

Abstract
The current study aimed to identify new breast and/or ovarian cancer predisposition genes. For that, whole‐exome sequencing (WES) was performed in the germline DNA of 52 non‐BRCA1/BRCA2/TP53 mutation carrier women at high‐risk for hereditary breast and ovarian cancer (HBOC). All variants were classified using information from population and disease specific databases, in silico prediction tools and the American College of Medical Genetics and Genomics (ACMG) criteria. Loss of heterozygosity (LOH) of tumor samples and segregation analyses were performed whenever possible. The variants identified were investigated in a second, independent cohort of 17 BC cases. Pathogenic/Likely Pathogenic variants were identified in known cancer genes such as CHEK2, MUTYH, PMS2, and RAD51C. Rare and potentially pathogenic variants were identified in DNA repair genes (FAN1, POLQ, and RAD54L) and other cancer‐related genes such as DROSHA and SLC34A2. Interestingly, the variant c.149T>G in the FAN1 gene was identified in two unrelated families, and exhibited LOH in the tumor tissue of one of them. In conclusion, this is the largest Brazilian WES study involving families at high‐risk for HBOC which has brought novel insights into the role of potentially new genetic risk factors for hereditary breast and ovarian cancer.
Keywords: BRCAX, breast cancer predisposition, hereditary breast and ovarian cancer predisposition syndrome, hereditary cancer, whole‐exome sequencing
The current study aimed to identify new breast and/or ovarian cancer predisposition genes. Rare and potentially pathogenic variants were identified in DNA repair genes (FAN1, POLQ, and RAD54L) and other cancer‐related genes such as DROSHA and SLC34A2. This is the largest Brazilian WES study involving families at high‐risk for hereditary breast and ovarian cancer which has brought novel insights into the role of potentially new genetic risk factors for hereditary breast and ovarian cancer.
1. INTRODUCTION
Germline variants in BRCA1/BRCA2 predispose to the hereditary breast and ovarian cancer (HBOC) syndrome and are responsible for approximately 25% of the familial breast cancer (BC) and ovarian cancer (OC) cases worldwide (Kast et al., 2016). In Brazil, our group recently reported similar findings, with 21.5% of the 349 index cases with clinical criteria for HBOC syndrome harboring BRCA1/BRCA2 germline variants (Fernandes et al., 2016). Genomic advances, such as next generation DNA sequencing platforms, allows the analysis of gene panels and the subsequent association of other high and moderated risk genes for HBOC syndrome with hereditary BC and OC development. These genes include, among others, ATM, BRIP1, CDH1, PALB2, PTEN, RAD51C, STK11, and TP53 (Lu et al., 2015). However, for a large proportion of HBOC families (50%–80%; Couch et al., 2014; Jones et al., 2017) the genetic cause associated with the BC and OC family history remains unknown.
Whole‐exome sequencing (WES) can be an effective diagnostic approach for individuals lacking classical pathogenic molecular alterations, especially for diseases with a high degree of genetic heterogeneity, which is the case of breast and ovarian cancer predisposition syndromes (Berberich et al., 2018). Recently, WES studies had identified new BC and OC susceptibility gene candidates, such as FANCC and BLM (Thompson et al., 2012), FANCM (Dicks et al., 2017; Kiiski et al., 2014), MDM1 and NBEAL1 (Glentis et al., 2019), RECQL (Cybulski et al., 2015; Sun et al., 2015), and RECQL5 (Tavera‐Tapia et al., 2019), providing further comprehension about the familial BC and OC susceptibility genes landscape.
The aim of the current study was to perform WES in 52 unrelated Brazilian women with high‐risk for BC and OC, previously tested negative for pathogenic BRCA1/BRCA2/TP53 germline variants, to identify driver genes of HBOC.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
The present study was approved by the Barretos Cancer Hospital (BCH) ethics committee (approval number: 916/2015), and all participants gave their written consent to participate.
2.2. Patients
Fifty‐two unrelated Brazilian women at‐risk for HBOC who attended the Oncogenetics Department of BCH (Palmero et al., 2016), lacking pathogenic variants in known breast/ovarian cancer susceptibility genes, were included in the study. All cases had personal and family history of BC and/or OC and fulfills the National Comprehensive Cancer Network (NCCN) criteria for Hereditary Breast and Ovarian Cancer. Analysis of the presence of germline variants in BRCA1/BRCA2/TP53 genes was conducted at the Center of Molecular Diagnosis of BCH as part of routine care through Sanger/panel NGS sequencing followed by MLPA (Multiplex Ligation‐dependent Probe Amplification Analysis) rearrangement analysis, as previously described (Fernandes et al., 2016). It is worth noting that 36 women, among the 52 included in this study, were previously analyzed in a 14‐gene NGS panel including high and moderate breast/ovarian cancer genes (ATM, BARD1, BRIP1, CDH1, CHEK2 MRE11, MUTYH, NBN, PALB2, PTEN, RAD50, RAD51C, TP53, and STK11; Grasel et al., 2020 accepted).
Clinical information was obtained through a detailed review of the patient's clinical chart. For the cancer family history, data were obtained from pedigrees. A study‐specific consent form was applied to all participants. Results were disclosed during genetic counseling sessions. For patients harboring pathogenic/likely pathogenic variants, point variant testing was offered to the interested relatives.
In addition, a selected group of candidate genes were analyzed in silico, in a second independent group of 17 breast cancer patients at risk for hereditary breast cancer, whose data were recently published (Torrezan et al., 2018).
2.3. Whole‐exome sequencing
Genomic DNA was isolated from peripheral blood lymphocytes (PBL) using the QIAamp DNA Blood Mini Kit (Qiagen) following the manufacturer's instructions. DNA concentration was determined using Qubit dsDNAHS Assay Kit (Thermo Fisher Scientific).
For the exome library preparation, 50 ng of PBL DNA of each sample was used with Nextera Rapid Capture Expanded Exome (Illumina), according to the manufacturer's recommendations. Quantified DNA library was loaded on flow cell for subsequent cluster generation. Samples were paired‐end sequenced on Illumina NextSeq 500 High Output Kit ‐ 300 cycles (Illumina).
2.3.1. Whole‐exome sequencing analysis
Briefly, reads were quality trimmed using the Trimmomatic v0.33 (Bolger et al., 2014), and then aligned with the reference genome (UCSC GRCh37/hg19) using the Burrows‐Wheeler Aligner v0.7.5a. PCR duplicates were removed using Picard v1.106 and BAM files were processed using the Genome Analysis Toolkit (GATK) v2.7.2 software. Realignment and search for indels were performed using GATK HaplotypeCaller and annotated using snpEFF v4.3 and SnpSift (Cingolani et al., 2012). A GEMINI v.0.19.1 database was created (Paila et al., 2013), and variants selected per functional rules. In addition, variants described by snpEFF/GEMINI as “low‐impact” were removed since they are assumed to have benign effects on DNA or protein functions.
2.3.2. In silico analysis workflow
The workflow analysis is illustrated in Figure 1. For a function‐based prioritization, variants leading to loss of function (LoF) “high‐impact” variants (frameshift, nonsense and canonical splice site variants) and missense variants (classified as “medium‐impact” variants) were selected. For quality filtering, variants with vertical coverage more than or equal to 10× and variant allele frequency more than or equal to 0.25 were selected. Next, a total of 2319 cancer‐associated genes were analyzed (“Cancer gene reference lists” described below). Variants present in the population database Genome Aggregation Database (gnomAD; Lek et al., 2016), with a minor allele frequency (MAF) less than or equal to 0.01 were maintained. Furthermore, Brazilian population‐specific variants were manually excluded (MAF > 0.01) using the publicly‐available AbraOM database, that contains WES data from 609 healthy individuals (Naslavsky et al., 2017). Loss of function variants were manually examined with Integrative Genomics Viewer (IGV; Thorvaldsdottir et al., 2013) looking for possible artifacts. In addition, to evaluate the potential impact of missense variant pathogenicity, we combined the CADD (score ≥ 20), REVEL (score ≥ 0.5), and M‐CAP (score ≥ 0.025) algorithms. Furthermore, variants classified as Benign/Likely Benign by Clinvar were excluded from further analysis (Figure 1). Finally, the remaining variants were classified according to the ACMG–AMP (American College of Medical Genetics and Genomics/Association for Molecular Pathology) guidelines (Kalia et al., 2017; Richards et al., 2015) as pathogenic, likely pathogenic, with uncertain significance, and likely benign or benign. Benign and Likely benign variants were excluded.
Figure 1.
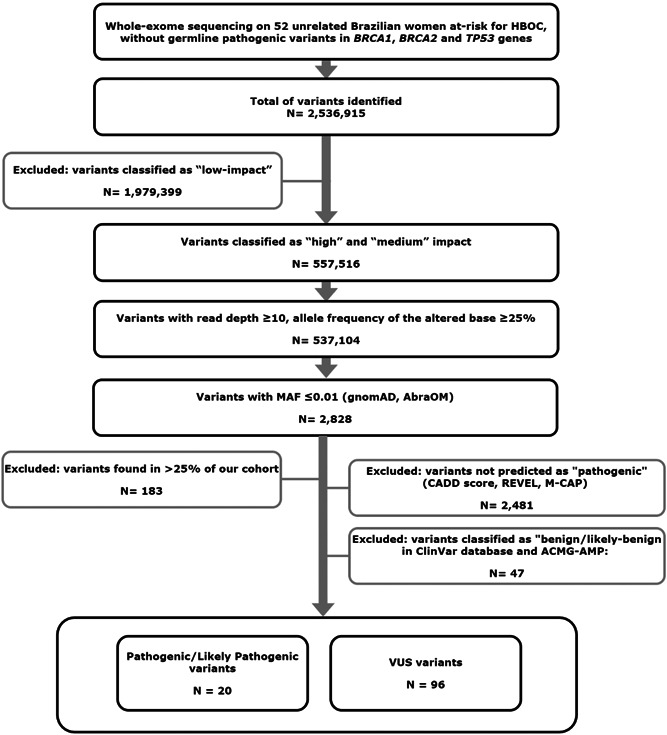
Variants selection workflow. Whole‐exome sequencing data from 52 unrelated Brazilian women at‐risk for HBOC, without germline pathogenic variants in BRCA1, BRCA2, and TP53 genes. Variants classified as “high‐impact” and “medium‐impact” by snpEFF/GEMINI were prioritized. Then, variants with base coverage more than or equal to 10× and variant allele frequency (VAF) more than or equal to 0.25 were selected, and those present in population databases with frequency less than or equal to 1% (MAF ≤ 0.01) were analyzed. The variants were also separated accordingly to ClinVar and ACMG classification. HBOC, hereditary breast and ovarian cancer
For in silico analysis of samples published by Torrezan et al. (2018), the VarSeq Software (Golden Helix) was used to filter and annotate rare (<1% in the Exome Aggregation Consortium) and possibly pathogenic variants in the candidate genes selected. Variants were then classified using the same criteria as in the discovery cohort.
2.3.3. Cancer gene reference lists
Three publicly available databases were used to generate a candidate list of genes, previously reported in association with any type of cancer: (i) The Cancer Gene Census v.86, a set of 719 genes manually curated by the Sanger Institute (Forbes et al., 2010), (ii) a query of DISEASES (Pletscher‐Frankild et al., 2015), a database of disease‐gene associations based largely on text‐mining approaches, and, (iii) UniprotKB (UniPr & ot: the universal protein knowledgebase, 2017), a manually curated database of protein functions (using the keyword‐terms “cancer,” “tumor‐suppressor gene,” “proto‐oncogene,” and “oncogene”). From these databases, a reference list of 2,319 genes was generated for prioritizing and characterizing gene variants (Table S1).
2.4. Confirmation and validation of results
2.4.1. Conventional sequencing
All variants classified, according to our pipeline as likely pathogenic or pathogenic were confirmed by conventional capillary Sanger sequencing. For this, the genomic DNAs were amplified by PCR, purified with the enzyme Exosap‐IT (USB) and Big Dye X terminator kit (Applied Biosystems) and sequenced bi‐directionally using the 3500XL platform (Applied Biosystems).
2.4.2. Segregation analysis
For the co‐segregation analysis, all families with class 4/5 germline variants were invited to participate. All index patient relatives, with or without cancer at any age, who were willing to participate in the study were tested.
2.4.3. Loss of heterozygosity
Loss of heterozygosity (LOH) analysis was performed for variants filtered through the prioritization pipeline for women with available tumor material by NGS sequencing (Ion Torrent Proton Platform). Data analysis was performed using Torrent Suite 5.10.1 software, and variants of interest were manually inspected with IGV. LOH of a wild‐type allele was considered when the variant allele had a frequency more than 60%.
3. RESULTS
3.1. Clinico‐pathological characterization
Of the 52 Brazilian women analyzed, 41 had BC, nine had OC and two were initially diagnosed with both OC and BC diagnosis (Table S2). The average age of BC and OC diagnoses were 43.2 years (21–59 years) and 41.0 years (20–60), respectively.
3.2. Family history
Besides BC and OC, other cancers associated with the HBOC spectrum, such as prostate (n = 14 relatives, 10 families) and pancreas (n = 4 relatives, four families) were observed (Table S3).
3.3. Germline variants by whole‐exome sequencing
WES identified a total of 2,536,915 variants in the 52 cases. To narrow down the analysis, a reference list of 2319 candidate genes previously associated with cancer was used. After applying the depicted workflow (Figure 1), our analysis identified 19 unique pathogenic/likely pathogenic variants (14 loss of function variants and 5 missense) in 18 genes, five of which affect DNA‐repair mechanisms (Figures 2 and 3 and Table 1). Besides, 92 unique variants of unknown significance were also identified (Table S4).
Figure 2.
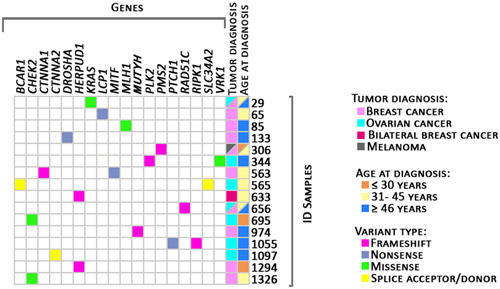
Likely pathogenic/pathogenic variants identified. In purple: frameshift variants; in blue: nonsense variants; in green: missense variants; in yellow: splice acceptor/donor variants. Information about tumor diagnosis: in light pink: breast cancer; in the dark pink: bilateral breast cancer; in greenish‐blue: ovarian cancer; in gray: melanoma. Information about age at diagnosis: in orange: diagnosis less than or equal to 30 years of age; in light yellow: 31–45 years of age; in blue: more than or equal to 46 years of age are represented
Figure 3.
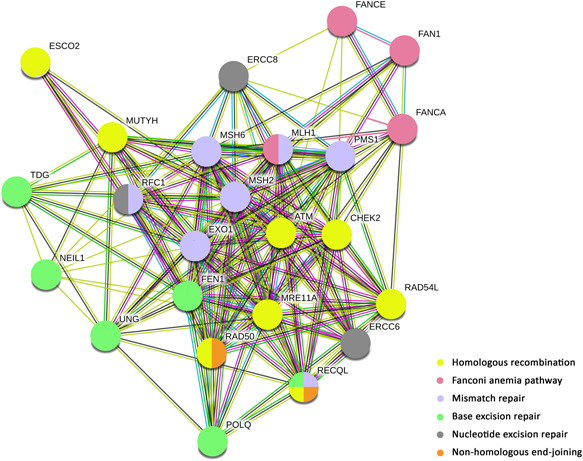
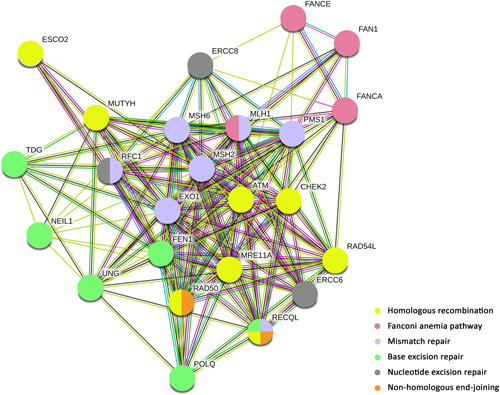
STRING pathway overview of DNA repair genes with pathogenic/likely pathogenic variants. Legends: in yellow: genes involved in homologous recombination, in light pink: genes involved Fanconi anemia, in blue: genes involved in mismatch repair, in green: genes involved in base excision repair, in orange: genes involved in nonhomologous end‐joining, and in gray: genes involved in nucleotide excision repair
Table 1.
Pathogenic/likely pathogenic variants identified
| ID | Diagnosis (Age) | BC | OC | Gene | Function | cDNA variant ‐ HGVS | Protein level | ACMG‐ | ClinVar | Transcript | gnomAD | gnomAD | ABraOM |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FH | FH | AMP | (all) | (NF) | |||||||||
| 29 | Ovarian (42), Breast (53) | Yes | Yes | KRAS | tumor‐associated | c.461A>G | p.(Asp154Gly) | LP | NR | NM_004985.4 | 0.000003 | 0.000000 | NR |
| 65 | Breast (36) | Yes | No | LCP1 | tumor‐associated | c.1122C>A | p.(Tyr374*) | P | NR | NM_002298.4 | NR | NR | NR |
| 85 | Breast (51) | Yes | No | MLH1 | MMR/FA | c.794G>A | p.(Arg265His) | LP | CI | NM_000249.4 | 0.000047 | 0.000070 | NR |
| 133 | Breast (46) | Yes | Yes | DROSHA | tumor‐associated | c.1498G>T | p.(Glu500*) | P | NR | NM_013235.4 | NR | NR | NR |
| 306 | Melanoma (26), Breast (36) | Yes | No | PMS2 | MMR | c.2182_2184delACTinsG | p.(Thr728fs) | P | P | NM_000535.7 | 0.000180 | 0.000155 | 0.006061 |
| 344 | Ovarian (47) | No | Yes | PLK2 | tumor‐associated | c.1004dupT | p.(Leu335fs) | P | NR | NM_006622.3 | NR | NR | NR |
| VRK1 | tumor‐associated | c.683C>T | p.(Thr228Met) | LP | VUS | NM_003384.2 | 0.000055 | 0.000087 | NR | ||||
| 563 | Breast (39) | Yes | No | CTNNA1 | tumor‐associated | c.1206_1207insCC | p.(Val403fs) | P | NR | NM_001290307.2 | NR | NR | NR |
| MITF | tumor‐associated | c.1A>G | p.(Met1?) | LP | NR | NM_000248.3 | NR | NR | NR | ||||
| 565 | Ovarian (43) | No | Yes | SLC34A2 | tumor‐associated | c.113‐2A>G | . | LP | NR | NM_006424.2 | 0.000031 | 0.000008 | NR |
| BCAR1 | tumor‐associated | c.12+2T>C | . | LP | NR | NM_014567.4 | 0.003987 | 0.001507 | 0.008079 | ||||
| 633 | Bilateral Breast (38) | Yes | No | HERPUD1 | tumor‐associated | c.584_585delTT | p.(Phe195fs) | P | NR | NM_014685.4 | 0.001551 | NR | 0.004105 |
| 656 | Ovarian (41), Breast (48) | No | Yes | RAD51C | HR/FA | c.890_899delTTGTTCCTGC | p.(Leu297fs) | P | P | NM_058216.3 | NR | NR | NR |
| 695 | Ovarian (21) | Yes | No | CHEK2 | HR | c.538C>T | p.(Arg180Cys) | LP | CI | NM_007194.4 | 0.000999 | 0.001316 | NR |
| 974 | Breast (46) | Yes | No | MUTYH | BER | c.1147delC | p.(Ala385fs) | LP | CI | NM_001048171.2 | NR | NR | NR |
| 1055 | Ovarian (57) | Yes | No | RIPK1 | tumor‐associated | c.1802_1805delGTGC | p.(Cys601fs) | P | NR | NM_003804.3 | NR | NR | NR |
| PTCH1 | tumor‐associated | c.1A>G | p.(Met1?) | P | NR | NM_001083607.1 | 0.000008 | 0.000000 | NR | ||||
| 1097 | Bilateral Ovarian (46) | Yes | No | CTNNA2 | tumor‐associated | c.103‐1G>A | . | P | NR | NM_001282597.2 | 0.016465 | 0.000512 | NR |
| 1294 | Breast (26) | Yes | No | HERPUD1 | tumor‐associated | c.584_585delTT | p.(Phe195fs) | P | NR | NM_014685.3 | 0.001551 | NR | 0.004105 |
| 1326 | Breast (35) | Yes | No | CHEK2 | HR | c.349A>G | p.(Arg117Gly) | LP | P/LP | NM_145862.2 | 0.000117 | 0.000188 | 0.000821 |
Abbreviations: BC, breast cancer; BER, base excision repair; CI, conflicting interpretations; FA, Fanconi anemia; FH, family history; HR, homologous recombination; LP, likely pathogenic; MMR, mismatch repair; NF, non‐Finish; NR, not reported; OC, ovarian cancer; P, pathogenic.
3.4. Loss of function variants
LoF variants in known hereditary/familial tumor‐associated genes, such as PMS2, RAD51C, and MUTYH, were identified in our high‐risk cases (Table 1; Figures S1 and S2). The PMS2 variant was identified in a patient (Figure S1) with melanoma and BC. Moreover, through segregation analysis, we observed the same variant in all three tested members (sisters), however, only one member was diagnosed with cancer (BC at 42 years of age and thyroid cancer at 43 years of age).
LoF variants were also identified in genes described in the COSMIC database as “hallmarks of cancer,” such as DROSHA and SLC34A2 (Figure 2).
A frameshift pathogenic alteration in HERPUD1 was found in two unrelated patients diagnosed with BC at very early ages (ID 1294: 26 years old, and ID 633: bilateral BC at 38 years of age).
Splicing variants were identified in patients diagnosed with OC at BCAR1 and CTNNA2 genes (ID 565 and ID 1097).
Variants affecting the start codon were identified in MITF and PTCH1 genes. However, it is important to mention that the MITF variant was identified in the patient where a frameshift variant at the CTNNA1 gene was also identified. Besides, a frameshift alteration at the RIPK1 gene was also identified in the patient with the PTCH1 variant.
3.5. Missense variants
As a result of our filtering pipeline, a total of five rare and potentially pathogenic missense variants were identified (Table 1, S2, and Figure 2).
One CHEK2 gene variant (c.349A>G) was considered as pathogenic/likely pathogenic by Clinvar submitters and by ACMG (Figure S3). The other variant in CHEK2 (c.538C>T) was identified in a patient with OC at 21 years of age (ID = 695), who reported a family history of BC and colorectal cancer.
Besides, likely pathogenic variants in KRAS (c.461A>G) and VRK1 (c.683C>T) were also identified. The patient with KRAS‐mutated (ID 29) had OC at 42 years of age and BC at 53 years of age (Table S3). The VRK1 gene variant was identified in a patient with OC, also carrying a PLK gene frameshift‐pathogenic variant.
3.6. Variants involved in DNA repair pathways
Among the 111 unique variants identified in our study, 29 (26.1%) were found in DNA‐repair genes (Table 1 and S4). According to KEGG and STRING, we found that homologous recombination was the most altered pathway, followed by mismatch repair, Fanconi anemia, base excision repair, nucleotide excision repair, and nonhomologous end‐joining pathways (Figure 3).
Two unrelated patients (ID 426 and ID 1264) with BC diagnosed at 38 and 27 years of age, respectively, carried the same FAN1 missense variant (c.149T>G), and although this variant has been described as probably benign by ClinVar (single submitter), functional data suggests that it confers pathogenicity (Lachaud et al., 2016). As both patients had tumor material available, NGS sequencing showed LOH, where loss of the wild‐type and retention of the variant FAN1 allele were observed in tumor DNA from one of the index patients (ID 1264; Figure S4).
Interestingly, two different missense variants in RAD54L were identified in two OC patients (ID 320: c.604C>T and ID 565: c.1094G>A). Both carriers reported a family history of OC (Figure S5). However, none of them showed RAD54L LOH.
Among the 23 altered DNA repair‐associated genes, seven were also found mutated in the independent group of 17 breast cancer patients at risk for hereditary breast cancer cohort published by Torrezan et al. (2018): ATM, EXO1, FANCA, FAN1, POLQ, RAD54L, and UNG. It is worthy to note the RAD54L c.604C>T, identified in the index patient ID 320, was also identified in one patient of this cohort (patient with BC at 29 years of age that reported ten BC cases in the family).
4. DISCUSSION
Sixteen patients carried a pathogenic/likely pathogenic variant (30.8%). Three of them with variants in known BC predisposition genes (RAD51C and CHEK2 genes). Besides, pathogenic/likely pathogenic variants were identified in the MMR genes MLH1 and PMS2.
Potentially pathogenic variants were identified in genes described as “hallmarks of cancer,” by COSMIC such as DROSHA and SLC34A2. DROSHA has been described as crucial in microRNA biogenesis and, more recently, in translational control and in the direct interaction with p53 effectors associated with RNA binding (UniProt: the universal protein knowledgebase, 2017). Interestingly, a genetic variant in the DROSHA gene (rs78393591), was reported in women of African ancestry and associated with increased BC risk (Qian et al., 2016). Concerning SLC34A2, we found an unreported splice acceptor variant identified in a woman with a personal and family history of OC. Similarly, a study by Kanchi et al. (2014) observed germline variants in SLC34A2 in patients with OC.
Although most of the known cancer predisposing genes are tumor suppressors, a small number of cancer predisposition syndromes are due to oncogene mutations, as is the case of MET in hereditary papillary renal, and RET in medullary thyroid carcinoma. In this study a pathogenic germline variant was identified at KRAS oncogene. Although germline variants on KRAS are associated with the development of RASophaties (Nissim et al., 2019), no clinical features compatible with any RASopathy were observed in our family.
Regarding variants of unknown significance, we would like to highlight five in particular, located in the POLQ, RAD54L, and FAN1 genes. A LoF POLQ variant was found in two unrelated patients with BC. Studies by Wang et al. (2008) and Brandalize et al. (2014) reported germline variants in POLQ in non‐BRCA1/BRCA2 mutated BC cases with a family history of BC. A case‐control study by Family et al. (2015) associated three missense variants in POLQ with an increased risk to BC. Thus, these findings suggest that POLQ variants may be involved in hereditary BC.
Two missense variants in RAD54L (c.604C>T and c.1094G>A), gene involved in the HR DNA repair pathway, were identified in patients with personal/family history of OC. We also identified a carrier of the c.604C>T variant in a BC patient from the independent cohort. Although the function of RAD54L in HBOC syndrome is still unclear, Matsuda et al. (1999) reported a carrier of RAD54L c.973G>A variant, in a patient diagnosed with BC at 63 years of age who had no family history of cancer. They also reported LOH in the tumor tissue, hypothesizing that RAD54L behaves as a suppressor tumor gene. In our study, LOH was not observed for either of the RAD54L variants in tumor tissue from carriers.
We identified three carriers of missense variants in FAN1, a gene involved in the Fanconi Anemia pathway. Two unrelated patients were carriers of FAN1 c.149T>G. This variant has also been reported to co‐segregate in two families with a personal and family history of pancreatic cancer (Smith et al., 2016). There have been independent reports of the association of other germline variants in the FAN1 with colorectal cancer (Gardenia Vargas et al., 2016; Segui et al., 2015). Lachaud et al. (2016) using in vitro assays involving the FAN1 c.149T>G, showed loss of protein function of the encoded isoform and suggested that this could be due to its location at a functional ubiquitin binding domain. LOH analysis of tumor tissue of both FAN1 variant carriers revealed loss of the normal allele and retention of variant allele in the tumor tissue of patient ID 1264. Further studies are required to elucidate a role of FAN1 variants in BC risk. One of the main limitations of this study is the WES inherent restriction of identifying potentially pathogenic variants located in intronic regions, putative regulatory elements, and long rearrangements. Besides, the lack of matched fresh tumor tissue did not allow the LOH analysis, or complementary approaches such as RNA‐seq, that could have brought insights into the consequences of the identified splicing variants, as well as the effect of the variants on the gene expression levels, providing additional evidence favoring or not the pathogenicity of the variants identified.
It is also important to mention that polygenic risk factors were not evaluated in this study, which could account for a proportion of the cases lacking pathogenic/likely pathogenic variants. In spite of that, this is the biggest Brazilian study evaluating women at high‐risk for hereditary breast and ovarian cancer and allowed the identification of pathogenic/likely pathogenic variants in almost one‐third of the patients evaluated. Candidate genes identified require further validation in Brazilian and other cancer cohorts. Besides, specific variants would also require in vitro analysis to investigate functional consequences on protein function.
5. CONCLUSION
In summary, the present study performed a characterization of germline variants identified in cancer‐associated genes, using WES and bioinformatic analyses in Brazilian non‐BRCA1/BRCA2/TP53 mutation‐carrier women with BC and/or OC. Our findings suggest that several cancer‐associated genes also may have a role in HBOC, such as RAD54L, FAN1, DROSHA, POLQ, and SLC34A2. In addition, the present study provides additional evidence for the association of moderate‐risk genes, such as CHEK2, RAD51C, and PMS2, to the development of familial BC/OC. Such advances will help with the molecular cataloging of breast/ovarian tumors in non‐BRCA1/BRCA2 mutation carrier patients.
CONFLICT OF INTERESTS
The authors declare no conflict of interest.
WEB Resources (Table S5)
SnpEff & SnpSift ‐ https://pcingola.github.io/SnpEff/
CADD ‐ https://cadd.gs.washington.edu/
REVEL ‐ https://sites.google.com/site/revelgenomics/
M‐CAP ‐ http://bejerano.stanford.edu/mcap/
gnomAD ‐ https://gnomad.broadinstitute.org/
AbraOM ‐ http://abraom.ib.usp.br/
GEMINI ‐ https://gemini.readthedocs.io/en/latest/
VARSOME‐ https://varsome.com/
CLINVAR ‐ https://www.ncbi.nlm.nih.gov/clinvar/
Supporting information
Supplementary Figure 1 ‐ Pedigree of index case (ID 306), PMS2 mutation carrier.
Supplementary Figure 2 ‐ Pedigree of index case (ID 656), RAD51C mutation carrier.
Supplementary Figure 3 ‐ Pedigree of index case (ID 1326), CHEK2 mutation carrier.
Supplementary Figure 4 ‐ Pedigree of index case FAN1 mutation carriers (A): ID 426, (B): 1264.
Supplementary Figure 5 ‐ Pedigree of index case (A): ID 565, RAD54L (c.1094 G > A) mutation carrier. (B): ID 320, RAD54L (c.604 C > T) mutation carrier.
Supporting information.
ACKNOWLEDGMENTS
We would like to thank all our patients and their families who contributed to this study. The authors are very grateful to Suzanna L. Arcand and Wejdan Alenezi for whole‐exome sequencing analysis and Claudia Andrade de Paula for LOH analysis.
Felicio PS, Grasel RS, Campacci N, et al. Whole‐exome sequencing of non‐BRCA1/BRCA2 mutation carrier cases at high‐risk for hereditary breast/ovarian cancer. Human Mutation. 2021;42:290–299. 10.1002/humu.24158
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The variants identified are publicly available (Clinvar database, available at https://www.ncbi.nlm.nih.gov/clinvar/). Accession numbers are SCV001438595 ‐ SCV001438703.
REFERENCES
- Berberich, A. J. , Ho, R. , & Hegele, R. A. (2018). Whole genome sequencing in the clinic: empowerment or too much information? Canadian Medical Association Journal/Journal de l'Association Medicale Canadienne, 190(5), E124–E125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandalize, A. P. , Schuler‐Faccini, L. , Hoffmann, J. S. , Caleffi, M. , Cazaux, C. , & Ashton‐Prolla, P. (2014). A DNA repair variant in POLQ (c.‐1060A > G) is associated to hereditary breast cancer patients: a case‐control study. BMC Cancer, 14, 850 10.1186/1471-2407-14-850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani, P. , Platts, A. , Wang le, L. , Coon, M. , Nguyen, T. , Wang, L. , & Ruden, D. M. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin), 6(2), 80–92. 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch, F. J. , Nathanson, K. L. , & Offit, K. (2014). Two decades after BRCA: Setting paradigms in personalized cancer care and prevention. Science, 343(6178), 1466–1470. 10.1126/science.1251827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski, C. , Carrot‐Zhang, J. , Kluźniak, W. , Rivera, B. , Kashyap, A. , Wokołorczyk, D. , Giroux, S. , Nadaf, J. , Hamel, N. , Zhang, S. , Huzarski, T. , Gronwald, J. , Byrski, T. , Szwiec, M. , Jakubowska, A. , Rudnicka, H. , Lener, M. , Masojć, B. , Tonin, P. N. , … Akbari, M. R. (2015). Germline RECQL mutations are associated with breast cancer susceptibility. [Research Support, Non‐U.S. Gov't]. Nature Genetics, 47(6), 643–646. 10.1038/ng.3284 [DOI] [PubMed] [Google Scholar]
- Dicks, E. , Song, H. , Ramus, S. J. , Oudenhove, E. V. , Tyrer, J. P. , Intermaggio, M. P. , & Pharoah, D. P. P. (2017). Germline whole exome sequencing and large‐scale replication identifies FANCM as a likely high grade serous ovarian cancer susceptibility gene. Oncotarget, 8(31), 50930–50940. 10.18632/oncotarget.15871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Family, L. , Bensen, J. T. , Troester, M. A. , Wu, M. C. , Anders, C. K. , & Olshan, A. F. (2015). Single‐nucleotide polymorphisms in DNA bypass polymerase genes and association with breast cancer and breast cancer subtypes among African Americans and Whites. Breast Cancer Research and Treatment, 149(1), 181–190. 10.1007/s10549-014-3203-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes, G. C. , Michelli, R. A. , Galvao, H. C. , Paula, A. E. , Pereira, R. , Andrade, C. E. , & Palmero, E. I. (2016). Prevalence of BRCA1/BRCA2 mutations in a Brazilian population sample at‐risk for hereditary breast cancer and characterization of its genetic ancestry. Oncotarget, 7(49), 80465–80481. 10.18632/oncotarget.12610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes, S. A. , Tang, G. , Bindal, N. , Bamford, S. , Dawson, E. , Cole, C. , & Futreal, P. A. (2010). COSMIC (the catalogue of somatic mutations in cancer): A resource to investigate acquired mutations in human cancer. Nucleic Acids Research, 38 Database issue, D652–D657. 10.1093/nar/gkp995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardenia Vargas, E. D. , Navarro, M. , Pons, T. , Mina, L. , Fernández, A. , Feliubadaló, L. , Solanes, A. , Iglesias, S. , Velasco, À. , Balmaña, J. , Ramón y Cajal, T. , Valencia, A. , Brunet, J. , Surrallés, J. , Lázaro, C. , Valle, L. , Pineda, M. , & Capellá, G. (2016). Identification of germline FAN1 variants in MSH2‐deficient Lynch‐like syndrome patients. European Hereditary Tumour Group.
- Glentis, S. , Dimopoulos, A. C. , Rouskas, K. , Ntritsos, G. , Evangelou, E. , Narod, S. A. , & Dimas, A. S. (2019). Exome sequencing in BRCA1‐ and BRCA2‐negative Greek families identifies MDM1 and NBEAL1 as candidate risk genes for hereditary breast cancer. Frontiers in Genetics, 10, 1005 10.3389/fgene.2019.01005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasel, R. S. , F. P., Paula, A. E. , Campacci, N. , Garcia, F. A. O. , Andrade, E. S. , Evangelista, A. F. , Fernandes, G. C. , Sabato, C. S. , De Marchi, P. , Souza, C. P. , Paula, C. A. A. , Torrezan, G. T. , Galvão, H. C. R. , Carraro, D. M. , & Palmero, E. I. (2020). Using Co‐segregation and loss of heterozygosity analysis to define the pathogenicity of unclassified variants in hereditary breast cancer patients. Frontiers in Oncology, 10 Advance online publication. 10.3389/fonc.2020.571330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, M. R. , Kamara, D. , Karlan, B. Y. , Pharoah, P. D. P. , & Gayther, S. A. (2017). Genetic epidemiology of ovarian cancer and prospects for polygenic risk prediction. Gynecologic Oncology, 147(3), 705–713. 10.1016/j.ygyno.2017.10.001 [DOI] [PubMed] [Google Scholar]
- Kalia, S. S. , Adelman, K. , Bale, S. J. , Chung, W. K. , Eng, C. , Evans, J. P. , & Miller, D. T. (2017). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genetics in Medicine, 19(2), 249–255. 10.1038/gim.2016.190 [DOI] [PubMed] [Google Scholar]
- Kanchi, K. L. , Johnson, K. J. , Lu, C. , McLellan, M. D. , Leiserson, M. D. , Wendl, M. C. , & Ding, L. (2014). Integrated analysis of germline and somatic variants in ovarian cancer. Nature Communications, 5, 3156 10.1038/ncomms4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kast, K. , Rhiem, K. , Wappenschmidt, B. , Hahnen, E. , Hauke, J. , Bluemcke, B. , & Engel, C. (2016). Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. [Research Support, Non‐U.S. Gov't]. Journal of Medical Genetics, 53(7), 465–471. 10.1136/jmedgenet-2015-103672 [DOI] [PubMed] [Google Scholar]
- Kiiski, J. I. , Pelttari, L. M. , Khan, S. , Freysteinsdottir, E. S. , Reynisdottir, I. , Hart, S. N. , & Nevanlinna, H. (2014). Exome sequencing identifies FANCM as a susceptibility gene for triple‐negative breast cancer. [Research Support, N.I.H., Extramural; Research Support, Non‐U.S. Gov't]. Proceedings of the National Academy of Sciences of the United States of America, 111(42), 15172–15177. 10.1073/pnas.1407909111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaud, C. , Moreno, A. , Marchesi, F. , Toth, R. , Blow, J. J. , & Rouse, J. (2016). Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science, 351(6275), 846–849. 10.1126/science.aad5634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , & MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, C. , Xie, M. , Wendl, M. C. , Wang, J. , McLellan, M. D. , Leiserson, M. D. , & Ding, L. (2015). Patterns and functional implications of rare germline variants across 12 cancer types. Nature Communications, 6, 10086 10.1038/ncomms10086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda, M. , Miyagawa, K. , Takahashi, M. , Fukuda, T. , Kataoka, T. , Asahara, T. , & Kamiya, K. (1999). Mutations in the RAD54 recombination gene in primary cancers. Oncogene, 18(22), 3427–3430. 10.1038/sj.onc.1202692 [DOI] [PubMed] [Google Scholar]
- Naslavsky, M. S. , Yamamoto, G. L. , de Almeida, T. F. , Ezquina, S. A. M. , Sunaga, D. Y. , Pho, N. , & Zatz, M. (2017). Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Human Mutation, 38(7), 751–763. 10.1002/humu.23220 [DOI] [PubMed] [Google Scholar]
- Nissim, S. , Leshchiner, I. , Mancias, J. D. , Greenblatt, M. B. , Maertens, O. , Cassa, C. A. , & Goessling, W. (2019). Mutations in RABL3 alter KRAS prenylation and are associated with hereditary pancreatic cancer. [Research Support, N.I.H., Extramural; Research Support, Non‐U.S. Gov't]. Nature Genetics, 51(9), 1308–1314. 10.1038/s41588-019-0475-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paila, U. , Chapman, B. A. , Kirchner, R. , & Quinlan, A. R. (2013). GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Computational Biology, 9(7), e1003153 10.1371/journal.pcbi.1003153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmero, E. I. , Galvao, H. C. , Fernandes, G. C. , Paula, A. E. , Oliveira, J. C. , Souza, C. P. , & Michelli, R. A. (2016). Oncogenetics service and the Brazilian public health system: the experience of a reference Cancer Hospital. Genetics and Molecular Biology, 39(2), 168–177. 10.1590/1678-4685-GMB-2014-0364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletscher‐Frankild, S. , Palleja, A. , Tsafou, K. , Binder, J. X. , & Jensen, L. J. (2015). Diseases: Text mining and data integration of disease‐gene associations. Methods, 74, 83–89. 10.1016/j.ymeth.2014.11.020 [DOI] [PubMed] [Google Scholar]
- Qian, F. , Feng, Y. , Zheng, Y. , Ogundiran, T. O. , Ojengbede, O. , Zheng, W. , & Huo, D. (2016). Genetic variants in microRNA and microRNA biogenesis pathway genes and breast cancer risk among women of African ancestry. Human Genetics, 135(10), 1145–1159. 10.1007/s00439-016-1707-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segui, N. , Mina, L. B. , Lazaro, C. , Sanz‐Pamplona, R. , Pons, T. , Navarro, M. , & Valle, L. (2015). Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology, 149(3), 563–566. 10.1053/j.gastro.2015.05.056 [DOI] [PubMed] [Google Scholar]
- Smith, A. L. , Alirezaie, N. , Connor, A. , Chan‐Seng‐Yue, M. , Grant, R. , Selander, I. , & Zogopoulos, G. (2016). Candidate DNA repair susceptibility genes identified by exome sequencing in high‐risk pancreatic cancer. Cancer Letters, 370(2), 302–312. 10.1016/j.canlet.2015.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, J. , Wang, Y. , Xia, Y. , Xu, Y. , Ouyang, T. , Li, J. , Wang, T. , Fan, Z. , Fan, T. , Lin, B. , Lou, H. , & Xie, Y. (2015). Mutations in RECQL gene are associated with predisposition to breast cancer. [Research Support, Non‐U.S. Gov't]. PLoS Genetics, 11(5), e1005228 10.1371/journal.pgen.1005228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavera‐Tapia, A. , de la Hoya, M. , Calvete, O. , Martin‐Gimeno, P. , Fernandez, V. , Macias, J. A. , & Osorio, A. (2019). RECQL5: Another DNA helicase potentially involved in hereditary breast cancer susceptibility. [Research Support, Non‐U.S. Gov't]. Human Mutation, 40(5), 566–577. 10.1002/humu.23732 [DOI] [PubMed] [Google Scholar]
- Thompson, E. R. , Doyle, M. A. , Ryland, G. L. , Rowley, S. M. , Choong, D. Y. , Tothill, R. W. , & Campbell, I. G. (2012). Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. [Research Support, Non‐U.S. Gov't]. PLoS Genetics, 8(9), e1002894 10.1371/journal.pgen.1002894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir, H. , Robinson, J. T. , & Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Briefings in Bioinformatics, 14(2), 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrezan, G. T. , de Almeida, F. , Figueiredo, M. C. P. , Barros, B. D. F. , de Paula, C. A. A. , Valieris, R. , & Carraro, D. M. (2018). Complex landscape of germline variants in Brazilian patients with hereditary and early onset breast cancer. Frontiers in Genetics, 9, 161 10.3389/fgene.2018.00161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt: the universal protein knowledgebase . (2017). UniProt: The universal protein knowledgebase. Nucleic Acids Research, 45(D1), D158–D169. 10.1093/nar/gkw1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Szabo, C. , Qian, C. , Amadio, P. G. , Thibodeau, S. N. , Cerhan, J. R. , & Couch, F. J. (2008). Mutational analysis of thirty‐two double‐strand DNA break repair genes in breast and pancreatic cancers. Cancer Research, 68(4), 971–975. 10.1158/0008-5472.CAN-07-6272 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 ‐ Pedigree of index case (ID 306), PMS2 mutation carrier.
Supplementary Figure 2 ‐ Pedigree of index case (ID 656), RAD51C mutation carrier.
Supplementary Figure 3 ‐ Pedigree of index case (ID 1326), CHEK2 mutation carrier.
Supplementary Figure 4 ‐ Pedigree of index case FAN1 mutation carriers (A): ID 426, (B): 1264.
Supplementary Figure 5 ‐ Pedigree of index case (A): ID 565, RAD54L (c.1094 G > A) mutation carrier. (B): ID 320, RAD54L (c.604 C > T) mutation carrier.
Supporting information.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The variants identified are publicly available (Clinvar database, available at https://www.ncbi.nlm.nih.gov/clinvar/). Accession numbers are SCV001438595 ‐ SCV001438703.