

Abstract
Cellular senescence is a complex stress response that induces an essentially permanent cell cycle arrest and a complex secretory phenotype termed the senescence‐associated secretory phenotype (SASP), which drives numerous aging pathologies. Characterization of the SASP can provide insights into aging and disease mechanisms, aging biomarker candidates, and targets for counteracting the deleterious effects of senescent cells. Here we describe a mass spectrometry (MS)‐compatible protocol to (1) generate senescent cells using different stimuli, (2) collect conditioned medium containing proteins secreted by senescent cells (i.e., SASP), and (3) prepare the SASP for quantitative proteomic analysis using data‐independent acquisition (DIA) MS. © 2021 The Authors.
Basic Protocol 1: Generating ionizing radiation‐induced senescent and control cells
Alternate Protocol 1: Generating doxorubicin‐induced senescent and control cells
Alternate Protocol 2: Generating oncogenic RAS‐induced senescent and control cells
Alternate Protocol 3: Generating mitochondrial dysfunction‐induced senescent and control cells
Alternate Protocol 4: Generating atazanavir/ritonavir‐induced senescent and control cells
Support Protocol: A multiple‐assay approach to confirm the phenotype of senescent cells
Basic Protocol 2: Generating conditioned medium from senescent cells cultured in low serum and quiescent control cells
Alternate Protocol 5: Generating conditioned medium from senescent cells cultured in complete medium and quiescent control cells
Basic Protocol 3: Quantitative proteomic analysis of the SASP
Keywords: aging, data‐independent acquisition, mass spectrometry, secretome, senescence, quantitative proteomic analysis
INTRODUCTION
Cellular senescence is a complex stress response that leads to an exit from the cell cycle that is essentially permanent, although it can in some cases be overcome by genetic or pharmacological manipulation in culture (Beausejour et al., 2003; Lee & Schmitt, 2019). It is not known whether senescence reversal occurs in vivo. Most dividing cell types can become senescent, and subsequently influence both physiological and pathological processes (Campisi & d'Adda di Fagagna, 2007; Gorgoulis et al., 2019). Mouse models and human cell and tissue experiments show that senescent cells can contribute to frailty and many age‐related conditions, including type II diabetes, pulmonary fibrosis, and cancer, making cellular senescence a key driver of aging (Campisi, 2013; Munoz‐Espin & Serrano, 2014). Many biological effects (both beneficial and detrimental) that senescent cells exert on organisms are triggered by their secretome, termed senescence‐associated secretory phenotype (SASP; Coppe et al., 2008).
The SASP is a complex mixture of biologically active molecules, including lipids, metabolites, and proteins (Basisty, Kale, Jeon, et al., 2020; Coppe et al., 2008; Schafer et al., 2020). Among the SASP factors are inflammatory cytokines, growth factors, hormones, proteases, hemostasis factors (Wiley et al., 2019), and many other still unknown components. Interestingly, senescent cells release not only soluble metabolites, lipids, and proteins, but also extracellular vesicles, including exosomes (Basisty, Kale, Jeon, et al., 2020; Borghesan et al., 2019; Jeon et al., 2019). Overall, the SASP is extremely heterogeneous and varies depending on cell type and the senescence‐inducing stimulus (Basisty, Kale, Jeon, et al., 2020; Ozcan et al., 2016).
To add even more complexity, the SASP is very dynamic and changes significantly with time after the initial senescence induction (Basisty, Kale, Jeon, et al., 2020; James, Bennett, & Parkinson, 2018). With newer technological developments, we are now able to use proteomic unbiased discovery workflows to gain deeper insights into highly complex SASP profiles (Basisty, Kale, Patel, Campisi, & Schilling, 2020) and are only beginning to appreciate their true heterogeneity. A better characterization of the SASP is of importance, as it will allow us to better understand the biological effects of cellular senescence, as well as offer new potential biomarkers and therapeutic targets that could be key tools in the fight against age‐related diseases.
Given the vast number and heterogeneity of protein components in the SASP, quantitative and comprehensive mass spectrometry (MS)‐based proteomic approaches, such as data‐independent acquisition (DIA) MS, are excellent techniques for unbiased profiling of the SASP. This protocol will guide researchers in the generation and validation of cultured senescent cells, collection and processing of conditioned medium containing the SASP, and discovery and quantification of SASP proteins using a comprehensive DIA‐MS‐based acquisition workflow (Collins et al., 2017; Gillet et al., 2012; Schilling, Gibson, & Hunter, 2017). These SASP isolation protocols are also compatible with other downstream quantitative mass spectrometric methods, such as data‐dependent acquisition (DDA) MS.
See Figure 1 for a schematic of the protocols. Basic Protocol 1 and its Alternate Protocols 1 to 4 describe in detail the preparation of senescent cells and appropriate control cells using some of the most common senescence inducers: ionizing radiation (IR; Basic Protocol 1), the chemotherapeutic drug doxorubicin (Alternate Protocol 1), oncogenic RAS overexpression (Alternate Protocol 2), mitochondrial disruption (Alternate Protocol 3), and the HIV protease inhibitor atazanavir (Alternate Protocol 4).
Figure 1.
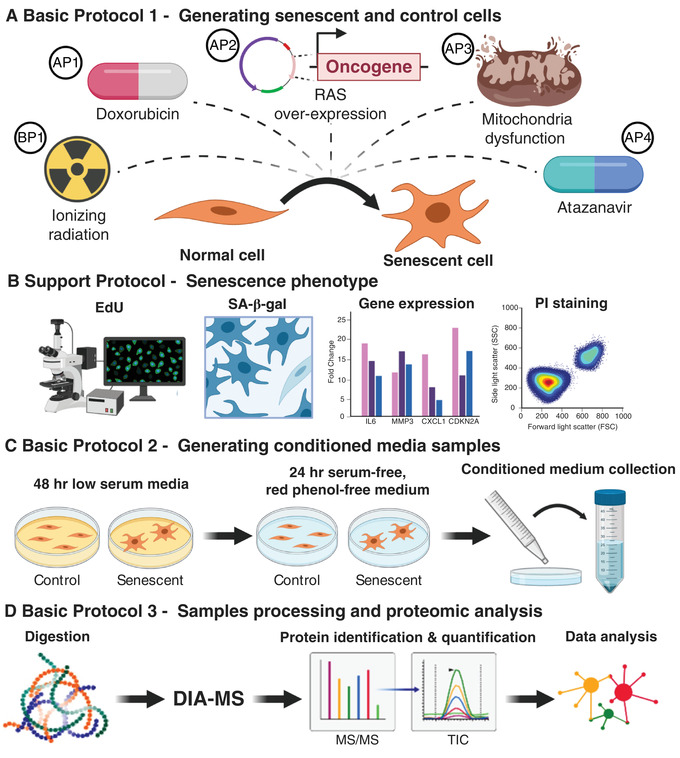
Overview of the senescence induction protocols, collection of conditioned medium, and proteomics analysis. Abbreviations: SA‐β‐Gal, senescence‐associated β‐galactosidase activity; PI, propidium iodide; DIA‐MS, data‐independent acquisition mass spectrometry.
Support Protocol describes assays to check for the presence of senescence‐associated markers and confirm successful senescence induction.
Basic Protocol 2 and Alternate Protocol 5 describe the procedure for generating and collecting conditioned medium (CM) from senescent and quiescent control cells.
Basic Protocol 3 describes the proteomic workflow that we recommend to identify and quantify the SASP proteins secreted by senescent cells in culture, including sample processing, mass spectrometric acquisition by DIA‐MS, and analysis of raw mass spectrometric data files for protein identification and quantification.
STRATEGIC PLANNING
Each researcher will choose a specific senescence inducer and version of Basic Protocol 1 to use based on the best models for the biological system and context under investigation. For example, if the objective is to identify SASP factors involved in the side effects of chemotherapy, then the doxorubicin‐induced senescence protocol below (Alternate Protocol 1) may be the most appropriate approach. Another important factor in deciding which senescence inducer to use is the availability of materials and/or instrumentation to perform senescence induction. For example, senescence induction using oncogenic RAS overexpression requires having cells harboring an inducible vector such as pLVX‐RASV12, while induction of senescence by IR requires access to an X‐ray generator. The same rationale applies to the decision about which cell type should be investigated. The selection of cell type will be based on what is most relevant to the tissue or disease of interest.
It should be noted that the protocols outlined here are optimized for IMR‐90 human fetal lung fibroblasts cultured at physiological O2 concentration (3% O2, 10% CO2 at 37°C), and the inducer doses and culture conditions should be individually tailored and optimized for other cell types. It should also be pointed out that the approaches described in Basic Protocol 1 and Alternate Protocols 1 through 4 are not the only means of senescence induction (Gorgoulis et al., 2019).
To generate SASP‐containing and quiescent control CM, Basic Protocol 2 should be followed. If after senescence induction the cell type investigated does not remain viable in low serum, then Alternate Protocol 5 should be used. Also, when using any combination of cell type and senescence induction for the first time, successful induction of senescence should be verified. Support Protocol describes several assays that are commonly used to confirm senescence induction. Note that other assays, such as assessing lack of proliferation through Ki67 staining or detecting increased lysosomal activity by staining for lipofuscin granules, could be used to confirm the presence of senescent cells (Gonzalez‐Gualda, Baker, Fruk, & Munoz‐Espin, 2020). However, the assays listed in Support Protocol are widely used, robust, and easy to perform.
Basic Protocol 1. GENERATING IONIZING RADIATION‐INDUCED SENESCENT AND CONTROL CELLS
The method described here allows for the generation of senescent cells by X‐rays (IR) and appropriate control cells. From these two cell populations, the researcher will later generate the SASP‐containing and control CM (see Basic Protocol 2 and Alternate Protocol 5). Cells are seeded at the desired density, cultured overnight, and irradiated the following day. After IR, medium is replaced with fresh complete medium. Then, medium is switched to fresh complete medium every 2 days. Three days before the irradiated cells reach a full senescent state, control cells are seeded and cultured overnight. These control cells are derived from the same stock culture used to seed the irradiated cells and have been cultured in parallel up to this point. The next day, control cells are mock irradiated. This protocol has been optimized for IMR‐90 primary human fetal lung fibroblasts and can serve as a guide for the use of other adherent human cells.
Materials
Desired cell type, e.g., human primary fibroblasts strain IMR‐90 (ATCC, cat. no. CCL‐186), WI‐38 (ATCC, cat. no. CCL‐75), or BJ (ATCC, cat. no. CRL‐2522)
Complete medium (CM) appropriate for the cell type used, e.g., complete medium composition for IMR‐90 fibroblasts: DMEM (Gibco brand, Thermo Fisher Scientific, cat. no. 12430‐054) supplemented with 100 U/ml penicillin‐streptomycin (Gibco brand, Thermo Fisher Scientific, cat. no. 15070063) and 10% FBS (Gibco brand, Thermo Fisher Scientific, cat. no. 2614079)
Serum‐free, phenol red‐free medium, e.g., when culturing IMR‐90 fibroblasts, use phenol red‐free DMEM (Gibco brand, Thermo Fisher Scientific, cat. no. 21063‐029) supplemented with 100 U/ml penicillin‐streptomycin (Gibco brand, Thermo Fisher Scientific, cat. no. 15070063) to avoid interference in subsequent protein concentration measurements, such as bicinchoninic acid (BCA) assays
Dulbecco's PBS (Gibco brand, Thermo Fisher Scientific, cat. no. 21600‐010)
Trypsin‐EDTA (Corning, cat. no. 25‐051‐CI)
Incubator set at optimal conditions of temperature and partial pressure of air gases to mimic physiological requirements of the cell type used, e.g., IMR‐90 cells are cultured at 37°C, 3% O2, and 10% CO2 in HERA CELL 240i CO2 incubators (Thermo Fisher Scientific, cat. no. 51026331)
T175 tissue culture flasks (Genesee Scientific, cat. no. 25‐211)
X‐ray generator (Polaris 320 kV X‐ray Generator Set; X‐Ray Associates)
Generate senescent cells
-
1
Seed three T175 flasks with 2 × 106 fibroblasts each (or other cell type of interest), using complete DMEM (or other medium optimized for the cell type used); culture overnight. These cells will be used to generate SASP‐containing CM. Seed (at least) three replicates per condition to ensure that changes observed in the MS data can be statistically analyzed.
The number of cells needed to obtain enough SASP protein for MS analysis may vary depending on the cell type and MS instrumentation available. Ideally, the total amount of secreted protein for each replicate should be >50 μg. If less protein is available, the protocol should be optimized for smaller protein amounts and tested. To estimate the level of protein secretion, conduct a pilot experiment by seeding the desired number of cells, allowing them to recover overnight, then wash them twice with PBS, add serum‐free, phenol red‐free medium, collect CM 24 hr later, and perform a BCA assay. This quantification will allow an estimation of the number of cells needed to obtain enough SASP protein for MS analysis. Culture medium used during this step must be free of serum and, as much as possible, of protein components and/or contaminants. This way, the protein concentration measured will reflect only the protein secreted by the cells. Also, the CM needs to be phenol red free because this compound interferes with the quantification of proteins using BCA.
-
2
Treat fibroblasts the next day with 15 Gy IR to induce senescence and then replace medium with fresh complete medium.
Fibroblasts will develop a full senescent phenotype ∼10 days later. By then, almost all fibroblasts should develop the enlarged, flattened morphology associated with senescence. The replacement of medium should not wash off cells as senescent fibroblasts tend to stick to plates more firmly than their control counterparts. The radiation dose and time needed may vary depending on the cell type.
-
3
Replace medium with fresh complete medium every 2 days until day 6 (including on day 6) after IR treatment. On day 8, proceed with Basic Protocol 2, step 1‐2 (or Alternate Protocol 5) to prepare cells for subsequent CM collection.
Generate control cells
-
4
Seed flasks with control fibroblasts 7 days after IR treatment of experimental cells (that is, 3 days before the IR‐treated fibroblasts reach a full senescent phenotype) and allow them to recover overnight.
These cells will be used to generate control CM.
-
5
On the next day, mock irradiate control cells by placing them into the X‐ray chamber without irradiating them. Leave control cells in the X‐ray chamber for the same amount of time that it took to give the irradiated cells their full dose of radiation.
-
6
After mock irradiation, proceed to Basic Protocol 2 (or Alternate Protocol 5).
Alternate Protocol 1. GENERATING DOXORUBICIN‐INDUCED SENESCENT AND CONTROL CELLS
This method allows for the generation of senescent cells upon treatment with the chemotherapeutic drug doxorubicin and appropriate control cells. From these two cell populations, the researcher will later generate the SASP‐containing and control CM (see Basic Protocol 2 and/or Alternate Protocol 5). Cells are seeded at the desired density, cultured overnight, and treated with doxorubicin for 24 hr. Thereafter, cells are washed with PBS, fresh medium is added, and cells are cultured until the development of a full senescent phenotype. Four days before the doxorubicin‐treated cells reach full senescence, the control cells are seeded and cultured overnight. These control cells are derived from the same stock culture used to seed the doxorubicin‐treated cells and have been cultured in parallel up to this point. The next day, control cells are treated with vehicle, dimethyl sulfoxide (DMSO), for 24 hr. This protocol has been optimized for IMR‐90 primary human fetal lung fibroblasts and can serve as a guide for the use of other adherent human cells.
Additional Materials (also see Basic Protocol 1)
Doxorubicin stock solution (2.5 mM in DMSO; see recipe)
DMSO (MilliporeSigma, cat. no. 67‐68‐5)
Generate senescent cells
-
1
Seed three T175 flasks with 2 × 106 fibroblasts each (or other cell type) using the appropriate complete medium and culture overnight. These cells will be used to generate SASP‐containing CM. Seed (at least) three replicates to ensure changes observed in the MS data can be statistically analyzed.
The number of cells needed to obtain enough SASP protein for MS analysis may vary depending on the cell type and MS instrumentation available. To verify that protein concentrations are sufficient for downstream MS analyses, see step 1 comment in Basic Protocol 1.
-
2
On the next day, dilute doxorubicin stock solution (2.5 mM doxorubicin in DMSO) in complete medium for a final concentration of 250 nM doxorubicin (dilution 1/10,000).
-
3
Remove culture medium from the flasks and then switch to doxorubicin‐containing medium. Incubate 24 hr to induce senescence.
Fibroblasts will develop a full senescent phenotype 10 days later. By then, almost all fibroblasts should develop the enlarged, flattened morphology associated with senescence. The dose of doxorubicin and time needed may vary depending on the cell type.
-
4
After doxorubicin treatment, wash cells twice with 25 ml PBS and add fresh complete medium. Then replace medium every 2 days until day 6 (including on day 6) after the beginning of doxorubicin treatment. On day 8, proceed with Basic Protocol 2, step 1‐2 (or Alternate Protocol 5) to prepare the cells for subsequent CM collection.
Generate control cells
-
5
Seed new flasks with fibroblasts 6 days after doxorubicin treatment (that is, 4 days before the doxorubicin‐treated cells reach senescence) and allow cells to recover overnight.
These cells will be used to generate quiescent control CM.
-
6
On the next day, treat control cells with vehicle (DMSO) for 24 hr.
-
7
After treatment with vehicle DMSO, proceed to Basic Protocol 2 (or Alternate Protocol 5) to prepare cells for subsequent CM collection.
Alternate Protocol 2. GENERATING ONCOGENIC RAS‐INDUCED SENESCENT AND CONTROL CELLS
The method described here allows for the generation of senescent cells via oncogenic RAS overexpression and appropriate control cells. From these cell populations, the researcher will later generate SASP‐containing and control CM (see Basic Protocol 2 and/or Alternate Protocol 5). Cells transduced with a pLVX‐RASV12 vector or green fluorescent protein (GFP) control vector are seeded at the desired density, allowed to recover overnight, then cultured with doxycycline to induce RAS or GFP overexpression. Medium is changed every 2 days with fresh complete medium containing doxycycline. Three days before the RAS‐overexpressing cells reach a full senescent phenotype, GFP‐overexpressing control cells are re‐seeded and cultured overnight. Until then, it is important to passage the control cells when appropriate (after reaching 80%‐90% confluency) because these cells keep proliferating. This protocol has been optimized for IMR‐90 primary human fetal lung fibroblasts and can serve as a guide for the use of other adherent human cells.
Additional Materials (also see Basic Protocol 1)
Desired cell type transduced with the inducible vector pLVX (Lenti‐X™ Tet On® Advanced Inducible Expression System; Takara Bio, cat. no. 632162) expressing either a constitutively active RASV12 oncogene or GFP (Basisty, Kale, Jeon, et al., 2020), e.g., IMR‐90 primary human fetal lung fibroblasts (ATCC, cat. no. CCL‐186) transduced with pLVX‐RASV12 or pLVX‐GFP (Basisty, Kale, Jeon, et al., 2020)
Tetracycline‐free FBS (tet‐free FBS; Takara Bio, cat. no. 631105)
Doxycycline hyclate (MilliporeSigma, cat. no. D9891‐5G)
Generate senescent and control cells
-
1
Seed three T175 flasks with 1.5 × 106 fibroblasts transduced with pLVX‐RASV12 (RAS cells) and three T175 flasks with 1.5 × 106 fibroblasts transduced with pLVX‐GFP (GFP cells). Use the appropriate complete medium and allow cells to recover overnight. Use medium containing tet‐free FBS to ensure that RASV12 or GFP is not expressed until the addition of doxycycline. These cell populations will be used to generate SASP‐containing or control CM, respectively. Seed (at least) three replicates for each condition to ensure changes observed in the MS data can be statistically analyzed.
The number of cells needed to obtain enough SASP protein for MS analysis may vary depending on the cell type and MS instrumentation available. To verify that protein secretion is sufficient for downstream MS analyses, see step 1 comment in Basic Protocol 1.
-
2
The next day, treat the two fibroblast populations with 1 μg/ml doxycycline, which will trigger senescence in RAS cells while serving as a control treatment for GFP cells.
Fibroblasts will develop a full senescent phenotype ∼7 days later. By then, almost all fibroblasts should develop the enlarged, flattened morphology associated with senescence. The time needed may vary depending on the cell type used.
-
3
Replace medium for both cell populations with fresh culture medium containing doxycycline every 2 days until day 4 (including on day 4) after the beginning of treatment. On day 5, proceed with Alternate Protocol 5, step 1‐2 (or Basic Protocol 2, step 1‐2) to prepare cells for subsequent CM collection.
Note that RAS cells will undergo a hyperproliferative phase before undergoing growth arrest. Importantly, IMR‐90 fibroblasts induced to senescence by oncogenic RAS overexpression are not viable in low serum. Therefore, after completing Alternate Protocol 2, we suggest using Alternate Protocol 5 (rather than Basic Protocol 2) when using this method of senescence induction in fibroblasts.
Note that when instructed to feed cells with fresh medium in Alternate Protocol 5 (or Basic Protocol 2), such medium must be supplemented with doxycycline. Only serum‐free medium given to both cell populations 24 hr before CM collection (see Collect CM sections in Alternate Protocol 5 or Basic Protocol 2) should be devoid of doxycycline.
Alternate Protocol 3. GENERATING MITOCHONDRIAL DYSFUNCTION‐INDUCED SENESCENT AND CONTROL CELLS
The method described here allows for generating senescent cells by mitochondrial dysfunction—mitochondrial dysfunction‐associated senescence (MiDAS)—and appropriate control cells. From these cell populations, the researcher will later generate the SASP‐containing and control CM (see Basic Protocol 2 and/or Alternate Protocol 5). Cells are seeded at the desired density, cultured overnight, and treated with either the mitochondrial complex III inhibitor Antimycin A or vehicle DMSO. Medium is replaced every 2 days with fresh complete medium containing either Antimycin A or DMSO. Three days before the Antimycin A‐treated cells reach a full senescent phenotype, DMSO treated cells are re‐seeded and cultured overnight. Until then, it is important to passage the DMSO‐treated cells when appropriate (after reaching 80%‐90% confluency) because these cells keep proliferating. This protocol has been optimized for IMR‐90 primary human fetal lung fibroblasts and can serve as a guide for other types of adherent human cells.
Additional Materials (also see Basic Protocol 1)
Antimycin A from Streptomyces species (MilliporeSigma, cat. no. A8674‐25MG)
DMSO (MilliporeSigma, cat. no. 67‐68‐5)
Generate senescent and control cells
-
1
Seed six T175 flasks (three for Antimycin A treatment and three for DMSO treatment) with 2 × 106 fibroblasts each using appropriate complete medium and culture overnight. These cells will be used to generate SASP‐containing or control CM. Seed (at least) three replicates for each condition to ensure that changes observed in the MS data can be statistically analyzed.
The number of cells needed to obtain enough SASP protein for MS analysis may vary depending on the cell type and MS instrumentation available. To verify that protein secretion is sufficient for downstream MS analyses, see step 1 comment in Basic Protocol 1.
-
2
The next day, treat fibroblasts with 250 nM Antimycin A or vehicle DMSO.
Cells treated with Antimycin A will become senescent and generate the SASP‐containing CM. Cells treated with DMSO will be used as quiescent controls to generate the control CM.
Fibroblasts will develop a full senescent phenotype ∼10 days later. By then, almost all fibroblasts should develop the enlarged, flattened morphology associated with senescence. The concentration of Antimycin A and time needed may vary depending on the cell type used.
-
3
Replace medium for both cell populations with fresh culture medium containing Antimycin A or DMSO every 2 days until day 6 (including on day 6) after the beginning of treatment. On day 8, proceed to the Basic Protocol 2, step 1‐2 (or Alternate Protocol 5, step 1‐2) to prepare cells for subsequent CM collection.
Note that DMSO‐treated cells will keep proliferating, so they must be passaged when appropriate (after reaching 80%‐90% confluency).
-
4
To generate control cells, re‐seed proliferating DMSO‐treated control cells 7 days after the beginning of vehicle treatment (that is, 3 days before Antimycin A‐treated cells reach a full senescent phenotype). By doing so, it will be possible to obtain similar numbers of senescent and control cells when collecting their CM.
-
5
Allow control DMSO‐treated cells to recover overnight and then, on the next day, proceed to Basic Protocol 2 (or Alternate Protocol 5) to prepare cells for subsequent CM collection.
Note that when instructed to feed cells with fresh medium in Basic Protocol 2 (or Alternate Protocol 5), medium must be supplemented with 250 nM Antimycin A or vehicle DMSO. Only serum‐free medium given to both cell populations 24 hr before CM collection (see Basic Protocol 2, step 3‐5) should be devoid of Antimycin A or DMSO.
Alternate Protocol 4. GENERATING ATAZANAVIR/RITONAVIR‐INDUCED SENESCENT AND CONTROL CELLS
The method described here allows for the generation of senescent cells upon treatment with the anti‐HIV drugs atazanavir and ritonavir (ATV/r), as well as appropriate control cells. From these cell populations, the researcher can generate SASP‐containing and control CM (see Basic Protocol 2 and/or Alternate Protocol 5). Cells are seeded at the desired density, cultured overnight, and treated with either ATV/r or vehicle (DMSO). Medium is changed every 2 days with fresh complete medium containing either ATV/r or DMSO. Three days before the ATV/r‐treated cells reach a full senescent phenotype, DMSO treated cells are re‐seeded and cultured overnight. Until then, it is important to passage the DMSO‐treated cells when appropriate (after reaching 80%‐90% confluency) because these cells keep proliferating. This protocol has been optimized for IMR‐90 primary human fetal lung fibroblasts and can serve as a guide for the use of other adherent human cells.
Additional Materials (also see Basic Protocol 1)
Atazanavir/ritonavir (ATV/r) stock solution (see recipe)
DMSO (MilliporeSigma, cat. no. 67‐68‐5)
Generate senescent and control cells
-
1
Seed six T175 flasks (three for ATV/r treatment and three for DMSO treatment) with 2 × 106 fibroblasts each using the appropriate complete medium and culture them overnight. These cells will be used to generate SASP‐containing or control CM. Seed (at least) three replicates for each condition to ensure that changes observed in the MS data can be statistically analyzed.
The number of cells needed to obtain enough SASP protein for MS analysis may vary depending on the cell type and MS instrumentation available. To verify that protein secretion is sufficient for downstream MS analyses, see step 1 comment in Basic Protocol 1.
-
2
On the next day, treat fibroblasts with 25 μM ATV/r or vehicle DMSO.
Cells treated with ATV/r will become senescent and generate SASP‐containing CM. Cells treated with DMSO will be used as quiescent controls to generate the control CM.
Fibroblasts will develop a full senescent phenotype ∼14 days later. By then, almost all fibroblasts should develop the enlarged, flattened morphology associated with senescence. The concentration of ATV/r and time needed may vary depending on the cell type.
-
3
Replace medium for both cell populations with fresh complete medium containing either ATV/r or DMSO every 2 days until day 10 (including on day 10), and on day 12 proceed with Basic Protocol 2, step 1‐2 (or Alternate Protocol 5, step 1‐2) to prepare the cells for subsequent CM collection.
Note that DMSO‐treated cells will keep proliferating, so they will have to be passaged when appropriate (after reaching 80%‐90% confluency).
-
4
Re‐seed proliferating DMSO‐treated control cells 11 days after the beginning of vehicle treatment (that is, 3 days before ATV/r‐treated cells reach full senescence). By doing so, it will be possible to obtain similar numbers of senescent and control cells when collecting CM.
-
5
Allow control DMSO‐treated cells to recover overnight and then proceed to Basic Protocol 2 (or Alternate Protocol 5) to prepare cells for subsequent CM collection.
Note that when instructed to feed cells with fresh medium in Basic Protocol 2 (or Alternate Protocol 5), such medium must be supplemented with ATV/r or DMSO for the culture of senescent or control cells, respectively. Only serum‐free medium given to both cell populations 24 hr before CM collection (Basic Protocol 2, step 3‐5) will be devoid of ATV/r and DMSO.
A MULTIPLE‐ASSAY APPROACH TO CONFIRM THE PHENOTYPE OF SENESCENT CELLS
This protocol describes multiple assays to verify that the senescence induction performed in Basic Protocol 1 (or Alternate Protocols 1 through 4) has been successful. Support Protocol is used to prove that the secreted proteins to be analyzed by MS are produced by senescent cells and therefore part of an SASP. This supporting protocol should be performed when the researcher uses any combination of a senescence inducer (Basic Protocol 1 or Alternate Protocols 1 through 4) and a desired cell type. These assays are not compatible with the generation of CM described in Basic Protocol 2 (or Alternate Protocol 5) because they require fewer cells compared to the number needed to analyze secreted proteins by MS. Therefore, cells should be seeded in the appropriate tissue culture dishes (specified in each of the corresponding assay protocols below) to perform the assays described and then the chosen senescence induction method (Basic Protocol 1 or Alternate Protocols 1 through 4) is performed. Once senescent and control cells are generated, the steps described below must be performed for each assay. The assays we recommend are used to verify reduced cell proliferation (see step 1‐8), upregulation of senescence‐associated β‐galactosidase (SA‐β‐gal) activity (see step 9‐14), and a gene transcription signature associated with senescence (see step 15‐21). We also recommend performing a viability assay (see step 22‐30) to verify that both senescent and control cells are healthy before generating CM for MS analysis.
Additional Materials (also see Basic Protocol 1 and Alternate Protocols 1, 2, 3, and 4)
Applied Biosystems MultiScribe Reverse Transcriptase Kit (Thermo Fisher Scientific, cat. no. 43‐112‐35)
Serum‐free medium: for IMR‐90 fibroblasts, use DMEM (Gibco brand, Thermo Fisher Scientific, cat. no. 12430‐054) supplemented with 100 U/ml penicillin‐streptomycin (Gibco brand, Thermo Fisher Scientific, cat. no. 15070063)
Click‐iT EdU Kit Alexa Fluor 488 HCS Assay (Thermo Fisher Scientific, cat. no. C10351)
Senescence Detection Kit (BioVision, cat. no. K320‐250)
X‐gal (Life Technologies, cat. no. 15520‐018)
ISOLATE II RNA Micro Kit (Bioline, cat. no. BIO‐52075)
Senescence‐associated human gene‐specific primers (Eurofins): Upregulated genes:
-
For CDKN1A (p21)
Forward: 5′‐TCACTGTCTTGTACCCTTGTGC‐3′
Reverse: 5′‐GGCGTTTGGAGTGGTAGAAA‐3′
-
For CDKN2A (p16)
Forward: 5′‐GAGCAGCATGGAGCCTTC‐3′
Reverse: 5′‐CGTAACTATTCGGTGCGTTG‐3′
-
For CDKN2B (p15)
Forward: 5′‐CTCCCGAAACGGTTGACTC‐3′
Reverse: 5′‐GCGGGGACTAGTGGAGAAG‐3′
-
For CXCL1
Forward: 5′‐GCTGAACAGTGACAAATCCAAC‐3′
Reverse: 5′‐CTTCAGGAACAGCCACCAGT‐3′
-
For CXCL10
Forward: 5′‐GAAAGCAGTTAGCAAGGAAAGGT‐3′
Reverse: 5′‐GACATATACTCCATGTAGGGAAGTGA‐3′
-
For IL1B
Forward: 5′‐CTGTCCTGCGTGTTGAAAGA‐3′
Reverse: 5′‐TTGGGTAATTTTTGGGATCTACA‐3′
-
For IL6
Forward: 5′‐GCCCAGCTATGAACTCCTTCT‐3′
Reverse: 5′‐GAAGGCAGCAGGCAACAC‐3′
-
For MMP3
Forward: 5′‐CAAAACATATTTCTTTGTAGAGGACAA‐3′
Reverse: 5′‐TTCAGCTATTTGCTTGGGAAA‐3′
-
For SERPINE1
Forward: 5′‐CCAGCTGACAACAGGAGGAG‐3′
Reverse: 5′‐CCCATGAGCTCCTTGTACAGAT‐3′
Senescence‐associated human gene‐specific primers (Eurofins): Downregulated genes:
-
For LAMNB1
Forward: 5′‐TTGGATGCTCTTGGGGTTC‐3′
Reverse: 5′‐AAGCAGCTGGAGTGGTTGTT‐3′
Propidium iodide (PI) staining solution (eBioscience, cat. no. 00‐6990‐50)
8‐well chamber slides Lab‐Tek II (MilliporeSigma, cat. no. C7057‐1CS)
Inverted fluorescence microscope (Olympus, IX70 Fluorescence Microscope)
6‐well cell culture plates (Genesee Scientific, cat. no. 25‐105)
T75 tissue culture flasks (Genesee Scientific, cat. no. 25‐211)
LightCycler® 480 Instrument II (Roche, cat. no. 05015278001)
Flow cytometer (BD™ LSR II with BD FACS Diva v8.0.2 acquisition software)
EdU incorporation
-
1
Follow steps listed in Basic Protocol 1 (or in one of Alternate Protocols 1 through 4) to generate senescent and control cells of the chosen cell type. For this immunostaining assay, seed 10,000 fibroblasts per well in 8‐well chamber slides for each replicate.
-
2
Wash senescent and control cells twice with PBS, then switch them to medium containing 0.2% FBS (instead of 10% FBS) and culture for 48 hr to induce quiescence by serum starvation in control cells while maintaining viability for a few days for both cell populations.
-
3
The following day (24 hr after switching to low serum‐containing medium), seed cells in new wells using complete medium and allow recovery overnight.
These cells will be used as a positive control for EdU staining (proliferating control cells).
Use complete medium (containing 10% FBS when using fibroblasts) to keep cells in a proliferative state.
-
4
The next day, remove half the volume of medium in each well and replace with fresh medium containing EdU (Click‐iT EdU kit) as instructed by the manufacturer's protocol. Use serum‐free medium for the senescent and quiescent cells. Use complete medium (10% FBS) for proliferating control cells.
For example, if each well of the 8‐well chamber slide contains 250 μl of medium, remove 125 μl and add 125 μl of the appropriate medium (serum free or complete medium) containing EdU (see manufacturer's protocol for the EdU concentration).
-
5
Incubate for 24 hr.
-
6
Twenty‐four hours later, follow the instructions (manufacturer's protocol) to fix, permeabilize, detect EdU incorporation, and stain cellular DNA with Hoechst 33342.
EdU staining is also compatible with DAPI DNA staining.
-
7
Remove all liquid from wells, remove well chambers, add mounting medium, and then apply coverslips onto the slides (avoid air bubbles).
-
8
Compare DNA staining with EdU staining to confirm that senescent and quiescent control populations have low numbers of EdU positive cells (whereas proliferating control cells should have high numbers of EdU positive cells; see Fig. 2).
Figure 2.
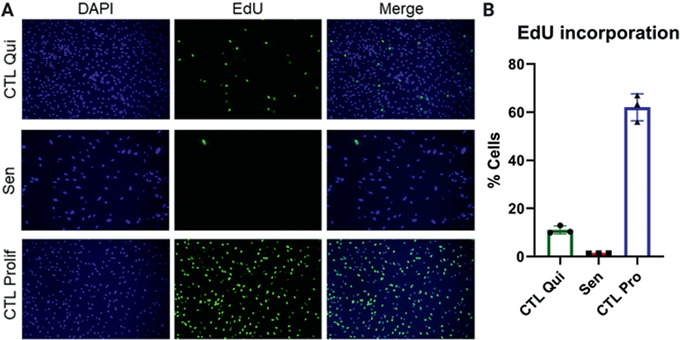
EdU incorporation. (A) Representative images of quiescent control (top), ionizing radiation (IR)‐induced senescent (center), and proliferating control IMR‐90 cells (bottom). DAPI staining is shown on the left and EdU staining in the middle. Merged images are shown on the right. (B) Quantification of EdU‐positive cells for quiescent control, IR‐induced senescent, and proliferating control cells. Data shown are means of 3 replicates ± SD.
Senescence‐associated β‐galactosidase
-
9
Follow steps listed in Basic Protocol 1 (or one of the Alternate Protocols 1 through 4) to generate senescent and control cells of the chosen cell type. For each replicate, seed 80,000 fibroblasts in a well of a 6‐well plate.
-
10
Wash senescent and control cells twice with PBS, then switch to medium containing 0.2% FBS (instead of 10% FBS) and culture for 48 hr.
This will induce quiescence by serum starvation in control cells while maintaining viability for a few days in both cell populations.
-
11
After 48 hr, wash cells twice with PBS, then switch to serum‐free medium and culture for 24 hr.
-
12
Fix and incubate cells with the X‐gal staining solution at 37°C overnight (as instructed in the manufacturer's protocol from the Senescence Detection Kit).
When preparing the staining solution as instructed by the manufacturer's protocol, we recommend using X‐gal from Life Technologies rather than X‐gal provided with the Senescence Detection Kit to optimize staining. Use the suggested solvent and concentration of X‐gal described in the Senescence Detection Kit manufacturer's manual.
-
13
On the next day, remove staining solution and leave cells in PBS.
The staining reaction might require incubations longer than the overnight suggested in the manufacturer's protocol. Cells should be monitored under a microscope before removing the staining solution. If senescent cells appear to be poorly stained, they can remain in the staining solution at 37°C for longer times. Observe the cells under a microscope about every 2 hr until the senescent cells appear clearly stained, while quiescent control cells should remain unstained. However, avoid extended incubation times as the quiescent cells might turn positive. We recommend acquiring images right after removing the staining solution and adding PBS, even though cells can be stored at 4°C for 1‐2 days before imaging.
-
14
Acquire bright‐field images using an inverted microscope and confirm that senescent cells have high SA‐β‐gal staining compared to quiescent control cells (see Fig. 3).
Figure 3.
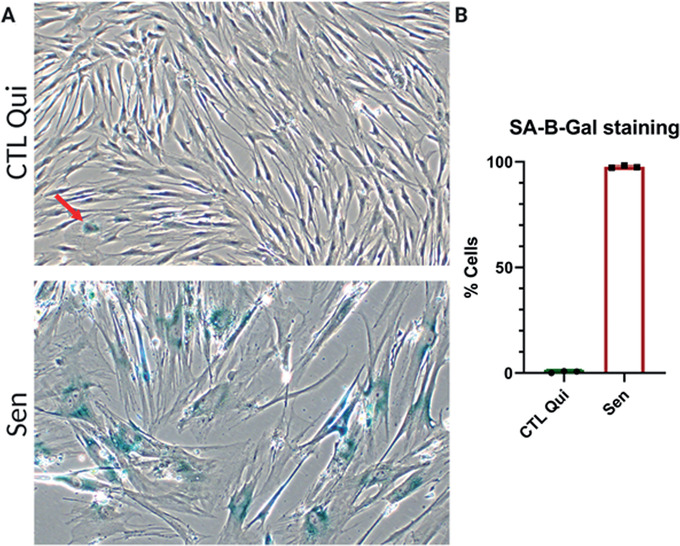
Senescence‐associated β‐galactosidase (SA‐β‐gal) staining. (A) Representative images of quiescent control (top) and ionizing radiation (IR) senescent IMR‐90 cells (bottom). (B) Quantification of SA‐β‐gal positive cells for quiescent control and senescent cells. Data shown are means of 3 replicates ± SD.
Senescence‐associated gene expression analysis by qPCR
-
15
Follow steps listed in Basic Protocol 1 (or in one of the Alternate Protocols 1 through 4) to generate senescent and control cells of the chosen cell type. For each replicate, seed 200,000 fibroblasts in a well of a 6‐well plate.
-
16
Wash both senescent and control cells twice with PBS, then switch them to medium containing 0.2% FBS (instead of 10% FBS) and culture for 48 hr to induce quiescence by serum starvation in control cells while maintaining viability for a few days in both cell populations.
-
17
After 48 hr, wash cells twice with PBS, then switch them to serum‐free medium and culture for 24 hr.
-
18
Twenty‐four hours later, remove medium from the wells and extract total RNA using the ISOLATE II RNA Micro Kit. We recommend adding Lysis Buffer RLY directly to the wells after removing the medium, making sure to detach all cells with the help of a cell scraper.
-
19
After RNA extraction, convert RNA samples into cDNA using the Applied Biosystems MultiScribe Reverse Transcriptase Kit.
-
20
Use the cDNA to analyze by quantitative polymerase chain reaction (qPCR) mRNA levels of the senescence‐associated genes.
See materials list for a list of genes that are significantly upregulated (all genes except LAMNB1) or downregulated (LAMNB1) upon senescence induction (Hernandez‐Segura et al., 2017). The primer sequences provided have been tested with the Universal Probe Library System. If using another probe system, the primer sequences listed are not guaranteed to work and primer sequence optimization might be needed.
-
21
Analyze qPCR data to compare the relative expression of senescence‐associated genes between senescent and quiescent control cells. Confirm that most genes have the same upregulation or downregulation trends shown in Figure 4.
Different cell types may express different complements of SASP factors. The senescence‐associated genes listed here are optimized for fibroblasts.
Figure 4.
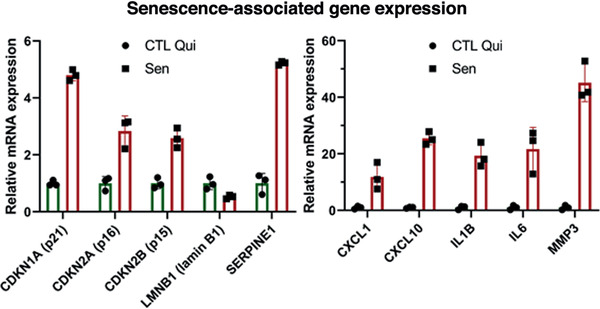
Gene expression analysis using qPCR. Relative mRNA levels of the indicated genes in senescent cells normalized to quiescent IMR‐90 cells. Data shown are means of 3 replicates ± SD.
Propidium iodide inclusion
-
22
Follow steps listed in Basic Protocol 1 (or Alternate Protocols 1 through 4) to generate senescent and control cells of the chosen cell type. Seed 1 × 106 fibroblasts in a T75 flask for each replicate.
-
23
Wash senescent and control cells twice with PBS, then switch to medium containing 0.2% FBS (instead of 10% FBS) and culture for 48 hr.
This will induce quiescence by serum starvation in control cells while maintaining viability for a few days in both cell populations.
-
24
After 48 hr, wash cells twice with PBS, then switch to serum‐free medium and culture them for 24 hr.
-
25
Wash cells with PBS and detach them from the substratum by incubation with trypsin/EDTA at 37°C for 5 min.
-
26
Resuspend cells in complete medium to inhibit trypsin, then wash cell suspensions twice with PBS.
To perform a PBS wash, pellet cells by centrifugation at 250 × g for 5 min, remove the supernatant, resuspend the pellet in PBS, centrifuge cells again at 250 × g for 5 min, and remove PBS.
-
27
Resuspend cell pellets in PBS to a final concentration of 1 × 106 to 1 × 107 cells/μl.
-
28
Aliquot 100 μl of each cell suspension in to a flow cytometry tube and add 5 μl PI staining solution.
-
29
Mix solution by pipetting and incubate 1 min at room temperature.
-
30
Analyze samples for PI staining by flow cytometry. Confirm that the number of PI positive cells is low in both senescent and control cells (see Fig. 5).
PI dye is impermeable to cells, so will not stain cells unless their membrane integrity is compromised (that is, the cells are dead or dying).
Figure 5.
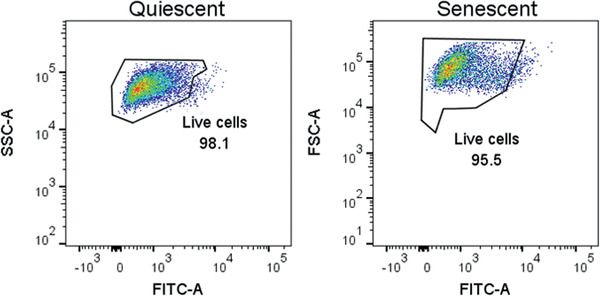
Propidium iodide (PI) staining for cell death. Percentage of PI‐positive cells in quiescent control (left) and ionizing radiation (IR)‐induced senescent (right) IMR‐90 cell populations. Data shown are representative of both cell populations.
Basic Protocol 2. GENERATING CONDITIONED MEDIUM FROM SENESCENT CELLS CULTURED IN LOW SERUM AND QUIESCENT CONTROL CELLS
With this method, SASP‐containing and control CM are generated from senescent and control cells, prepared following Basic Protocol 1 (or one of the Alternate Protocols 1 through 4). Both senescent and control cells are cultured in low‐serum medium (0.2% FBS) for 48 hr, which allows control cells to become quiescent. Then, cells are cultured in phenol red‐free, serum‐free medium for 24 hr. CM from senescent and control quiescent cells are collected and the cells are counted, which will allow normalization for proteomic analysis. Protein levels normalized to cell count accounts for changes due to differences in the number of cells and represent a more meaningful measure of “proteins secreted per cell.” This protocol has been optimized for IMR‐90 primary human fetal lung fibroblasts and can serve as a guide for the use of other adherent human cells.
Additional Materials (also see Basic Protocol 1)
Senescent and control cells generated using Basic Protocol 1 or Alternate Protocols 1‐4
Low‐serum medium: for IMR‐90 fibroblasts, use DMEM (Gibco brand, Thermo Fisher Scientific, cat. no. 12430‐054) supplemented with 100 U/ml penicillin‐streptomycin (Gibco brand, Thermo Fisher Scientific, cat. no. 15070063) and 0.2% FBS (Gibco brand, Thermo Fisher Scientific, cat. no. 2614079)
Phenol red‐free, serum‐free medium: for IMR‐90 fibroblasts, use phenol‐red free DMEM (Gibco brand, Thermo Fisher Scientific, cat. no. 21063‐029) supplemented only with 100 U/ml penicillin‐streptomycin (Gibco brand, Thermo Fisher Scientific, cat. no. 15070063)
Beckman Coulter Z1 Particle Counter (Beckman, cat. no. 6605698)
Swing Rotor Centrifuge 5804 (Eppendorf, cat. no. 022622501)
Generate CM samples
-
1
Aspirate culture medium from senescent and control cells, then wash cells twice by adding PBS and subsequently aspirate it. After washes, switch to low‐serum medium for both senescent and control cells and culture for 48 hr.
Using low‐serum medium induces quiescence in control cells by serum starvation, while maintaining viability for a few days in both cell populations.
-
2
After 48 hr, aspirate culture medium, then wash senescent and quiescent control cells twice by adding PBS and subsequently aspirate it. After washes, switch to serum‐free and phenol red‐free medium and incubate 24 hr.
The collected CM needs to be phenol red free because this compound interferes with quantification of protein using BCA. Also, culture medium used during this step must be free of serum and, as much as possible, of protein components/contaminants. Abundant exogenous protein contamination can limit the identification and quantification of secreted proteins. High concentrations of proteins present in serum and other cell culture supplements interfere with and suppress the ionization of secreted proteins during MS analysis. If for some reason, the culture medium contains protein components, these proteins must be excluded from later proteomic analysis.
Collect CM
-
3
After the 24 hr incubation, collect CM from each replicate and perform cell counts.
Right after collection, keep the CM on ice to minimize protein degradation during cell counting. To further minimize protein degradation, we recommend moving on to step 4 as soon as cell counting is completed. Cell counts are important to normalize the levels of secreted proteins during proteomic analysis (see Basic Protocol 3). Senescent fibroblasts tend to attach more firmly to plates, so a slightly longer incubation (1‐2 min) with trypsin‐EDTA might be required for the senescent cells to detach.
-
4
Centrifuge collected CM at 10,000 × g for 15 min to pellet cell debris and transfer supernatant into a new tube.
-
5
Proceed to Basic Protocol 3 or store the samples at −80°C for later processing.
Alternate Protocol 5. GENERATING CONDITIONED MEDIUM FROM SENESCENT CELLS CULTURED IN COMPLETE MEDIUM AND QUIESCENT CONTROL CELLS
If after senescence induction the cell type investigated does not remain viable in low‐serum medium, then Alternate Protocol 5 should be used to generate CM for MS analysis. Some indicators of poor viability are loss of cell number and cell detachment. Loss of viability can be quantitatively confirmed by increasing cell death, as measured by cell viability assays (for example, see Support Protocol, step 22‐30). In this protocol, CM containing the SASP are collected from senescent cells that are cultured in complete medium up to 24 hr before CM collection. Control cells are still cultured in low serum to induce quiescence. However, when comparing these two conditions, it is difficult to determine whether differences are due to a senescent versus non‐senescence state or due to culturing in complete medium versus low‐serum medium. This protocol is optimized for primary lung fibroblasts, which remain viable in low serum under control conditions. If the cell type under investigation is not viable under control conditions, we recommend optimization of culture conditions appropriate to the cell type under investigation. A possible alternative approach may be to compare CM collected from senescent cells cultured in complete medium versus CM collected from non‐senescent cells cultured in complete medium. However, under these conditions, one cannot distinguish if changes in protein secretion are the result of comparing proliferating cells versus non‐proliferating cells, differences in cell density between control and senescent conditions, or differences between senescent and non‐senescent cells. Also see Cell cultures in low‐serum medium before CM collection section under Critical Parameters and Troubleshooting.
Materials
See Basic Protocol 2
Generate CM samples
-
1
Aspirate culture medium from senescent and quiescent control cells, then wash cells twice by adding PBS, and subsequently aspirate it. After the washes, switch control cells to low‐serum medium and add complete medium to senescent cells. Culture both cell populations for 48 hr.
Using low‐serum medium induces quiescence in control cells by serum starvation, while maintaining viability for a few days.
-
2
After 48 hr, remove culture medium by aspiration, then wash senescent and quiescent control cells twice by adding PBS, and subsequently aspirate it. After washes, add serum‐free and phenol red‐free medium, and incubate 24 hr.
The CM needs to be phenol red free because this compound interferes with the quantification of protein using BCA. Also, culture medium used during this step must be free of serum and, as much as possible, of protein components/contaminants. Abundant exogenous protein contamination can limit the identification and quantification of secreted proteins. High concentrations of proteins present in the serum and other cell culture supplements interfere with and suppress the ionization of secreted proteins during MS analysis. If for some reason, the culture medium contains protein components, these proteins must be excluded from later MS data analysis.
Collect CM
-
3
For the collection of CM, follow Basic Protocol 2, steps 3 through 5.
Basic Protocol 3. QUANTITATIVE PROTEOMIC ANALYSIS OF THE SASP
This protocol describes a comprehensive unbiased MS‐based approach to identify and quantify the secreted proteins of cultured cells. CM prepared in Basic Protocol 2 (or Alternate Protocol 5) is concentrated, digested, and desalted. The processed samples are analyzed using a liquid chromatography‐mass spectrometry (LC‐MS) method, specifically a DIA workflow. The protocol illustrates in detail how to use an LC‐MS system composed of a nano‐LC 2D HPLC coupled to a TripleTOF 6600 high‐resolution mass spectrometer. The DIA quantitative proteomics analysis software Spectronaut (Biognosys) is used to perform relative quantification of protein levels and create reports of protein abundance, fold changes, and statistics.
Materials
Senescence‐associated secretory phenotype (SASP) conditioned medium (CM) and control CM (CTL CM) generated in Basic Protocol 2 (or Alternate Protocol 5)
Ammonium bicarbonate (NH4HCO3; MilliporeSigma, cat. no. A 6141)
Dithiothreitol (DTT; MilliporeSigma, cat. no. D9779)
Iodoacetamide (IAA; MilliporeSigma, cat. no. I1149)
Sequencing grade trypsin (Promega, cat. no. V5113)
Acetic acid (MilliporeSigma, cat. no. 695092‐100ML)
Formic acid (MilliporeSigma, cat. no. 94318)
Acetonitrile (ACN; Burdick & Jackson, cat. no. AH015)
Indexed retention time (iRT) peptide standards (Biognosys, cat. no. Ki‐3002)
Bicinchoninic Acid (BCA) Kit (Thermo Fisher Scientific, cat. no. PI23225)
Amicon® Ultra centrifugal filters with 3 kDa molecular weight cutoff (MilliporeSigma, cat. no. UFC9003)
Litmus strips (VWR International, cat. no. EM1095350007)
Heating Shaking Drybath (Thomas Scientific, cat. no. 1199A66)
Benchtop microcentrifuge (Thermo Fisher Scientific, cat. no. 75002401)
Oasis HLB solid‐phase extraction (SPE) cartridges, 10 mg Sorbent per Cartridge, 30 µm (Waters, cat. no. 186006339)
SpeedVac concentrator (Savant™ SPD131DDA; Thermo Fisher Scientific)
Ultrasonic Bath Sonicator (Thomas Scientific)
Autosampler vials for HPLC (Agilent Technologies, cat. no. 5190‐3155)
Nano‐LC 2D HPLC system (Eksigent Ultra Plus; Eksigent)
cHiPLC system (Eksigent)
200 µm × 0.4 mm ChromXP C18‐CL chip, 3 µm, 120 Å (SCIEX)
75 µm × 15 cm ChromXP C18‐CL chip, 3 µm, 120 Å (SCIEX)
High‐resolution mass spectrometer TripleTOF 6600 System (SCIEX) or other high‐resolution mass spectrometer
Spectronaut™ software (Biognosys, Schlieren)
Concentrate CM
-
1
Transfer CM to Amicon ultrafilter tubes, without exceeding the maximum volume capacity (see manufacturer instructions).
It is likely that the volumes of CM samples are higher than the ultrafilter tubes’ capacity. If so, use multiple centrifugations until the volume of every CM sample has been reduced to <0.5 ml (see next step).
-
2
Following manufacturer instructions, buffer exchange protein samples into 50 mM NH4HCO3 buffer. After initially concentrating protein samples in the columns, we recommend performing one buffer exchange with a maximum volume of 50 mM NH4HCO3, followed by a buffer exchange with a maximum volume of 8 M urea in 50 mM NH4HCO3. After the addition of 8 M urea in 50 mM NH4HCO3, centrifuge samples until reaching a final volume <0.5 ml in NH4HCO3 buffer.
-
3
Buffer exchanged samples can be stored up to 1 year at −80°C.
In‐solution proteolytic digestion
-
4
Quantify secreted protein concentrations by BCA assay.
Use at least a 1/3 dilution of sample in milliQ water for the BCA assay. The BCA assay is compatible with urea concentrations up to 3 M.
-
5
Aliquot 50 µg of each sample into new 1.5‐ml tubes.
If sample yields are lower than 50 µg, we recommend aliquoting at least 20 µg of secreted proteins due to sample losses during subsequent steps. Still, sample amounts as low as 10 µg can be used.
-
6
Bring all samples to equal volumes with 8 M urea in 50 mM NH4HCO3.
-
7
Vortex to mix.
-
8
Add DTT (1 M in 50 mM NH4HCO3 buffer stock solution) to a final concentration of 20 mM (in 50 mM NH4HCO3 buffer) to each sample to reduce disulfide bridges.
-
9
Incubate samples 30 min at 37°C with shaking and/or agitation.
-
10
Allow samples to cool to room temperature.
-
11
Add IAA (200 mM in 50 mM NH4HCO3 buffer stock solution) to a final concentration of 40 mM IAA (in 50 mM NH4HCO3 buffer) to each sample to alkylate reduced thiols.
It is important to use an IAA concentration at least double that of DTT to ensure that all the reduced thiols are alkylated.
-
12
Incubate samples in the dark at room temperature for 30 min.
-
13
Dilute all samples 1/6 in 50 mM NH4HCO3 to reduce the concentration of urea for trypsin digestion.
-
14
Verify that the pH of samples is between 7.0 and 8.5 by pipetting small volumes (<1 µl) onto litmus strips. Adjust pH as needed.
-
15
Resuspend lyophilized trypsin in 50 mM acetic acid to a final concentration of 0.1 mg/ml.
For example, 20 μg of lyophilized trypsin are resuspended in 200 µl of solution at pH 8, for a final concentration of 0.1 μg/μl trypsin.
-
16
Digest protein samples by adding trypsin at a protease‐to‐substrate protein ratio of 1:50 (w/w). Incubate samples overnight at 37°C on a shaking dry bath.
-
17
Quench protein digestion by adding formic acid to a final concentration of 1% by volume from a 10% formic acid (in water) stock.
Stock concentrations of formic acid ≤10% can be pipetted safely with plastic pipet tips. Higher concentrations may dissolve plasticware and should be handled with glass to avoid polymer contamination of samples.
-
18
Spin sample at 5,000 × g for 15 min at room temperature to pellet insoluble material. The supernatants, which contain peptides, are desalted in the next steps.
Desalt samples
-
19
Desalt samples using commercially available SPE cartridges. The next steps describe a desalting process using Oasis HLB SPE cartridges.
-
20
Wet each HLB SPE cartridge twice with 800 µl 50% ACN in 0.2% formic acid in water.
-
21
Equilibrate each cartridge three times with 800 µl 0.2% formic acid in water.
-
22
Load peptide samples onto HLB SPE cartridges.
-
23
Wash each cartridge three times with 800 µl 0.2% formic acid in water.
-
24
Elute peptides once with 800 µl 50% ACN in 0.2% formic acid in water and once with 400 µl 50% ACN in 0.2% formic acid in water in the same tube.
-
25
Dry samples completely in a SpeedVac concentrator.
If desired, one can pause sample processing; dried peptide pellets can be safely stored up to 1 year at −80°C.
-
26
Resuspend dry pellets in 0.2% formic acid to a final concentration of 1 µg/µl.
Calculate the final peptide concentration based on the initial protein sample mass aliquoted for digestion. For example, if the initial aliquot contained 50 μg of protein, the dried pellet will be resuspended in 50 µl of 0.2% formic acid.
-
27
Sonicate samples in a water bath sonicator 5 min.
-
28
Vortex samples at 4°C for 10 min.
-
29
Centrifuge samples at 15,000 × g for 15 min.
-
30
Transfer supernatants to autosampler vials.
-
31
Add retention time standard peptides to each autosampler vial.
For example, use iRT peptide standards at a 1/20 dilution.
-
32
Centrifuge samples in autosampler vials at 5,000 × g in a clinical centrifuge for 1 min to remove bubbles.
Mass spectrometry acquisition
The following steps describe in detail the use of a nano‐LC 2D HPLC coupled to a TripleTOF 6600. The protocol can be adjusted depending on the available MS instruments or preference.
-
33
Transfer autosampler vials into the autosampler tray with cooling set at 4°C.
-
34
Create loading, injection, and analytical gradient methods with the following settings:
Load a total of 1 µg sample into the autosampler loop.
After injection, transfer peptide mixtures onto a C18 pre‐column chip (200 µm × 0.4 mm ChromXP C18‐CL chip, 3 µm, 120 Å; or column) and desalt by washing with aqueous mobile phase A at 2 µl/min for 10 min. Then, transfer peptides to an analytical chip (75 µm × 15 cm ChromXP C18‐CL chip, 3 µm, 120 Å; or column) and elute at a flow rate of 300 nl/min with a gradient method using mobile phases A (aqueous, composed of 97.9% water, 2% acetonitrile, and 0.1% formic acid) and B (organic, composed of 97.9% acetonitrile, 2% water, and 0.1% formic acid). Apply a linear gradient from 5% mobile phase B to 35% mobile phase B over 120 min.
Ramp the mobile phase B to 80% over 5 min, then hold at 80% B for 8 min before returning to 5% B for a 25 min re‐equilibration.
-
35
Build a DIA MS instrument method and define the following instrument scan experiments:
Experiment 1: Perform MS1 precursor ion scan from m/z 400 to 1,250 (accumulation time of 250 ms).
Experiments 2‐65: Perform MS/MS product ion scans for 64 variable window segments with a MS2 scan range from m/z 100 to 1,500 (accumulation time of 45 ms per each of the 64‐product ion scans per cycle). Set collision energy spread to CES = 10, then select the “high sensitivity product ion scan mode.”
Use the 64 variable window DIA acquisition strategy as described by Schilling et al. (2017) for a total cycle time of ∼3.2 s. In this acquisition, a series of variable width isolation windows (5‐90 m/z) is used to step over the full mass range (m/z 400‐1,250 over 64 SWATH segments, each with a 45‐ms accumulation time, yielding a cycle time of 3.2 s, which includes one MS1 scan with a 250‐ms accumulation time).
The variable window width is adjusted according to the complexity of the typical MS1 ion current observed within a certain m/z range using a variable window calculator algorithm (more narrow windows are chosen in “busy” m/z ranges, wide windows in m/z ranges with few eluting precursor ions). On other MS instrument platforms, other DIA window strategies (isolation schemes) may be implemented.
-
36
Create a sample queue/batch for all biological replicate samples.
To obtain consistent and reliable data, regular LC‐MS system suitability assessments need to be performed before and during the entire SWATH study. Initially, use pre‐digested β‐galactosidase standards and/or more complex human HeLa cell digests and perform quality control (QC) acquisitions or mass calibration acquisitions typically used in your laboratory or proteomics core. Also, randomize study samples to avoid systematic errors; block randomization of biologically different samples is often applied in proteomics studies.
Assign file names for each sample.
Set injection volumes corresponding to 1 μg of sample (for example, 1 μl of a 1 μg/μl sample).
Assign acquisition method generated above.
-
37
Submit samples for MS acquisition.
MS data analysis
Here, we describe a workflow using the commercially available software Spectronaut (alternative software packages can also be used). The procedure for entering settings and processing data will vary depending on the analysis software.
-
38
To analyze and quantify protein levels, open DIA analysis software Spectronaut.
-
39
To start the Quantification Analysis, select “Pipeline” tab, click “Set up a DIA Analysis from File,” and open the MS DIA raw files of interest for relative quantification.
-
40
Select “Assign Spectral Library” and select “Pan Human Library,” click “load” | “next.”
The Pan Human Spectral Library (Rosenberger et al., 2014) should be used only for human samples (however, other spectral library approaches can also be applied).
-
41
Select “BGS Factory Settings” analysis schema. Verify that the peptide FDR settings are set to Q‐value <0.01 with sparse identifications and click “next”.
-
42
Select the appropriate human database FASTA file (the default UniProt FASTA file assigned to the Pan Human Library) and click “next”.
-
43
Define the condition setup (e.g., “senescent” and “non‐senescent”).
-
44
In the condition setup form, assign to each sample a correction factor that is equal to the inverse of its cell count (1 divided by the cell count) and click “next”.
It is critical to assign correction factors based on cell counts to account for differences in protein secretion levels due to differences in the number of cells in each cell culture flask.
-
45
Select “goa_human” as the gene annotation (ontology) file and click “next”.
-
46
Review the analysis overview (summary of the experiment setup) and select “output directory” to assign an output directory. Click “finish”.
-
47
Click “Run Pipeline” to perform label‐free quantitative analysis.
-
48
Review results in the “output directory.”
The Spectronaut DIA Quantitative Analysis Software automatically performs FDR analysis, generates heat maps, and volcano plots (Fig. 6). It generates lists of identified and quantified peptides and proteins and provides Q‐values along with relative fold changes comparing different conditions in a “candidates.tsv” file.
Expected results: The elevation of several SASP factors is expected in a successful proteomic analysis, including GDF15, MMP1, STC1, CXCL1 (these are known “core” SASP factors for fibroblasts). The number of proteins identified in an experiment with five replicates per each condition (five senescent and five controls) is typically ∼1,000 proteins identified in the secretome. The number of significantly changed proteins (SASP proteins) will typically vary depending on the senescence inducer or cell type used. For example, we have previously reported 548 proteins significantly increased in the SASP of irradiated senescent fibroblasts but 332 proteins in the SASP of ATV/r‐treated senescent fibroblasts, and 180 proteins in the SASP of irradiated epithelial cells (Basisty, Kale, Jeon, et al., 2020). Significant SASP factors change heterogeneously with senescence induction (and cell type). In senescent fibroblasts, it is typically expected that the majority of significant protein changes are increases in protein secretion. In parallel, it is important to independently verify senescence in the cells using the Support Protocol assays described above.
Figure 6.

Senescence‐associated secretory phenotype (SASP) proteomic analysis figures generated by the Spectronaut software. (A) Heatmap depicting the abundance of all proteins identified in the conditioned medium of oncogenic RAS‐induced senescent (n = 6) and control (n = 6) IMR‐90 cells. (B) Volcano plot of the log 2 fold changes in abundance of proteins secreted by senescent versus control cells. Red dots represent fold changes >1.5‐fold, and p‐values >.05. (C) Line plot showing the abundance of a classical SASP protein, MMP1, in the secretomes of senescent and non‐senescent cells.
REAGENTS AND SOLUTIONS
Doxorubicin stock solution
Prepare a stock solution of doxorubicin by dissolving doxorubicin hydrochloride (Tocris, cat. no. 2252) in DMSO (MilliporeSigma, cat. no. 67‐68‐5) to obtain a final concentration of 2.5 mM.
Aliquot and store at −20°C for up to 1 month or at −80°C for up to 1 year.
Atazanavir/ritonavir (ATV/r) stock solution
Prepare a stock solution of ATV/r by mixing atazanavir sulfate (MedChem Express, cat. no. HY‐17367A) with ritonavir (MedChem Express, cat. no. HY‐90001; w/w ratio atazanavir/ritonavir of 4:1) and dissolving in DMSO (MilliporeSigma, cat. no. 67‐68‐5) to obtain a concentration of 8 mg/ml and 2 mg/ml, respectively.
For example, mix 32 mg of atazanavir with 8 mg of ritonavir and dissolve in 4 ml DMSO. In a sterile environment, filter solution using a sterile 0.2‐μm filter compatible with DMSO (Fisherbrand Syringe Filters Sterile, Thermo Fisher Scientific, cat. no. 09‐719C). After filtration, keep solution sterile.
Store stock solution at −20°C for up to 1 month or at −80°C for up to 1 year.
The working concentration of ATV/r described in Alternate Protocol 4 (25 μM) is calculated considering only the atazanavir sulfate concentration in the stock solution (that is 8 mg/ml, ∼10 mM). Therefore, to prepare medium with a final concentration of 25 μM ATV/r, dilute stock solution 1/400 in medium.
COMMENTARY
Background Information
Although senescent cells participate in several physiological functions, they are known to contribute to frailty as well as many age‐related diseases and therefore are considered a major driver of aging and age‐related diseases (Campisi & d'Adda di Fagagna, 2007; Gorgoulis et al., 2019). The secretome of senescent cells, termed the senescence‐associated secretory phenotype (SASP), is known to cause many of these effects (both beneficial and detrimental; Campisi, 2013; Munoz‐Espin & Serrano, 2014). For example, the SASP is linked to optimal wound healing (Demaria et al., 2014) but also to pathological states such as osteoarthritis, tumor growth, and metastasis (Coppe, Desprez, Krtolica, & Campisi, 2010; Jeon, David, Campisi, & Elisseeff, 2018). The SASP is extremely heterogeneous and dynamic, varying with the cell type and senescence‐inducing stimulus, as well as how much time has passed since senescence was induced (Hernandez‐Segura et al., 2017). Characterization of the SASP on a molecular level may provide novel mechanistic insights into how cellular senescence drives aging, potentially leading to the discovery of aging biomarkers and targets for preventing or counteracting the deleterious effects of senescent cells (Basisty, Kale, Jeon, et al., 2020).
Previously, antibody arrays were used to study the SASP protein composition (Coppe et al., 2008). However, antibody arrays can detect only a pre‐selected number of proteins. This method is therefore both biased and limited. Another approach to study the SASP is unbiased gene expression analysis of senescent cells (Hernandez‐Segura et al., 2017). This approach enables researchers to analyze a much larger number of genes compared to a small number of proteins using antibody arrays. Nevertheless, gene expression analysis cannot directly provide information about proteins secreted by cells. Recently, more comprehensive and unbiased mass spectrometric studies have been used to characterize and quantify SASP protein profiles directly (Basisty, Kale, Jeon, et al., 2020; Schafer et al., 2020; Wiley et al., 2019). A study by Basisty, Kale, Jeon, et al. (2020) found the SASP to be much larger than previously thought and extremely heterogeneous, depending on the cell type and senescence inducer. Interestingly, many of the SASP proteins overlapped with the plasma protein signature of aging identified in the Baltimore Longitudinal Study of Aging (BLSA). This overlap demonstrates the extent to which this approach not only expands our understanding of the SASP but also permits the identification of promising candidate protein biomarkers of organismal senescent cell burden in a biofluid that is easily accessible. The step‐by‐step procedure described here is meant as a guide to (1) generate senescent cells, (2) collect SASP‐containing conditioned media, and (3) perform a quantitative, unbiased analysis of the SASP using MS with a DIA workflow.
Critical Parameters and Troubleshooting
The methods for senescence induction described in this protocol collection have been developed and optimized for human IMR‐90 fibroblasts. Therefore, some adjustments might be required when using other cell strains. Nevertheless, these protocols have been successfully used in our laboratory to induce senescence in multiple human primary cell strains with little to no modification. Hence, we are confident that they can be applied as they are, or with little optimization, to many human primary cell strains.
For compatibility with downstream MS analysis, there are several important general considerations. First, the conditioned medium in which the final collection of secreted proteins takes place should be free of highly abundant protein additives and free of serum because abundant exogenously added proteins will interfere with the detection of secreted proteins. Secondly, all buffers and reagents used during the sample processing for MS should be MS grade. Thirdly, during all steps of sample processing, the sample should be completely free of detergents and chemicals that are incompatible with MS analysis.
Senescence Induction
Observation: cells do not develop a senescent phenotype.
Possible causes and solutions:
The dose of senescence inducer is too low. The cell strain or line used might be resistant to the senescence inducer. Increase the dose and perform a dose titration checking for senescence induction using only a couple of easily performed assays, such as SA‐β‐gal activity and cell counts as a proxy for growth arrest. Once a promising dose is identified, perform Support Protocol to more thoroughly confirm that the cells developed a senescent phenotype.
Observation: significant cell death upon treatment with the senescence inducer.
Possible causes and solutions:
The senescence inducer chosen is toxic to the cell type used. Reduce the dose (perform a dose titration) and check for senescence induction using only a couple of easily performed assays, as suggested above. Alternatively, use another senescence inducer. Once a promising inducer and dose are identified, perform Support Protocol to more thoroughly confirm that the cells developed a senescent phenotype.
The senescence‐inducing compound was not sterile. Ensure compounds given to cells are sterile by filtering the solutions with 0.2‐μm filters compatible with the compound solvent.
Cell cultures in low‐serum medium before CM collection
Observation: significant death of senescent cells (viability ≤80%, as measured by PI inclusion assay) when cultured in low‐serum medium for a prolonged period.
Possible causes and solutions:
The cell strain or line is highly sensitive to low‐serum medium upon senescence induction. Increase the percentage of serum in the medium; perform a titration to determine the desired percent increase. Check that the concentration at which senescent cells remain viable still induces quiescence in the control cells. Otherwise, do not culture the senescent cells in low‐serum medium (except for the last 24 hr before CM collection) as described in Alternate Protocol 5.
Observation: significant death in control cells when cultured in low‐serum medium for a prolonged period.
Possible causes and solutions:
The cell strain or line is highly sensitive to low‐serum medium. Increase the percentage of serum in the medium; perform a titration to determine the desired percent increase. Check that the concentration at which control cells remain viable still induces quiescence. Otherwise, try to induce quiescence by other means, such as contact inhibition. If quiescence cannot be achieved in the control cells, then culture both control and senescent cells in complete medium (except for the last 24 hr before CM collection) and compare the SASP‐containing CM to CM generated from these proliferating control cells. However, under these conditions, one cannot distinguish if changes in protein secretion are the result of comparing proliferating cells versus non‐proliferating cells, major differences in cell density between control and senescent conditions, or differences between senescent and non‐senescent cells.
Sample processing and MS analysis
Observation: High background signal or poor standard curve in BCA results.
Possible causes and solutions:
Residual phenol red or incompletely buffer‐exchanged samples. Ensure phenol red‐free medium is used for the collection of CM containing secreted proteins.
Buffer exchange was not complete. Ensure the buffer exchange is followed according to the manufacturer protocol, making sure to follow multiple rounds of exchanging buffer with 50 mM NH4HCO3.
Observation: Albumin is the only protein detected.
Possible causes and solutions:
The final CM is contaminated with residual serum. Take care to wash cells two times or more with PBS before addition of serum‐free medium for collection of CM.
Observation: Unexpected direction of protein changes between senescent and control cells, or unexpectedly high biological variation among samples.
Possible causes and solutions:
MS analysis is not corrected for cell count. Verify that correction factors are properly assigned in the MS software before sample acquisition. To ensure protein levels reflect changes in secretion levels, rather than differences in cell numbers, it is critical to correct for cell counts.
Understanding Results
It is important to confirm that the desired cell line develops a senescent phenotype following senescence induction. To this end, multiple senescence‐associated markers should be tested.
EdU incorporation
Senescent cells are characterized by an essentially irreversible growth arrest. Because the senescent cells do not divide, they should not incorporate nucleotides in DNA. The Click‐iT EdU Kit detects nucleotide incorporation by supplementing cells with a nucleotide analog (EdU) that can later be tagged by a selective chemical reaction. The tag contains a fluorophore and thus incorporation into DNA can be detected with a fluorescence microscope. Senescent cells, as well as quiescent control cells, should have low EdU incorporation (Fig. 2). Conversely, non‐senescent, proliferating cells should have high EdU staining.
SA‐β‐gal activity
Senescent cells increase lysosomes and lysosomal β‐galactosidase (β‐gal) activity. Therefore, the β‐gal substrate X‐gal is broken down at a higher rate in senescent cells and at a suboptimal pH compared to control cells. The breakdown of X‐gal causes the formation of blue indole crystals, visible by light microscopy. Senescent cells should have high SA‐β‐gal staining, whereas the opposite should be true for control cells (Fig. 3). Note that if cells are close to confluency, staining can increase significantly, leading to false SA‐β‐gal positivity in control cells.
Senescence‐associated gene expression
The transcriptome of senescent cells changes significantly compared to that of non‐senescent cells. While the transcriptome is very heterogeneous—dependent on cell strain and senescence inducer—there is a transcription signature that associates with the senescent phenotype. This signature includes upregulated (e.g., p16INK4a) and downregulated (e.g., LaminB1) genes. These changes in transcription can be detected by qPCR. Figure 4 shows a panel of selected genes and their expected relative mRNA levels in senescent cells compared to quiescent control cells. Depending on the cell type and senescence inducer, the expression of some of these genes might remain unchanged or trend in the opposite direction. However, most of the genes in Figure 4 should show similar results, regardless of the cell type and inducer.
Propidium iodide inclusion
Cells used to generate SASP and control CM should be viable and healthy when producing their secretome, otherwise proteins from dying cells might significantly contaminate the CM and falsely be assigned to the SASP. To verify that the cells are viable and healthy when producing CM, staining for dead or unhealthy cells, for example using propidium iodide, can be used with flow cytometry. Both senescent and control cells should have a low percentage of positive cells.
Time Considerations
Characterizing the SASP of a desired cell type using a chosen senescence inducer will require ∼2 months of dedicated effort. The procedure can be divided into sequential and independent stages.
Validation of senescence induction: Generating senescent and control cells and checking for senescence markers takes about 2 weeks. It might be necessary to invest more time if the senescence induction protocol requires optimization.
Production of CMs: The generation of senescent and control cells and collection of CMs should take 10 to 14 days.
Processing of CMs for MS analysis: Proteins in CM samples can be concentrated, digested, and desalted in 2 to 3 days.
LC‐MS analysis: Running one sample takes ∼1.5 hr. When planning experiments, keep in mind that the LC‐MS in your laboratory/MS core might not be able to process your samples immediately. Data analysis and identification of SASP proteins can be done in as little as a few hours to a few days, depending on the complexity of the MS data.
Conflicts of Interest
J.C. is a founder and shareholder of Unity Biotechnology that develops senolytic drugs. All other authors declare no conflict of interest.
Author Contributions
Francesco Neri: Formal analysis; Methodology; Visualization; writing‐original draft. Nathan Basisty: Formal analysis, Methodology; writing‐review & editing. Pierre‐Yves Desprez: Methodology; writing‐review & editing. Judith Campisi: Conceptualization; Funding acquisition; Investigation; Methodology; writing‐review & editing. Birgit Schilling: Conceptualization; Funding acquisition; Methodology; writing‐review & editing.
Acknowledgments
This work was supported by the National Institutes of Health under award numbers U01 AG060906‐02 and U01 AG060906‐02S1 (Principal Investigator: Schilling), P01 AG017242 and R01 AG051729 (Principal Investigator: Campisi), K99 AG065484 (Principal Investigator: Basisty), and 1S10 OD016281 (Buck Institute).
Neri, F. , Basisty, N. , Desprez, P.‐Y. , Campisi, J. , & Schilling, B. (2021). Quantitative proteomic analysis of the senescence‐associated secretory phenotype by data‐independent acquisition. Current Protocols, 1, e32. doi: 10.1002/cpz1.32
Literature Cited
- Basisty, N. , Kale, A. , Jeon, O. H. , Kuehnemann, C. , Payne, T. , Rao, C. , … Schilling, B. (2020). A proteomic atlas of senescence‐associated secretomes for aging biomarker development. PLoS Biology, 18, e3000599. doi: 10.1371/journal.pbio.3000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basisty, N. , Kale, A. , Patel, S. , Campisi, J. , & Schilling, B. (2020). The power of proteomics to monitor senescence‐associated secretory phenotypes and beyond: Toward clinical applications. Expert Reviews on Proteomics, 17, 297–308. doi: 10.1080/14789450.2020.1766976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausejour, C. M. , Krtolica, A. , Galimi, F. , Narita, M. , Lowe, S. W. , Yaswen, P. , & Campisi, J. (2003). Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO Journal, 22, 4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borghesan, M. , Fafian‐Labora, J. , Eleftheriadou, O. , Carpintero‐Fernandez, P. , Paez‐Ribes, M. , Vizcay‐Barrena, G. , … O'Loghlen, A. (2019). Small extracellular vesicles are key regulators of non‐cell autonomous intercellular communication in senescence via the interferon protein IFITM3. Cell Reports, 27, 3956–3971.e6. doi: 10.1016/j.celrep.2019.05.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi, J. (2013). Aging, cellular senescence, and cancer. Annual Review of Physiology, 75, 685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi, J. , & d'Adda di Fagagna, F. (2007). Cellular senescence: When bad things happen to good cells. Nature Reviews Molecular Cell Biology, 8, 729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- Collins, B. C. , Hunter, C. L. , Liu, Y. , Schilling, B. , Rosenberger, G. , Bader, S. L. , … Aebersold, R. (2017). Multi‐laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH‐mass spectrometry. Nature Communication, 8, 291. doi: 10.1038/s41467-017-00249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe, J. P. , Desprez, P. Y. , Krtolica, A. , & Campisi, J. (2010). The senescence‐associated secretory phenotype: The dark side of tumor suppression. Annual Review of Pathology, 5, 99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe, J. P. , Patil, C. K. , Rodier, F. , Sun, Y. , Munoz, D. P. , Goldstein, J. , … Campisi, J. (2008). Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biology, 6, 2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria, M. , Ohtani, N. , Youssef, S. A. , Rodier, F. , Toussaint, W. , Mitchell, J. R. , … Campisi, J. (2014). An essential role for senescent cells in optimal wound healing through secretion of PDGF‐AA. Developmental Cell, 31, 722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet, L. C. , Navarro, P. , Tate, S. , Rost, H. , Selevsek, N. , Reiter, L. , … Aebersold, R. (2012). Targeted data extraction of the MS/MS spectra generated by data‐independent acquisition: A new concept for consistent and accurate proteome analysis. Molecular & Cell Proteomics, 11, O111.016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Gualda, E. , Baker, A. G. , Fruk, L. , & Munoz‐Espin, D. (2020). A guide to assessing cellular senescence in vitro and in vivo. FEBS Journal. doi: 10.1111/febs.15570. [DOI] [PubMed] [Google Scholar]
- Gorgoulis, V. , Adams, P. D. , Alimonti, A. , Bennett, D. C. , Bischof, O. , Bishop, C. , … Demaria, M. (2019). Cellular senescence: Defining a path forward. Cell, 179, 813–827. doi: 10.1016/j.cell.2019.10.005. [DOI] [PubMed] [Google Scholar]
- Hernandez‐Segura, A. , de Jong, T. V. , Melov, S. , Guryev, V. , Campisi, J. , & Demaria, M. (2017). Unmasking transcriptional heterogeneity in senescent cells. Current Biology, 27, 2652–2660.e2654. doi: 10.1016/j.cub.2017.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James, E. N. L. , Bennett, M. H. , & Parkinson, E. K. (2018). The induction of the fibroblast extracellular senescence metabolome is a dynamic process. Scientific Reports, 8, 12148–12148. doi: 10.1038/s41598-018-29809-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon, O. H. , David, N. , Campisi, J. , & Elisseeff, J. H. (2018). Senescent cells and osteoarthritis: A painful connection. Journal of Clinical Investigation, 128, 1229–1237. doi: 10.1172/JCI95147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon, O. H. , Wilson, D. R. , Clement, C. C. , Rathod, S. , Cherry, C. , Powell, B. , … Elisseeff, J. H. (2019). Senescence cell‐associated extracellular vesicles serve as osteoarthritis disease and therapeutic markers. JCI Insight, 4(7), e125019. doi: 10.1172/jci.insight.125019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , & Schmitt, C. A. (2019). The dynamic nature of senescence in cancer. Nature Cell Biology, 21, 94–101. doi: 10.1038/s41556-018-0249-2. [DOI] [PubMed] [Google Scholar]
- Munoz‐Espin, D. , & Serrano, M. (2014). Cellular senescence: From physiology to pathology. Nature Reviews of Molecular Cell Biology, 15, 482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- Ozcan, S. , Alessio, N. , Acar, M. B. , Mert, E. , Omerli, F. , Peluso, G. , & Galderisi, U. (2016). Unbiased analysis of senescence associated secretory phenotype (SASP) to identify common components following different genotoxic stresses. Aging, 8, 1316–1329. doi: 10.18632/aging.100971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger, G. , Koh, C. C. , Guo, T. , Rost, H. L. , Kouvonen, P. , Collins, B. C. , … Aebersold, R. (2014). A repository of assays to quantify 10,000 human proteins by SWATH‐MS. Science Data, 1, 140031. doi: 10.1038/sdata.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer, M. J. , Zhang, X. , Kumar, A. , Atkinson, E. J. , Zhu, Y. , Jachim, S. , … LeBrasseur, N. K. (2020). The senescence‐associated secretome as an indicator of age and medical risk. JCI Insight, 5(12), e133668. doi: 10.1172/jci.insight.133668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling, B. , Gibson, B. W. , & Hunter, C. L. (2017). Generation of high‐quality SWATH((R)) acquisition data for label‐free quantitative proteomics studies using TripleTOF((R)) mass spectrometers. Methods in Molecular Biology, 1550, 223–233. doi: 10.1007/978-1-4939-6747-6_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, C. D. , Liu, S. , Limbad, C. , Zawadzka, A. M. , Beck, J. , Demaria, M. , … Kapahi, P. (2019). SILAC analysis reveals increased secretion of hemostasis‐related factors by senescent cells. Cell Reports, 28, 3329–3337.e3325. doi: 10.1016/j.celrep.2019.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Internet Resource
The data regarding SASP profiles obtained by Basisty, Kale, Jeon et al., 2020 (see above) is freely available on the SASP Atlas website.