

Summary
The heterogeneous nature of lotic habitats plays an important role in the complex ecological and evolutionary processes that structure the microbial communities within them. Due to such complexity, our understanding of lotic microbial ecology still lacks conceptual frameworks for the ecological processes that shape these communities. We explored how bacterial community composition and underlying ecological assembly processes differ between lotic habitats by examining community composition and inferring community assembly processes across four major habitat types (free‐living, particle‐associated, biofilm on benthic stones and rocks, and sediment). This was conducted at 12 river sites from headwater streams to the main river in the River Thames, UK. Our results indicate that there are distinct differences in the bacterial communities between four major habitat types, with contrasting ecological processes shaping their community assembly processes. While the mobile free‐living and particle‐associated communities were consistently less diverse than the fixed sediment and biofilm communities, the latter two communities displayed higher homogeneity across the sampling sites. This indicates that the relative influence of deterministic environmental filtering is elevated in sediment and biofilm communities compared with free‐living and particle‐associated communities, where stochastic processes play a larger role.
Introduction
Lotic habitats (i.e. flowing water) are characterized by high spatiotemporal variability caused by natural factors such as variations in hydrology, temperature, water chemistry and resource availability, including nutrients and both particulate and dissolved organic matter. In addition, anthropogenic activity has extensively modified many river catchments, via changes in land use, channel modification, urban run‐off and sewage discharge (Gessner and Chauvet, 1994; Dodds et al., 2000; Biggs et al., 2005; Mulholland et al., 2008; Boyero et al., 2011; Tatariw et al., 2013; Atashgahi et al., 2015). The longitudinal, lateral and vertical movement of water provides the means for a diverse range of both abiotic and biotic matter to disperse, mix and interact, facilitating complex ecological and evolutionary processes that govern the communities that live within them. Unlike soil bacterial communities where pH and organic matter have frequently been identified as the dominant drivers (Fierer et al., 2007), our understanding of lotic microbial ecology still lacks conceptual frameworks for the ecological processes and drivers that shape these communities.
River catchments represent a continuum of interlinked habitats that change considerably in abiotic and biotic character longitudinally, along the river continuum from source to sea (Vannote et al., 2008), laterally, via exchanges with the floodplain, and vertically, from free‐flowing water through to the sediment–water interface and the saturated hyporheic zone (Stegen et al., 2016). Despite a recent increase in the number of studies of lotic bacteria, many of them have focussed only on individual habitat types and the communities within them, including free‐living (planktonic), particle‐associated planktonic, biofilm or sediment dwelling (Lindström and Bergström, 2005; Battin et al., 2007; Crump et al., 2007; Wang et al., 2013; Gibbons et al., 2014; Liu et al., 2018; Wisnoski and Lennon, 2020). Consequently, there remain fundamental unanswered questions as to how similar communities found in these contrasting habitats are, what taxa are responsible for any similarities, and what processes are involved in structuring these communities. A previous meta‐analysis comparing studies on lotic habitats indicated that bacterial communities exhibit remarkable consistency within habitat types (Zeglin, 2015). However, lotic habitats are also found along a gradient of contrasting physicochemical conditions, from headwaters to downstream rivers (Vannote et al., 2008). As such, it is still unknown whether such consistency is preserved across this river continuum, and whether the processes that govern their community composition are also consistent along this gradient.
Understanding the underlying processes driving their bacterial community assemblages and spatial distribution is becoming increasingly important as the relevance of lotic microbes is recognized in processes such as nutrient recycling (Tatariw et al., 2013), global flux of organic carbon into the atmosphere (Battin et al., 2008; Raymond et al., 2013), persistence and transport of pathogens (Marti et al., 2013), biogeochemical processes implicated with climate change (Butman and Raymond, 2011; Raymond et al., 2013; Stanley et al., 2016), pollution attenuation (Peterson et al., 2001; Mulholland et al., 2008; Valett et al., 2008) and as reservoirs and disseminators of antibiotic resistance (Marti et al., 2014).
Two major processes are known to influence how communities are assembled: neutrality‐based stochastic and niche‐based deterministic process (Leibold et al., 2004; Lindström and Langenheder, 2012). Neutrality‐based stochastic processes emphasize that communities are neutrally assembled by probabilistic or random dispersal, immigration, or ecological drift. In contrast, niche‐based deterministic processes emphasize that communities are structured by biotic filtering processes such as competition, facilitation and predation, and abiotic filtering where functional differences between individual species play a central role (Nemergut et al., 2013). It has been widely recognized that both stochastic and deterministic processes play a role in shaping community assemblages, but their relative contribution varies depending on ecological and environmental conditions (Chase, 2007; Chase and Myers, 2011; Myers et al., 2013; Tripathi et al., 2018). While recent studies have looked into the relative influence of these underlying processes in different lotic habitat types such biofilms and groundwater (Stegen et al., 2012; Wang et al., 2013; Veach et al., 2016), we addressed this question by examining a range of interacting lotic niches at locations across a river catchment.
In this study, we explore how bacterial community composition and underlying ecological processes differ between four major lotic habitat types (free‐living and particle‐associated plankton in the water column, biofilm on benthic stones and rocks, and sediment) and across a single river catchment (the River Thames in United Kingdom). Bacterial communities across 12 sites, from headwater streams to the main river, alongside a wide range of biotic and abiotic environmental conditions, were investigated with regards to their community composition and structure, as well as the relative contributions of ecological processes that influence the assemblages, ultimately setting a foundation for conceptual frameworks for future research on this topic.
Methods
Sampling sites and procedure
All samples were collected in August 2015 from 12 selected sites across the River Thames basin in southern United Kingdom (Fig. 1, Supplementary Table 7). These sites form part of the UK Centre for Ecology and Hydrology's Thames Initiative research platform (Bowes et al., 2018). The selected study sites include three sites along the main channel of the upper and middle River Thames, and nine tributaries representing a wide range of the lotic ecosystem in terms of distance from source, flow, land use, sewage input and cover much of the basin above the tidal limit.
Fig 1.
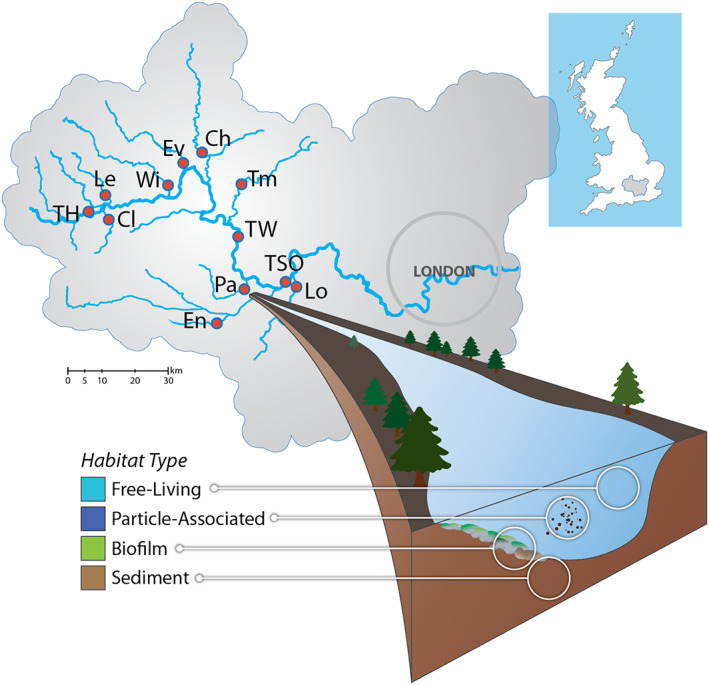
Sampling points and illustrating four habitats in lotic environment. A total of 12 water samples, 111 biofilm samples (between 8 and 10 per site), 60 sediment samples (five per site) were collected (the water samples were divided into free‐living and particle‐associated communities by filtration).
A single water sample was collected from each site using a sterile bucket lowered into the centre of the channel. Samples were transferred to a 1000 ml autoclaved polypropylene bottle and stored in a darkened icebox at 4°C until returned to the laboratory for filtering on the same day. Samples were filtered sequentially through 1.7 μm glass fibre GF/A filters (Whatman, Buckinghamshire, UK) to obtain particle‐associated microbes, and then through a 0.22 μm Durapore membrane filter (Merck Millipore, Watford, UK) to collect free‐living bacteria. Filters were stored at −20°C for DNA extraction. To examine biofilm communities, 8–10 submerged stones exposed to open‐canopy at depths between 10 and 40 cm were randomly collected from each site by hand using sterile gloves. The stones were placed in sterile Whirl‐Pak™ bags and stored in an icebox at 4°C until returned to the laboratory for processing. For each stone, approximately a 5 cm × 5 cm area exposed to the water column was thoroughly brushed with 40 ml of deionized water to remove the biofilm, using a disposable toothbrush. These had been pre‐sterilized in 10% sodium hypochlorite and rinsed in MQ water. After brushing, the liquid was poured into a 50 ml centrifuge tube and centrifuged at 5000g for 10 min. The supernatant was discarded, and the pellet was retained and stored at −20°C for DNA extraction. Five sediment sample cores were collected from each site using a Corning™ Falcon 50 ml Conical tube. The samples were put into an icebox in an upright position and were immediately placed in a −80°C freezer when returned to the laboratory. Frozen cores were then cut with a hacksaw (sterilized with 10% sodium hypochlorite and flame) to remove the top 2 cm of the sediment to ensure that biofilm exposed to water column was excluded. From the remaining top layer, 0.25 g of sediment was used for the subsequent DNA extraction.
DNA extraction and sequencing
DNA was extracted using the PowerSoil‐htp 96 Well Soil DNA Isolation Kit (Mobio Laboratories, Carlsbad, USA) using the manufacturer's protocol. Polymerase chain reaction (PCR) negative controls consisting of extraction and PCR blanks were also processed to remove likely potential contaminants in subsequent data processing (Salter et al., 2014). Approximately 20–30 ng of template DNA was amplified using Q5® high‐fidelity DNA polymerase (New England Biolabs, Hitchin, UK) each with a unique dual‐index barcode primer combination (Kozich et al., 2013). Individual PCR reactions employed 25 cycles of an initial 30 s, 98°C denaturation step, followed by annealing phase for 30 s at 53°C and final extension step lasting 90 s at 72°C. Primers were based upon the universal primer pair 341F (5′‐CCTACGGGAGGCAGCAG‐3′) (Muyzer et al., 1993) and 806R (5′‐GGACTACHVGGGTWTCTAAT‐3′) (Caporaso et al., 2011). An amplicon library consisting of approximately 550 bps amplicons spanning the V3 to V4 hypervariable regions of the 16S rRNA gene was sequenced at a concentration of 5.4 pM with a 0.6 pM addition of PhiX on an Illumina MiSeq platform (V3 chemistry, 600 cycles).
Data processing
Sequenced paired‐end reads were joined using PEAR v.0.9.10 (Zhang et al., 2014), quality filtered using FASTX tools v.0.0.14 (fastq_quality_filter, kept reads that have at least 80% of bases with a quality score of 30 or more) (Gordon and Hannon, 2010) and chimeras were identified and removed with VSEARCH_UCHIME_REF v.2.1.2 (Rognes et al., 2016) using Greengenes Release 13_8 (97%) (Larsen et al., 2006). Singletons were removed and the resulting sequences were clustered into operational taxonomic units (OTUs) with VSEARCH_CLUSTER_FAST v.2.1.2 (Rognes et al., 2016) at 97% sequence identity (Tindall et al., 2010). Representative sequences for the OTUs were taxonomically assigned with the RDP Classifier v.2.12 with the bootstrap threshold of 0.8 or greater (Wang et al., 2007) using the Greengenes Release 13_8 (FULL) (Larsen et al., 2006) as a reference. Unless stated otherwise, default parameters were used for the steps listed. Where multiple replicate samples were collected from a site, ≥75% prevalence filter was used to select OTUs that best represent the site and their abundances were then averaged into a single sample. The OTU table was subsequently rarefied down to 24 007 sequences per sample to address variability in sampling depth (Weiss et al., 2015) and to ensure that the number of reads used for each of the 48 samples is the same for downstream analysis. Low‐abundance OTUs below 0.01% abundance (Bokulich et al., 2013) and those that belong to ‘chloroplasts’ were removed. The representative sequences for the OTUs were aligned using SSU‐ALIGN v.0.1 (Nawrocki, 2009) and FastTree v.2.1.9 (Price et al., 2009) with a GTR + G substitution model used on the resulting alignment to generate a phylogenetic tree to infer phylogenetic relationships among the OTUs.
Statistical analysis
To assess differences in community compositions, the Bray–Curtis dissimilarities were determined which quantify compositional differences based on relative abundances of OTUs. One‐way ANOVA and Tukey HSD (Honestly Significant Difference) tests, Shannon index, Pielou's evenness index, Whittaker's beta diversity, distances of group members to the group centroid, non‐metric multidimensional scaling (Bray–Curtis dissimilarity with 999 randomisations) and permutational multivariate analysis of variance test (PERMANOVA) were performed using the package Vegan v.2.4‐1 (Oksanen et al., 2007). Values were Bonferroni‐corrected where applicable.
SPEC‐OCCU plots
The 500 most abundant OTUs were selected from the OTU table for each habitat, and from the table, specificity and occupancy (or otherwise known as fidelity) were calculated as per Dufrene and Legendre (1997). Specificity is defined as the mean abundance of species (S) in the samples of habitat (H); and occupancy is defined as the relative frequency of occurrence of S in the samples of H (See Eq. 1).
| (1) |
NindividualS,H is the mean number of individual OTU S across all samples of habitat H, while NindividualS is the sum of the mean number of individual S over all habitats; NsitesS,H is the number of samples in H where S is present, while NsitesH is the total number of samples in H (Dufrene and Legendre, 1997). These two metrics were subsequently used as the axes in SPEC‐OCCU plots.
Phylogenetic turnover
We implemented a previously developed null modelling approach to infer community assembly processes (Stegen et al., 2013; Wang et al., 2013; Dini‐Andreote et al., 2015). We used Phylocom v.4.2 (Webb et al., 2008) to compare phylogenetic turnover between the habitats by calculating the β‐Nearest Taxon Index (βNTI) from the phylogenetic tree. For each pair of communities within each habitat, βNTI was generated using the independent swap null model (Gotelli and Entsminger, 2003) to generate null communities (999 permutations). The βNTI is a standardized effect size measure that expresses the difference between observed between‐community mean‐nearest‐taxon‐distance and the mean of null distribution in units of standard deviation (Stegen et al., 2012; Stegen et al., 2015). A mean βNTI value less than −2 or greater +2 across all pairwise comparisons suggests deterministic processes while a value between −2 and + 2 is considered to signify the influence of stochastic assembly (Stegen et al., 2012; Zhou and Ning, 2017).
Results
Sequences and OTUs
The 195 samples from the 12 sites across the Thames basin generated a total of 22 292 615 paired‐end raw sequences, which were reduced to 20 252 671 post‐processed sequences with an average of 77 895 sequences per sample. After the rarefaction, 4,608,494 sequences remained that clustered into 15 962 OTUs (3564, 5330, 8606 and 10 124 OTUs for free‐living, particle‐associated, biofilm and sediment bacterial communities respectively).
Bacterial community diversity and composition
An NMDS ordination plot highlighted a distinct segregation between communities from all four habitat types (Fig. 2A, PERMANOVA, p < 0.005). There was greater segregation between the planktonic (free‐living and particle‐associated) and benthic (biofilm and sediment) communities (see Supplementary Table S1). Diversity (Fig. 2B) and evenness (Fig. 2C) also varied along these habitats, where both indices increased from free‐living, particle‐associated, through biofilm to sediment (ANOVA, Shannon: DF = 3, F (Larsen et al., 2006; Tatariw et al., 2013) = 127.03, p < 0.0001; Evenness: DF = 3, F (Larsen et al., 2006; Tatariw et al., 2013) = 62.167, p < 0.0001; A post hoc Tukey's HSD p < 0.01—see Supplementary Tables S2a & S2b). There was a significant difference in beta diversity between the planktonic and benthic communities, where the benthic communities were much more homogenous than the planktonic communities (Fig. 2D).
Fig 2.
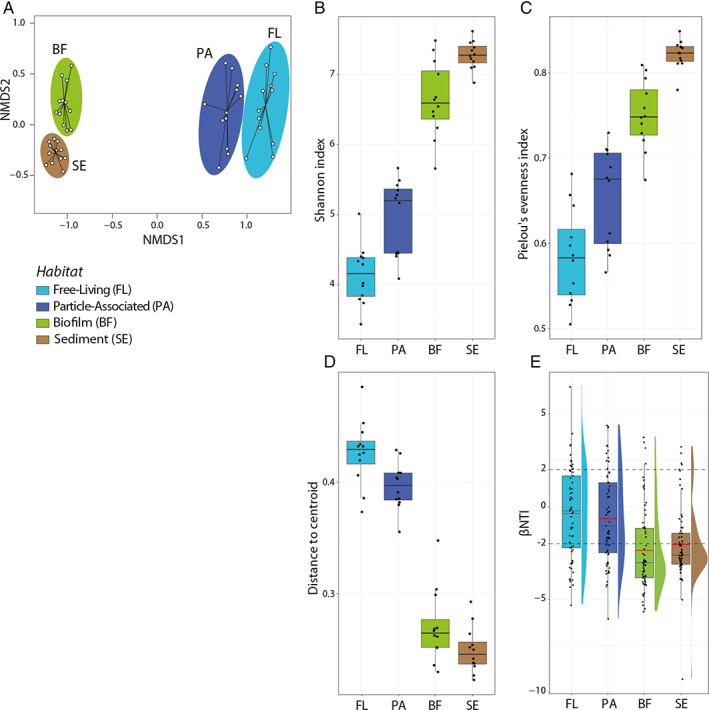
A. Non‐metric multidimensional scaling (NMDS) resulting from Bray–Curtis dissimilarity matrices exhibited distinct differences in community composition between all four habitat types (PERMANOVA between all pairs: p‐value <0.005).
B. Increase in Shannon diversity index from FL, PA and through BF and SE.
C. Increase in Pielou's evenness indices again from FL to SE.
D. Distance to centroid in multivariate homogeneity of group variance analysis for lotic bacterial communities in four habitat types (A post hoc Tukey's HSD: marginal significance between FL and PA, p = 0.038; between BF and SE, p = 0.225, all other pairs were significant, p < 0.001; See Supplementary Table S2c).
E. β‐Nearest Taxon Index (βNTI) for all pairs of communities within each habitat. A mean βNTI (red lines: −0.3819, −0.6356, −2.353, −2.034 for free‐living, particle‐associated, biofilm and sediment habitats respectively) across all pairwise comparisons that is greater than +2 or less than −2 (dotted lines) as are seen in benthic communities (biofilm and sediment) suggests the influence of deterministic processes in microbial assembly.
Bacterial community composition and structure at the phylum level
At the phylum level, the most abundant and common taxa across all habitat types were Bacteroidetes, Proteobacteria, Actinobacteria and Verrucomicrobia (Fig. 3A). The proportion of Bacteroidetes was significantly higher (a post hoc Tukey's HSD p < 0.000—see Supplementary Table S3a) in planktonic habitats (free‐living: 38.9%, particle‐associated: 47.5%) than the benthic habitats (biofilm: 14.1%, sediment: 13.3%), and Actinobacteria were relatively more abundant in free‐living. Although Verrucomicrobia were present in similar relative abundance in all habitats, a greater variance in the planktonic communities was observed. Cyanobacteria were significantly more abundant in biofilm samples (a post hoc Tukey's HSD p < 0.000—see Supplementary Table S3a). Notably, the proportion of Acidobacteria and Chloroflexi showed an increasing trend from free‐living, particle‐associated, biofilm to sediment. OD1 was statistically more abundant in the free‐living habitat (a post hoc Tukey's HSD p < 0.000—see Supplementary Table S3b). While Firmicutes and WS3 were not abundant in all communities they showed a clear increasing trend in relative abundance from free‐living, particle‐associated, biofilm to sediment (Fig. 3B).
Fig 3.
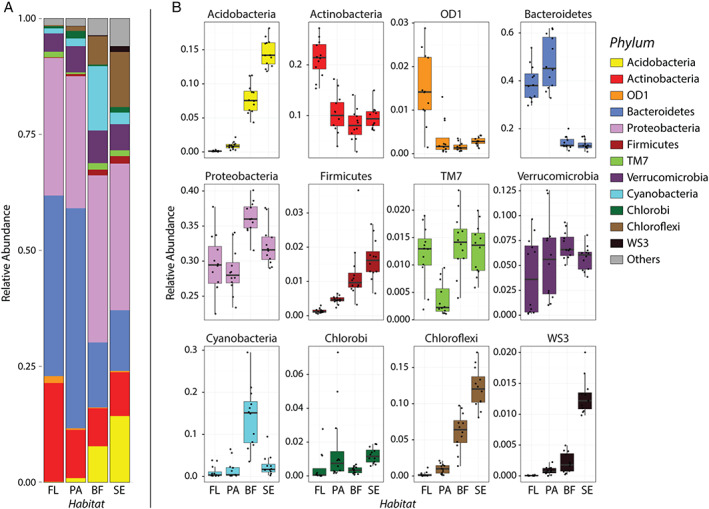
(A) Relative abundances of bacterial phyla in the four lotic habitat types.
B. Relative abundances of 12 most abundant bacterial phyla.
Distribution and overlap of the abundant OTUs across lotic locations
To inspect how OTUs from each habitat type are spread across sites and also how specific they are to their habitat, specificity and occupancy were calculated for each OTU (Eq. 1) which were then projected onto a plot (SPEC‐OCCU plot, Fig. 4A). As indicated by the spread of OTUs across the x‐axis (occupancy), OTUs from free‐living communities displayed highly varied occupancy (i.e. fewer were present in all sites), while the majority of OTUs from sediment communities exhibit remarkably homogenous occupancy at sites across the river catchment. To find specialist species attributable to each habitat type, we selected OTUs with specificity and occupancy greater or equal to 0.7 (dotted boxes in Fig 3A and B), i.e. they are specific to a habitat and common in their habitat in most sites. The number of these specialist OTUs differed significantly between the habitats with an increasing trend in terms of richness from free‐living (51 OTUs represent), particle‐associated (86 OTUs), biofilm (275 OTUs) to sediment (611 OTUs) representing 5.4%, 5.4%, 8.5% and 7.4% of the total sequences respectively. Proteobacteria and Bacteroidetes were found in all four habitat specialist groups while notable free‐living specialists included OTUs from OD1 and TM7. Particle‐associated specialists were mostly Bacteroidetes and Proteobacteria, with no Actinobacteria found to be both specific and common in this habitat type. Notable biofilm specialists were Cyanobacteria, and sediment specialists included OTUs from Acidobacteria, Chloroflexi and WS3.
Fig 4.
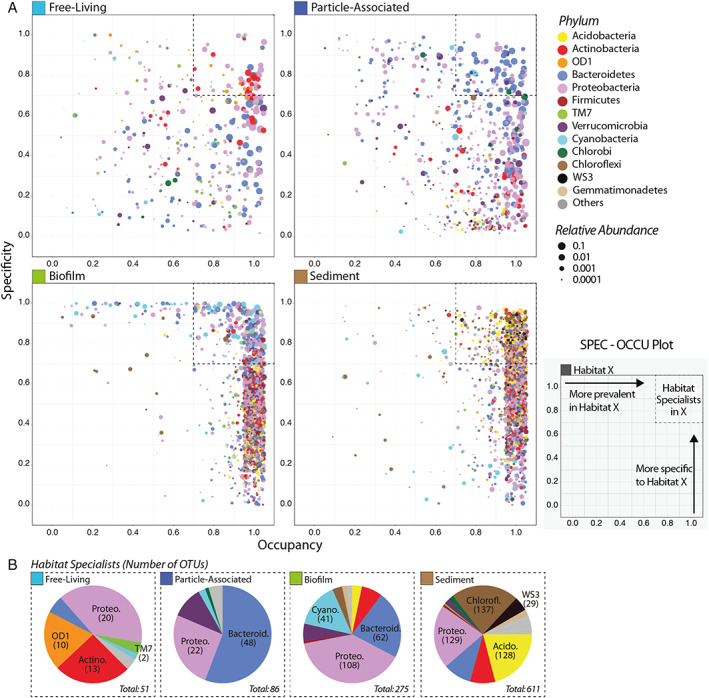
(A) The SPEC‐OCCU plots show 500 most abundant OTUs in each habitat type; the x‐axis represents occupancy, i.e. how well an OTU is distributed across all 12 sites; and the y‐axis represents specificity, i.e. whether they are also found in other habitats.
B. Pie charts showing the number of OTUs representing specialists in each habitat (See Supplementary Table S6 for the list of these specialists).
The number and taxonomic composition of the OTUs shared between different sets of habitats were also compared (Fig. S1). Overall, 5735 OTUs were found in a single habitat (3045 OTUs in sediment, 1419 OTUs in biofilm, 646 OTUs in free‐living and 625 OTUs in particle‐associated) while 1279 OTUs occurred in all four habitats. The habitats with the highest number of shared OTUs were biofilm and sediment (3103 OTUs). Sediment, free‐living and particle‐associated communities had the least shared OTUs (145 OTUs), followed by sediment and free‐living (150 OTUs), highlighting the distinctness of these habitats.
The influence of deterministic or stochastic processes
To infer the relative influence of deterministic and stochastic processes in each habitat type, we examined βNTI. The mean βNTI values were −0.3819, −0.6356, −2.353 and −2.034 for free‐living, particle‐associated, biofilm and sediment habitats respectively (Fig. 2E). This result suggests that the relative influence of deterministic processes in community dynamics are much higher in benthic habitat types (sediment in particular) compared with planktonic habitats, where stochastic processes play a more significant role.
Discussion
Recently there has been increased interest in the processes that structure lotic microbial communities (Read et al., 2015; Ruiz‐González et al., 2015; Savio et al., 2015; Niño‐García et al., 2016; Henson et al., 2018), partly due to the recognition that streams and rivers act as ‘active pipes’ that process nutrients and organic matter rather than passively channelling them from land to sea (Cole et al., 2007). As a result, lotic environments are now seen as sites of intense biological activity that play a major role in global biogeochemical cycles (Raymond et al., 2013; Tranvik et al., 2018). As microbes are primarily responsible for these cycles, understanding the ecological controls over community composition is a necessary first step to developing a more complete understanding of both the ecology and the biogeochemistry of flowing water. However, this is complicated by the fact that lotic environments are made up of a number of distinct habitats that occur across a gradient of physicochemical conditions from headwaters to downstream, and our knowledge of how these communities are structured relative to one another and how much ecological exchange occurs between these habitats is lacking.
Community composition across habitats
Our results demonstrate that there is a distinct segregation in bacterial community composition between habitat types (free‐living, particle‐associated, biofilm and sediment), and this pattern is consistent across the catchment (from tributaries to the main river channel or the River Thames) despite considerable variation in water physicochemical water quality conditions between sites (Supplementary Table S1). Habitat types were distinct from one another, not only in overall community composition but also in diversity, evenness and variance in community composition within a habitat. These results suggest that, despite the extensive physical and chemical environmental gradients between sites across the river catchment, each habitat type provides sufficiently unique conditions to facilitate communities to be assembled that are consistent in composition and structure according to their habitat type.
Our observations of the community composition of each habitat are largely in agreement with previous studies focussing on individual lotic habitats. For example, across all sites, OTUs belonging to the phyla Actinobacteria and OD1 (also referred to as Parcubacteria) were significantly enriched in the free‐living or planktonic community (Fig. 2B), agreeing with previous findings examining lotic bacterioplankton (Peura et al., 2012; Jackson et al., 2014; Read et al., 2015; Savio et al., 2015), where these taxa, along with the Proteobacteria are frequently characteristic of freshwater free‐living planktonic communities. Furthermore, it has been suggested that members of the OD1 exhibit either a free‐living or symbiotic lifestyle due to their reduced genome size and lack of biosynthetic capabilities (Nelson and Stegen, 2015). If symbiosis is the predominant lifestyle in lotic OD1, the symbiotic relationship is likely to be with small commensal/symbiotic/parasitic organisms, present as free‐living plankton and able to pass through a 1.7 μm filter (the ‘free‐living’ fraction in this study). Future studies would need correlative techniques such as visualization with FISH or SEM, or flow sorting and genomic investigation to give a further insight into these interactions. The second characteristic group of the free‐living fraction, the Actinobacteria, have frequently been identified as a dominant member of free‐living freshwater communities, both in lakes (Allgaier and Grossart, 2006; Neuenschwander et al., 2018) and in the lower reaches of rivers (Read et al., 2015; Savio et al., 2015; Henson et al., 2018). It has been suggested that they are well suited to a free‐living planktonic lifestyle due to their small size, metabolic versatility and the presence of rhodopsin pigments that can be used to harvest sunlight for energetic and photosensory functions (Newton et al., 2007; Sharma et al., 2009; Ghai et al., 2014).
Suspended particles play a crucial role in freshwater environments as microhabitats for cell attachment and as sources of labile carbon (Rieck et al., 2015; Cordero et al., 2016). Bacterial communities found on particles have previously been shown to have a high degree of specialization (Crump et al., 1999; Rösel et al., 2012), and whilst the particles are planktonic, the attached lifestyle of particle‐associated microbes allows for higher levels of cell–cell interactions between cells (Amalfitano et al., 2017) and rates of activity (Ghiglione et al., 2009). Our results confirm that communities on suspended particles were distinct from the planktonic, biofilm and sediment communities (Fig. 3A) in community composition, and also in diversity, evenness and variance. In contrast to Niederdorfer et al. (2016) who found that experimentally manipulated communities associated with suspended aggregates had a higher richness and Shannon's Index when compared with biofilms, we found that the mobile free‐living and particle‐associated communities were consistently less diverse than the fixed sediment and biofilm communities. The reasons for this are not clear, although it may be due to the different lotic systems studied, or reflect the difficulties in using experiments to represent natural systems (Carpenter, 1996). Particle‐associated communities in the River Thames were characterized by members of the phylum Bacteroidetes, which have previously been highlighted as being particle‐associated in aquatic environments (Fernández‐Gómez et al., 2013; Milici et al., 2017), and have been shown to play an important role in the degradation of complex biopolymers (Kirchman, 2002).
Biofilm communities in the Thames catchment were enriched in Acidobacteria, Proteobacteria, Chloroflexi and Cyanobacteria when compared with the planktonic habitats, which is consistent with previous studies on the major constituents of freshwater biofilms (Battin et al., 2016). Chloroflexi have previously been identified as playing a critical role in carbon cycling, and vary in metabolic lifestyle including photoautotrophs, fermentative species, organohalide respiring species and aerobic thermophiles (Hug et al., 2013). Biofilms are considered to be a major component of lotic food webs, as key sites of enzymatic activity that play a role in organic matter cycling, ecosystem respiration and primary production (Battin et al., 2016). Their position at the interface between the water and sediment means that they have the potential to mediate both the exchange of chemicals as well as the exchange of cells between these habitats. Biofilm communities were significantly different in composition to those found in the free‐living and particle‐associated fractions, as well as containing a more diverse community that was more even in terms of species abundance and homogeneous across sites.
Sediment communities were characterized by OTUs from the phyla Acidobacteria, Chloroflexi and Proteobacteria. Acidobacteria have previously been shown to be particularly abundant in soils and sediments (López‐García et al., 2003; Janssen, 2006; Barns et al., 2007) and recent genomic and metagenomics studies indicate that their successful adaptation to the sediment environment could be due to their ability to produce high‐affinity transporters, exploit a wide variety of carbohydrates as substrate and produce secondary metabolites as well as being resistant to antibiotics (Ward et al., 2009; Kielak et al., 2016). It is worth noting that the sediment community features a unique phylum WS3, also known as the candidate phylum ‘Latescibacteria’. This is consistent with previous studies on freshwater sediment (Morrison et al., 2017; Guan et al., 2018). Another study derived from single‐cell amplified genomes of WS3 indicates their heterotrophic and strictly fermentative lifestyle and suggested that it may play a crucial role in the turnover of algal detritus (Farag et al., 2017).
One group, the Proteobacteria, was present in a high abundance across all lotic habitats (Fig. 3) and habitat specialists were found from this group in all habitats (Fig. 4). The ubiquity of Proteobacteria in freshwater has previously been identified by Graham et al. (2017), in a study of free‐living and attached microbes in the hyporheic zone of the Columbia River in the United States. Proteobacteria contain members with diverse energy requirements, and the ability to assimilate a range of substrates such as sulfate, nitrate, and inorganic C, as well as diverse organic C compounds (Gupta, 2000; Kersters et al., 2006). This suggests that they may have members who can thrive in the varying conditions found in both planktonic and sessile lifestyles.
OTU‐level habitat specificity and overlap
Since lotic ecosystems are characterized by constantly mixing water, the opportunities for the exchange of taxa between habitats are high. Due to this unique characteristic of lotic environments, it has been speculated that many aquatic bacteria possess a complex lifestyle with the ability to alternate between different stages to take advantage of available nutrients or other environmental factors such as light and symbionts (Grossart, 2010). We found many OTUs with low specificity to a single habitat type; for example, 1279 OTUs were found across all habitats, and the highest number of OTUs (3103) were found exclusively in both biofilm and sediment (Supplementary Fig. 1). However, there are also OTUs that are both highly specific to one habitat type and common across all sites within the catchment (dotted box in Fig. 4A). The sediment had the highest number of habitat specialist OTUs (611 OTUs), while biofilm, particle‐associated and free‐living habitat types had 275, 86 and 51 OTUs respectively.
Within habitat beta‐diversity
Planktonic communities, both free‐living and particle‐associated, are generally expected to be heterogeneous in both space and time, as continuous inflow of water from a wide range of terrestrial sources allows a diverse pool of source communities to be assembled (Ruiz‐González et al., 2015). Despite this, a number of recent catchment‐scale studies have highlighted that bacterial communities can exhibit a predictable gradient in community composition from the headwaters to the downstream river, driven by the relative importance of terrestrial inputs and species sorting along the river continuum (Read et al., 2015; Savio et al., 2015; Niño‐García et al., 2016; Henson et al., 2018). Although this study was not designed to capture the full headwater to downstream gradient, sites were chosen to encompass a wide range of physicochemical parameters and hydrology types across the catchment (Supplementary Table S1). We observed that although the free‐living community had the lowest diversity and evenness it also had the highest variance across the sites (Figs 2B–D), possibly reflecting the relative importance of immigration or ‘mass effects’ (Leibold et al., 2004) from the surrounding catchment in structuring these communities (Ruiz‐González et al., 2015). Due to the importance of mass effects in structuring lotic bacterioplankton it is not clear which members of the community are simply ‘passing through’ and play no role in the functioning of the ecosystem (other than as a potential source of nutrients) and which members are proliferating and adapted to the freshwater niche.
Compared with the planktonic habitats, both the biofilm and sediment communities show significantly higher homogeneity (Fig 2A and D) across the sampling sites. This is apparent in the SPEC‐OCCU plots (Fig. 4A), where the majority of the OTUs have high occupancy, meaning that most of them are also found in all other sites across the catchment. These results indicate that the sessile (biofilm and sediment) environments are highly selective for a large and stable core community, despite differences in the overlaying water conditions (Supplementary Table S4). It is not immediately obvious how the biofilm and sediment communities are able to display such spatial homogeneity. We did not investigate the physical and chemical characteristics of the sediment or biofilms themselves, so further work is needed to find out if these are responsible for buffering the varying nutrient levels in the overlaying water, and thus assembling similar communities, or whether other factors, possibly related to an attached lifestyle, are responsible.
Community assembly processes
Aquatic systems, and the communities they contain, are dynamic, and the movement of microbes along river networks, as well as between habitats within the river channel, represents a challenge to developing an understanding of the processes responsible for community assembly in these systems. The deterministic theory asserts that biotic and abiotic filtering play a major role in structuring species composition, whereas stochastic theory emphasizes that communities are neutrally assembled by probabilistic dispersal, ecological drift or historical inertia and neglects the functional differences between species (Hubbell, 2001). Both indices indicate deviations from null model expectations across temporal and spatial environmental gradients and allowed us to infer the relative influences of stochastic and deterministic processes between habitats and samples. The resulting βNTI distributions and their means (Fig. 2E) suggest that the relative influence of deterministic environmental filtering is elevated in sediment and biofilm communities compared with free‐living and particle‐associated communities, where stochastic processes play a larger role. We speculate that deterministic processes such as niche filtering are responsible for creating consistent communities in the sediment and biofilm habitats (Fig. 2A and D). This is supported by the higher degree of habitat specialization observed among the OTUs in sediment and biofilm communities, indicating that the unique properties of these environments (and potentially lower rates of mass effects) are responsible for structuring these communities. Previous studies have indicated that biofilms have structural resilience and possess resistance to environmental and chemical influences (Mah and O'Toole, 2001; Donlan and Costerton, 2002; Hall‐Stoodley et al., 2004). Sediment has also been demonstrated to contain resilient structures kept intact by internal adhesives such as biofilms and supracellular ropes (Garcia‐Pichel and Wojciechowski, 2009; Vignaga et al., 2013; Battin et al., 2016), potentially contributing to a high degree of environmental filtering in this environment.
Both the planktonic habitats had a mean βNTI between −2 and +2, indicating that the stochastic process plays an important role in structuring these environments. This is perhaps unsurprising, given that surface water originates from a variety of sources, including groundwater, surface runoff, sewage inputs and connections to manmade water sources such as canals and reservoirs. A study in a relatively un‐impacted catchment identified a clear trend in the influence of terrestrial inputs (Ruiz‐González et al., 2015), thus highlighting the important role of the surrounding terrestrial landscape in shaping freshwater ecology (Reid et al., 2018).
Conclusions
Our results indicate that there are distinct differences in community structure and composition between the four major lotic habitat types, with substantial contrast in ecological processes shaping their community assemblages. The high dispersal and invasion ability of bacteria in a uniform environment can lead to the ubiquity of communities across space and time (Van der Gucht et al., 2007; Urban and De Meester, 2009). We speculate that the constant supply of migrating individuals in the water column, alongside the sufficiently stable habitats of sediment and biofilm environments, results in the remarkably heterogeneous microbial communities found in each habitat type.
Author Contributions
HSG and DSR conceived and designed the study. HSG, HLM, MJBo and DSR performed the fieldwork. HSG performed the molecular work in the laboratory. AEO performed DNA quantification and sequencing. HSG performed the sequence analysis, statistical analyses, interpreted the statistical findings and wrote the manuscript. MJBa, MCA and DSR contributed to the preparation of the manuscript.
Supporting information
Supplementary Figure 1 Upset chart showing overlap in all OTUs identified in each habitat type. Numbers of OTUs shared between different sets of habitats are indicated in the top bar chart and the specific habitats in each set are indicated with solid points below the bar chart. Total number of OTUs for each habitat is indicated on the left. Figure generated using Upset R package (Lex et al., 2014). Do note that the bar chart is not showing relative abundances.
Supplementary Table 1 PERMANOVA analyses produces a p‐value for significance and the R2 value, which indicates the amount of variation attributed to a specific treatment within a model. The R2 values indicate greater differences between the planktonic communities (Free‐Living and Particle‐Associated) and benthic communities (Biofilm and Sediment).
Supplementary Table 2 Tukey's HSD on diversity indices (Tukey multiple comparisons of means, 95% family‐wise confidence level)
Supplementary Table 3 Tukey's HSD on the differences in relative abundances of selected phyla between four different habitats (Tukey multiple comparisons of means, 95% family‐wise confidence level)
Supplementary Table 4 Supporting information
Supplementary Table 5 Supporting information
Supplementary Table 6 Supporting information
Supplementary Table 7 Supporting information
Acknowledgements
DSR was supported by the Natural Environment Research Council award number NE/R016429/1 as part of the UK‐SCAPE programme delivering National Capability. The Thames Initiative is supported by the Natural Environment Research Council via UK‐SCAPE (NE/R016429/1) and Achieving Sustainable Agricultural Systems (ASSIST) (NE/N018125/1). We gratefully acknowledge David Nicholls and Linda Armstrong for generating the Thames Initiative nutrient data.
Contributor Information
Hyun S. Gweon, Email: h.s.gweon@reading.ac.uk.
Daniel S. Read, Email: dasr@ceh.ac.uk.
Data Availability Statement
The raw sequence data reported in this study have been deposited in the European Nucleotide Archive under the accession number PRJEB29699. The relevant barcode information for each sample is shown in Supplementary Table S5.
References
- Allgaier, M. , and Grossart, H.P. (2006) Diversity and seasonal dynamics of Actinobacteria populations in four lakes in northeastern Germany. Appl Environ Microbiol 72: 3489–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amalfitano, S. , Corno, G. , Eckert, E. , Fazi, S. , Ninio, S. , Callieri, C. , et al (2017) Tracing particulate matter and associated microorganisms in freshwaters. Hydrobiologia 800: 145–154. [Google Scholar]
- Atashgahi, S. , Aydin, R. , Dimitrov, M.R. , Sipkema, D. , Hamonts, K. , Lahti, L. , et al (2015) Impact of a wastewater treatment plant on microbial community composition and function in a hyporheic zone of a eutrophic river. Sci Rep 5: 17284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barns, S.M. , Cain, E.C. , Sommerville, L. , and Kuske, C.R. (2007) Acidobacteria phylum sequences in uranium‐contaminated subsurface sediments greatly expand the known diversity within the phylum. Appl Environ Microbiol 73: 3113–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battin, T.J. , Besemer, K. , Bengtsson, M.M. , Romani, A.M. , and Packmann, A.I. (2016) The ecology and biogeochemistry of stream biofilms. Nat Rev Microbiol 14: 251–263. [DOI] [PubMed] [Google Scholar]
- Battin, T.J. , Kaplan, L.A. , Findlay, S. , Hopkinson, C.S. , Marti, E. , Packman, A.I. , et al (2008) Biophysical controls on organic carbon fluxes in fluvial networks. Nat Geosci 1: 95–100. [Google Scholar]
- Battin, T.J. , Sloan, W.T. , Kjelleberg, S. , Daims, H. , Head, I.M. , Curtis, T.P. , et al (2007) Microbial landscapes: new paths to biofilm research. Nat Rev Microbiol 5: 76–81. [DOI] [PubMed] [Google Scholar]
- Biggs, B.J.F. , Nikora, V.I. , and Snelder, T.H. (2005) Linking scales of flow variability to lotic ecosystem structure and function. River Res Appl 21: 283–298. [Google Scholar]
- Bokulich, N.A. , Subramanian, S. , Faith, J.J. , Gevers, D. , Gordon, I. , Knight, R. , et al (2013) Quality‐filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10: 57–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowes, M.J. , Armstrong, L.K. , Harman, S.A. , Wickham, H.D. , Nicholls, D.J.E. , Scarlett, P.M. , et al (2018) Weekly water quality monitoring data for the river Thames (UK) and its major tributaries (2009‐2013): the Thames initiative research platform. Earth Syst Sci Data 10: 1637–1653. [Google Scholar]
- Boyero, L. , Pearson, R.G. , Gessner, M.O. , Barmuta, L.A. , Ferreira, V. , Graça, M.A.S. , et al (2011) A global experiment suggests climate warming will not accelerate litter decomposition in streams but might reduce carbon sequestration. Ecol. Lett. 14: 289–294. [DOI] [PubMed] [Google Scholar]
- Butman, D. , and Raymond, P.A. (2011) Significant efflux of carbon dioxide from streams and rivers in the United States. Nat Geosci 4: 839–842. [Google Scholar]
- Caporaso, J.G. , Lauber, C.L. , Walters, W.A. , Berg‐Lyons, D. , Lozupone, C.A. , Turnbaugh, P.J. , et al (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108: 4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter, S.R. (1996) Microcosm experiments have limited relevance for community and ecosystem ecology. Ecology 77: 677–680. [Google Scholar]
- Chase, J.M. (2007) Drought mediates the importance of stochastic community assembly. Proc Natl Acad Sci U S A 104: 17430–17434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase, J.M. , and Myers, J.A. (2011) Disentangling the importance of ecological niches from stochastic processes across scales. Philos Trans R Soc B Biol Sci 366: 2351–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, J.J. , Prairie, Y.T. , Caraco, N.F. , McDowell, W.H. , Tranvik, L.J. , Striegl, R.G. , et al (2007) Plumbing the global carbon cycle: integrating inland waters into the terrestrial carbon budget. Ecosystems 10: 171–184. [Google Scholar]
- Cordero, O.X. , Stocker, R. , Otto, X.C. , and Stocker, R. (2016) A particularly useful system to study the ecology of microbes. Environ Microbiol Rep 7: 472–485. 10.1111/j.1462-2920.2005.00803.x/abstract/npapers3://publication/uuid/9A507F51-5388-4350-998F-B3BC7A43DDC0. [DOI] [PubMed] [Google Scholar]
- Crump, B.C. , Adams, H.E. , Hobbie, J.E. , and Kling, G.W. (2007) Biogeography of bacterioplankton in lakes and streams of an arctic tundra catchment. Ecology 88: 1365–1378. [DOI] [PubMed] [Google Scholar]
- Crump, B.C. , Armbrust, E.V. , and Baross, J.A. (1999) Phylogenetic analysis of particle‐attached and free‐living bacterial communities in the Columbia River, its estuary, and the adjacent coastal ocean. Appl Environ Microbiol 65: 3192–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufrene, M. , Legendre, P. (1997). Species Assemblages and Indicator Species: The Need for a Flexible Asymmetrical Approach. Ecological Monographs, 67: 345. [Google Scholar]
- Dini‐Andreote, F. , Stegen, J.C. , van Elsas, J.D. , and Salles, J.F. (2015) Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc Natl Acad Sci U S A 112: 1326–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds, W.K. , Evans‐White, M.A. , Gerlanc, N.M. , Gray, L. , Gudder, D.A. , Kemp, M.J. , et al (2000) Quantification of the nitrogen cycle in a prairie stream. Ecosystems 3: 574–589. [Google Scholar]
- Donlan, R.M. , and Costerton, J.W. (2002) Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev 15: 167–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farag, I.F. , Youssef, N.H. , and Elshahed, M.S. (2017). Global Distribution Patterns and Pangenomic Diversity of the Candidate Phylum “Latescibacteria” (WS3). Applied and Environmental Microbiology, 83(10), e00521‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Gómez, B. , Richter, M. , Schüler, M. , Pinhassi, J. , Acinas, S.G. , González, J.M. , and Pedrós‐Alió, C. (2013) Ecology of marine bacteroidetes: a comparative genomics approach. ISME J 7: 1026–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierer, N. , Bradford, M.A. , and Jackson, R.B. (2007) Toward an ecological classification of soil bacteria. Ecology 88: 1354–1364. [DOI] [PubMed] [Google Scholar]
- Garcia‐Pichel, F. , and Wojciechowski, M.F. (2009) The evolution of a capacity to build supra‐cellular ropes enabled filamentous cyanobacteria to colonize highly erodible substrates. PLoS One 4: e7801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessner, M.O. , and Chauvet, E. (1994) Importance of stream microfungi in controlling breakdown rates of leaf litter. Ecology 75: 1807–1817. [Google Scholar]
- Ghai, R. , Mizuno, C.M. , Picazo, A. , Camacho, A. , and Rodriguez‐Valera, F. (2014) Key roles for freshwater Actinobacteria revealed by deep metagenomic sequencing. Mol Ecol 23: 6073–6090. [DOI] [PubMed] [Google Scholar]
- Ghiglione, J.F. , Conan, P. , and Pujo‐Pay, M. (2009) Diversity of total and active free‐living vs. particle‐attached bacteria in the euphotic zone of the NW Mediterranean Sea. FEMS Microbiol Lett 299: 9–21. [DOI] [PubMed] [Google Scholar]
- Gibbons, S.M. , Jones, E. , Bearquiver, A. , Blackwolf, F. , Roundstone, W. , Scott, N. , et al (2014) Human and environmental impacts on river sediment microbial communities. PLoS One 9: e97435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon A, Hannon GJ. Fastx‐toolkit. FASTQ/A short‐reads pre‐processing tools. Unpublished, http://hannonlab.cshl.edu/fastx_toolkit/.
- Gotelli Nicholas J., Entsminger Gary L. (2003). Swap algorithms in null model analysis. Ecology 84(2): 532–535. [DOI] [PubMed] [Google Scholar]
- Graham, E.B. , Crump, A.R. , Resch, C.T. , Fansler, S. , Arntzen, E. , Kennedy, D.W. , et al (2017) Deterministic influences exceed dispersal effects on hydrologically‐connected microbiomes. Environ Microbiol 19: 1552–1567. [DOI] [PubMed] [Google Scholar]
- Grossart, H.P. (2010) Ecological consequences of bacterioplankton lifestyles: changes in concepts are needed. Environ Microbiol Rep 2: 706–714. [DOI] [PubMed] [Google Scholar]
- Guan, Y. , Jia, J. , Wu, L. , Xue, X. , Zhang, G. , and Wang, Z. (2018) Analysis of bacterial community characteristics, abundance of antibiotics and antibiotic resistance genes along a pollution gradient of Ba River in Xi'an, China. Front Microbiol 9: 3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, R.S. (2000) The phylogeny of proteobacteria: relationships to other eubacterial phyla and eukaryotes. FEMS Microbiol Rev 24: 367–402. [DOI] [PubMed] [Google Scholar]
- Hall‐Stoodley, L. , Costerton, J.W. , and Stoodley, P. (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2: 95–108. [DOI] [PubMed] [Google Scholar]
- Henson, M.W. , Hanssen, J. , Spooner, G. , Fleming, P. , Pukonen, M. , Stahr, F. , and Thrash, J.C. (2018) Nutrient dynamics and stream order influence microbial community patterns along a 2914 kilometer transect of the Mississippi River. Limnol Oceanogr 63: 1837–1855. [Google Scholar]
- Hubbell, S.P. (2001) The Unified Neural Theory of Biodiversity and Biogeography. Princeton, NJ: Princeton University Press. [Google Scholar]
- Hug, L.A. , Castelle, C.J. , Wrighton, K.C. , Thomas, B.C. , Sharon, I. , Frischkorn, K.R. , et al (2013) Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, C.R. , Millar, J.J. , Payne, J.T. , and Ochs, C.A. (2014) Free‐living and particle‐associated bacterioplankton in large rivers of the Mississippi river basin demonstrate biogeographic patterns. Appl Environ Microbiol 80: 7186–7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen, P.H. (2006) Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ. Microbiol. 72: 1719–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersters, K. , De Vos, P. , Gillis, M. , Swings, J. , Vandamme, P. , and Stackebrandt, E. (2006) Introduction to the Proteobacteria. Prokaryotes 5: 3–37. [Google Scholar]
- Kielak, A.M. , Barreto, C.C. , Kowalchuk, G.A. , van Veen, J.A. , and Kuramae, E.E. (2016) The ecology of Acidobacteria: moving beyond genes and genomes. Front Microbiol 7: 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchman, D.L. (2002) The ecology of Cytophaga‐Flavobacteria in aquatic environments. FEMS Microbiol Ecol 39: 91–100. [DOI] [PubMed] [Google Scholar]
- Kozich, J.J. , Westcott, S.L. , Baxter, N.T. , Highlander, S.K. , and Schloss, P.D. (2013) Development of a dual‐index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl Environ Microbiol 79: 5112–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen, N. , Brodie, E.L. , Andersen, G.L. , Huber, T. , DeSantis, T.Z. , Keller, K. , et al (2006) Greengenes, a chimera‐checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibold, M.A. , Holyoak, M. , Mouquet, N. , Amarasekare, P. , Chase, J.M. , Hoopes, M.F. , et al (2004) The metacommunity concept: a framework for multi‐scale community ecology. Ecol Lett 7: 601–613. [Google Scholar]
- Lindström, E.S. , and Bergström, A.K. (2005) Community composition of bacterioplankton and cell transport in lakes in two different drainage areas. Aquat Sci 67: 210–219. [Google Scholar]
- Lindström, E.S. , and Langenheder, S. (2012) Local and regional factors influencing bacterial community assembly. Environ Microbiol Rep 4: 1–9. [DOI] [PubMed] [Google Scholar]
- Liu, T. , Zhang, A.N. , Wang, J. , Liu, S. , Jiang, X. , Dang, C. , et al (2018) Integrated biogeography of planktonic and sedimentary bacterial communities in the Yangtze River. Microbiome 6: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐García, P. , Duperron, S. , Philippot, P. , Foriel, J. , Susini, J. , and Moreira, D. (2003) Bacterial diversity in hydrothermal sediment and epsilonproteobacterial dominance in experimental microcolonizers at the Mid‐Atlantic Ridge. Environ Microbiol 5: 961–976. [DOI] [PubMed] [Google Scholar]
- Mah, T.F.C. , and O'Toole, G.A. (2001) Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 9: 34–39. [DOI] [PubMed] [Google Scholar]
- Marti, E. , Jofre, J. , and Balcazar, J.L. (2013) Prevalence of antibiotic resistance genes and bacterial community composition in a river influenced by a wastewater treatment plant. PLoS One 8: e78906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti, E. , Variatza, E. , and Balcazar, J.L. (2014) The role of aquatic ecosystems as reservoirs of antibiotic resistance. Trends Microbiol 22: 36–41. [DOI] [PubMed] [Google Scholar]
- Milici, M. , Vital, M. , Tomasch, J. , Badewien, T.H. , Giebel, H.A. , Plumeier, I. , et al (2017) Diversity and community composition of particle‐associated and free‐living bacteria in mesopelagic and bathypelagic Southern Ocean water masses: evidence of dispersal limitation in the Bransfield Strait. Limnol Oceanogr 62: 1080–1095. [Google Scholar]
- Morrison, J.M. , Baker, K.D. , Zamor, R.M. , Nikolai, S. , Elshahed, M.S. , and Youssef, N.H. (2017) Spatiotemporal analysis of microbial community dynamics during seasonal stratification events in a freshwater Lake (Grand Lake, OK, USA). PLoS One. 12: e0177488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland, P.J. , Helton, A.M. , Poole, G.C. , Hall, R.O. , Hamilton, S.K. , Peterson, B.J. , et al (2008) Stream denitrification across biomes and its response to anthropogenic nitrate loading. Nature 452: 202–205. [DOI] [PubMed] [Google Scholar]
- Muyzer, G. , De Waal, E.C. , and Uitterlinden, A.G. (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction‐amplified genes coding for 16S rRNA. Appl Environ Microbiol 59: 695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, J.A. , Chase, J.M. , Jiménez, I. , Jørgensen, P.M. , Araujo‐Murakami, A. , Paniagua‐Zambrana, N. , and Seidel, R. (2013) Beta‐diversity in temperate and tropical forests reflects dissimilar mechanisms of community assembly. Ecol Lett 16: 151–157. [DOI] [PubMed] [Google Scholar]
- Nawrocki EP. Structural RNA homology search and alignment using covariance models. Ph.D. thesis. 2009.
- Nelson, W.C. , and Stegen, J.C. (2015) The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Front Microbiol 6: 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemergut, D.R. , Schmidt, S.K. , Fukami, T. , O'Neill, S.P. , Bilinski, T.M. , Stanish, L.F. , et al (2013) Patterns and processes of microbial community assembly. Microbiol Mol Biol Rev 77: 342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuenschwander, S.M. , Ghai, R. , Pernthaler, J. , and Salcher, M.M. (2018) Microdiversification in genome‐streamlined ubiquitous freshwater Actinobacteria. ISME J. 12: 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton, R.J. , Jones, S.E. , Helmus, M.R. , and McMahon, K.D. (2007) Phylogenetic ecology of the freshwater Actinobacteria acI lineage. Appl Environ Microbiol 73: 7169–7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederdorfer, R. , Peter, H. , and Battin, T.J. (2016) Attached biofilms and suspended aggregates are distinct microbial lifestyles emanating from differing hydraulics. Nat Microbiol 1: 16178. [DOI] [PubMed] [Google Scholar]
- Niño‐García, J.P. , Ruiz‐González, C. , and del Giorgio, P.A. (2016) Landscape‐scale spatial abundance distributions discriminate core from random components of boreal lake bacterioplankton. Ecol Lett 19: 1506–1515. 10.1111/ele.12704. [DOI] [PubMed] [Google Scholar]
- Oksanen J, Kindt R, Legendre P, O'Hara RB, MHH Stevens, Oksanen MJ, et al The vegan package. Community Ecol Package. 2007.
- Peterson, B.J. , Wollheim, W.M. , Mulholland, P.J. , Webster, J.R. , Meyer, J.L. , Tank, J.L. , et al (2001) Control of nitrogen export from watersheds by headwater streams. Science 292: 86–90. [DOI] [PubMed] [Google Scholar]
- Peura, S. , Eiler, A. , Bertilsson, S. , Nykänen, H. , Tiirola, M. , and Jones, R.I. (2012) Distinct and diverse anaerobic bacterial communities in boreal lakes dominated by candidate division OD1. ISME J 6: 1640–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, M.N. , Dehal, P.S. , and Arkin, A.P. (2009) Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26: 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond, P.A. , Hartmann, J. , Lauerwald, R. , Sobek, S. , McDonald, C. , Hoover, M. , et al (2013) Global carbon dioxide emissions from inland waters. Nature 503: 355–359. [DOI] [PubMed] [Google Scholar]
- Read, D.S. , Gweon, H.S. , Bowes, M.J. , Newbold, L.K. , Field, D. , Bailey, M.J. , et al (2015) Catchment‐scale biogeography of riverine bacterioplankton. ISME J. 9: 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid, A.J. , Carlson, A.K. , Creed, I.F. , Eliason, E.J. , Gell, P.A. , Johnson, P.T.J. , et al (2018) Emerging threats and persistent conservation challenges for freshwater biodiversity. Biol Rev 94: 849–873. [DOI] [PubMed] [Google Scholar]
- Rieck, A. , Herlemann, D.P.R. , Jürgens, K. , and Grossart, H.P. (2015) Particle‐associated differ from free‐living bacteria in surface waters of the Baltic Sea. Front Microbiol 6: 1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognes, T. , Flouri, T. , Nichols, B. , Quince, C. , and Mahé, F. (2016) VSEARCH: a versatile open source tool for metagenomics. PeerJ 4: e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rösel, S. , Allgaier, M. , and Grossart, H.P. (2012) Long‐term characterization of free‐living and particle‐associated bacterial communities in Lake Tiefwaren reveals distinct seasonal patterns. Microb Ecol 64: 571–583. [DOI] [PubMed] [Google Scholar]
- Ruiz‐González, C. , Niño‐García, J.P. , and del Giorgio, P.A. (2015) Terrestrial origin of bacterial communities in complex boreal freshwater networks. Ecol Lett 18: 1198–1206. [DOI] [PubMed] [Google Scholar]
- Salter, S.J. , Cox, M.J. , Turek, E.M. , Calus, S.T. , Cookson, W.O. , Moffatt, M.F. , et al (2014) Reagent and laboratory contamination can critically impact sequence‐based microbiome analyses. BMC Biol 12: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savio, D. , Sinclair, L. , Ijaz, U.Z. , Parajka, J. , Reischer, G.H. , Stadler, P. , et al (2015) Bacterial diversity along a 2600 km river continuum. Environ Microbiol 17: 4994–5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, A.K. , Sommerfeld, K. , Bullerjahn, G.S. , Matteson, A.R. , Wilhelm, S.W. , Jezbera, J. , et al (2009) Actinorhodopsin genes discovered in diverse freshwater habitats and among cultivated freshwater Actinobacteria. ISME J 3: 726–737. [DOI] [PubMed] [Google Scholar]
- Stanley, E.H. , Casson, N.J. , Christel, S.T. , Crawford, J.T. , Loken, L.C. , and Oliver, S.K. (2016) The ecology of methane in streams and rivers: patterns, controls, and global significance. Ecol Monogr 86: 146–171. [Google Scholar]
- Stegen, J.C. , Fredrickson, J.K. , Wilkins, M.J. , Konopka, A.E. , Nelson, W.C. , Arntzen, E.V. , et al (2016) Groundwater‐surface water mixing shifts ecological assembly processes and stimulates organic carbon turnover. Nat Commun 7: 11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegen, J.C. , Lin, X. , Fredrickson, J.K. , Chen, X. , Kennedy, D.W. , Murray, C.J. , et al (2013) Quantifying community assembly processes and identifying features that impose them. ISME J 7: 2069–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegen, J.C. , Lin, X. , Fredrickson, J.K. , and Konopka, A.E. (2015) Estimating and mapping ecological processes influencing microbial community assembly. Front Microbiol 6: 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegen, J.C. , Lin, X. , Konopka, A.E. , and Fredrickson, J.K. (2012) Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J 6: 1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatariw, C. , Chapman, E.L. , Sponseller, R.A. , Mortazavi, B. , and Edmonds, J.W. (2013) Denitrification in a large river: consideration of geomorphic controls on microbial activity and community structure. Ecology 94: 2249–2262. [DOI] [PubMed] [Google Scholar]
- Tindall, B.J. , Rosselló‐Móra, R. , Busse, H.J. , Ludwig, W. , and Kämpfer, P. (2010) Notes on the characterization of prokaryote strains for taxonomic purposes. Int J Syst Evol Microbiol 60: 249–266. [DOI] [PubMed] [Google Scholar]
- Tranvik, L.J. , Cole, J.J. , and Prairie, Y.T. (2018) The study of carbon in inland waters‐from isolated ecosystems to players in the global carbon cycle. Limnol Oceanogr Lett 3: 41–48. [Google Scholar]
- Tripathi, B.M. , Stegen, J.C. , Kim, M. , Dong, K. , Adams, J.M. , and Lee, Y.K. (2018) Soil pH mediates the balance between stochastic and deterministic assembly of bacteria. ISME J 12: 1072–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban, M.C. , and De Meester, L. (2009) Community monopolization: local adaptation enhances priority effects in an evolving metacommunity. Proc R Soc B Biol Sci 276: 4129–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valett, H.M. , Thomas, S.A. , Mulholland, P.J. , Webster, J.R. , Dahm, C.N. , Fellows, C.S. , et al (2008) Endogenous and exogenous control of ecosystem function: N cycling in headwater streams. Ecology 89: 3515–3527. [DOI] [PubMed] [Google Scholar]
- Van der Gucht, K. , Cottenie, K. , Muylaert, K. , Vloemans, N. , Cousin, S. , Declerck, S. , et al (2007) The power of species sorting: local factors drive bacterial community composition over a wide range of spatial scales. Proc Natl Acad Sci 104: 20404–20409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannote, R.L. , Minshall, G.W. , Cummins, K.W. , Sedell, J.R. , and Cushing, C.E. (2008) The river continuum concept. Can J Fish Aquat Sci 37: 130–137. [Google Scholar]
- Veach, A.M. , Stegen, J.C. , Brown, S.P. , Dodds, W.K. , and Jumpponen, A. (2016) Spatial and successional dynamics of microbial biofilm communities in a grassland stream ecosystem. Mol Ecol 25: 4674–4688. [DOI] [PubMed] [Google Scholar]
- Vignaga, E. , Sloan, D.M. , Luo, X. , Haynes, H. , Phoenix, V.R. , and Sloan, W.T. (2013) Erosion of biofilm‐bound fluvial sediments. Nat Geosci 6: 770–774. [Google Scholar]
- Wang, J. , Shen, J. , Wu, Y. , Tu, C. , Soininen, J. , Stegen, J.C. , et al (2013) Phylogenetic beta diversity in bacterial assemblages across ecosystems: deterministic versus stochastic processes. ISME J 7: 1310–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G.M. , Tiedje, J.M. , and Cole, J.R. (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, N.L. , Challacombe, J.F. , Janssen, P.H. , Henrissat, B. , Coutinho, P.M. , Wu, M. , et al (2009) Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl Environ Microbiol 75: 2046–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb, C.O. , Ackerly, D.D. , and Kembel, S.W. (2008) Phylocom: software for the analysis of phylogenetic community structure and trait evolution. Bioinformatics 24: 2098–2100. [DOI] [PubMed] [Google Scholar]
- Weiss, S.J. , Xu, Z. , Amir, A. , Peddada, S. , Bittinger, K. , Gonzalez, A. , et al (2015) Effects of library size variance, sparsity, and compositionality on the analysis of microbiome data. PeerJ PrePrints 3: e1157v1. [Google Scholar]
- Wisnoski, N.I. , and Lennon, J.T. (2020) Microbial community assembly in a multi‐layer dendritic metacommunity. Oecologia. [DOI] [PubMed] [Google Scholar]
- Zeglin, L.H. (2015) Stream microbial diversity in response to environmental changes: review and synthesis of existing research. Front Microbiol 6: 454 10.1007/s00442-020-04767-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Kobert, K. , Flouri, T. , and Stamatakis, A. (2014) PEAR: a fast and accurate Illumina paired‐end reAd mergeR. Bioinformatics 30: 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , and Ning, D. (2017) Stochastic community assembly: does it matter in microbial ecology? Microbiol Mol Biol Rev 81: e00002‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Upset chart showing overlap in all OTUs identified in each habitat type. Numbers of OTUs shared between different sets of habitats are indicated in the top bar chart and the specific habitats in each set are indicated with solid points below the bar chart. Total number of OTUs for each habitat is indicated on the left. Figure generated using Upset R package (Lex et al., 2014). Do note that the bar chart is not showing relative abundances.
Supplementary Table 1 PERMANOVA analyses produces a p‐value for significance and the R2 value, which indicates the amount of variation attributed to a specific treatment within a model. The R2 values indicate greater differences between the planktonic communities (Free‐Living and Particle‐Associated) and benthic communities (Biofilm and Sediment).
Supplementary Table 2 Tukey's HSD on diversity indices (Tukey multiple comparisons of means, 95% family‐wise confidence level)
Supplementary Table 3 Tukey's HSD on the differences in relative abundances of selected phyla between four different habitats (Tukey multiple comparisons of means, 95% family‐wise confidence level)
Supplementary Table 4 Supporting information
Supplementary Table 5 Supporting information
Supplementary Table 6 Supporting information
Supplementary Table 7 Supporting information
Data Availability Statement
The raw sequence data reported in this study have been deposited in the European Nucleotide Archive under the accession number PRJEB29699. The relevant barcode information for each sample is shown in Supplementary Table S5.