

Abstract
Key points
Heart failure is characterised by limb and respiratory muscle impairments that limit functional capacity and quality of life. However, compared with heart failure with reduced ejection fraction (HFrEF), skeletal muscle alterations induced by heart failure with preserved ejection fraction (HFpEF) remain poorly explored.
Here we report that obese‐HFpEF induces multiple skeletal muscle alterations in the rat hindlimb, including impaired muscle mechanics related to shortening velocity, fibre atrophy, capillary loss, and an impaired blood flow response to contractions that implies a perfusive oxygen delivery limitation.
We also demonstrate that obese‐HFpEF is characterised by diaphragmatic alterations similar to those caused by denervation – atrophy in Type IIb/IIx (fast/glycolytic) fibres and hypertrophy in Type I (slow/oxidative) fibres.
These findings extend current knowledge in HFpEF skeletal muscle physiology, potentially underlying exercise intolerance, which may facilitate future therapeutic approaches.
Abstract
Peripheral skeletal muscle and vascular alterations induced by heart failure with preserved ejection fraction (HFpEF) remain poorly identified, with limited therapeutic targets. This study used a cardiometabolic obese‐HFpEF rat model to comprehensively phenotype skeletal muscle mechanics, blood flow, microvasculature and fibre atrophy. Lean (n = 8) and obese‐HFpEF (n = 8) ZSF1 rats were compared. Skeletal muscles (soleus and diaphragm) were assessed for in vitro contractility (isometric and isotonic properties) alongside indices of fibre‐type cross‐sectional area, myosin isoform, and capillarity, and estimated muscle PO2. In situ extensor digitorum longus (EDL) contractility and femoral blood flow were assessed. HFpEF soleus demonstrated lower absolute maximal force by 22%, fibre atrophy by 24%, a fibre‐type shift from I to IIa, and a 17% lower capillary‐to‐fibre ratio despite increased capillary density (all P < 0.05) with preserved muscle PO2 (P = 0.115) and isometric specific force (P > 0.05). Soleus isotonic properties (shortening velocity and power) were impaired by up to 17 and 22%, respectively (P < 0.05), while the magnitude of the exercise hyperaemia was attenuated by 73% (P = 0.012) in line with higher muscle fatigue by 26% (P = 0.079). Diaphragm alterations (P < 0.05) included Type IIx fibre atrophy despite Type I/IIa fibre hypertrophy, with increased indices of capillarity alongside preserved contractile properties during isometric, isotonic, and cyclical contractions. In conclusion, obese‐HFpEF rats demonstrated blunted skeletal muscle blood flow during contractions in parallel to microvascular structural remodelling, fibre atrophy, and isotonic contractile dysfunction in the locomotor muscles. In contrast, diaphragm phenotype remained well preserved. This study identifies numerous muscle‐specific impairments that could exacerbate exercise intolerance in obese‐HFpEF.
Keywords: blood flow, diaphragm, heart failure, HFpEF, muscle atrophy, muscle contraction, skeletal muscle
Key points
Heart failure is characterised by limb and respiratory muscle impairments that limit functional capacity and quality of life. However, compared with heart failure with reduced ejection fraction (HFrEF), skeletal muscle alterations induced by heart failure with preserved ejection fraction (HFpEF) remain poorly explored.
Here we report that obese‐HFpEF induces multiple skeletal muscle alterations in the rat hindlimb, including impaired muscle mechanics related to shortening velocity, fibre atrophy, capillary loss, and an impaired blood flow response to contractions that implies a perfusive oxygen delivery limitation.
We also demonstrate that obese‐HFpEF is characterised by diaphragmatic alterations similar to those caused by denervation – atrophy in Type IIb/IIx (fast/glycolytic) fibres and hypertrophy in Type I (slow/oxidative) fibres.
These findings extend current knowledge in HFpEF skeletal muscle physiology, potentially underlying exercise intolerance, which may facilitate future therapeutic approaches.
Introduction
Increasing prevalence of heart failure with preserved ejection fraction (HFpEF), in the absence of recognised pharmaceutical treatments, presents one of the biggest challenges to modern cardiology (Butler et al. 2014; Sharma & Kass, 2014; Fukuta et al. 2016). While the primary pathology of HFpEF is of cardiac origin, there is a poor correlation between heart dysfunction and the main symptom of exercise intolerance (Haykowsky & Kitzman, 2014), and many clinical trials have shown cardiac‐orientated drugs are not associated with beneficial outcomes (Shah et al. 2016). Recent investigations, therefore, have suggested non‐cardiac ‘peripheral’ factors as major mechanisms limiting functional capacity and quality of life in patients with HFpEF, with skeletal muscle abnormalities receiving much attention (Adams et al. 2017; Poole et al. 2018; Zamani et al. 2020). For example, animal and human studies have shown that HFpEF is associated with various skeletal muscle impairments that are closely associated with exercise intolerance and lower quality of life, including lower skeletal muscle mass and strength (Bekfani et al. 2016), generalised fibre atrophy (Bowen et al. 2018), fat infiltration (Haykowsky et al. 2014; Zamani et al. 2020), reduced global capillary‐to‐fibre ratio (Kitzman et al. 2014; Bowen et al. 2018), reduced mitochondrial function and content (Bowen et al. 2015; Molina et al. 2016; Bowen et al. 2017b), disrupted high‐energy phosphate metabolism (Bhella et al. 2011b; Weiss et al. 2017), and impaired O2 extraction (Dhakal et al. 2015; Houstis et al. 2018; Zamani et al. 2020).
Despite recent progress, our current understanding of the skeletal muscle pathophysiology in HFpEF at both the structural and functional level is in its infancy, with only limited and often conflicting experimental data available (Poole et al. 2018). For example, it remains controversial whether leg muscle arterial blood flow (i.e. perfusive O2 transport) is impaired during exercise in HFpEF due to a lack of direct measurements (Hundley et al. 2007; Haykowsky et al. 2013; Lee et al. 2016a; Weavil et al. 2020), with key studies dependent upon systemic blood sampling reporting contrasting findings regarding whether a perfusive or diffusive O2 transport limitation impairs muscle O2 extraction and thus exercise intolerance in HFpEF (Dhakal et al. 2015; Houstis et al. 2018; Zamani et al. 2020). As such, there remains a lack of clarity on whether functional indices related to leg blood flow and perfusive O2 delivery are constrained in HFpEF. Likewise, we also have a limited understanding of the muscle and microvascular structural phenotype that occurs with HFpEF. For example, while studies have shown that a global fibre atrophy is present alongside a loss of capillaries‐per‐fibre (Kitzman et al. 2014; Bowen et al. 2015, 2017b, 2018; Schauer et al. 2020), the degree of atrophy and capillary rarefaction quantified locally across all fibre isoforms which can be further harnessed to provide novel estimates of muscle PO2 remains undefined. Furthermore, a knowledge gap still exists in relation to potential sites of skeletal muscle dysfunction in HFpEF that include the contribution of neuromuscular transmission vs. excitation–contraction failure and/or the impact on more physiologically relevant mechanical measures (i.e. shortening velocity and power), with previous studies performed using only in vitro isometric contractions under direct muscle stimulation (Bowen et al. 2015, 2017b, 2018; Schauer et al. 2020).
Beyond this, the majority of experimental work has been directed towards characterising the locomotor muscles despite key evidence showing that respiratory muscle dysfunction is linked to exercise intolerance in HFpEF, as shown by non‐invasive patient measures (Lavietes et al. 2004; Yamada et al. 2016) and direct diaphragm contractility measures in experimental models (Bowen et al. 2015, 2017b). Similar to limb muscle, however, detailed quantification of diaphragm fibre‐type morphology, capillarity, PO2, and clinically relevant functional measurements during cyclical length changes (i.e. as occurs during breathing) remain largely undefined in HFpEF and are unlikely to follow a similar response to the locomotor muscles.
The present study, therefore, aimed to provide a more comprehensive assessment of the skeletal muscle phenotype in HFpEF, by applying in vitro, in situ and in silico approaches to a validated obese cardiometabolic rat model, where ex vivo magnetic resonance imaging was used to characterise the degree of cardiac remodelling. Specifically, using hindlimb (soleus/EDL) and respiratory (diaphragm) muscle, we performed global and local fibre‐type specific phenotyping of cross‐sectional area, isoform, and capillarity alongside estimated muscle PO2. In parallel, we also directly assessed key functional measures during rest and contractions, including hindlimb blood flow as well as neural‐ and direct‐muscle stimulated contractile mechanics. We reasoned that a better understanding of the skeletal muscle phenotype in HFpEF across multiple‐system levels would provide important insights for better understanding the pathophysiology of exercise intolerance in this disease and help direct future patient experiments and therapeutic development in this field.
Methods
Ethical approval
All procedures and experiments were performed in accordance with the UK Scientific Procedures (Animals) Act 1986 and local approval was given by the University of Leeds Animal Welfare and Ethical Review Committee. All work conforms to the ethical requirements outlined by The Journal of Physiology (Grundy, 2015).
Animals
Twenty‐week‐old male obese (n = 8) and lean (n = 8) diabetic Zucker fatty/spontaneously hypertensive heart failure F1 hybrid (ZSF1) rats (Charles River Laboratories) were used in this study. While both lean and obese ZSF1 rats inherit the hypertension gene, only the obese ZSF1 rats inherit a mutation in the leptin receptor gene (LeprfaLeprcp/Crl) that drives weight gain and the metabolic impairments associated with typical signs of HFpEF developing as early as 10 weeks of age (Schauer et al. 2020) and well established after 20 weeks (Leite et al. 2015; Franssen et al. 2016; van Dijk et al. 2016; Bowen et al. 2017b). Lean ZSF1 rats served as controls. All rats were maintained in a 12 h light/dark cycle, with standard chow diets (RM1 chow, SDS) and water provided ad libitum.
Cardiometabolic function
Cardiometabolic impairments were confirmed by measures of body mass, mean arterial pressure (via an implanted carotid catheter (PP10)) with a blood pressure transducer (BP transducer, AD Instruments, UK) and blood glucose levels (via a commercial blood glucose meter (FreeStyle Mini Meter), while hearts were perfused and immersion fixed ex vivo using low osmolality Karnovsky's fixative and subsequently imaged using a diffusion‐weighted fast spin echo sequence at a resolution of 120 μm isotropic for cardiac phenotyping (Teh et al. 2016). These data were used to calculate mean thicknesses of the left and right ventricular free walls and the septum, for tissue located in the middle third of the distance between the base and apex of the appropriate ventricular cavity. Myocyte helix angles (quantifying myocyte inclination with respect to the short axis of the heart) were extracted from regions in the left and right ventricular free walls and the septum as previously described (Benson et al. 2011); myocyte disarray in these regions was quantified using the R 2 of a 5th order polynomial fit to the helix angles plotted as a function of transmural distance (Benson et al. 2008). All DT‐MRI analyses were carried out using in‐house software.
In situ muscle performance and femoral artery blood flow
In situ measurements of muscle function and blood flow were made under surgical anaesthesia, which was induced with isoflurane (4% in 100% oxygen) and maintained throughout experiments by constant syringe pump infusion (30–35 mg kg−1 h−1) of Alfaxalone (Jurox, Crawley, UK) delivered via an implanted jugular vein catheter. In situ functional assessment of muscle performance was determined as previously described (Egginton & Hudlicka, 1999; Tickle et al. 2020). In brief, extensor digitorum longus (EDL) isometric twitch force was recorded via a lever arm force transducer (305B‐LR: Aurora Scientific, Aurora, ON, Canada) following surgical extirpation of the overlying synergist tibialis anterior muscles. Electrical stimulation of the EDL (0.3 ms pulse width) was accomplished via electrodes placed adjacent to the popliteal nerve (Hudlická et al. 1977), with initial electrical pulses (1 Hz) delivered to determine optimal muscle length and supramaximal current delivery. Simultaneous measurement of bilateral blood flow was facilitated by placement of perivascular flow probes (0.7PSB; Transonic, Ithaca, NY, USA) on the proximal portion of the femoral artery, adjacent to the profunda femoris bifurcation (Tickle et al. 2020). Quantification of resting and end‐stimulation blood flows enabled determination of the functional hyperaemia recruited during stimulation. Blood flow data are provided (ml min−1) and after normalisation for blood pressure variation, vascular conductance (ml min−1 mm Hg−1). All data were recorded via PowerLab and LabChart software (AD Instruments, UK).
EDL twitch and maximal tetanic force, as well as fatigue resistance, were also assessed. Fatigue resistance was quantified by monitoring isometric force throughout a period of continuous 10 Hz stimulation for 3 min. A fatigue index was then calculated as: (end‐stimulation twitch tension/peak twitch tension) x 100. An average of five consecutive twitches was used to quantify end stimulation and peak EDL tension. Differences in the magnitude of the absolute tetanic force generated between groups were taken into account by employing a second bout of fatigue stimulation, such that absolute forces in HFpEF were initially similar to those attained in controls (i.e. matched initial force) (Ferreira et al. 2010). This protocol is relevant for clinical translation of muscle fatigue, where daily tasks in patients are often dependent upon the absolute rather than relative force being sustained that may involve an increase in firing frequency of motor neurons to achieve task completion (Weavil et al. 2020). Thus, by adjusting the stimulation frequency in HFpEF rats to around 25 Hz, tetanic force was increased and matched to the level recorded in the lean group, with fatigue allowed to proceed over 3 min. In addition, tetanic force production was quantified by 200 Hz stimulation (200 ms duration) after a minimum of 10 min recovery from fatigue, as determined by restoration of pre‐fatigue resting blood flow. All protocols were performed in exactly the same order for each rat, thus minimising any effects of methodological variation. Force is presented in absolute units (g) and normalised to wet mass (g mg−1).
In vitro functional assessment
Immediately following killing, the soleus and diaphragm were excised and prepared in a Krebs–Henseleit solution (117 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 24.8 NaHCO3, 2.5 CaCl2, 11.1 glucose; in mmol l−1) at 4°C equilibrated with 95% O2/5% CO2. For the soleus, silk sutures (4.0) attached to tendons at either end were used to suspend the muscle vertically in a buffer‐filled organ bath between a hook and a length‐controlled lever system (305C, Aurora Scientific, Aurora, Canada). In vitro field stimulation using platinum electrodes was provided via a high power bipolar stimulator (701C, Aurora Scientific) outputting supramaximal current (700 mA; 1 s train duration; 0.25 ms pulse width). After optimal contractile length (L0) was determined, the muscle was thermoequilibrated in a Krebs–Henseleit solution for 15 min at ∼21°C (Bowen et al., 2017a). For the diaphragm, a bundle of muscle fascicles (∼2–3 mm wide) was removed from the medial section of the left costal diaphragm leaving two ribs and a section of the central tendon intact. The muscle bundle was transferred to a flow‐through muscle chamber and anchored between a base and ergometer (series 300B‐LR, Aurora Scientific Inc.) and stimulated through parallel platinum electrodes using a stimulus isolation unit (0.2 ms pulse width; UISO model 236, Hugo Sachs Elektronik). After L0 was determined, the diaphragm bundle was thermoequilibrated in a Krebs–Henseleit solution for at least 15 min at 37°C. Each muscle was circulated with oxygenated (95% O2 /5% CO2) Krebs–Henseleit solution throughout each experiment.
The soleus underwent two protocols: isometric force–frequency and isotonic force–velocity. The force–frequency relationship was determined in response to pulses at 1, 15, 30, 50, 80, 120 and 150 Hz, with 1 min of recovery between contractions. After a 5 min period in which muscle length was measured using digital callipers, the soleus was subjected to a series of afterloaded‐isotonic contractions to determine the force–velocity relationship, where the muscle was allowed to shorten against external loads (80 – ∼5% of the maximal tetanic force; each separated by 1 min for the soleus or 5 min for the diaphragm) after being stimulated at 150 Hz for 300 ms. Shortening velocity was determined 10 ms after the first change in length and on the linear section of the transient (605A DMA software, Aurora Scientific). For the diaphragm, maximal isometric twitch and tetanic (250 ms train at 150 Hz), isotonic force–velocity (as above), and work loop protocols were performed. Muscle performance assessed using the work loop technique included simulating performance in vivo, by subjecting the muscle to cyclical length changes and phasic stimulation (Josephson, 1985). A sinusoidal length change at a range of cycle frequencies (1–15 Hz) and strain amplitude of 0.065 L 0 was imposed on the muscle and, for each cycle frequency, the timing and duration of stimulation were optimised to maximise net work. Isometric tetanic contractions and cyclical contractions at 5 Hz were performed periodically to monitor any decline in the preparation, assessed by expressing isometric stress (isotonic contractions) and net work relative to maximal values. A linear decline in performance was assumed in correcting data for preparation decline. A period of 5 min was allowed following isotonic and work loop contractions for recovery. To assess the muscle's ability to sustain work, a fatigue test was carried out by subjecting the muscle to a series of cyclical contractions (cycle frequency 2 Hz, strain amplitude 0.065 L 0, phase −20 ms relative to peak length, 210 ms stimulation duration). Custom‐written software was used to control muscle length and stimulation and to acquire length and force data (CEC Testpoint version 7) via a D/A data acquisition card (DAS1802AO, Keithley Instruments). Data were acquired at a sample frequency of 10 kHz (isometric and isotonic) or 1000 x cycle frequency (work loops).
At the end of each experiment, the muscle was blotted on paper tissue and wet mass recorded. Force (N) was normalised to muscle cross‐sectional area (CSA; cm2) after dividing muscle mass (g) by the product of L0 (cm) and estimated muscle density (1.06 g/cm3) to allow specific force (i.e. stress) in N/cm2 to be calculated (Close, 1972). Shortening velocity was normalised to optimal muscle length (in L0/s), while power was calculated as the product of shortening velocity and force normalised to muscle mass (in W kg−1). Twitch properties (i.e. peak force; time‐to‐peak tension; half relaxation time) as well as maximal isometric tetanic force (i.e.) were calculated. A hyperbolic‐linear relationship was fit to the force–velocity data to determine the maximum shortening velocity (), peak isotonic power (), and the power ratio (; i.e., a measure of the curvature of the force–velocity relationship (Marsh and Bennet, 1986).
Histological analysis
Mid‐portions of the right costal diaphragm and left soleus muscle were mounted in optimal cutting temperature embedding medium (Thermo Scientific, Loughborough, UK), frozen in liquid nitrogen‐cooled isopentane and stored at −80°C. To identify muscle fibre types, sections (10 μm thick) were fixed for 2 min in 2% paraformaldehyde, washed in phosphate‐buffered saline (PBS; P4417, Sigma‐Aldrich, St Louis, MO) and blocked for 10 min in 1% bovine serum albumin (A6003, Sigma‐Aldrich, St Louis, MO). Sections were then incubated for 60 min with monoclonal‐myosin heavy chain antibodies BA‐D5 (IgG2B, 1:1000) and SC‐71 (IgG1, 1:500) for Type I (oxidative) and Type IIa (fast oxidative, glycolytic) fibres, respectively (Developmental Studies Hybridoma Bank, Iowa City, IA, USA). The remaining unstained fibres were considered to be Type IIb/IIx, as previously described (Kissane et al. 2018). After washing in PBS, sections were incubated for 60 min with secondary antibodies Alexa Fluor 555 (conjugated goat anti‐mouse IgG, 1:1000, A‐21422, Thermo Fisher Scientific, Waltham, MA) and Alexa Fluor 488 (conjugated rabbit anti‐mouse IgG, 1:1000, A11059, Thermo Fisher Scientific, Waltham, MA). Muscle fibre boundaries were labelled with a rabbit anti‐laminin antibody (1:200; L9393, Sigma‐Aldrich, St Louis, MO), an extracellular matrix glycoprotein within the basement membrane. Finally, capillaries were stained with a carbohydrate‐binding protein (lectin) specific to rodent endothelial cells, Griffonia simplicifolia lectin I (Vector Labs, Peterborough, UK; FL‐1101). Slides were then imaged at magnifications of x10 (soleus) and x20 (diaphragm) using the Nikon Eclipse E600 (Nikon, Tokyo, Japan) optical microscope attached to a digital camera (QIMAGING, MicroPublisher 5.0 RTV, Surrey, BC, Canada). Subsequent image analysis with the stand‐alone graphic user interface, DTect, and a MATLAB‐based oxygen transport modeller (The MathWorks, Cambridge, United Kingdom; (Al‐Shammari et al. 2019)) enabled calculation of fibre type‐specific cross‐sectional area (FCSA), capillary‐to‐fibre (C:F) ratio, capillary density (CD), capillary domain area (CDA), local capillary‐to‐fibre ratio (LCFR), local capillary density (LCD) and estimated tissue oxygen tension (PO2). Multiple regions of interest of each muscle (three for the diaphragm and two for the soleus) were randomly assigned to establish an unbiased counting frame, taking into account the regional heterogeneity across muscles (Kissane et al. 2018). In general, each region of interest of the soleus muscle contained ∼155 fibres and the diaphragm ∼70 fibres.
In silico muscle PO2 modelling
Our model applied mathematical and computational frameworks to generate theoretical predictions of the cross‐sectional distribution of PO2 in the soleus and diaphragm using a custom MATLAB ‘oxygen transport modeller’, as previously described (Al‐Shammari et al. 2019). Briefly, using digitised images of muscle cryosections, individual fibre boundaries were identified, a phenotype assigned, and capillary locations defined. A computational framework was then established allowing a mathematical mesh of equations to be superimposed on realistic geometry. Tissue PO2 measurements were then derived by incorporating estimates (applied similarly in each group) of capillary radius (1.8−2.5 × 10−4 cm), muscle oxygen consumption (15.7 × 10−5 ml O2 ml−1 s−1), myoglobin concentration (10.2 × 10−3 ml O2 ml−1), O2 solubility (3.89 × 10−5 ml O2 ml−1 mmHg−1) and diffusivity (1.73 × 10−7 cm2 s−1) as detailed elsewhere (Al‐Shammari et al. 2019), with direct measurement of these specific parameters beyond the scope of the present study (Tickle et al. 2020). As such, any differences between groups in terms of mitochondrial function (or other assumed variables) were not accounted for in our model. Relevant biophysical parameters affecting O2 diffusion from reputable sources were used in the mathematical model to generate predictions of the cross‐sectional distribution of PO2 in a muscle biopsy under simulated resting and maximal oxygen consumption conditions. As with all biological models, inherent limitations prevent full characterisation of the wide myriad of interacting variables; however, relative changes within a given tissue were the key output and are likely robust. In line with former studies (Al‐Shammari et al. 2019), compensation for differences in many parameters, e.g. myoglobin saturation, have relatively small effects on the documented outcomes due to the dominant effect of capillary supply and fibre size on peripheral O2 transport.
Statistical analyses
Following appropriate checks of normality, between‐group differences were assessed by unpaired two‐tailed Student's t tests. Contractile relationships were analysed as two‐way repeated measures ANOVA followed by the Bonferroni post hoc test, where appropriate. Analyses were performed in GraphPad Prism v.8. Data are presented as means ± SD, and the level of significance was set at P < 0.05 for all analyses.
Results
Cardiometabolic phenotype
As previously noted (Leite et al. 2015; Franssen et al. 2016; van Dijk et al. 2016; Bowen et al. 2017b; Schauer et al. 2020), by 20 weeks of age obese‐ZSF1 rats have developed typical metabolic signs associated with HFpEF including obesity (P < 0.001; Fig. 1 A), hyperglycaemia (P < 0.001; Fig. 1 B) and hypertension (P = 0.012; Fig. 1 C). In addition, obese rats developed cardiac remodelling typically associated with obese‐HFpEF that included right ventricular (RV) hypertrophy (P = 0.034; Fig. 1 D), although left ventricular (LV) and septal wall enlargement was not observed at this time point (P = 0.719; Fig. 1 E and P = 0.849; Fig. 1 F). Further, myocyte organisation/disarray was not significantly deteriorated either (RV: P = 0.971; LV: P = 0.13; septum: P = 0.064; Fig. 1 G–H).
Figure 1. Cardiometabolic characteristics.
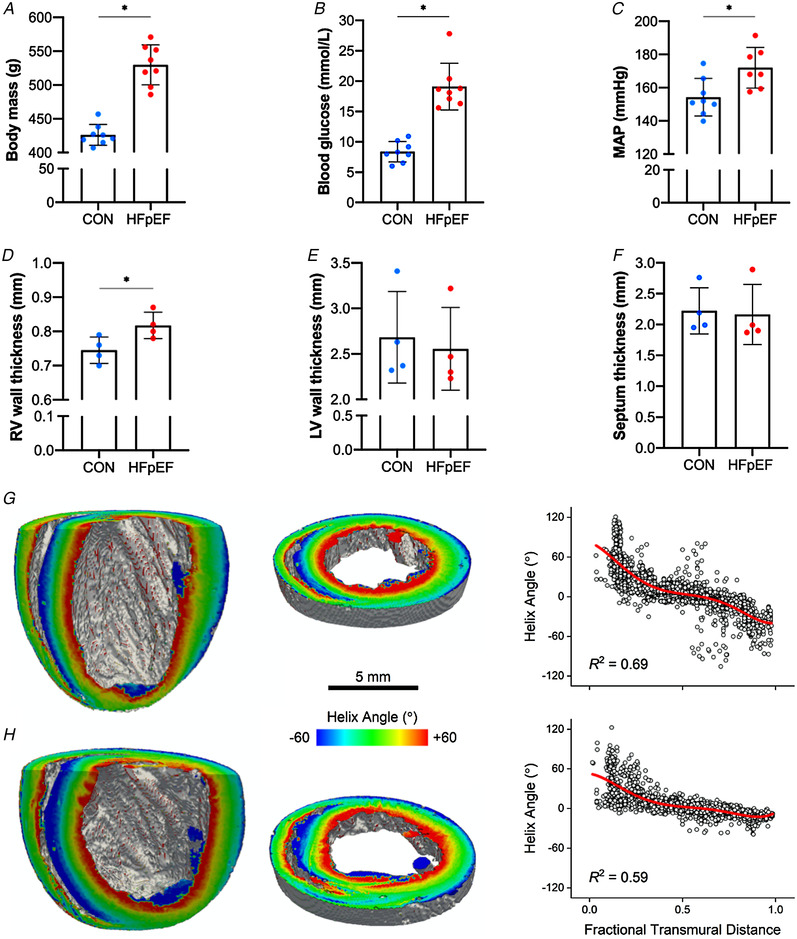
At 20 weeks of age, HFpEF rats developed obesity (426.25 ± 15.41 vs. 529.88 ± 29.56 g P < 0.001) (A), hyperglycaemia (8.38 ± 1.69 vs. 19.10 ± 3.83 mmol l−1; P < 0.001) (B) and hypertension (154.26 ± 11.28 vs. 172.03 ± 12.26 mmHg; P = 0.012) (C). Compared with lean controls, obese‐HFpEF rats also showed increased right ventricular (RV) wall thickness (0.74 ± 0.04 vs. 0.82 ± 0.04 mm; P = 0.034) (D); however, left ventricular (LV) wall and the septum thickness were not different between groups (2.68 ± 0.51 vs. 2.55 ± 0.46 mm; P = 0.719 and 2.22 ± 0.38 vs. 2.16 ± 0.49 mm; P = 0.849, respectively) (E–F). Left and middle panels: long axis cuts (left) and short axis slices (middle) of representative lean (G) and obese (H) hearts, with myocyte helix (inclination) angle colour coded on the cut surfaces. Right panel: the helix angle in the RV free wall plotted as a function of fractional transmural distance (0.0, endocardium; 1.0, epicardium) for representative lean (G) and obese (H) hearts. The red continuous line is a 5th order polynomial fit to the data. Myocyte disarray is quantified by the R 2 of this fit. [Color figure can be viewed at wileyonlinelibrary.com]
Histological and in vitro functional characteristics of the soleus muscle
As shown in representative muscle sections (Fig. 2 A–B), soleus from obese‐HFpEF rats demonstrated clear atrophy with a 26% lower wet mass (P < 0.001; Fig. 2 C) and a 23% lower CSA of both Type I (P < 0.001) and Type IIa fibres (P = 0.001; Fig. 2 D) when compared with lean controls. No Type IIx/b fibres were detected in either group. HFpEF rats also had a lower numerical and areal composition of Type I fibres (P = 0.002; Fig. 2 E and P = 0.043; Fig. 2 F, respectively), whereas these were higher in Type IIa fibres (P = 0.002 and P = 005, respectively). In addition, HFpEF rats had a lower C:F ratio (P = 0.002; Fig. 2 G) but a higher CD, indicating that atrophy proceeded at a greater rate than capillary loss (P = 0.027; Fig. 2 H), while CDA did not differ significantly (P = 0.059; Fig. 2 I). Analyses of local capillary distribution revealed that HFpEF rats had lower LCFR in Type I fibres (P = 0.011), and while a similar trend was found in Type IIa fibres this did not reach significance (P = 0.154; Fig. 2 J). In contrast, LCD in Type I fibres was higher in HFpEF rats (P = 0.029), and again while a similar trend was found in Type IIa fibres this did not reach significance (P = 0.196; Fig. 2 K). To understand whether HFpEF influenced muscle PO2, we simulated muscle oxygen tension (PO2) under resting (Fig. 3 A–C) and maximal demand (Fig. 3 D–F). No differences between groups were found after the calculation of muscle oxygenation at either rest (Type I fibres: P = 0.099; Type IIa: P = 0.167; all fibres: P = 0.102; Fig. 3 C) or maximal rate of oxygen consumption (Type I: P = 0.109; Type IIa: P = 0.177; all fibres: P = 0.115; Fig. 3 F).
Figure 2. Histological features of the soleus muscle.
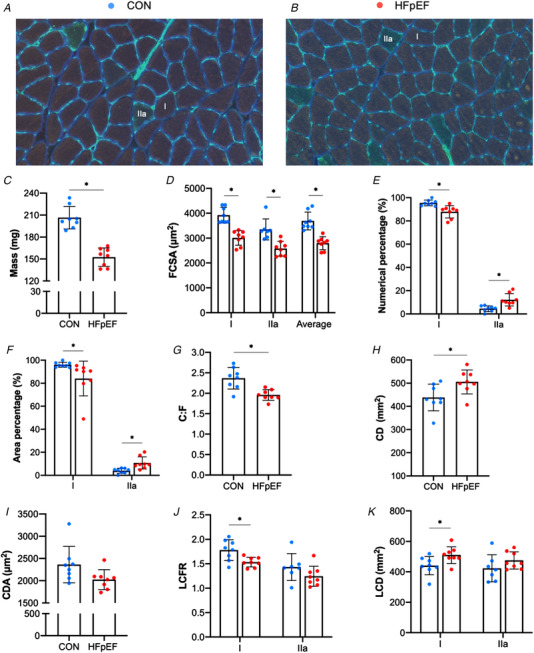
Representative soleus sections from control (A) and obese‐HFpEF (B). Obese‐HFpEF showed atrophy in the soleus muscle, with reduced wet muscle mass (206.54 ± 15.48 vs. 152.50 ± 12.55 mg; P < 0.001) (C) and reduced cross‐sectional area (CSA) in both Type I (3921.88 ± 316.51 vs. 3009.75 ± 298.03 μm2; P < 0.001) and Type IIa fibres (3351.43 ± 422.09 vs. 2575.69 ± 296.86 μm2; P = 0.001) (D). HFpEF rats also had a lower numerical and areal composition of Type I fibres (95.49 ± 2.45 vs. 87.84 ± 5.32%, P = 0.002 and 96.06 ± 2.17 vs. 84.06 ± 15.14 %, P = 0.043, respectively), whereas these were higher in Type IIa fibres (4.51 ± 2.45 vs. 12.16 ± 5.32 %, P = 0.002 and 3.94 ± 2.17 vs. 10.74 ± 5.31 %, P = 0.005, respectively) (E‐F). Moreover, compared with lean controls, obese rats had reduced capillary‐to‐fibre (C:F) ratio (2.37 ± 0.26 vs. 1.96 ± 0.13; P = 0.002) (G), whereas capillary density (CD), was increased (438.23 ± 57.66 vs. 505.5 ± 51.53 mm−2; P = 0.027) (H) with no change in capillary domain area (CDA) (2363.28 ± 410.48 vs. 2024.48 ± 223.24 μm2; P = 0.059) (I). Finally, local analyses of capillary distribution showed that HFpEF rats had lower local capillary‐to‐fibre ratio (LCFR) in Type I fibres (1.78 ± 0.22 vs. 1.53 ± 0.10; P = 0.011), although this was unchanged in Type IIa fibres (1.43 ± 0.27 vs. 1.24 ± 0.21; P = 0.154) (J). In contrast, local capillary density (LCD) in Type I fibres was increased in HFpEF rats (440.41 ± 59.71 vs. 510.15 ± 54.9 mm−2; P = 0.029), with no changes in Type IIa fibres (423.20 ± 88.64 vs. 474.73 ± 56.17 mm−2; P = 0.196) (K). [Color figure can be viewed at wileyonlinelibrary.com]
Figure 3. Modelling of soleus muscle oxygen tension.
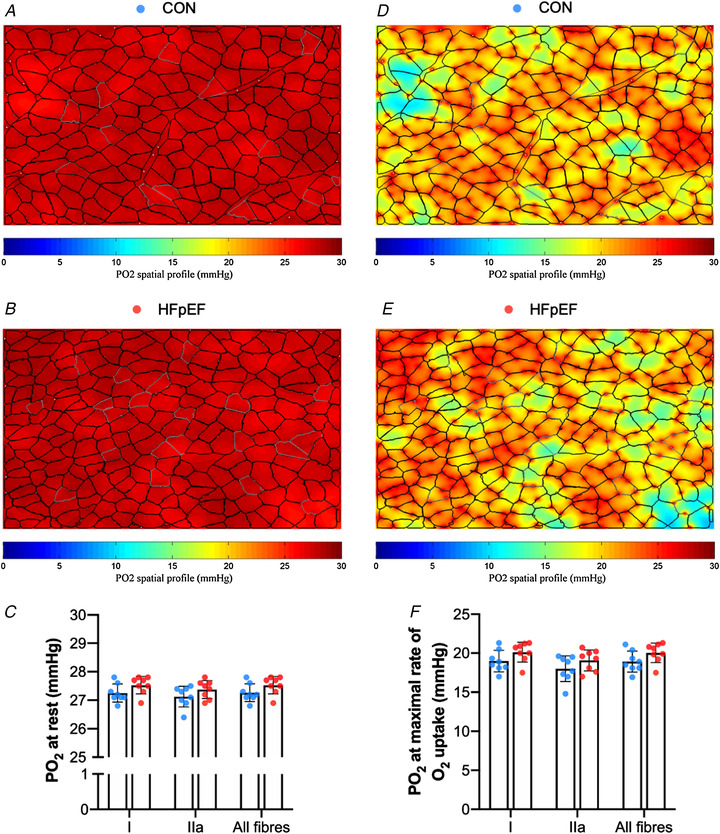
Simulation of muscle PO2 at rest (A–B) and maximal rate of oxygen consumption (D–E) in representative images. There were no significant differences in simulations of muscle PO2 at rest (Type I fibres: 27.26 ± 0.33 vs. 27.54 ± 0.30 mmHg, P = 0.099; Type IIa: 27.12 ± 0.39 vs. 27.37 ± 0.31 mmHg, P = 0.16; all fibres: 27.25 ± 0.33 vs. 27.52 ± 0.29 mmHg, P = 0.102) (C) or at maximal rate of oxygen consumption (Type I: 18.97 ± 1.38 vs. 20.11 ± 1.27 mmHg, P = 0.109; Type IIa: 18.00 ± 1.64 vs. 19.07 ± 1.36 mmHg, P = 0.177; all fibres: 18.92 ± 1.36 vs. 20.02 ± 1.24 mmHg, P = 0.115) (F). Areas of muscle hypoxia (PO2 < 0.5 mmHg) are highlighted in blue. [Color figure can be viewed at wileyonlinelibrary.com]
The soleus generated lower absolute twitch and maximal forces in HFpEF than control rats (P < 0.001; Fig. 4 A and P = 0.005; Fig. 4 B, respectively), consistent with muscle atrophy, although after adjustment for muscle cross‐sectional area there was no difference between groups in specific forces (P = 0.056, Fig. 4 C; and P = 0.557, Fig. 4 D, respectively). Similarly, twitch characteristics of half relaxation time (P = 0.603; Fig. 4 E) and time‐to‐peak tension (P = 0.474; Fig. 4 F) remained unchanged, as did the maximal twitch:tetanus ratio between control and HFpEF (0.16 ± 0.03 vs. 0.14 ± 0.02; P = 0.140). However, HFpEF rats demonstrated impairments to both shortening velocity (range 10–17%) and mechanical power (range 14–22%) when measured across various percentages of their maximal force (P < 0.05; Fig. 4 G–H), suggesting obese‐HFpEF reduces intrinsic soleus contractile function related to muscle shortening rather than specific force. Interestingly, however, while V max was not different between groups (1.16 ± 0.52 vs. 0.88 ± 0.33 L 0/s; P = 0.307), there was a tendency for the force–velocity curvature to be 25% greater in HFpEF than controls (i.e. a lower power ratio: 0.06 ± 0.02 vs. 0.08 ± 0.02; P = 0.086).
Figure 4. In vitro skeletal muscle function.
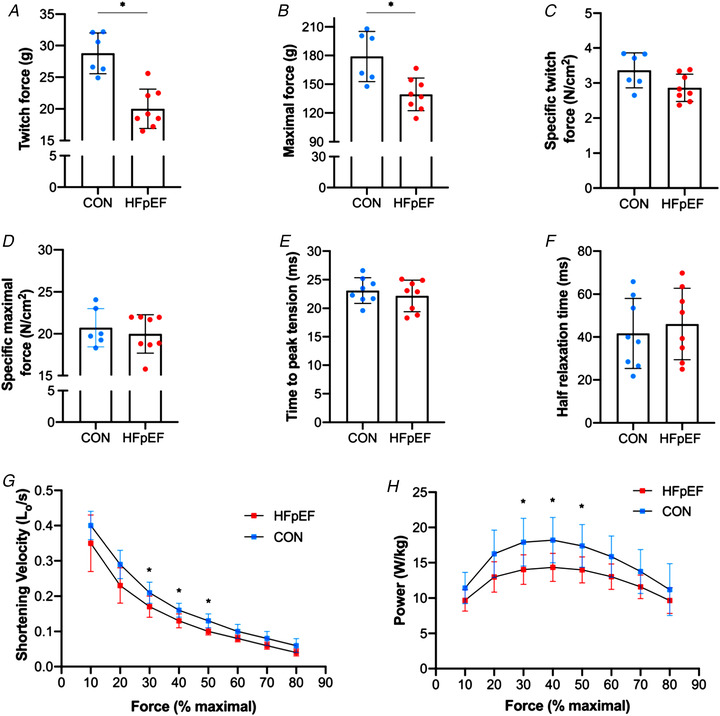
The soleus of HFpEF rats showed lower absolute twitch force (28.79 ± 3.23 vs. 20.01 ± 3.10 g; P < 0.001) (A) and absolute maximal tetanic force (178.95 ± 26.18 vs. 139.37 ± 17.05 g; P = 0.005) (B), although mass‐specific twitch and maximal forces were similar between groups (3.36 ± 0.50 vs. 2.86 ± 0.39 N/cm2; P = 0.056 and 20.72 ± 2.28 vs. 19.98 ± 2.29 N/cm2; P = 0.557, respectively) (C–D). Similarly, time‐to‐peak tension and half relaxation time remained unchanged (23.10 ± 2.25 vs. 22.18 ± 2.75 ms; P = 0.474 and 41.68 ± 16.33 vs. 46.08 ± 16.70 ms; P = 0.603, respectively) (E–F). However, HFpEF rats showed impairments in shortening velocity and muscle power when measured across different percentages of their maximal force (30, 40 and 50%) (P < 0.05) (G–H). [Color figure can be viewed at wileyonlinelibrary.com]
In situ muscle function and femoral artery blood flow
EDL muscle wet mass was 26% lower in HFpEF rats (P < 0.001; Fig. 5 A). This corresponded to lower absolute twitch and maximal forces of 27% and 33%, respectively (P = 0.016; Fig. 5 B and P = 0.030; Fig. 5 C, respectively). When normalised to muscle mass, twitch and maximal specific forces were not different between groups (P = 0.968; Fig. 5 D and P = 0.675; Fig. 5 E, respectively), while relative fatigability was unaffected (P = 0.325; Fig. 5 F). However, HFpEF rats tended to be around 26% more fatigable during the force‐matched protocol (P = 0.079; Fig. 5 G). While HFpEF rats had higher levels of resting femoral artery blood flow (P = 0.039; Fig. 5 H), they showed a severely blunted hyperaemic response of 73% to repeated contractions (P = 0.012; Fig. 5 I). Similarly, impairments in the hyperaemia calculated using vascular conductance was found in HFpEF (P = 0.004; Fig. 5 J). Overall, this suggests that while obese‐HFpEF does not induce muscle dysfunction related to neuromuscular transmission failure, a severe decrement to increase leg blood flow in response to contractions is apparent.
Figure 5. In situ EDL contractile function and femoral artery blood flow.
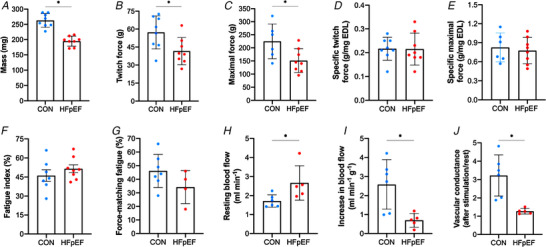
Absolute twitch and maximal tetanic forces of the EDL muscle were lower in HFpEF rats than in controls (57.24 ± 13.82 vs. 41.62 ± 11.31 g, P = 0.016 and 224.64 ± 66.36 vs. 151.38 ± 45.45 g, P = 0.030, respectively) (B–C). However, when normalised to muscle mass, which was reduced in HFpEF rats (262.88 ± 22.47 vs. 194.73 ± 15.35 mg; P < 0.001) (A), these were not significantly affected (0.22 ± 0.05 vs. 0.22 ± 0.07 g mg−1 EDL, P < 0.968 and 0.82 ± 0.23 vs. 0.77 ± 0.21 g mg−1 EDL, P = 0.675, respectively) (D–E). The fatigue index was similar between groups (0.46 ± 0.12 vs. 0.51 ± 0.09 %; P = 0.325) (F). However, HFpEF rats tended to be more fatigable during the force‐matched protocol (45.96 ± 11.96 vs. 34.35 ± 12.08; P = 0.079) (G). Resting femoral artery blood flow was augmented in HFpEF rats (1.71 ± 0.33 vs. 2.66 ± 0.90 ml min−1; P = 0.039) (H). In contrast, HFpEF rats showed an impaired increase in muscle‐specific EDL blood flow during stimulation (2.59 ± 1.30 vs. 0.69 ± 0.37 ml min−1 g−1; P = 0.012) (I). Moreover, a reduction in the functional hyperaemic scope was also found in HFpEF (3.22 ± 1.12 vs. 1.27 ± 0.15; P = 0.004) (J). [Color figure can be viewed at wileyonlinelibrary.com]
Histological and functional characteristics of the diaphragm
Representative diaphragm sections from control and HFpEF rats are presented in Fig. 6 A–B. Average FCSA was similar between groups (P = 0.609; Fig. 6 C). However, compared with lean controls HFpEF increased FCSA in both Type I and Type IIa fibres by 46% (P < 0.001) and 26% (P = 0.005), respectively (Fig. 6 C), but reduced Type IIb/IIx FCSA by 22% (P = 0.004). Additionally, HFpEF rats had a higher numerical percentage of Type I fibres (P = 0.003; Fig. 6 D) and a higher area percentage of Type I fibres (P < 0.001; Fig. 6 E).
Figure 6. Histological features of the diaphragm.
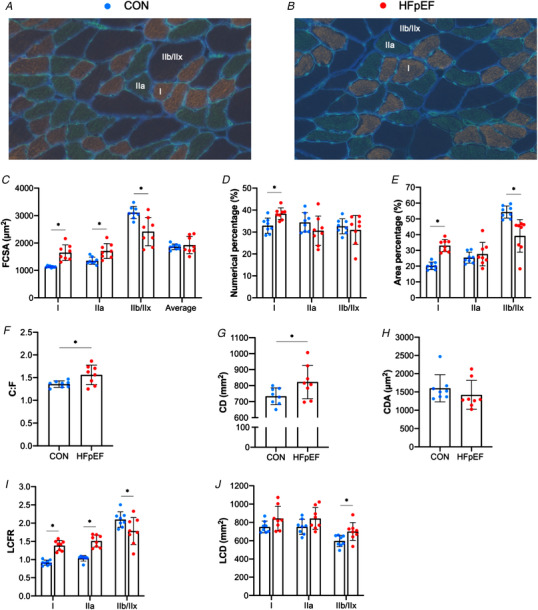
Representative diaphragm sections from control (A) and obese‐HFpEF (B). Compared with lean controls, HFpEF rats had increased cross‐sectional area (CSA) in Type I (1130.67 ± 30.45 vs. 1647.83 ± 286.52 μm2; P < 0.001) and Type IIa fibres (1352.19 ± 133.46 vs. 1709.33 ± 273.24 μm2; P = 0.005), whereas CSA of Type IIb/IIx fibres was reduced (3109.90 ± 222.49 vs. 2418.50 ± 514.36 μm2; P = 0.004) (C). HFpEF rats also had a higher numerical percentage of Type I fibres (32.93 ± 3.46 vs. 38.38 ± 2.64 %; P = 0.003), although this remained unchanged in Type IIa (34.41 ± 4.39 vs. 30.65 ± 6.67 %; P = 0.203) and IIb/IIx fibres (32.63 ± 3.43 vs. 31.00 ± 6.63 %; P = 0.545) (D). Additionally, HFpEF rats showed a higher area percentage of Type I fibres (20.26 ± 2.37 vs. 33.12 ± 3.85 %; P < 0.001), whereas this was unchanged in Type IIa fibres (25.38 ± 3.50 vs. 27.66 ± 7.47 %; P = 0.449) and reduced in Type IIb/IIx fibres (54.36 ± 3.83 vs. 39.23 ± 10.33; P = 0.002) (E). HFpEF rats also showed general and local alterations in capillary distribution. General changes included increased capillary‐to‐fibre (C:F) ratio (1.96 ± 0.12 vs. 2.26 ± 0.28; P = 0.015) (F) and capillary density (CD) (733.42 ± 51.94 vs. 822.30 ± 104.43 mm2; P = 0.049) (G), although capillary domain area (CDA) remained unchanged (1600.57 ± 371.08 vs. 1420.58 ± 392.25 μm2; P = 0.362) (H). Local changes included increased LCFR in Type I (0.92 ± 0.08 vs. 1.39 ± 0.14; P < 0.001) and Type IIa fibres (1.05 ± 0.04 vs. 1.51 ± 0.16; P < 0.001) and reduced local capillary‐to‐fibre ratio (LCFR) in glycolytic/Type IIb/IIx fibres (2.13 ± 0.21 vs. 1.78 ± 0.37; P = 0.040) (I). In contrast, however, HFpEF rats had increased local capillary density (LCD) in Type IIb/IIx fibres (615.85 ± 45.69 vs. 700.85 ± 97.37 mm2; P = 0.042), with no changes in Type I (765.26 ± 60.76 vs. 843.27 ± 132.38 mm2; P = 0.152) and Type IIa fibres (763.78 ± 68.71 vs. 842.75 ± 119.64 mm2; P = 0.128) (J). [Color figure can be viewed at wileyonlinelibrary.com]
Global and local alterations were also observed in capillary distribution, with C:F ratio and CD increased in HFpEF rats (P = 0.015; Fig. 6 F and P = 0.049; Fig. 6 G, respectively) but CDA did not differ significantly (P = 0.362; Fig. 6 H). HFpEF rats had increased LCFR in Type I (P < 0.001) and Type IIa fibres (P < 0.001), whereas this was reduced in Type IIb/IIx fibres (P = 0.040; Fig. 6 I). In contrast, LCD was higher in HFpEF for Type IIb/IIx fibres (P = 0.042), with no changes in Type I (P = 0.152) and Type IIa fibres (P = 0.128; Fig. 6 J). We next estimated diaphragm PO2 levels (Fig. 7 A–B) and found HFpEF elevated resting muscle oxygen tension (Type I fibres: P = 0.043; Type IIa: P = 0.019; Type IIb/IIx: P = 0.006; all fibres: P = 0.009; Fig. 7 C) and at maximal metabolic rates (Type I: P = 0.045; Type IIa: P = 0.018; Type IIb/IIx: P = 0.004; all fibres: P = 0.006; Fig. 7 F), indicating improved muscle oxygenation in obese‐HFpEF.
Figure 7. Modelling of diaphragm oxygen tension.
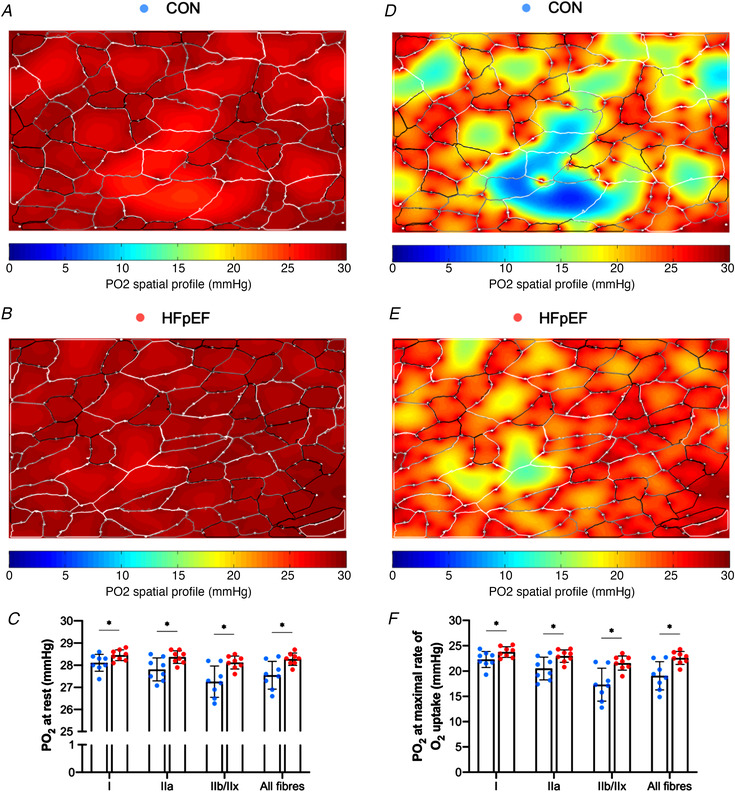
Simulation of muscle PO2 at rest (A–B) and maximal rate of oxygen consumption (D–E) in representative images. Compared with lean controls, HFpEF rats showed higher muscle oxygen tension at rest (Type I fibres: 28.09 ± 0.39 vs. 28.45 ± 0.24 mmHg, P = 0.043; Type IIa: 27.80 ± 0.52 vs. 28.35 ± 0.27 mmHg, P = 0.019; Type IIb/IIx: 27.26 ± 0.70 vs. 28.13 ± 0.29 mmHg, P = 0.006; all fibres: 27.55 ± 0.61 vs. 28.28 ± 0.27, P = 0.009) (C) or at maximal rate of oxygen consumption (Type I: 22.29 ± 1.57 vs. 23.76 ± 1.06 mmHg, P = 0.045; Type IIa: 20.52 ± 2.27 vs. 22.95 ± 1.22 mmHg, P = 0.018; Type IIb/IIx: 17.33 ± 3.24 vs. 21.58 ± 1.39 mmHg, P = 0.004; all fibres: 19.07 ± 2.78 vs. 22.58 ± 1.26 mmHg, P = 0.006) (F). [Color figure can be viewed at wileyonlinelibrary.com]
Isometric twitch and maximal tetanic stress of the diaphragm were similar between groups (P = 0.254; Fig. 8 A and P = 0.225; Fig. 8 B, respectively). However, analysis of twitch kinetics demonstrated that HFpEF rats had a slower time‐to‐peak tension (P = 0.006; Fig. 8 C) while half relaxation time remained unchanged (P = 0.170; Fig. 8 D) as did the maximal twitch:tetanus ratio between control and HFpEF (0.31 ± 0.06 vs. 0.32 ± 0.05; P = 0.630). Similarly, there were no differences in isotonic properties as assessed by maximal shortening velocity or maximal isotonic power between groups (P = 0.756; Fig. 8 E and P = 0.670; Fig. 8 F), with the power ratio also not different between groups (0.11 ± 0.01 vs. 0.11 ± 0.01; P = 0.253). During the cyclical contractions, there were no differences in the net power recorded at any given frequency and no difference in the cycle frequency that yielded maximum net power (i.e. 5 Hz for both groups) (all P > 0.05; Fig. 8 G). However, during repeated cyclical contractions the ability of the diaphragm to sustain work and power relative to the unfatigued state was reduced in HFpEF compared with controls (P = 0.001) and this occurred after relatively few cycles of work (cycles 6–12; P < 0.05; Fig. 8 H).
Figure 8. Functional properties of the diaphragm.
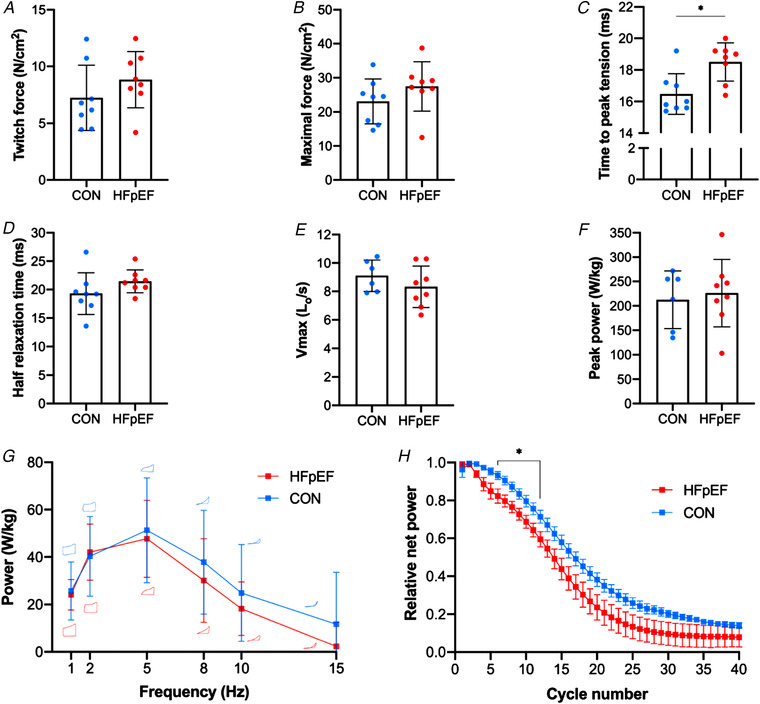
Isometric twitch and tetanic stress of the diaphragm were not different between groups (7.24 ± 2.87 vs. 8.83 ± 2.47 N/cm2, P = 0.254 and 23.09 ± 6.56 vs. 27.46 ± 7.22 N/cm2, P = 0.225, respectively) (A–B). In contrast, HFpEF rats showed slowed time‐to‐peak tension (16.48 ± 1.29 vs. 18.50 ± 1.21 ms; P = 0.006) (C), although half relaxation time was not significantly affected (19.30 ± 3.68 vs. 21.45 ± 2.02 ms; P = 0.170) (D). There were no differences in maximal shortening velocity (Vmax) (9.10 ± 1.11 vs. 8.32 ± 1.46 Lo/s; P = 0.278) (E) or peak isotonic power (212.56± 59.28 vs. 226.11 ± 69.36 W kg−1; P = 0.701) (F) between groups. During cyclical contractions, while the net power‐cycle frequency relationship remained unaltered between groups (P > 0.05; typical work loops are shown at each cycle frequency for each group) (G), relative fatigue was greater in HFpEF (P < 0.001) under cycles 6–12 (H). [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
This study has identified novel skeletal muscle impairments in obese‐HFpEF that likely predispose towards the pathophysiology of exercise intolerance. The main findings from this study are:
Limb muscle weakness was closely associated with fibre atrophy in HFpEF, but isometric contractile properties were not impaired under neural‐ or direct‐muscle assessments, indicating preserved isometric neuromuscular function.
In contrast, limb isotonic muscle properties including shortening velocity and mechanical power were impaired in HFpEF.
An abnormal leg blood flow response to contractions alongside fibre type‐specific structural capillary loss was found in HFpEF, indicating perfusive O2 transport limitations.
Significant remodelling of the diaphragm occurred in HFpEF including divergent fibre‐type hypertrophy/atrophy, higher capillarity/PO2, and a Type II‐to‐I fibre‐type shift, with preserved muscle mechanics.
Impact of HFpEF on limb muscle function
A reduction in skeletal muscle mass in patients with HFpEF is strongly associated with reduced muscle strength and poor quality of life (Bekfani et al. 2016). Muscle weakness is generally underpinned by either a reduction in muscle mass (i.e. atrophy) and/or intrinsic contractile dysfunction. In this study, we used in vitro (i.e. direct muscle) and in situ (i.e. peripheral nerve) stimulation approaches to assess isometric contractile properties in limb muscle. This allowed various sites in the muscle contractile process to be evaluated for dysfunction in HFpEF, including neuromuscular transmission and excitation–contraction coupling. Consistent with previous data where absolute maximal soleus force was reduced by ∼20% in HFpEF rats vs. controls (Bowen et al. 2018; Schauer et al. 2020), we observed that absolute twitch and maximal forces in both the soleus and EDL were lower in HFpEF rats. However, limb muscle weakness was closely associated with fibre atrophy as, after normalising for muscle mass, specific forces were not different between groups independent of whether the neural and blood supply remained intact. This is important, as it indicates that neuromuscular transmission and excitation–contraction coupling is likely preserved under isometric contractions in obese‐HFpEF.
However, most daily activities require the muscle to shorten against different loads to generate mechanical power and thus perform work. Therefore, assessment of muscle isotonic properties such as shortening velocity and power, which remained undefined in HFpEF, provides a more relevant assessment in relation to daily patient activities. Here, we observed that HFpEF rats had impairments to both shortening velocity and mechanical power in the soleus. This functional loss in HFpEF cannot be explained by a simple shift towards more Type I fibres (i.e. typically associated with slower shortening velocities where myosin heavy chain isoform is a key determinant (Bottinelli et al. 1991)), as we observed a higher proportion of Type IIa fibres in HFpEF (i.e., a Type I‐to‐II fibre‐type shift). As such, while this rules out a Type I fibre‐type shift as a potential mechanism underlying the slower shortening velocities observed in HFpEF, shortening velocity is also thought to be limited by the rate of ADP dissociation from actomyosin (Nyitrai et al. 2006). Thus, our data suggest obese‐HFpEF rats develop slowed rates of cross‐bridge detachment through impaired ADP release, potentially due to post‐translational modifications of myosin related to oxidative stress or glycation, as previously reported in HFrEF (Coirault et al. 2007) and ageing (Ramamurthy et al. 1999). In further support, slowed cross‐bridge kinetics have previously been reported in Type I and IIa fibres of vastus lateralis biopsies from patients with HFrEF (Miller et al. 2010), although other mechanisms such as impaired sarcoplasmic reticulum calcium pumping cannot be ruled out. Interestingly, we also found a tendency in the soleus for the curvature of the force–velocity relationship to be greater in HFpEF than the controls (i.e. where curvature is the inverse of the a/ ratio in the Hill equation), which is in line with previous studies highlighting significant power loss during fatiguing exercise when the curvature is greater (or a/ lower) (Jones, 2010). As such, a greater curvature of the force–velocity relationship may be another potential mechanism contributing to the loss of power observed in HFpEF. Overall, therefore, the significant loss of absolute limb force associated with muscle atrophy in obese‐HFpEF alongside intrinsic impairments related to lower shortening velocity and increased curvature would be predicted to severely reduce mechanical power and thus predispose towards exercise intolerance.
Impact of HFpEF on limb skeletal muscle morphology
Despite skeletal muscle morphological alterations being well investigated in HFrEF (Kennel et al. 2015), little information is available on HFpEF from either animal or patient studies. Previous data from patients with HFpEF indicated the vastus lateralis Type I to Type II fibre‐type shift and a lower global C:F ratio, which is associated with reduced V̇O2peak (Kitzman et al. 2014). Animal models (hypertensive or cardiometabolic) have shown in the soleus/EDL a significant fibre atrophy and a lower global C:F ratio (Bowen et al. 2015, 2017b, 2018; Schauer et al. 2020). Consistent with this, in the present study we found that soleus muscle from HFpEF rats exhibited a fibre atrophy of 24% and a Type IIa fibre‐type shift alongside a lower global C:F ratio of 17%. In contrast, however, we also provide new evidence that CD in the soleus was higher in HFpEF rats by 15% vs. controls, and by using novel local measures of capillarity (i.e. LCRF, LCD), we identified that while a similar trend was found in both fibre types, only Type I reached statistical significance. These additional local indices allowed us to further conclude that the lower C:F ratio observed in HFpEF was the result of a greater contribution from Type I fibres (i.e. LCRF), suggesting that slow‐ rather than fast‐twitch fibres tended to be more susceptible to microvascular alterations in this disease. However, the capillary supply per cross‐sectional area of Type I fibres (LCD) was in fact higher in HFpEF muscle. These global and local measures of capillarity where the C:F ratio was lower but CD higher in HFpEF are likely explained by the observed fibre atrophy, as CD is highly dependent upon fibre size (Egginton, 2011). This indicates that the degree of fibre atrophy exceeded the rate of capillary loss in HFpEF, thus increasing the CD, which has been suggested as an adaptive process to reduce diffusion distances across muscle fibres (Al‐Shammari et al. 2019), helping to discriminate structural from functional consequences of microvascular remodelling. A reduced distance for oxygen diffusion could be the result of an adaptation to changes in leg blood flow, as we observed. In HFpEF, this may also be a compensatory mechanism to preserve O2 flux from capillary to myocyte and thus maintain a better PO2 status across the muscle, not only in the face of capillary loss but also in response to the reported deficits in mitochondrial O2 utilisation (Bowen et al. 2015, 2017b; Molina et al. 2016).
In this regard, data collected from patients with HFpEF have identified clear impairments in the ability to widen arterial–venous O2 content (ΔAVO2) and augment peripheral oxygen extraction during exercise compared with HFrEF or controls (Haykowsky et al. 2011; Bhella et al. 2011a; Dhakal et al. 2015; Houstis et al. 2018; Zamani et al. 2020). It remains unclear whether the capillary loss we observed contributes to abnormal skeletal muscle O2 extraction at peak exercise in HFpEF, with a peripheral O2 diffusive limitation postulated as a major mechanism underpinning exercise intolerance in HFpEF (Dhakal et al. 2015; Houstis et al. 2018). To expand current knowledge (Dhakal et al. 2015; Houstis et al. 2018; Zamani et al. 2020), we therefore used in silico modelling to provide the first fibre‐type specific estimates of microvascular contribution to muscle oxygenation in HFpEF. We found muscle PO2 was similar between HFpEF and controls during simulated rest and at maximal exercise, which indicates adequate oxygenation is maintained in HFpEF with no evidence for enhanced tissue hypoxia. A recent patient study using forearm exercise found estimated peripheral O2 diffusion was not different between HFpEF and controls (Zamani et al. 2020), but this contrasts with previous cycling studies where O2 diffusion was significantly lower based on systemic haemodynamic, blood gas and pulmonary gas exchange measurements (Dhakal et al. 2015; Houstis et al. 2018). It should be noted that these patient studies did not measure leg/muscle ΔAVO2, fibre size, capillarity, or microvascular distribution, which can all influence peripheral O2 diffusion. While the current data do not allow us to directly confirm whether O2 diffusion was impaired in obese‐HFpEF, our data suggest that fibre morphology and capillary distribution are unlikely to contribute to O2 diffusive limitations.
Interestingly, and in contrast to HFrEF or controls, patients with HFpEF are unable to lower venous PO2 during exercise and therefore demonstrate a blunted peripheral O2 extraction response (Dhakal et al. 2015; Houstis et al. 2018; Poole et al. 2018; Zamani et al. 2020). The extent of this impaired muscle O2 extraction in HFpEF is likely explained, at least in part, by the significant mitochondrial abnormalities reported in patients with HFpEF (Molina et al. 2016). In our study, estimated muscle PO2 in HFpEF was not lower than controls at simulated maximal exercise, and while not discounting the potential for a muscle O2 diffusion limitation (Poole et al. 2018), this is consistent with maintenance of a high muscle oxygenation across the muscle in HFpEF to support more optimally functioning mitochondria (Al‐Shammari et al. 2019). Clearly, more studies are warranted to clarify the role of limitations to muscle O2 diffusion in HFpEF. However, our present data of a reduced leg blood flow (see below) implicate a perfusive O2 delivery limitation as one key mechanism that could potentially blunt O2 extraction in obese‐HFpEF.
Impaired muscle blood flow response in HFpEF
Up until now, direct measures of leg (muscle) arterial blood flow had not been assessed in HFpEF, limiting mechanistic understanding. In the present study, using perivascular flow probes (the gold standard for measuring volumetric blood flow in animal studies), we directly demonstrated that the functional hyperaemic response to contractions was blunted in HFpEF rats. These data support the concept that the peripheral response to exercise is impaired in HFpEF (Houstis et al. 2018), and are consistent with non‐invasive measurements in patients with HFpEF during knee‐extensor exercise where impaired leg blood flow and vascular conductance occurred independently of limitations to heart rate, stroke volume and cardiac output (Lee et al. 2016b; Weavil et al. 2020) but contrast with recent isometric forearm data which did not find any change (Zamani et al. 2020). However, some (Maréchaux et al. 2016; Lee et al. 2016b; Kishimoto et al. 2017; Weavil et al. 2020) but not others (Hundley et al. 2007; Haykowsky et al. 2013; Lee et al. 2016a; Zamani et al. 2020) report an abnormal blood flood response to exercise in HFpEF patients when compared with controls, which is probably related to differences in the muscle studied, non‐invasive and different measurement techniques, and patient heterogeneity. Overall, our data support the potential for perfusive, feed‐artery O2 transport limitations in HFpEF, which likely contributes to exercise intolerance in this disease (Poole et al. 2018; Weavil et al. 2020).
Although the mechanisms underlying abnormal limb blood flow response to exercise in HFpEF remain unclear, endothelial function is impaired (Schmederer et al. 2018) and postulated as a central mechanism underlying disease progression (Paulus & Tschöpe, 2013; Gevaert et al. 2017; Schmederer et al. 2018). Beyond this, upstream central impairments related to cardiac output likely play a major role (Wolsk et al. 2019). While the functional significance of an impairment to limb blood flow during exercise in HFpEF is not known, it may exacerbate the degree of muscle fatigue experienced and limit daily activities performed by patients where the ability to repeatedly sustain absolute forces rather than relative forces becomes crucial, as recently demonstrated in patients with HFpEF (Weavil et al. 2020). This is supported by our employed matched‐initial force fatigue protocol, where HFpEF rats showed a trend to be 26% more fatigable than controls. These data provide important clinical relevance, as patients with muscle weakness such as HFpEF are often required to increase motor firing frequencies to perform certain daily activities that induce early fatigue (Ferreira et al. 2010; Weavil et al. 2020), although it is important to note this may not necessarily reflect direct differences in relation to muscle fatigue properties. Our data therefore indicate that skeletal muscle arterial blood delivery is potentially constrained during exercise in HFpEF. As such, at least in the context of this animal model and in support of recent patient findings (Weavil et al. 2020), a perfusive O2 delivery limitation could play a key role limiting O2 extraction and thus exacerbating exercise intolerance in obese‐HFpEF (Poole et al. 2018; Zamani et al. 2020). Interestingly, a lower leg blood flow during contractions coupled with a higher muscle CD in obese‐HFpEF could result in an increased red blood cell transit time to mediate a greater O2 extraction. However, this may not transpire in HFpEF due to the significant mitochondrial abnormalities developed in patients (Molina et al. 2016), thus preventing any significant widening of the ΔAVO2.
Impact of HFpEF on diaphragm remodelling and muscle mechanics
Inspiratory (i.e. diaphragm) muscle weakness is evident and closely associated with symptoms of dyspnoea and poor prognosis in patients with HFpEF (Lavietes et al. 2004; Hamazaki et al. 2020). Multiple alterations to the diaphragm have been reported in HFpEF, including in vitro muscle weakness and fatigue alongside a Type II‐to‐I fibre‐type shift, fibre atrophy, and impaired in situ mitochondrial respiration in a hypertensive rat model (Bowen et al. 2015). In contrast, we show remodelling of the HFpEF diaphragm that is reminiscent of exercise‐training including fibre hypertrophy, increased mitochondrial content, and preserved fatigue resistance, although evidence for mitochondrial uncoupling and a mild isometric contractile dysfunction have been noted (Bowen et al. 2017b). The disparity in findings between models is likely explained by the co‐morbidity of obesity and its associated chronic respiratory loading, which can act as a training stimulus to increase fibre size, mitochondrial function/content, and fatigue resistance (Farkas et al. 1994; Powers et al. 1996). Whether similar findings are observed between obese vs. lean patients with HFpEF remains unknown.
Given that approximately 80% of HFpEF patients are obese (Shah et al. 2016) and a recent distinct obese‐HFpEF phenotype has been established (Obokata et al. 2017), the present study contributes novel and highly relevant data in relation to diaphragm plasticity. Until now, only limited data have been available with respect to fibre‐type structure, isoform, and microvasculature of the diaphragm in HFpEF (Bowen et al. 2015, 2017b). In the present study, we identified three major fibre types (Types I, IIa and IIb/IIx) to provide new evidence of a divergent hypertrophy/atrophy fibre remodelling in obese‐HFpEF alongside increased indices of global and local capillarity (i.e. C:F ratio and CD), and estimated levels of fibre oxygenation at rest and maximal exercise. Specifically, compared with controls, obese‐HFpEF rats had increased Type I/IIa FCSA, and reduced Type IIb/IIx FCSA, indicating compensatory adaptations in slow‐twitch fibres. This observation corresponds to the morphometric alterations following unilateral denervation of rat diaphragm muscle, where hypertrophy in Type I fibres but atrophy in Type IIb/IIx fibres occurred (Aravamudan et al. 2006). In support, albeit in the limb gastrocnemius, denervation has been demonstrated to induce fibre atrophy at a relatively higher rate than capillary rarefaction to mediate a higher CD (Paudyal et al. 2018), which is in line with our findings from the diaphragm in HFpEF. Nevertheless, it remains unclear whether obese‐HFpEF induces partial diaphragm muscle denervation in fast‐twitch Type IIb/x as reported in other conditions such as ageing (Elliott et al. 2016).
Overall the obese‐HFpEF diaphragm demonstrates improved indices of oxygen transport (increased capillarity and PO2 distribution) that likely supports the observed shift towards an oxidative phenotype (i.e. higher proportion of Type I fibres, mitochondrial content, and antioxidative enzyme capacity (Bowen et al. 2015, 2017b). This suggests that any functional diaphragm impairments developed in obese‐HFpEF were likely offset by morphological adaptations. To decipher this potential trade‐off, we performed in vitro isometric, isotonic and cyclical contractions on the diaphragm in one of the most detailed functional assessments in HFpEF to date. Our data indicate that impaired muscle mechanics and intrinsic diaphragm dysfunction generally do not develop early in the time course of obese‐HFpEF (∼20 weeks), with only a mild increase found compared with controls in terms of fatigability. Again, these data conflict with previous experimental data where a significant reduction in diaphragm force during repeated isometric contractions was measured in rats with more advanced hypertensive‐induced HFpEF (Bowen et al. 2015). In the present study we also simulated in vivo respiratory muscle mechanics, by applying cyclical length changes and phasic stimulation to the muscle to generate cycles of work, using the in vitro work loop technique (Josephson, 1985). Similar to our isolated isometric and isotonic measures, net power output during unfatigued cyclical contractions in obese‐HFpEF was unaffected in the diaphragm, but we did observe a mild increase in fatigue during repeated cyclical contractions. Collectively, these data suggest that respiratory muscle dysfunction is unlikely to be a key player in the pathogenesis of exercise intolerance in obese‐HFpEF, at least during early disease progression.
Study limitations
We did not use echocardiography or invasive haemodynamics to quantify the extent of LV diastolic function and ejection fraction. However, this rat model has been validated and consistently develops key features of HFpEF as early as 10–15 weeks of age (Schauer et al. 2020), including impaired diastolic function, preserved ejection fraction, myocardial remodelling, and exercise intolerance (Hamdani et al. 2013; Leite et al. 2015; Franssen et al. 2016; van Dijk et al. 2016; Bowen et al. 2017b). Instead, we used MRI to confirm the presence of cardiac remodelling which occurred in the RV and this is known to be closely associated with HFpEF development in obese patients (Obokata et al. 2017) and one of the strongest predictors of poor prognosis (Burke et al. 2014). We also compared groups at a relatively early time point in the progression of HFpEF, which may limit translation of our findings to more advanced stages of the disease. In addition, our experiments were performed in male rats only such that it remains unclear whether similar findings would be observed in females, although a recent study confirmed that a similar time course in disease progression is observed in females (Schauer et al. 2020). Further, while differences in physical activity levels between groups were not measured and cannot be ruled out as having an influence on our experimental measures, it is well established that disuse alone fails to account for the skeletal muscle impairments developed during heart failure (Simonini et al. 1996; Miller et al. 2009). While we saw a trend for fatigue to be higher in HFpEF under our matched‐initial force protocol, this was only performed in n = 4 and thus may have been underpowered to detect statistical, rather than biologically meaningful, differences. Furthermore, as with all biological models, inherent limitations should be considered and our estimated muscle PO2 values from this study were based on a number of assumptions (as detailed in Methods) and may not be directly comparable to humans due to allometric scaling and/or fibre‐type differences.
Conclusions
Obese‐HFpEF rats have a blunted hindlimb blood flow response to contractions alongside microvascular structural remodelling, fibre atrophy, and isotonic contractile dysfunction, which may be important factors underlying exercise intolerance in this disease. In contrast, diaphragm phenotype was largely preserved, indicating a more prominent role for limb rather than respiratory muscle abnormalities in obese‐HFpEF.
Additional information
Competing interests
None declared.
Author contributions
E.E.G. performed the histological staining and image analyses and drafted the manuscript. P.G.T. performed in situ experiments and helped draft the manuscript. G.N.A. performed the in vitro diaphragm experiments and helped draft the manuscript. R.K. performed the in vitro diaphragm experiments and helped draft the manuscript. A.P.B. performed the cardiac imaging experiments and helped draft the manuscript. S.E. contributed to conception and design of the experiments, and interpretation of the data. T.S.B. contributed to conception and design of the study, performed muscle experiments, helped interpret data and draft the manuscript. All authors approved the final version of the manuscript.
Funding
E.E.G. is a recipient of the Doctoral Fellowship from the Mexican National Council of Science and Technology (CONACYT). A.P.B. was supported by British Heart Foundation grant PG/16/74/32374. T.S.B. is a recipient of a Medical Research Council (MRC) New Investigator Research Grant (NIRG) (MR/S025472/1) and also received a Research Grant from The Physiological Society to support this work.
Supporting information
Statistical Summary Document
Acknowledgements
We thank Dr Sarah Calaghan, University of Leeds, for her kind assistance in helping to organise the logistics regarding the cardiac imaging experiments.
Biography
Ever Espino‐Gonzalez received his Undergraduate and Master of Science degrees from the Autonomous University of Chihuahua. He is currently a PhD student in Biomedical Sciences funded by CONACYT at the University of Leeds under the supervision of Dr Scott Bowen and Professor Stuart Egginton. His research focuses on mechanisms and treatments of skeletal muscle weakness in heart failure with preserved ejection fraction (HFpEF). He uses in situ and in vitro functional analyses, histological assays of fibre‐type distribution and capillarity, alongside mitochondrial respiration in rodents and patients in order to provide new insights into HFpEF‐induced skeletal muscle impairments.

Edited by: Scott Powers & Bruno Grassi
Linked articles: This article is highlighted in a Perspectives article by Hendrickse & Degens. To read this article, visit https://doi.org/10.1113/JP281204.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Adams V, Linke A & Winzer E (2017). Skeletal muscle alterations in HFrEF vs. HFpEF. Curr Heart Fail Rep 14, 489–497. [DOI] [PubMed] [Google Scholar]
- Al‐Shammari AA, Kissane RWP, Holbek S, Mackey AL, Andersen TR, Gaffney EA, Kjaer M & Egginton S (2019). Integrated method for quantitative morphometry and oxygen transport modeling in striated muscle. J Appl Physiol 126, 544–557. [DOI] [PubMed] [Google Scholar]
- Aravamudan B, Mantilla CB, Zhan WZ & Sieck GC (2006). Denervation effects on myonuclear domain size of rat diaphragm fibers. J Appl Physiol 100, 1617–1622. [DOI] [PubMed] [Google Scholar]
- Bekfani T, Pellicori P, Morris DA, Ebner N, Valentova M, Steinbeck L, Wachter R, Elsner S, Sliziuk V, Schefold JC, Sandek A, Doehner W, Cleland JG, Lainscak M, Anker SD & von Haehling S (2016). Sarcopenia in patients with heart failure with preserved ejection fraction: Impact on muscle strength, exercise capacity and quality of life. Int J Cardiol 222, 41–46. [DOI] [PubMed] [Google Scholar]
- Benson AP, Bernus O, Dierckx H, Gilbert SH, Greenwood JP, Holden AV, Mohee K, Plein S, Radjenovic A, Ries ME, Smith GL, Sourbron S & Walton RD (2011). Construction and validation of anisotropic and orthotropic ventricular geometries for quantitative predictive cardiac electrophysiology. Interface Focus 1, 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson AP, Gilbert SH, Li P, Newton SM & Holden AV (2008). Reconstruction and quantification of diffusion tensor imaging‐derived cardiac fibre and sheet structure in ventricular regions used in studies of excitation propagation. Math Model Nat Phenom 3, 101–130. [Google Scholar]
- Bhella PS, Prasad A, Heinicke K, Hastings JL, Arbab‐Zadeh A, Adams‐Huet B, Pacini EL, Shibata S, Palmer MD, Newcomer BR & Levine BD (2011a). Abnormal haemodynamic response to exercise in heart failure with preserved ejection fraction. Eur J Heart Fail 13, 1296–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhella PS, Prasad A, Heinicke K, Hastings JL, Arbab‐Zadeh A, Adams‐Huet B, Pacini EL, Shibata S, Palmer MD, Newcomer BR & Levine BD (2011b). Abnormal haemodynamic response to exercise in heart failure with preserved ejection fraction. Eur J Heart Fail 13, 1296–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottinelli R, Schiaffino S & Reggiani C (1991). Force‐velocity relations and myosin heavy chain isoform compositions of skinned fibres from rat skeletal muscle. J Physiol 437, 655–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen TS, Adams V, Werner S, Fischer T, Vinke P, Brogger MN, Mangner N, Linke A, Sehr P, Lewis J, Labeit D, Gasch A & Labeit S (2017a). Small‐molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J Cachexia Sarcopenia Muscle 8, 939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen TS, Brauer D, Rolim NPL, Baekkerud FH, Kricke A, Ormbostad Berre AM, Fischer T, Linke A, da Silva GJ, Wisloff U & Adams V (2017b). Exercise training reveals inflexibility of the diaphragm in an animal model of patients with obesity‐driven heart failure with a preserved ejection fraction. J Am Heart Assoc 6, e006416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen TS, Herz C, Rolim NPL, Berre AO, Halle M, Kricke A, Linke A, da Silva GJ, Wisloff U & Adams V (2018). Effects of endurance training on detrimental structural, cellular, and functional alterations in skeletal muscles of heart failure with preserved ejection fraction. J Card Fail 24, 603–613. [DOI] [PubMed] [Google Scholar]
- Bowen TS, Rolim NP, Fischer T, Baekkerud FH, Medeiros A, Werner S, Bronstad E, Rognmo O, Mangner N, Linke A, Schuler G, Silva GJ, Wisloff U, Adams V & Optimex Study G (2015). Heart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. Eur J Heart Fail 17, 263–272. [DOI] [PubMed] [Google Scholar]
- Burke MA, Katz DH, Beussink L, Selvaraj S, Gupta DK, Fox J, Chakrabarti S, Sauer AJ, Rich JD, Freed BH & Shah SJ (2014). Prognostic importance of pathophysiologic markers in patients with heart failure and preserved ejection fraction. Circ Heart Fail 7, 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler J, Fonarow GC, Zile MR, Lam CS, Roessig L, Schelbert EB, Shah SJ, Ahmed A, Bonow RO, Cleland JG, Cody RJ, Chioncel O, Collins SP, Dunnmon P, Filippatos G, Lefkowitz MP, Marti CN, McMurray JJ, Misselwitz F, Nodari S, O'Connor C, Pfeffer MA, Pieske B, Pitt B, Rosano G, Sabbah HN, Senni M, Solomon SD, Stockbridge N, Teerlink JR, Georgiopoulou VV & Gheorghiade M (2014). Developing therapies for heart failure with preserved ejection fraction: current state and future directions. JACC Heart Fail 2, 97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close RI (1972). Dynamic properties of mammalian skeletal muscles. Physiol Rev 52, 129–197. [DOI] [PubMed] [Google Scholar]
- Coirault C, Guellich A, Barbry T, Samuel JL, Riou B & Lecarpentier Y (2007). Oxidative stress of myosin contributes to skeletal muscle dysfunction in rats with chronic heart failure. Am J Physiol Heart Circ Physiol 292, H1009‐H1017. [DOI] [PubMed] [Google Scholar]
- Dhakal BP, Malhotra R, Murphy RM, Pappagianopoulos PP, Baggish AL, Weiner RB, Houstis NE, Eisman AS, Hough SS & Lewis GD (2015). Mechanisms of exercise intolerance in heart failure with preserved ejection fraction: the role of abnormal peripheral oxygen extraction. Circ Heart Fail 8, 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egginton S (2011). Physiological factors influencing capillary growth. Acta Physiol 202, 225–239. [DOI] [PubMed] [Google Scholar]
- Egginton S & Hudlicka O (1999). Early changes in performance, blood flow and capillary fine structure in rat fast muscles induced by electrical stimulation. J Physiol 515, 265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott JE, Greising SM, Mantilla CB & Sieck GC (2016). Functional impact of sarcopenia in respiratory muscles. Respir Physiol Neurobiol 226, 137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas GA, Gosselin LE, Zhan WZ, Schlenker EH & Sieck GC (1994). Histochemical and mechanical properties of diaphragm muscle in morbidly obese Zucker rats. J Appl Physiol 77, 2250–2259. [DOI] [PubMed] [Google Scholar]
- Ferreira LF, Moylan JS, Gilliam LA, Smith JD, Nikolova‐Karakashian M & Reid MB (2010). Sphingomyelinase stimulates oxidant signaling to weaken skeletal muscle and promote fatigue. Am J Physiol Cell Physiol 299, C552‐C560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschope C, Leite‐Moreira AF, Musters R, Niessen HW, Linke WA, Paulus WJ & Hamdani N (2016). Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail 4, 312–324. [DOI] [PubMed] [Google Scholar]
- Fukuta H, Goto T, Wakami K & Ohte N (2016). Effects of drug and exercise intervention on functional capacity and quality of life in heart failure with preserved ejection fraction: A meta‐analysis of randomized controlled trials. Eur J Prev Cardiol 23, 78–85. [DOI] [PubMed] [Google Scholar]
- Gevaert AB, Lemmens K, Vrints CJ & Van Craenenbroeck EM (2017). Targeting endothelial function to treat heart failure with preserved ejection fraction: the promise of exercise training. Oxid Med Cell Longev 2017, 1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamazaki N, Kamiya K, Matsuzawa R, Nozaki K, Ichikawa T, Tanaka S, Nakamura T, Yamashita M, Maekawa E, Noda C, Yamaoka‐Tojo M, Matsunaga A, Masuda T & Ako J (2020). Prevalence and prognosis of respiratory muscle weakness in heart failure patients with preserved ejection fraction. Respir Med 161, 105834. [DOI] [PubMed] [Google Scholar]
- Hamdani N, Franssen C, Lourenço A, Falcão‐Pires I, Fontoura D, Leite S, Plettig L, López B, Ottenheijm CA, Becher PM, González A, Tschöpe C, Díez J, Linke WA, Leite‐Moreira AF & Paulus WJ (2013). Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail 6, 1239–1249. [DOI] [PubMed] [Google Scholar]
- Haykowsky MJ, Brubaker PH, John JM, Stewart KP, Morgan TM & Kitzman DW (2011). Determinants of exercise intolerance in elderly heart failure patients with preserved ejection fraction. J Am Coll Cardiol 58, 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haykowsky MJ, Herrington DM, Brubaker PH, Morgan TM, Hundley WG & Kitzman DW (2013). Relationship of flow‐mediated arterial dilation and exercise capacity in older patients with heart failure and preserved ejection fraction. J Gerontol A Biol Sci Med Sci 68, 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haykowsky MJ & Kitzman DW (2014). Exercise physiology in heart failure and preserved ejection fraction. Heart Fail Clin 10, 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haykowsky MJ, Kouba EJ, Brubaker PH, Nicklas BJ, Eggebeen J & Kitzman DW (2014). Skeletal muscle composition and its relation to exercise intolerance in older patients with heart failure and preserved ejection fraction. Am J Cardiol 113, 1211–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houstis NE, Eisman AS, Pappagianopoulos PP, Wooster L, Bailey CS, Wagner PD & Lewis GD (2018). Exercise intolerance in heart failure with preserved ejection fraction: diagnosing and ranking its causes using personalized O(2) pathway analysis. Circulation 137, 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudlická O, Brown M, Cotter M, Smith M & Vrbová G (1977). The effect of long‐term stimulation of fast muscles on their blood flow, metabolism and ability to withstand fatigue. Pflügers Archiv 369, 141–149. [DOI] [PubMed] [Google Scholar]
- Hundley WG, Bayram E, Hamilton CA, Hamilton EA, Morgan TM, Darty SN, Stewart KP, Link KM, Herrington DM & Kitzman DW (2007). Leg flow‐mediated arterial dilation in elderly patients with heart failure and normal left ventricular ejection fraction. Am J Physiol Heart Circ Physiol 292, H1427‐H1434. [DOI] [PubMed] [Google Scholar]
- Jones, DA (2010). Changes in the force vecloity realtionhsip of fatigue muscle: implications for power production and possible causes. J Physiol 588: 2977–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephson RK (1985). Mechanical power output from striated muscle during cyclic contraction. J Exp Biol 114, 493–512. [Google Scholar]
- Kennel PJ, Mancini DM & Schulze PC (2015). Skeletal muscle changes in chronic cardiac disease and failure. Compr Physiol 5, 1947–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto S, Kajikawa M, Maruhashi T, Iwamoto Y, Matsumoto T, Iwamoto A, Oda N, Matsui S, Hidaka T, Kihara Y, Chayama K, Goto C, Aibara Y, Nakashima A, Noma K & Higashi Y (2017). Endothelial dysfunction and abnormal vascular structure are simultaneously present in patients with heart failure with preserved ejection fraction. Int J Cardiol 231, 181–187. [DOI] [PubMed] [Google Scholar]
- Kissane RWP, Egginton S & Askew GN (2018). Regional variation in the mechanical properties and fibre‐type composition of the rat extensor digitorum longus muscle. Exp Physiol 103, 111–124. [DOI] [PubMed] [Google Scholar]
- Kitzman DW, Nicklas B, Kraus WE, Lyles MF, Eggebeen J, Morgan TM & Haykowsky M (2014). Skeletal muscle abnormalities and exercise intolerance in older patients with heart failure and preserved ejection fraction. Am J Physiol Heart Circ Physiol 306, H1364‐H1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavietes MH, Gerula CM, Fless KG, Cherniack NS & Arora RR (2004). Inspiratory muscle weakness in diastolic dysfunction. Chest 126, 838–844. [DOI] [PubMed] [Google Scholar]
- Lee JF, Barrett‐O'Keefe Z, Garten RS, Nelson AD, Ryan JJ, Nativi JN, Richardson RS & Wray DW (2016a). Evidence of microvascular dysfunction in heart failure with preserved ejection fraction. Heart 102, 278–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JF, Barrett‐O'Keefe Z, Nelson AD, Garten RS, Ryan JJ, Nativi‐Nicolau JN, Richardson RS & Wray DW (2016b). Impaired skeletal muscle vasodilation during exercise in heart failure with preserved ejection fraction. Int J Cardiol 211, 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite S, Oliveira‐Pinto J, Tavares‐Silva M, Abdellatif M, Fontoura D, Falcao‐Pires I, Leite‐Moreira AF & Lourenco AP (2015). Echocardiography and invasive hemodynamics during stress testing for diagnosis of heart failure with preserved ejection fraction: an experimental study. Am J Physiol Heart Circ Physiol 308, H1556‐H1563. [DOI] [PubMed] [Google Scholar]
- Maréchaux S, Samson R, van Belle E, Breyne J, de Monte J, Dédrie C, Chebai N, Menet A, Banfi C, Bouabdallaoui N, Le Jemtel TH & Ennezat PV (2016). Vascular and Microvascular Endothelial Function in Heart Failure With Preserved Ejection Fraction. J Card Fail 22, 3–11. [DOI] [PubMed] [Google Scholar]
- Marsh RL & Bennett AF (1986). Thermal dependence of contractile properties of skeletal muscle from the lizard Sceloporus occidentalis with comments on methods for fitting and comparing force‐velocity curves. J Exp Biol 126, 63–77. [DOI] [PubMed] [Google Scholar]
- Miller MS, VanBuren P, LeWinter MM, Braddock JM, Ades PA, Maughan DW, Palmer BM & Toth MJ (2010). Chronic heart failure decreases cross‐bridge kinetics in single skeletal muscle fibres from humans. J Physiol 588, 4039–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MS, Vanburen P, Lewinter MM, Lecker SH, Selby DE, Palmer BM, Maughan DW, Ades PA & Toth MJ (2009). Mechanisms underlying skeletal muscle weakness in human heart failure: alterations in single fiber myosin protein content and function. Circ Heart Fail 2, 700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina AJ, Bharadwaj MS, Van Horn C, Nicklas BJ, Lyles MF, Eggebeen J, Haykowsky MJ, Brubaker PH & Kitzman DW (2016). Skeletal muscle mitochondrial content, oxidative capacity, and Mfn2 expression are reduced in older patients with heart failure and preserved ejection fraction and are related to exercise intolerance. JACC Heart Fail 4, 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyitrai M, Rossi R, Adamek N, Pellegrino MA, Bottinelli R & Geeves MA (2006). What limits the velocity of fast‐skeletal muscle contraction in mammals? J Mol Biol 355, 432–442. [DOI] [PubMed] [Google Scholar]
- Obokata M, Reddy YNV, Pislaru SV, Melenovsky V & Borlaug BA (2017). Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation 136, 6–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paudyal A, Slevin M, Maas H & Degens H (2018). Time course of denervation‐induced changes in gastrocnemius muscles of adult and old rats. Exp Gerontol 106, 165–172. [DOI] [PubMed] [Google Scholar]
- Paulus WJ & Tschöpe C (2013). A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62, 263–271. [DOI] [PubMed] [Google Scholar]
- Poole DC, Richardson RS, Haykowsky MJ, Hirai DM & Musch TI (2018). Exercise limitations in heart failure with reduced and preserved ejection fraction. J Appl Physiol 124, 208–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Farkas GA, Demirel H, Coombes J, Fletcher L, Hughes MG, Hodge K, Dodd SL & Schlenker EH (1996). Effects of aging and obesity on respiratory muscle phenotype in Zucker rats. J Appl Physiol 81, 1347–1354. [DOI] [PubMed] [Google Scholar]
- Ramamurthy B, Höök P & Larsson L (1999). An overview of carbohydrate‐protein interactions with specific reference to myosin and ageing. Acta Physiol Scand 167, 327–329. [DOI] [PubMed] [Google Scholar]
- Schauer A, Draskowski R, Jannasch A, Kirchhoff V, Goto K, Männel A, Barthel P, Augstein A, Winzer E, Tugtekin M, Labeit S, Linke A & Adams V (2020). ZSF1 rat as animal model for HFpEF: Development of reduced diastolic function and skeletal muscle dysfunction. ESC Heart Fail 7, 2123‐2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmederer Z, Rolim N, Bowen TS, Linke A, Wisloff U & Adams V (2018). Endothelial function is disturbed in a hypertensive diabetic animal model of HFpEF: Moderate continuous vs. high intensity interval training. Int J Cardiol 273, 147–154. [DOI] [PubMed] [Google Scholar]
- Shah SJ, Kitzman DW, Borlaug BA, van Heerebeek L, Zile MR, Kass DA & Paulus WJ (2016). Phenotype‐specific treatment of heart failure with preserved ejection fraction: A multiorgan roadmap. Circulation 134, 73–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K & Kass DA (2014). Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ Res 115, 79–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonini A, Long CS, Dudley GA, Yue P, McElhinny J & Massie BM (1996). Heart failure in rats causes changes in skeletal muscle morphology and gene expression that are not explained by reduced activity. Circ Res 79, 128–136. [DOI] [PubMed] [Google Scholar]
- Teh I, McClymont D, Burton RA, Maguire ML, Whittington HJ, Lygate CA, Kohl P & Schneider JE (2016). Resolving fine cardiac structures in rats with high‐resolution diffusion tensor imaging. Sci Rep 6, 30573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tickle PG, Hendrickse PW, Degens H & Egginton S (2020). Impaired skeletal muscle performance as a consequence of random functional capillary rarefaction can be restored with overload‐dependent angiogenesis. J Physiol 598, 1187–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk CG, Oosterhuis NR, Xu YJ, Brandt M, Paulus WJ, van Heerebeek L, Duncker DJ, Verhaar MC, Fontoura D, Lourenco AP, Leite‐Moreira AF, Falcao‐Pires I, Joles JA & Cheng C (2016). Distinct endothelial cell responses in the heart and kidney microvasculature characterize the progression of heart failure with preserved ejection fraction in the obese ZSF1 rat with cardiorenal metabolic syndrome. Circ Heart Fail 9, e002760. [DOI] [PubMed] [Google Scholar]
- Weavil JC, Thurston TS, Hureau TJ, Gifford JR, Kithas PA, Broxterman RM, Bledsoe AD, Nativi JN, Richardson RS & Amann M (2020) Heart failure with preserved ejection fraction diminishes peripheral hemodynamics and accelerates exercise‐induced neuromuscular fatigue. Am J Physiol Heart Circ Physiol. 10.1152/ajpheart.00266.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss K, Schär M, Panjrath GS, Zhang Y, Sharma K, Bottomley PA, Golozar A, Steinberg A, Gerstenblith G, Russell SD & Weiss RG (2017). Fatigability, exercise intolerance, and abnormal skeletal muscle energetics in heart failure. Circ Heart Fail 10, e004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolsk E, Kaye D, Komtebedde J, Shah SJ, Borlaug BA, Burkhoff D, Kitzman DW, Lam CSP, van Veldhuisen DJ, Ponikowski P, Petrie MC, Hassager C, Møller JE & Gustafsson F (2019). Central and peripheral determinants of exercise capacity in heart failure patients with preserved ejection fraction. JACC Heart Fail 7, 321–332. [DOI] [PubMed] [Google Scholar]
- Yamada K, Kinugasa Y, Sota T, Miyagi M, Sugihara S, Kato M & Yamamoto K (2016). Inspiratory muscle weakness is associated with exercise intolerance in patients with heart failure with preserved ejection fraction: a preliminary study. J Card Fail 22, 38–47. [DOI] [PubMed] [Google Scholar]
- Zamani P, Proto EA, Mazurek JA, Prenner SB, Margulies KB, Townsend RR, Kelly DP, Arany Z, Poole DC, Wagner PD & Chirinos JA (2020). Peripheral determinants of oxygen utilization in heart failure with preserved ejection fraction: central role of adiposity. JACC Basic Transl Sci 5, 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistical Summary Document
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.