

Abstract
KBG syndrome is a rare genetic disease characterized mainly by skeletal abnormalities, distinctive facial features, and intellectual disability. Heterozygous mutations in ANKRD11 gene, or deletion of 16q24.3 that includes ANKRD11 gene are the cause of KBG syndrome. We describe two patients presenting with short stature and partial facial features, whereas no intellectual disability or hearing loss was observed in them. Two ANKRD11 variants, c.4039_4041del (p. Lys1347del) and c.6427C > G (p. Leu2143Val), were identified in this study. Both of them were classified as variants of uncertain significance (VOUS) by ACMG/AMP guidelines and were inherited from their mothers. ANKRD11 could enhance the transactivation of p21 gene, which was identified to participate in chondrogenic differentiation. In this study, we demonstrated that the knockdown of ANKRD11 could reduce the p21‐promoter luciferase activities while re‐introduction of wild type ANKRD11, but not ANKRD11 variants (p. Lys1347del or p. Leu2143Val), could restore the p21 levels. Thus, our study report two loss‐of‐function ANKRD11 variants which might provide new insight on pathogenic mechanism that correlates ANKRD11 variants with the short stature phenotype of KBG syndrome.
Keywords: ANKRD11, KBG syndrome, p21, short stature
1. INTRODUCTION
“KBG” represents the surname initials of the first families diagnosed with the disorder. KBG syndrome (OMIM 148050) is a rare genetic disease commonly manifested macrodontia of the upper central incisors, distinctive facial features, short stature, skeletal anomalies, hearing loss, developmental delay, and intellectual disability(Herrmann, Pallister, Tiddy, & Opitz, 1975). Heterozygous pathogenic variant in the ANKRD11 gene (OMIM 611192) or deletion of 16q24.3 that includes ANKRD11 gene will cause KBG syndrome, and the transmission of this disease follows an autosomal dominant pattern (Isrie et al., 2012; Willemsen et al., 2010).
Zhang et al. had demonstrated that ANKRD11 contains a transcriptional activation domain and two transcriptional repression domains(Zhang, Li, & Chen, 2007). ANKRD11 interacts with both HDAC corepressors and p160 coactivators to antagonize transcriptional activation(Gallagher et al., 2015). ANKRD11 also acts as a p53 coactivator, enhancing the transactivation of p53 target genes such as p21(Neilsen et al., 2008a). Cell lines and animal model studies indicated that ANKRD11 functions as a vital chromatin regulator that controls histone acetylation and gene expression during neural development(Gallagher et al., 2015), which provides a likely explanation for the intellectual disability occurring in KBG patients. Intriguingly, similar reduced body size was observed in Yoda mice burdened with a point mutation in the Ankrd11 gene(Barbaric et al., 2008), and earlier studies had shown that chondrocyte differentiation is an essential process for endochondral ossification(Kronenberg, 2003). According to previous study, p21 is involved in early chondrogenic differentiation (Negishi et al., 2001) and ANKRD11 interacts with p21(Neilsen et al., 2008a). A rational suspicion was raised in this study that the ANKRD11 variant might impair or even significantly annihilate its ability to interact with p21, leading to a negative effect on bone development.
In this study, two ANKRD11 variants were found in patients presenting with short stature and partial facial features. Further in vitro studies were performed to investigate the molecular effect of the two variants on p21 signaling.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical
Two Chinese patients were recruited here and none of them was previously reported. Six individuals were included as healthy controls. This study was approved by the Institutional Review Board of the Rui‐Jin Hospital. The informed consent was obtained from each participant.
2.2. Molecular investigations
DNA was extracted from peripheral blood leukocytes using DNA extraction kit (Qiagen, Hilden, Germany). Variants were identified by next generation sequencing (NGS) on custom gene panel for short stature, which contains 187 genes including ANKRD11, and the result was confirmed with sanger sequencing. Obtained sequences were compared to NCBI entry of ANKRD11 (NM_013275.6). We followed the ACMG/AMP guidelines for variant pathogenicity assessment (Nykamp et al., 2017).
2.3. Plasmids construction
The cDNA containing ANKRD11, which was purchased from Sino Biological (HG20681‐UT, Beijing, China), was used as template, and inserted into pCDNA3.0 plasmids with 5'flag tag. Variants of ANKRD11 were introduced by site‐directed mutagenesis as previously reported(Li et al., 2020a). The p21 gene promoter region (−2000 to −1 bp) was amplified from human genome DNA and inserted into pGL3‐basic plasmid. The shRNAs for ANKRD11 were designed and inserted into pLKO.1 plasmid, which was purchased from SIGMA‐ALDRICH (Merck, Germany), and their specific sequences are provided in Table 1.
TABLE 1.
Sequences of shRNAs for ANKRD11
| shRNAs for ANKRD11 | Target site sequence |
|---|---|
| Scramble | GCGCGATAGCGCTAATAATTT |
| shANKRD11‐1 | CCGCGTGGTTTGTATCTTCTT |
| shANKRD11‐2 | CCGTATTGAAATGGAGTCAAA |
2.4. Cell culture and transfection
Human HEK293T cell line was kindly provided by Professor Ronggui Hu (Chinese Academy of Sciences, Shanghai, China) and cultured in Dulbecco's modified Eagle medium (DMEM, Life Technologies) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin (all from Gibco, Layola,) in a 37°C humidified atmosphere of 5% CO2. Plasmids were transfected into HEK293T cells using a Lipofectamine 2000 (Life Technologies, Carlsbad) according to the manufacturer's instructions.
2.5. Reverse transcription‐quantitative PCR
Blood samples were obtained from antecubital vein of participants in test tubes containing 50 mM EDTA anticoagulant. Blood was centrifuged at 2,100 × g at 4°C for 10 min to collect serum. TRIzol RNA extraction kit (Sangon Biotech, Shanghai, China) was used to extract total RNA from peripheral blood, and nucleic acid quantification detector was used to measure RNA concentration. Total RNA kit (Tiangen, Beijing, China) was used to extract total RNA from HEK293T cells. Complementary DNA (cDNA) was synthesized using ReverTra Ace qPCR RT Master Mix (Toyobo, Osaka, Japan). The RT‐qPCR assay was performed using the SYBR Green Master Mix (Toyobo, Osaka, Japan) with the CFX96 real‐time PCR system (Bio‐Rad Laboratories, CA), according to the manufacturer's protocol(Xu et al., 2018). The relative abundances of the specified genes were normalized to that of GAPDH, using the 2△△Ct method. The specific sequences of primers used in this study are listed in Table 2. All the data were obtained from three independent experiments and data were analyzed using one‐way ANOVA with Tukey's post hoc test.
TABLE 2.
Sequences of the primers used in RT‐qPCR
| Primers for RT‐PCR | ||
|---|---|---|
| Target gene | Forward primer (5′‐3′) | Reverse primer (5′‐3′) |
| GAPDH | ATTTGGTCGTATTGGGCGCCTG | CCTTGCCCACAGCCTTGGCAG |
| ANKRD11 | GCAGGGCCCTGAGCGGAAGAGG | CTGGGCTGTTGGCAGACTCCTC |
| p21 | TCCGTCAGAACCCATGCGGC | CAGGCGAAGTCACCCTCCAGTG |
Abbreviation: RT‐qPCR, reverse transcription‐quantitative polymerase chain reaction.
2.6. Immunoblotting
Cell lysates were subjected to SDS‐PAGE and transferred to a PVDF membrane (Bio‐Rad) as previously reported(Li et al., 2019). The membrane was incubated with the appropriate antibodies against GAPDH (1:5000, 60,004‐1‐Ig, Proteintech, China), ANKRD11 (1:500, 55,364‐1‐AP, Proteintech), Flag (1:1000, 20,543‐1‐AP, Proteintech) or p21(1:1000, 10,355‐1‐AP, Proteintech). Secondary antibodies were labeled with HRP, and the signals were visualized using Tanon 5,200 Imaging System (Tanon, China).
2.7. Luciferase reporter assays
HEK293T cells were seeded at 0.5 × 105 cells/well into 24‐well plates. After overnight culture, cells were transiently transfected with p21‐promoter‐Luc, PRL‐TK together with other vectors (shRNAs for ANKRD11, wild type ANKRD11 or its indicated mutants). The 48 hrs after transfection, the cells were harvested, lysed with 5× passive buffer, and subjected to Dual‐Luciferase Reporter assay according to the manufacturer's instruction (Li et al., 2020b) (Promega). Three independent experiments were performed and data were analyzed using one‐way ANOVA with Tukey's post hoc test.
3. RESULTS
3.1. Identification of ANKRD11 variants
Pedigrees of the families with ANKRD11 variants and results of mutation analysis by direct DNA sequencing were shown in Figure 1. Proband 1 had a c.4039_4041del mutation (p. Lys1347del, rs757750566) in ANKRD11 and Proband 2 had a c.6427C > G mutation (p. Leu2143Val, rs750499484) in the same gene. The c.4039_4041del mutation was found in gnomAD browser in one East Asian out of 18,394 alleles and c.6427C > G mutation was found in one East Asian out of 18,086 alleles. Both of them can be classified as variants of uncertain significance (VOUS) by ACMG/AMP guidelines and were inherited from their mothers.
FIGURE 1.
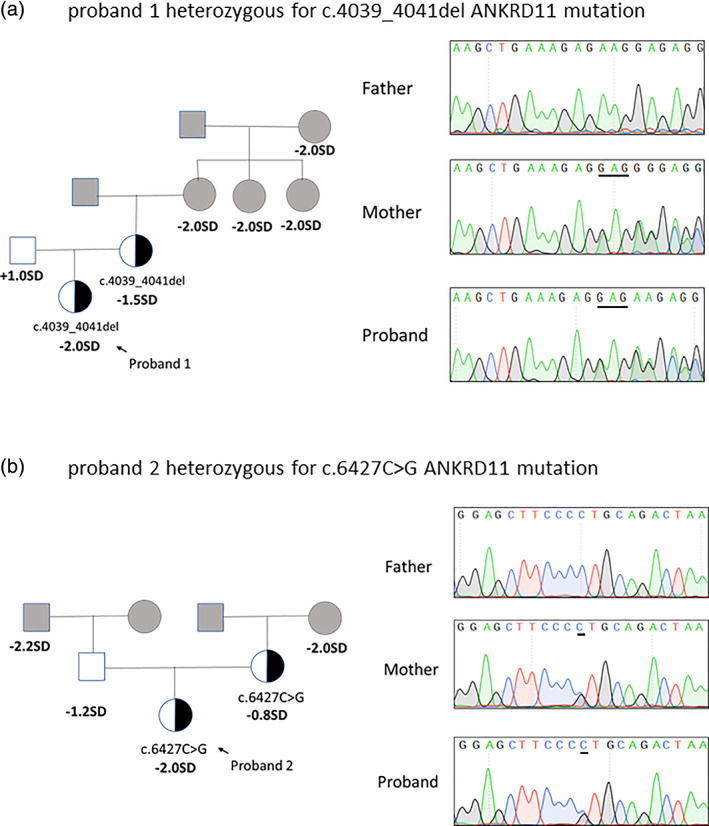
The pedigrees of two families with ANKRD11 variants. (a) Pedigree of the family of proband 1 with the c.4039_4041del mutation in ANKRD11 gene, and partial sequencing chromatographs of the proband and the parents. (b) Pedigree of the family of proband 2 with a c.6427C > G mutation in ANKRD11 gene, and partial sequencing chromatographs of the proband and the parents. Squares and circles in gray indicate family members who had not received mutation analysis [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Clinical characteristics of two probands
Proband 1 was born at term following an uneventful pregnancy. Birth weight and length were normal at birth. At the age of 6.5 years old, short stature was noted (height:112 cm, ‐2SD) and became the first reason to seek medical assistance. She had no history of feeding difficulties and sleep disorder. Physical examination showed a triangular face, large cheek bone, low bridge of the nose and normal teeth (Figure. 2(a)‐1). She had no report of intellectual disability or developmental delay. Arginine GH test results showed that the patient had no absence of growth hormone for a peak GH level as 14.330 ng/ml. Insulin‐like growth factor 1 level was 215 ng/ml (normal range: 74‐388 ng/ml), and thyroid function had no abnormalities. X‐ray examination showed a delayed bone age (by the Greulich‐Pyle method) (Figure. 2(a)‐2). Idiopathic short stature was clinically diagnosed and the patient had no additional medical treatment. When the patient was 10 years and 8 months old, her bone age was advanced to 12 years old (by the Greulich‐Pyle method) (Figure. 2(a)‐3). A GnRH stimulation test showed a peak LH as 17.32 mUI/ml and a peak FSH as 8.60 mUI/ml. A gonadotropin‐releasing hormone agonist treatment due to medical necessity was started at that time (height ‐2SD) because of an advanced bone age compared with calendar age and a threat of short adult stature (<150 cm). After a regular follow‐up, growth curves of proband 1 was recorded and depicted in Figure. 2(a)‐4. The ANKRD11 mutation was transmitted from her mother who presented a final height of 153 cm (−1.5SD) (Figure. 1(a)).
FIGURE 2.
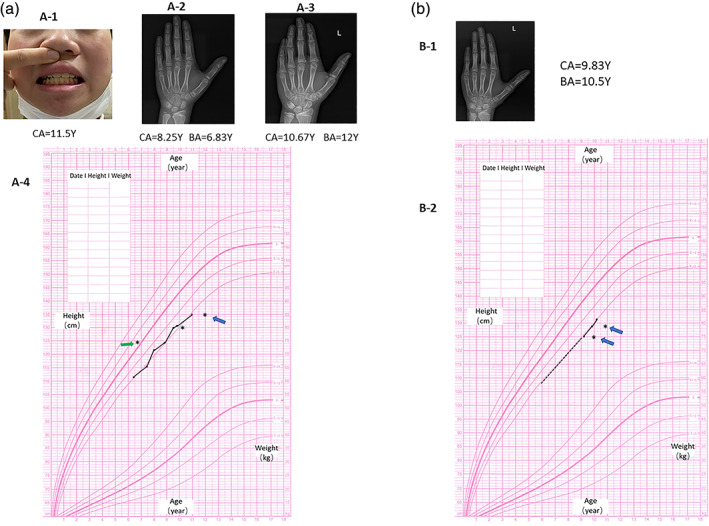
Clinical characteristics of two probands with ANKRD11 variants. (a‐1) Proband 1 had a triangular face, large cheek bone, low bridge of the nose and normal teeth. (a‐2) X‐ray examination of proband 1 at the age of 8 years and 4 months showing a delayed bone age (6.83 years) (by the Greulich‐Pyle method). (a‐3) X‐ray examination of proband 1 at the age of 10 years and 8 months showing an advanced bone age (12 years) (by the Greulich‐Pyle method). (a‐4) Growth curves of proband 1 showed an unideal growth tendency. Green arrow: delayed bone age; Blue arrow: advanced bone age. (b‐1) X‐ray examination showed an advanced bone age of proband 2 (by the Greulich‐Pyle method). (b‐2) Growth curves of proband 2 showed an unideal growth tendency. Approximate growth tendency from 6 years old to 9 years old was indicated by dotted line. Blue arrow: advanced bone age [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 3.
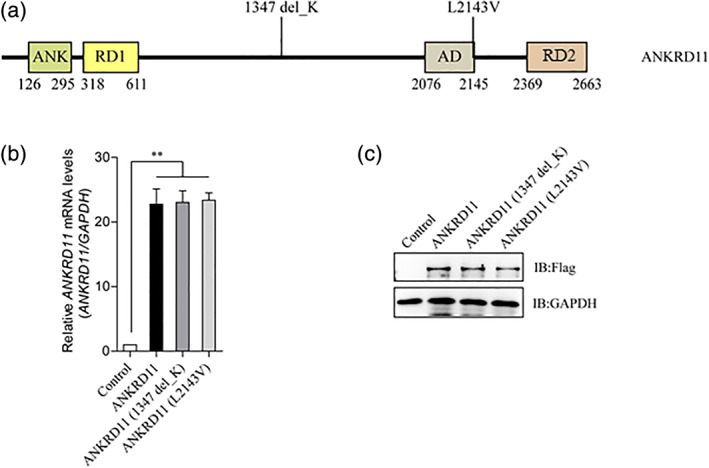
The variants of ANKRD11 have no effect on the expression of its protein and mRNA. (a) Schematic view of human ANKRD11 protein showing the location of the two variants involved in our study. ANK, Ankyrin repeats; RD1, Repression domain 1; AD, Activation domain; RD2, Repression domain 2. (b) Detection of the relative mRNA levels of wild type ANKRD11 and its mutants by RT‐qPCR. Empty vector, wild type ANKRD11, ANKRD11(p. Lys1347del) or ANKRD11 (p. Leu2143Val) were transfected into HEK293T cells. Total RNA was extracted, complementary DNA (cDNA) was synthesized, and then the relative mRNA levels of ANKRND11 were detected by RT‐qPCR. Data are expressed as mean ± SD. **p < 0.01, very significantly difference. No mRNA level changes of ANKRND11 were observed. (c) Detection of the protein levels of wild type ANKRD11 and its mutants by immunoblotting. Empty vector, or Flag tagged wild type ANKRD11/ANKRD11 (p. Lys1347del)/ANKRD11 (p. Leu2143Val) were transfected into HEK293T cells, and the protein levels of Flag tagged protein and GAPDH were detected by immunoblotting. No protein level changes of ANKRD11 were observed [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 4.
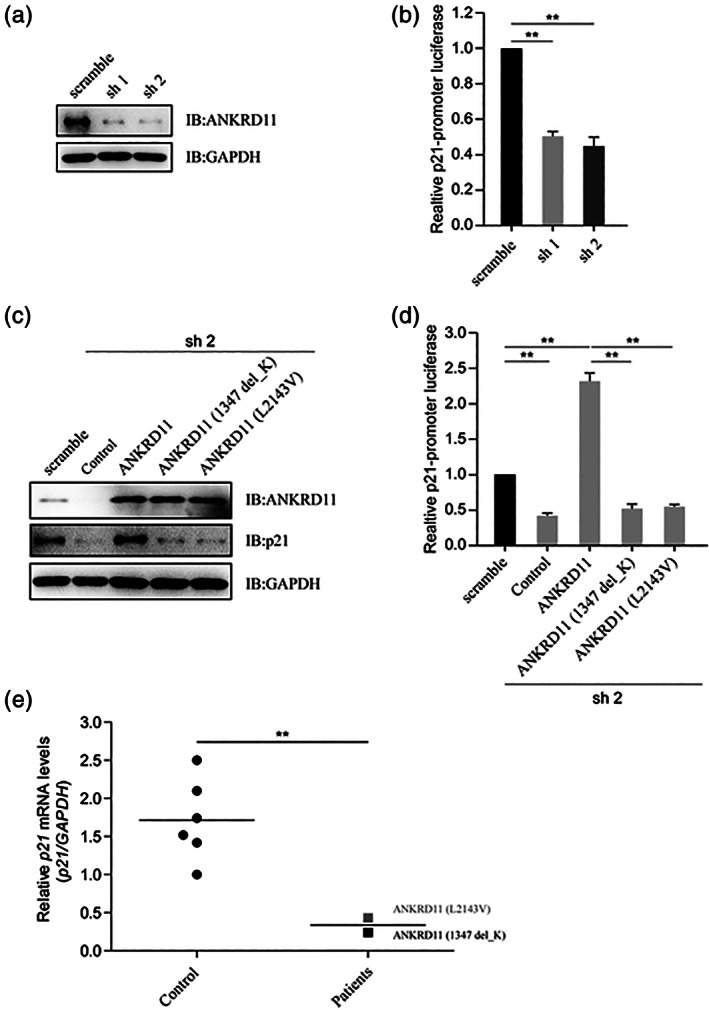
The variants of ANKRD11 attenuate p21. (a) Knockdown efficiency of shRNAs for ANKRD11 by immunoblotting. HEK293T cells were transfected with shRNAs (scramble, sh1 or sh2) for ANKRD11, and the protein levels of ANKRD11 and GAPDH were detected by immunoblotting. Sh1 and sh2 reduced the expression of ANKRD11 protein, respectively. (b) Luciferase reporter assay to detect the relative activities of human p21‐promoter. HEK293T cells were transfected with shRNAs for ANKRD11, as well as with p21‐promoter luciferase reporter and PRL‐TK. The cell lysates subjected to Dual‐Luciferase Reporter assay 48 hrs later. Data are expressed as mean ± SD. **p < 0.01, very significantly difference. With sh1 or sh2, the knockdown of ANKRD11 reduced the p21‐promoter luciferase activities. (c) Detection of the protein levels of p21 by immunoblotting. HEK293T cells were transfected with shRNA (sh 2) for ANKRD11, and reintroduced with wild type ANKRD11, ANKRD11 (p. Lys1347del) or ANKRD11 (p. Leu2143Val). The protein levels of ANKRD11, p21 and GAPDH were detected by immunoblotting. ANKRD11 knockdown reduced the p21 protein expression, while the re‐introduction of wild type ANKRD11 could restore the p21 levels, but not those two ANKRD11 mutants. (d) The variants of ANKRD11 comprised its activating effect on p21 promoter activity. HEK293T cells were transfected with shRNA (sh 2) for ANKRD11, as well as with p21‐promoter luciferase reporter and PRL‐TK, and reintroduced with wild type ANKRD11, ANKRD11 (p. Lys1347del) or ANKRD11 (p. Leu2143Val). The cell lysates subjected to Dual‐Luciferase Reporter assay 48 hrs later. Data are expressed as mean ± SD. **p < 0.01, very significantly difference. Wild type ANKRD11 activated the p21‐promoter luciferase, while those two ANKRD11 mutants lost this function. (e) Comparison of the relative p21 mRNA levels in two patients and healthy controls. The peripheral blood of two patients and six healthy controls were collected. Total RNA was extracted and complementary DNA (cDNA) was synthesized. The relative mRNA levels of p21 were detected by RT‐qPCR. Data are expressed as mean ± SD. **p < 0.01, very significantly difference. Compared with healthy controls, the relative p21 mRNA levels of the two patients were much lower
Proband 2 was born at 37 WG after an uncomplicated pregnancy and delivery. At birth, her weight and length were 2.5 kg (‐2SD) and 50 cm (−0.2SD), respectively. She manifested a slow rate of growth (growth velocity < 4 cm/year) since she was 6 years old, but her parents did not seek medical intervention. She was referred to the hospital at 9 years old for bilateral mammary development meanwhile an unideal height below−2 SD (height: 125 cm) had also attracted the attention of endocrinologist. Physical examination showed a low bridge of the nose, wide eye distance and normal teeth (picture not available). She had no report of intellectual disability or developmental delay. Arginine GH test results showed a peak GH level as 9.310 ng/ml. IGF‐1 was 221.5 ng/ml (normal range: 74‐388 ng/ml). Plasma cortisol, fasting insulin and thyroid function were within normal range. Basic value of LH and FSH were 0.16 mIU/ml and 2.09 mIU/ml, respectively. The concentration of E2 was less than 10 pg/ml. X‐ray examination showed she had an advanced bone age (by the Greulich‐Pyle method) (Figure. 2(b)‐1). Pelvic ultrasound revealed a start‐up pubertal status. After 1 year of follow‐up with no additional treatment, growth curves of proband 2 showed the height always being below ‐2SD (Figure. 2(b)‐2). The ANKRD11 mutation was inherited from her mother who presented a final height as 157 cm (−0.8SD) (Figure. 1(b)).
Other family members of the two probands reported a history of short stature: members from the maternal side of Proband 1 and members from both maternal and paternal sides of Proband 2. (Figure. 1(a) and (b)).
3.3. The variants of ANKRD11 involved in our study attenuate the p21 signaling
Two variants of ANKRD11 ‐ ANKRD11 (p. Lys1347del) and ANKRD11 (p. Leu2143Val), were involved in this study (Figure. 3(a)). After transfection, no differences between wild type and mutated ANKRD11 isoforms were observed both at mRNA and protein levels (Figure. 3(b) and (C)). Two shRNAs targeting the UTR (untranslated region) of ANKRD11 were designed, and the knockdown efficiency was detected (Figure. 4(a)). These two shRNAs worked well and used for further studies. The p21 gene is directly regulated by ANKRD11(Neilsen et al., 2008b), and the knockdown of ANKRD11 significantly reduced the p21‐promoter luciferase activity (Figure. 4(b)). ANKRD11 knockdown reduced the p21 protein expression, while the re‐introduction of wild type ANKRD11 could restore the p21 levels, but not the two ANKRD11 variants (Figure. 4(C)). Luciferase reporter assay indicated that wild type ANKRD11 significantly activated the p21‐promoter luciferase, while the two ANKRD11 variants lost this function (Figure. 4(D)). The p21 mRNA level in blood of control group was more than five times higher than that of patient group (Figure. 4(e)). These data suggest that ANKRD11 (p. Lys1347del) and ANKRD11 (p. Leu2143Val) are two loss‐of‐function variants.
4. DISCUSSION
Approximately 200 KBG patients had been reported to date and several diagnostic criteria were proposed by different investigators based on their own cohorts (Ockeloen et al., 2015; Skjei, Martin, & Slavotinek, 2007). The latest diagnostic criterion is that a patient with developmental delay/learning difficulties, speech delay or significant behavioral issues with at least two major criteria or one major and two minor criteria should be clinically diagnosed as KBG syndrome(Low et al., 2016). Although two patients in this study showed atypical phenotype (short stature) without intellectual disability or hearing loss, molecular analysis disclosed two rare ANKRD11 variants with an unclear pathogenic relevance, suggesting a KBG syndrome diagnosis. It's worth noting the two ANKRD11 mutations were transmitted from their mothers, who also showed limited phenotype such as unsatisfied final height (−1.5SD, −0.8SD, respectively). It is reported that short stature (below the third centile) is prevalent in affected cohorts and has been identified in 40%–77% of KBG patients(Butler, Rafi, Hossain, Stephan, & Manzardo, 2015). As far as we know, patients with KBG syndrome had been rarely reported in China or even in East Asian. A recent case reported by physicians in China was referred to hospital for idiopathic short stature without other complaints and abnormal signs(Kang et al., 2019). Perhaps other factors could theoretically confound genotype–phenotype correlation, such as specific mutation condition, ethnic differences, environmental factors and so on. Besides, other variants might contribute to the phenotype in addition with ANKRD11 variants and perhaps an exome sequencing is needed in the future to rule out this possibility. It's noteworthy that the final heights of the patients' mothers did not fit the diagnosis of short stature although both mothers harbored the loss‐of‐function mutations. An incomplete penetrance of loss‐of‐function mutation, a theory that had been documented before(Ropers & Wienker, 2015), might explain this discrepancy.
Available knowledge on molecular mechanism of short stature phenotype in KBG syndrome has not been investigated yet. In this study, we examined changes in the mRNA and protein levels of mutated ANKRD11 by RT‐qPCR and immunoblotting studies. The results demonstrated that the two ANKRD11 variants did not disturb mRNA and protein expression (Figure 3(b) and (C)), thus an indirect impact of the variant on bone development was considered. Since ANKRD11 could enhance p21 expression, we investigated whether ANKRD11 knockdown was able to weaken the transcriptional activity of p21. Luciferase reporter assay indeed showed that shRNA‐mediated silencing of endogenous ANKRD11 had diminution effects on the transcription of p21 (Figure.4(b)). The observation that restoration of wild type ANKRD11 could renew the protein level and transcriptional activity of p21 further provided evidence to support an enhancement role for ANKRD11 as a p21 regulator. On the other hand, the reintroduction of two ANKRD11 variants (p. Lys1347del and p. Leu2143Val) failed to up‐regulate p21 expression (Figure 4(C) and (D)), indicating that these two variants lost or partially lost its function. Consistent with the results in vitro studies, reduced relative p21 mRNA levels was observed in blood of affected patients compared with that of healthy controls, which provides a powerful evidence to support the ANKRD11‐p21 pathway (Figure.4(e)).
Bone formation can be explained as a process named endochondral bone formation(Kronenberg, 2003). As noted earlier, the process of chondrogenic differentiation requires critical regulation by p21 protein: Negishi and his colleagues had established a cell line expressing antisense RNA of the p21 gene. They had not solely observed the down‐regulation of p21 but also identified inhibited progression of early differentiation in chondrocytes. In contrast, after inducing the differentiation of mouse ATDC5 prechondrocytes, cells which will differentiate to proliferating chondrocytes, investigators had observed the increased expression of p21 mRNA. What's more, they noted that the expression of p21 mRNA preceded the up‐regulation of type II collagen gene(Negishi et al., 2001), an early‐chondrogenic marker(Wang et al., 2019).
Besides p21, multiple molecules participating in bone metabolism had been reported to interact with ANKRD11, such as SOX6 (Smits, Dy, Mitra, & Lefebvre, 2004), Rela (Kobayashi et al., 2016),Notch (Zanotti & Canalis, 2016),FOXP1(Zhao et al., 2015), ATF5(Leong et al., 2012), SPRY1(Joo et al., 2016). Future studies will be necessary to test these relationships.
Taken together, our study report two ANKRD11 variants and two atypical KBG patients manifested mainly with short stature and partial facial features. Although our patients did not meet the criteria needed for clinical diagnosis, molecular studies demonstrated that the two variants could impair the ANKRD11 function to up‐regulate p21 expression. Moreover, ANKRD11‐p21 pathway turned out to be a likely mechanism to interpret short stature phenotype in KBG syndrome.
ACKNOWLEDMENTS
We are grateful to all the patients and their families who participated in this study. We also express our gratitude to all of the pediatricians, who provided patients' clinical data.
CONFLICT OF INTEREST
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTIONS
Tingting Zhang: Methodology (equal), Formal analysis (equal), Writing—original draft preparation (lead). Yun yang: Methodology (equal), Formal analysis (equal). Xueling Yin and Xueqing Wang: Validation (equal). Jihong Ni: Resources (equal). Zhiya Dong: Supervision (lead). Wenli Lu: Conceptualization (equal), Resources (equal), Writing—review and editing (equal), Project administration (lead), Funding acquisition (lead). Chuanyin Li: Writing—review and editing (equal), Funding acquisition (supporting), Visualization (equal). All authors have read and agreed to the published version of the manuscript.
Zhang T, Yang Y, Yin X, et al. Two loss‐of‐function ANKRD11 variants in Chinese patients with short stature and a possible molecular pathway. Am J Med Genet Part A. 2021;185A:710–718. 10.1002/ajmg.a.62024
Tingting Zhang and Yun Yang contributed equally to this work.
Funding information shanghai science & technology committee, Grant/Award Number: 14411958600; Shanghai Talent Development Fund, Grant/Award Number: 2017120; the National Natural Science Foundation for Young Scientists of China, Grant/Award Number: 31900804
Contributor Information
Chuanyin Li, Email: lichuanyin2013@sibcb.ac.cn.
Wenli Lu, Email: lwl204059@126.com.
DATA AVAILABILITY STATEMENT
All datasets generated and analyzed for this study are included in the article/Supplementary Material.
REFERENCES
- Barbaric, I. , Perry, M. J. , Dear, T. N. , Rodrigues Da Costa, A. , Salopek, D. , Marusic, A. , & Brown, S. D. (2008). An ENU‐induced mutation in the Ankrd11 gene results in an osteopenia‐like phenotype in the mouse mutant Yoda. Physiological Genomics, 32(3), 311–321. 10.1152/physiolgenomics.00116.2007 [DOI] [PubMed] [Google Scholar]
- Butler, M. G. , Rafi, S. K. , Hossain, W. , Stephan, D. A. , & Manzardo, A. M. (2015). Whole exome sequencing in females with autism implicates novel and candidate genes. International Journal of Molecular Sciences, 16(1), 1312–1335. 10.3390/ijms16011312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher, D. , Voronova, A. , Zander, M. A. , Cancino, G. I. , Bramall, A. , Krause, M. P. , & Miller, F. D. (2015). Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Developmental Cell, 32(1), 31–42. 10.1016/j.devcel.2014.11.031 [DOI] [PubMed] [Google Scholar]
- Herrmann, J. , Pallister, P. D. , Tiddy, W. , & Opitz, J. M. (1975). The KBG syndrome‐a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Original Artical Series, 11(5), 7–18. [PubMed] [Google Scholar]
- Isrie, M. , Hendriks, Y. , Gielissen, N. , Sistermans, E. A. , Willemsen, M. H. , Peeters, H. , & Van Esch, H. (2012). Haploinsufficiency of ANKRD11 causes mild cognitive impairment, short stature and minor dysmorphisms. European Journal of Human Genetics, 20(2), 131–133. 10.1038/ejhg.2011.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo, A. , Long, R. , Cheng, Z. , Alexander, C. , Chang, W. , & Klein, O. D. (2016). Sprouty2 regulates endochondral bone formation by modulation of RTK and BMP signaling. Bone, 88, 170–179. 10.1016/j.bone.2016.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, Y. , He, D. , Li, Y. , Zhang, Y. , Shao, Q. , Zhang, M. , & Ban, B. (2019). A heterozygous point mutation of the ANKRD11 (c.2579C>T) in a Chinese patient with idiopathic short stature. Molecular Genetics & Genomic Medicine, 7(12), e988 10.1002/mgg3.988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi, H. , Chang, S. H. , Mori, D. , Itoh, S. , Hirata, M. , Hosaka, Y. , & Saito, T. (2016). Biphasic regulation of chondrocytes by Rela through induction of anti‐apoptotic and catabolic target genes. Nature Communications, 7, 13336 10.1038/ncomms13336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg, H. M. (2003). Developmental regulation of the growth plate. Nature, 423(6937), 332–336. 10.1038/nature01657 [DOI] [PubMed] [Google Scholar]
- Leong, D. T. , Abraham, M. C. , Gupta, A. , Lim, T. C. , Chew, F. T. , & Hutmacher, D. W. (2012). ATF5, a possible regulator of osteogenic differentiation in human adipose‐derived stem cells. Journal of Cellular Biochemistry, 113(8), 2744–2753. 10.1002/jcb.24150 [DOI] [PubMed] [Google Scholar]
- Li, C. , Han, T. , Guo, R. , Chen, P. , Peng, C. , Prag, G. , & Hu, R. (2020a). An integrative synthetic biology approach to interrogating cellular ubiquitin and Ufm signaling. International Journal of Molecular Sciences, 21(12), 4231 10.3390/ijms21124231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. Y. , Lu, W. L. , Yang, L. G. , Li, Z. W. , Zhou, X. Y. , Guo, R. , & Hu, R. G. (2020b). MKRN3 regulates the epigenetic switch of mammalian puberty via ubiquitination of MBD3. National Science Review, 7(3), 671–685. 10.1093/nsr/nwaa023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. X. , Liang, Z. Q. , Yang, J. , Wang, D. , Wang, H. B. , Zhu, M. Y. , & Xu, E. Y. (2019). DAZL is a master translational regulator of murine spermatogenesis. National Science Review, 6(3), 455–468. 10.1093/nsr/nwy163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low, K. , Ashraf, T. , Canham, N. , Clayton‐Smith, J. , Deshpande, C. , Donaldson, A. , & Smithson, S. (2016). Clinical and genetic aspects of KBG syndrome. American Journal of Medical Genetics. Part A, 170(11), 2835–2846. 10.1002/ajmg.a.37842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi, Y. , Ui, N. , Nakajima, M. , Kawashima, K. , Maruyama, K. , Takizawa, T. , & Endo, H. (2001). p21Cip‐1/SDI‐1/WAF‐1 gene is involved in chondrogenic differentiation of ATDC5 cells in vitro. The Journal of Biological Chemistry, 276(35), 33249–33256. 10.1074/jbc.M010127200 [DOI] [PubMed] [Google Scholar]
- Neilsen, P. M. , Cheney, K. M. , Li, C. W. , Chen, J. D. , Cawrse, J. E. , Schulz, R. B. , & Callen, D. F. (2008a). Identification of ANKRD11 as a p53 coactivator. Journal of Cell Science, 121(Pt 21), 3541–3552. 10.1242/jcs.026351 [DOI] [PubMed] [Google Scholar]
- Nykamp, K. , Anderson, M. , Powers, M. , Garcia, J. , Herrera, B. , Ho, Y. Y. , & Topper, S. (2017). Sherloc: A comprehensive refinement of the ACMG‐AMP variant classification criteria. Genetics in Medicine, 19(10), 1105–1117. 10.1038/gim.2017.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ockeloen, C. W. , Willemsen, M. H. , Munnik, S. , Bon, B. W. , Leeuw, N. , Verrips, A. , & Kleefstra, T. (2015). Further delineation of the KBG syndrome phenotype caused by ANKRD11 aberrations. European Journal of Human Genetics, 23(9), 1176–1185. 10.1038/ejhg.2014.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropers, H. H. , & Wienker, T. (2015). Penetrance of pathogenic mutations in haploinsufficient genes for intellectual disability and related disorders. European Journal of Medical Genetics, 58(12), 715–718. 10.1016/j.ejmg.2015.10.007 [DOI] [PubMed] [Google Scholar]
- Skjei, K. L. , Martin, M. M. , & Slavotinek, A. M. (2007). KBG syndrome: Report of twins, neurological characteristics, and delineation of diagnostic criteria. American Journal of Medical Genetics. Part A, 143a(3), 292–300. 10.1002/ajmg.a.31597 [DOI] [PubMed] [Google Scholar]
- Smits, P. , Dy, P. , Mitra, S. , & Lefebvre, V. (2004). Sox5 and Sox6 are needed to develop and maintain source, columnar, and hypertrophic chondrocytes in the cartilage growth plate. The Journal of Cell Biology, 164(5), 747–758. 10.1083/jcb.200312045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R. , Zheng, C. , Jiang, W. , Xie, X. , Liao, R. , & Zhou, G. (2019). Neuropeptide W regulates proliferation and differentiation of ATDC5: Possible involvement of GPR7 activation, PKA and PKC‐dependent signalling cascades. Journal of Cellular and Molecular Medicine, 23(3), 2093–2102. 10.1111/jcmm.14118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen, M. H. , Fernandez, B. A. , Bacino, C. A. , Gerkes, E. , Brouwer, A. P. , Pfundt, R. , & Kleefstra, T. (2010). Identification of ANKRD11 and ZNF778 as candidate genes for autism and variable cognitive impairment in the novel 16q24.3 microdeletion syndrome. European Journal of Human Genetics, 18(4), 429–435. 10.1038/ejhg.2009.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X. , Li, C. , Gao, X. , Xia, K. , Guo, H. , Li, Y. , & Hu, R. (2018). Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Research, 28(1), 48–68. 10.1038/cr.2017.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti, S. , & Canalis, E. (2016). Notch signaling and the skeleton. Endocrine Reviews, 37(3), 223–253. 10.1210/er.2016-1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, A. , Li, C. W. , & Chen, J. D. (2007). Characterization of transcriptional regulatory domains of ankyrin repeat cofactor‐1. Biochemical and Biophysical Research Communications, 358(4), 1034–1040. 10.1016/j.bbrc.2007.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, H. , Zhou, W. , Yao, Z. , Wan, Y. , Cao, J. , Zhang, L. , & Guo, X. (2015). Foxp1/2/4 regulate endochondral ossification as a suppresser complex. Developmental Biology, 398(2), 242–254. 10.1016/j.ydbio.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All datasets generated and analyzed for this study are included in the article/Supplementary Material.