D. A., J. Am. Chem. Soc. 1992, 114, 3235–3241; [Google Scholar]
- 34b. Kawasumi K., Wu T., Zhu T., Chae H. S., Van Voorhis T., Baldo M. A., Swager T. M., J. Am. Chem. Soc. 2015, 137, 11908–11911; [DOI] [PubMed] [Google Scholar]
- 34c. Talipov M. R., Navale T. S., Rathore R., Angew. Chem. Int. Ed. 2015, 54, 14468–14472; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14676–14680; [Google Scholar]
- 34d. Baumgärtner K., Hoffmann M., Rominger F., Elbert S. M., Dreuw A., Mastalerz M., J. Org. Chem. 2020, 85, 15256–15272. [DOI] [PubMed] [Google Scholar]
- 35. Kuck D., Chem. Rev. 2006, 106, 4885–4925. [DOI] [PubMed] [Google Scholar]
- 36.
- 36a. Menke E. H., Lami V., Vaynzof Y., Mastalerz M., Chem. Commun. 2016, 52, 1048–1051; [DOI] [PubMed] [Google Scholar]
- 36b. Kirschbaum T., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2020, 59, 270–274; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 276–280; [Google Scholar]
- 36c. Lv L., Roberts J., Xiao C., Jia Z., Jiang W., Zhang G., Risko C., Zhang L., Chem. Sci. 2019, 10, 4951–4958; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36d. Takase M., Narita T., Fujita W., Asano M. S., Nishinaga T., Benten H., Yoza K., Müllen K., J. Am. Chem. Soc. 2013, 135, 8031–8040. [DOI] [PubMed] [Google Scholar]
- 37. Bredas J.-L., Mater. Horiz. 2014, 1, 17–19. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary