

Abstract
Defects in the functions of RNA binding proteins (RBPs) are at the origin of many diseases; however, targeting RBPs with conventional drugs has proven difficult. PROTACs are a new class of drugs that mediate selective degradation of a target protein through a cell's ubiquitination machinery. PROTACs comprise a moiety that binds the selected protein, conjugated to a ligand of an E3 ligase. Herein, we introduce RNA‐PROTACs as a new concept in the targeting of RBPs. These chimeric structures employ small RNA mimics as targeting groups that dock the RNA‐binding site of the RBP, whereupon a conjugated E3‐recruiting peptide derived from the HIF‐1α protein directs the RBP for proteasomal degradation. We performed a proof‐of‐concept demonstration with the degradation of two RBPs—a stem cell factor LIN28 and a splicing factor RBFOX1—and showed their use in cancer cell lines. The RNA‐PROTAC approach opens the way to rapid, selective targeting of RBPs in a rational and general fashion.
Keywords: Lin28, oligonucleotides, PROTAC, proteasomal degradation, RNA-binding proteins
RNA‐PROTACs are new chimeric structures that mediate the proteasomal degradation of RNA‐binding proteins (RBP). They comprise an E3‐recruiting peptide conjugated to a short modified RNA, which is iso‐sequential with the native binding element to which the RBP binds in cells. RNA‐PROTACs offer a fast, rational and general approach to target an entire class of protein (RBPs) that has proven difficult until today to target pharmacologically.
Introduction
RNA binding proteins (RBPs) constitute a large fraction of a cell's proteome. [1] Over 1500 RBPs are known and their genes are evolutionally conserved and transcribed into splicing variants with unique functions. RBPs bind to RNAs in a dynamic, coordinative and sequence‐selective manner to form ribonucleoprotein (RNP) complexes that play key roles in RNA‐dependent processes. [2] Several diseases are caused by genetic alterations in RBPs, which affect their binding to RNA. [3]
Here we introduce a new concept in targeting RBPs using a novel class of chimeric structure, which we have termed RNA‐PROTACs. Conventional PROTACs (PROteolysis‐TArgeting Chimeras) comprise three components: a target‐binding ligand, a ubiquitin E3 ligase‐recruiting ligand and a connecting linker group. [4] The function of the E3 binding moiety is to recruit E3‐dependent factors, whereby formation of a ternary complex between the PROTAC and the protein of interest (POI) via the targeting ligand, activates ubiquitination of the latter, marking it for degradation through the proteasome pathway. [5] This results in lowered levels of the target, and a corresponding loss of its function. Following a rapid phase of development, the first PROTACs have now entered clinical trials. [6]
The targeting ligand of a conventional PROTAC is a drug‐like small molecule that binds selectively to the POI. Ligands have been reported for a variety of proteins, including enzymes and receptors. [4] They are typically identified by high‐throughput screening or rational design, based upon the natural ligands of the target proteins. [7]
Intuitively, the RNA binding site of an RBP represents a viable target site for drugs, since it is functionally important and structural data for RBPs is often available. However, with the exception of a few natural products inhibiting splicing, (ref. [8] and refs therein) examples of small‐molecule ligands that target RBPs are rare. [9] This may be due to several reasons: RBP binding pockets are intrinsically disordered and change conformation upon RNA binding; [10] compound‐screening assays are challenging to implement; [11] and RBPs share homologous domains that recognize short, degenerate sequence motifs. [12]
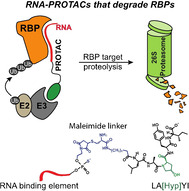
We employed short oligonucleotides that are iso‐sequential with the RNA consensus binding element (RBE) of an RBP for the first time as the targeting moiety for the PROTAC (Figure 1 A). The oligonucleotide competes with native RNAs for binding to the RBP in cells. We describe here a proof‐of‐concept for this approach with the design, synthesis and characterization of an RNA‐PROTAC targeting LIN28 (Lin28A), a small RBP comprising a C‐terminal CCHC‐type zinc knuckle domain (ZKD). [13]
Figure 1.
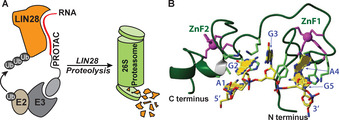
RNA‐PROTACs bind RBPs and direct them for degradation. A) An RNA‐PROTAC comprising a short oligonucleotide binds the RNA‐binding domain of the RBP and mediates its ubiquitination and degradation. B) The Lin28 zinc finger domain binding to its consensus sequence AGGAGAU (adapted from Ref. [13b], PDB: 2LI8).
To target Lin28, we used 5′‐AGGAGAU‐3′ (L28RBE), a conserved sequence present in microRNAs, to which Lin28 binds (Figure 1 B). We modified L28RBE with 2′‐O‐methoxyethyl (MOE) ribonucleotides and conjugated it to a E3‐recruiting peptide derived from the HIF‐1α protein. This RNA‐PROTAC binds selectively to Lin28 in vitro, and suppressed levels of Lin28A in two cancer cell lines in ubiquitin‐dependent fashion. Taken together, these findings pave the way to a new class of rationally designed inhibitors for RBPs, a protein family that until now has been difficult to address with conventional drugs.
Results and Discussion
A short oligoribonucleotide corresponding to the consensus RBE was a conceptually natural choice for the targeting element of an RNA PROTAC. Short oligonucleotides have been investigated previously as antisense [14] and anti‐miRNA reagents, [15] but have not been pursued into clinical development to our knowledge. Oligoribonucleotides require structural modifications to protect them against ubiquitous nucleases in vivo. Therefore, we adopted a similar strategy employed for recently approved oligonucleotide therapeutics: replacement of the phosphodiester backbone by diastereoisomeric phosphorothioate (PS) linkages [16] and alkylation of the 2′‐hydroxyl group. PS‐linkages endow oligonucleotides with three essential properties for use in vivo: enhanced nuclease resistance, an ability to enter cells and a weak non‐specific binding to proteins that slows renal excretion and allows systemic circulation. [17] Especially, the alkylation of the ribose 2′‐OH with 2′‐O‐methoxyethyl (MOE) substituents [18] brings further nuclease stability, [19] and adds a hydration layer to the structure that improves biodistribution and tolerability in vivo. [20]
The oligonucleotide element of an RNA‐PROTAC binds its target RBP via interactions at the riboses and the backbone, as well as the nucleobases. Therefore, it was important to show first that the MOE and PS groups would not disturb these interactions. We showed previously using NMR spectroscopy that Lin28_ZKD recognizes the Watson–Crick faces of G2 and G5 in 5′‐A1G2G3A4G5A6U7‐3′ (Figure 1 B), and that the two zinc finger domains are necessary and sufficient to recognize selectively the GNNG sequence of its partner RNAs.[ 13b , 21 ] Thus, we used the same method to examine how various ribose modifications of L28RBE (Table 1, Figure S1) might affect binding of an RNA‐PROTAC to Lin28_ZKD. Pleasingly, we observed similar chemical shift perturbations for these analogs as for wild‐type RNA ORN1 (Figure 2 A). This suggested that the modified oligonucleotides adopted a similar mode of binding to Lin28_ZKD. In the case of ORN2, ORN3 and ORN7 (Figure 2 B, Figure S2) some of the NMR signals broadened or disappeared (precipitation at higher concentrations was also observed). This may have been at least partly caused by the presence of PS diastereoisomers.
Table 1.
Oligonucleotide sequences used in this study and corresponding IC 50 values determined by fluorescence polarization (ORN5 is a randomized negative control sequence).
|
Name |
L28RBE‐containing sequences (5′ to 3′) |
Backbone chemistry |
IC 50 [nM] |
|---|---|---|---|
|
Let7FAM |
FAM_GGGGUAGUGAUUU UACCCUGUUCAGGAGAU |
PO‐RNA |
117.4±17.9 |
|
ORN1 |
AGGAGAU |
PO‐RNA |
n.d. |
|
ORN2 |
AGGAGAU |
PS‐RNA |
58.1±19.8 |
|
ORN3 |
AGGAGAU |
PS‐MOE |
192.2±34.8 |
|
ORN4 |
AGGAGAU |
PS‐OMe |
55.4±13.3 |
|
ORN5 |
UUUAUUG |
PS‐MOE |
1384.5±268.0 |
|
ORN6 |
AGGAGAU |
PO‐OMe |
– |
|
ORN7 |
AGGAGAUAACU |
PS‐MOE |
162±38 |
|
ORN8 |
AGGAG |
PS‐MOE |
1289±339.4 |
[a] n.d.=not determined; ORN6 was not measured.
Figure 2.
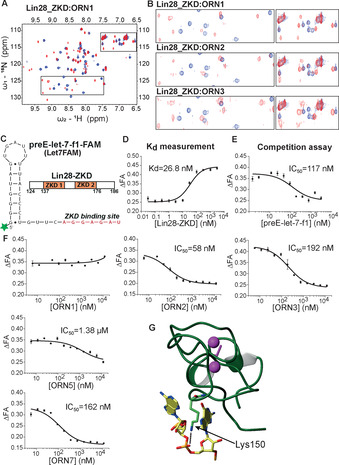
Fluorescence polarization competition assay with Lin28‐binding RNA mimics. A) 1H–15 N HSQC spectra of Lin28_ZKD, free (blue) and bound (red) to ORN1 at a 1:1 ratio. B) Overlay of selected 1H–15 N HSQC spectra of Lin28_ZKD, free (blue) and bound (red) to ORN1, ORN2 and ORN3 at 1:1 ratios. C) Sequence of FAM labelled preE‐let‐7f‐1 used in this study; domain structures of Lin28 (ZKD: orange; RNA residues that bind the ZKD: red). D) Fluorescence polarization assay of Lin28_ZKD binding to preE‐let‐7f‐1‐FAM. E) Competition assay using unlabeled preE‐let‐7f‐1. F) Competition fluorescence polarization assays with ORN1, ‐2, ‐3, ‐5 and ‐7. Wild‐type RNA (ORN1) is a weak competitor to preE‐let‐7f‐1. (n=3 replicates) G) The solution structure of Lin28_ZKD bound to 5′‐AGGAGAU‐3. Dotted line identifies possible interaction between Lys150 and the phosphorothioate linkage between residues A4/G5 (adapted from Ref. [13b], PDB: 2LI8).
In cells, RNA‐PROTACs must compete with native RNAs for binding the target RBP. In general, RNA‐RBP binding interactions are complex (sometimes multimeric) and often extend outside the RBE. [12] RNA secondary structure may also facilitate the interactions. Therefore, in order to determine whether short, modified derivatives of L28RBE could compete with native RNAs for binding to Lin28 in cells, we adapted a competition binding assay that was originally developed to screen for small‐molecule inhibitors of Lin28 (Figure S3).[ 13c , 22 ]
PreE‐let‐7f‐1 RNA is a 30‐nucleotide (nt) stretch of structured sequence present in let‐7 precursors to which Lin28_ZKD binds with nanomolar affinity.[ 13c , 22 ] Thus, preE‐let‐7f‐1 was synthesized with a 6‐fluoroscein (FAM) label at its 5′ terminus (let7FAM; Figure S1, Figure 2 C). It was then incubated with graded concentrations of human recombinant Lin28_ZKD [13b] in order to determine an equilibrium dissociation constant for the Lin28_ZKD/let7FAM interaction. This yielded a K D of 27 nM, in agreement with previously recorded values [22] (Figure 2 D). Based on this, 40 nM concentrations of Lin28_ZKD were subsequently used for testing L28RBE analogs.
Incubation of Lin28_ZKD/let7FAM with unlabeled preE‐let‐7f‐1 decreased the fluorescence polarization signal with an IC 50 of 117 nM (Figure 2 E), consistent with displacement of let7FAM from the protein. In contrast, the 7‐mer ORN1 (unmodified L28RBE RNA) was inactive (Figure 2 F), consistent with observations of Wang et al. [22] The stark difference between the activities of the 7‐mer and the 30‐mer RNAs in this assay may indicate that Lin28_ZKD can make contacts with elements outside of the 7‐nt consensus sequence in native RNAs. Surprisingly, the fully phosphorothioated analog ORN2 yielded an IC 50 of 58 nM in the assay (Figure 2 F). ORN4 and ORN3, which have PS‐OMe and PS‐MOE modifications, respectively, yielded IC 50 values of 56 and 192 nM, respectively. A randomized sequence (negative control) of L28RBE (ORN5) and a 5‐nt analog of ORN3 (ORN8) showed about 20‐fold and 7‐fold weaker activities than ORN4 and ORN3, respectively.
To provide insight on why the short PS‐oligonucleotides displaced let7FAM so readily from Lin28_ZKD, we measured binding affinities of selected PO‐ and PS‐ L28RBE analogs to Lin28_ZKD by surface plasmon resonance spectroscopy (SPR). Lin28_ZKD was immobilized to a chip and incubated with graded concentrations of selected ORNs, as well as positive and negative controls.
A let‐7 precursor, pre‐let‐7 g bound Lin28_ZKD with the highest affinity (K d approx. 10 nM; Figure S4). ORN1, ORN2 and ORN3 bound Lin28_ZKD with K ds of 25.8 μM, 550 nM and 580 nM, respectively. We re‐examined the NMR‐structure of Lin28_ZKD/L28RBE in an effort to explain the enhanced binding/activity of the PS‐oligonucleotides. Interestingly, the phosphodiester group between A4/G5 of ORN1 contacts Lys150 of Lin28_ZKD (Figure 2 G). Thus, a phosphorothioate group at this position might positively enhance this interaction and partly explain the superior activity of the PS‐analogs. Based on the results from these independent assays, three L28RBE analogs (ORN3, ORN4 and ORN7), together with the sequence‐randomized control (ORN5) were selected for further study.
Various E3 systems have been employed as PROTACs to mediate degradation of POIs.[ 5c , 23 ] Empirical testing is generally used to identify that combination of linker and E3 system that yields the most potent PROTAC reagent. [24] One of the earliest PROTAC systems was an ALAPYIP‐containing peptide derived from the HIF‐1α (hypoxia‐inducible factor 1α) transcription factor. When its central proline is hydroxylated, [25] this peptide recruits VHL (von Hippel‐Lindau) into the VBC‐Cul2 E3 ubiquitin ligase complex. [26] A shortened segment of the peptide (comprising the hydroxylated proline; LA[Hyp]YI) still retains ubiquitinating activity and was selected for this PoC study, although recent work has uncovered non peptide‐based E3‐ligands that might also be used. [27] LA[Hyp]YI is cell‐permeable, [28] although it seemed likely that the uptake of RNA‐PROTACs into cells would be dominated by the properties of the oligonucleotide.
Single‐stranded PS oligonucleotides are internalized after prolonged incubation into many types of cultured cells via gymnosis. Uptake leads to endosomal‐lysosomal accumulation, with a slow leakage into the cytosol, from where the oligonucleotides distribute to other parts of the cell, including the nucleus. [29] The PS‐groups are crucial for the uptake, and the most efficient uptake is typically seen with longer oligonucleotides. [30] We examined the uptake of FAM‐conjugated oligonucleotides ORN3 (7‐nt) and ORN7 (11‐nt) into human immortalized myelogenous leukemia line K562 cells using fluorescence microscopy. Both ORN3 and ORN7 were visible in the cytosol and the nuclei of cells, with the uptake of the longer ORN7 being higher than that of ORN3 (Figure S5). Taken together, the data suggested that an RNA‐PROTAC comprising a 7‐mer PS‐MOE sequence would reach the cell cytoplasm and therefore be able to elicit its effect.
L28RBE analogs (ORN3, ORN4, ORN5, ORN7) were conjugated in solution to LA[Hyp]YI via the peptide N‐terminus (Figure 3 A, Figures S1, S6). The PS‐MOE derivative ORN3 was prioritized for study since PS‐MOE oligonucleotide drugs distribute widely after administration in vivo, [17b] are metabolically stable and are safe for patients. [31] Masked maleimide‐oligonucleotides were prepared on solid support. They were activated prior to coupling with peptide P1 (LA[Hyp]YI) or a control sequence Pcon, in which norleucine was exchanged for the hydroxyproline (Figure 3 A). In all, ten oligonucleotide‐peptide conjugates were synthesized and purified (Table 2, Figure S1). Several of the conjugates were tested in the Lin28_ZKD competition binding assays to ensure that the addition of the linker/peptide did not adversely affect binding of the RNA‐PROTAC to Lin28 (Figure 3 B, Figure S7). ORN3P1 and ORN7P1 competed slightly more strongly for binding to Lin28_ZKD than their parent unconjugated oligonucleotides, presumably due to contacts between LA[Hyp]YI and the protein (Figure 3 B, Figure S7). Consistent with this, the sequence control ORN5P1 showed a weak effect on Lin28_ZKD (Figure S7).
Figure 3.

Synthesis of RNA‐PROTACs and their mechanism of action. A) Synthetic route to RNA‐PROTACs and controls. B) Fluorescence polarization assay for ORN3P1 and negative seq‐control ORN5P1. C,D) RNA‐PROTAC‐mediated ubiquitination of myc‐Lin28A in 22Rv1 cells co‐transfected with HA‐Ubiquitin (HA‐Ub) and pCMV‐myc‐Lin28A, or pDEST‐Myc‐QKI with either vehicle (OptiMEM) or 500 nM ORN3P1 (C) or ORN7P1 (D). cMyc‐immunoprecipitated lysates were separated by SDS‐PAGE followed by western blots detecting HA (Ub). Slow‐migrating smears in right‐hand lanes represent ubiquitin‐conjugated cMyc‐Lin28A. WCL=whole‐cell lysate input.
Table 2.
RNA‐PROTACs synthesized and used in this study.
|
Name |
Sequence |
Peptide[a] |
Mass obs. [g mol−1] |
Mass calc. [g mol−1] |
|---|---|---|---|---|
|
ORN3P1 |
AGGAGAU |
C[Ahx]LA[Hyp]YI |
3810.8 |
3811.5 |
|
ORN5P1 |
UUUAUUG |
C[Ahx]LA[Hyp]YI |
3742.8 |
3743.5 |
|
ORN3Pcon |
AGGAGAU |
C[Ahx]LA[Nle]YI |
3810.8 |
3811.6 |
|
ORN4P1 |
AGGAGAU |
C[Ahx]LA[Hyp]YI |
3488.5 |
3489.2 |
|
ORN4Pcon |
AGGAGAU |
C[Ahx]LA[Nle]YI |
3488.5 |
3489.2 |
|
ORN7P1 |
AGGAGAUAACU |
C[Ahx]LA[Hyp]YI |
5404.9 |
5404.2 |
|
ORN7Pcon |
AGGAGAUAACU |
C[Ahx]LA[Nle]YI |
5404.9 |
5404.3 |
|
ORN9P1 |
UGCAUGU |
C[Ahx]LA[Hyp]YI |
3766.7 |
3767.5 |
|
ORN10P1 |
GCCAUCU |
C[Ahx]LA[Hyp]YI |
3739.8 |
3740.5 |
|
ORN9Pcon |
UGCAUGU |
C[Ahx]LA[Nle]YI |
3766.8 |
3767.6 |
[a] Ahx=6‐(Fmoc‐amino)hexanoic acid, Nle=Norleucine, Hyp=l‐hydroxyproline (mutated negative control peptide).
Next, we investigated the ability of ORN3P1 and ORN7P1 to mediate ubiquitination of Lin28 in cells, using a previously described protocol. [32] Cells from the prostate cancer cell line 22RV1, which express very low levels of Lin28, were co‐transfected with two plasmids expressing pCMV‐myc‐Lin28A and HA‐tagged polyubiquitin (HA‐Ub).
After 48 h of incubation with ORN3P1 and ORN7P1, the tagged Lin28 (and the control QKI) was immunoprecipitated using an anti‐Myc antibody. The isolated protein was then analyzed by western blotting using the anti‐HA and anti‐c‐Myc antibodies. Consistent with other previously described PROTAC reagents,[ 32 , 33 ] ORN3P1‐ and ORN7P1‐treated cells showed a smear on the gel towards higher molecular weight (Figure 3 C,D, Figure S8), consistent with conjugation of poly‐Ub to the myc‐Lin28A fusion protein. Vehicle‐treated cells, or cells transfected with a plasmid that expresses an alternative RBP as negative control (Quaking homolog, QKI; pDEST‐Myc‐QKI) instead of pCMV‐myc‐Lin28A, showed no such effects.
Humans express two paralogs of the protein, LIN28 (Lin28A) and LIN28B (Lin28B), that play key roles in development, metabolism, and pluripotency. [34] Lin28 proteins are abundantly expressed during embryonic development, as well as in several tumors and tumor‐derived cell lines. [35] They bind with nanomolar affinity to let‐7 precursors and the 3′ untranslated regions (UTRs) of mRNAs involved in cell proliferation. In order to confirm that RNA‐PROTACs bind to endogenous Lin28 in cells, we performed a pull‐down RNA ELISA assay that we have previously described. [36] Thus, we conjugated a biotin group to the 3′ ends of ORN3P1 and ORN7P1 and added these reagents separately on to myelogenous leukemia K562 cells. We incubated lysates from treated cells on streptavidin‐coated plates so as to capture the RNA‐PROTACs present in the cell lysates. We then assayed for the presence of Lin28A in the plates using an anti‐Lin28A antibody and a negative control anti‐FUS antibody. Lysates from both ORN3P1‐ and ORN7P1‐treatments yielded a strong signal for Lin28A protein, in comparison to lysates from mock‐treated cells, or compared to measurement using the FUS antibody (Figure 4 B, Figure S9). The data confirmed that RNA‐PROTACs entered cells and engaged their intended target.
Figure 4.
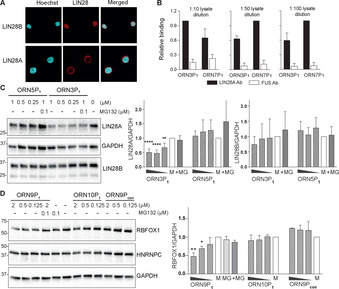
RNA‐PROTACs mediate Lin28A and RBFOX1 degradation in cells. A) Lin28A and Lin28B localization in K562 cells; Hoechst staining indicates location of nuclei. B) Pull‐down of Lin28A onto streptavidin‐coated microtiter plates from the lysates of K562 cells treated with biotin‐labeled ORN3P1 and ORN7P1. Relative binding is normalized to mock‐treated cells. C) Western blot and quantification of Lin28A from K562 cells incubated with ORN3P1 and negative seq‐control ORN5P1 for 48 h. D) RNA‐PROTACs mediate degradation of RBFOX1 in HEK293 cells. HEK293 cells were incubated with ORN9P1, ORN10P1 and negative peptide‐control ORN9Pcon for 48 h. Data are shown as mean ±SD of three independent experiments for (C) and two for (D). M=cells treated with mock solutions, MG=cells treated with 100 nM MG132, +MG=cells co‐treated with RNA‐PROTACs and 100 nM MG132. Error bars indicate standard deviations. Asterisks denote statistical significance compared to 0 nM dose assessed by 1‐way ANOVA test whereas: ns P>0.05, * P≤0.05, ** P≤0.01, *** P≤0.001, **** P≤0.0001. Uncropped western blots are shown in Figure S14,15.
Finally, we tested RNA‐PROTACs for their ability to degrade their target in two cell lines: the embryonic NT2/D1 cell line, which expresses high levels of Lin28A, [13a] and K562 cells which express Lin28A and Lin28B. NT2/D1 cells treated with 2 μM concentrations of ORN3P1 or ORN7P1 showed an approximate 50 % reduction of Lin28A, whereas the negative control peptide ORN3Pcon was inactive (Figure S10). Suppression of the target was not quantitative, as is often observed from testing PROTAC reagents without an optimization of the targeting ligand, the linker and the E3 system. Furthermore, there was no statistical difference in the activity of the 7‐mer and 11‐mer reagents. In K562 cells, ORN3P1 suppressed 50 % of Lin28A at the highest concentration, whereas the negative control ORN5P1 was inactive, confirming a sequence‐selective action of the RNA‐PROTAC (Figure 4 C). The reagents had no discernible effect on cell toxicity (Figure S11). Lin28A and Lin28B have identical zinc finger domains,[ 13c , 21 ] however ORN3P1 had little effect on Lin28B (Figure 4 C), despite its presence in the nucleus. This may have been in part, because Lin28B is mainly localized in the nucleus where proteasomal degradation might be less effective or require additional steps or factors (Figure 4 A, Figure S12).[ 35b , 37 ] Finally, we co‐treated K562 cells with ORN3P1 and MG132, a reversible inhibitor of the 26S proteasome. This largely attenuated Lin28A suppression by the RNA‐PROTAC, strongly suggesting that ORN3P1 elicited its effects through the proteasome, as intended (Figure 4 C). In order to explore the versatility of RNA‐PROTACs with non peptide‐based VHL ligands, we conjugated small‐molecule VH032 [38] to ORN3 via an amino linker to yield ORN3VH032 (Figure S1). In K562 cells, ORN3VH032 also reduced levels of Lin28A after 24 h in a way that could be rescued by addition of MG132 (Figure S13).
In order to demonstrate that RNA‐PROTACs can be generated against other RBPs using their respective consensus RNA binding elements as a targeting ligand, we turned to RBFOX1. RBFOX1 (A2BP1) [39] is an alternative splicing factor expressed in neuronal tissues, [40] muscle and heart. [41] It binds with nanomolar affinity to 5′‐UGCAUGU‐3′ through its RNA recognition motif (RRM). We synthesized a PS‐MOE‐modified variant of the RBFOX1 RBE (FOXRBE) and conjugated it to C[Ahx]LA[Hyp]YI (ORN9P1) using the same protocol as for the Lin28 RNA‐PROTACs. In parallel, we prepared two negative control reagents: ORN10P1 comprises a random oligonucleotide sequence, whereas ORN9Pcon has the FOXRBE conjugated to the mutated peptide (Table 2).
Human embryonic kidney cells (HEK293T) cells were treated with the three reagents at increasing concentrations, under similar conditions for the Lin28 RNA‐PROTACs. Western blotting of protein isolated from treated cells showed an approximate 50 % reduction of RBFOX1 protein at 2 μM (Figure 4 D). ORN9Pcon and ORN10P1 did not affect levels of RBFOX1 protein, suggesting that ORN9P1 is selective for its target. In order to demonstrate further the selectivity of ORN9P1 for RBFOX1, the blot was probed with an antibody against heterogeneous nuclear ribonucleoproteins HNRNPC. HNRNPC contains an amino‐terminal sequence that is known to bind strongly uridine (U)‐rich sequences, and therefore provided a strict test of selectivity for the FOXRBE analogs. [42] Happily, none of the treatments with ORN9P1, ORN10P1 and ORN9Pcon affected significantly the level of endogenous HNRNPC protein (Figure 4 D).
Conclusion
Conventional drugs can be classified into distinct structural types, such as small‐molecules, therapeutic proteins and oligonucleotides. Single‐stranded oligonucleotide drugs have cellular RNA as their common target, with which they hybridize according to Watson–Crick rules. Amongst them, the most prevalent groups are the antisense‐ and splice‐switching‐oligonucleotides that target mRNAs. However, new targeting mechanisms mediated by anti‐miRNA and decoy oligonucleotides are also under investigation (see refs. [43] and [44] for examples).
Here we describe RNA‐PROTACs, a new class of chimeric oligonucleotides that are rationally designed to target RBPs, that is, proteins. Many groups have conjugated oligonucleotides to peptides, usually however, for the peptide to transport the oligonucleotide to its target RNA in vitro/vivo. [45] Here, the roles of the peptide and the oligonucleotide are reversed; the oligonucleotide delivers the peptide to its target site. RNA‐PROTACs dock into the RNA binding site of an RBP via the structurally modified oligoribonucleotide that is sequence‐identical with the native RNA‐binding element of the RBP. The first RNA‐PROTAC targets the Lin28 protein, a stem cell factor and oncoprotein of high interest as a potential drug target for several diseases. Using a structure‐based approach, we designed a 7‐nt PS‐MOE oligonucleotide analogue that binds tightly to the zinc finger domain of Lin28A. A VHL‐recruiting peptide that is conjugated to the 5′‐end of the oligonucleotide then mediates degradation of the target in cells via the ubiquitination pathway. This proof‐of‐concept represents a new means of degrading and thereby inhibiting RNA‐binding proteins, a target class that until now has proven difficult to address pharmacologically.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank A. Behera and E. Lekka for the c‐Myc‐Lin28, c‐Myc‐QKI plasmids and for the helpful discussions. We thank Harry Towbin for valuable insights and K.‐H. Altmann for reading the manuscript. We thank D. Neri and M. Catalano for access to the spectrofluorometer and suggestions. We thank J. C. Leroux and D. Carradori for the access to the fluorescence microscope and discussions. We thank Verena Schösser for preliminary experiments. This work was supported by a grant from the Swiss National Science Foundation: the NCCR RNA & Disease to J.H. and F.H.T.A.
A. Ghidini, A. Cléry, F. Halloy, F. H. T. Allain, J. Hall, Angew. Chem. Int. Ed. 2021, 60, 3163.
References
- 1.
- 1a. Ghosh P., Sowdhamini R., Mol. BioSyst. 2016, 12, 532–540; [DOI] [PubMed] [Google Scholar]
- 1b. Hentze M. W., Castello A., Schwarzl T., Preiss T., Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341; [DOI] [PubMed] [Google Scholar]
- 1c. Wang Z.-L., Li B., Luo Y.-X., Lin Q., Liu S.-R., Zhang X.-Q., Zhou H., Yang J.-H., Qu L.-H., Cell Rep. 2018, 22, 286–298. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Müller-McNicoll M., Neugebauer K. M., Nat. Rev. Genet. 2013, 14, 275–287; [DOI] [PubMed] [Google Scholar]
- 2b. Quinones-Valdez G., Xiao X., Tran S. S., Xiao X., Jun H.-I., Bahn J. H., Yang E.-W., Brummer A., Xiao X., Zhan L., Wei X., Graveley B. R., Van N. E. L., Pratt G. A., Yeo G. W., Van N. E. L., Pratt G. A., Yeo G. W., Pratt G. A., Yeo G. W., Xiao X., Xiao X., Commun. Biol. 2019, 2, 19; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Gerstberger S., Hafner M., Tuschl T., Nat. Rev. Genet. 2014, 15, 829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Wang E. T., Matthew T. J., Lee J.-A., Sudhakaran I. P., Rossoll W., Gross C., Moss K. R., Bassell G. J., J. Neurosci. 2016, 36, 11418–11426; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Hong S., J. Cancer Prev. 2017, 22, 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burslem G. M., Crews C. M., Cell 2020, 181, 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Amm I., Sommer T., Wolf D. H., Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 182–196; [DOI] [PubMed] [Google Scholar]
- 5b. Huang X., Dixit V. M., Cell Res. 2016, 26, 484–498; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Edmondson S. D., Yang B., Fallan C., Bioorg. Med. Chem. Lett. 2019, 29, 1555–1564; [DOI] [PubMed] [Google Scholar]
- 5d. Konstantinidou M., Li J., Zhang B., Wang Z., Shaabani S., Ter B. F., Essa K., Domling A., Expert Opin. Drug Discovery 2019, 14, 1255–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Neklesa T., Snyder L. B., Willard R. R., Vitale N., Pizzano J., Gordon D. A., Bookbinder M., Macaluso J., Dong H., Ferraro C., Wang G., Wang J., Crews C. M., Houston J., Crew A. P., Taylor I., J. Clin. Oncol. 2019, 37, 259–259; [Google Scholar]
- 6b. Cancer Discovery 2018, 8, 377–378; [DOI] [PubMed]
- 6c.I. Arvinas Clinical Trial of ARV-471 in Patients With ER+/HER2- Locally Advanced or Metastatic Breast Cancer. https://ClinicalTrials.gov/show/NCT04072952;
- 6d.I. Arvinas A Phase 1 Clinical Trial of ARV-110 in Patients With Metastatic Castration-resistant Prostate Cancer. https://ClinicalTrials.gov/show/NCT03888612.
- 7. Scheepstra M., Hekking K. F. W., van Hijfte L., Folmer R. H. A., Comput. Struct. Biotechnol. J. 2019, 17, 160–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Kaida D., Motoyoshi H., Tashiro E., Nojima T., Hagiwara M., Ishigami K., Watanabe H., Kitahara T., Yoshida T., Nakajima H., Tani T., Horinouchi S., Yoshida M., Nat. Chem. Biol. 2007, 3, 576–583; [DOI] [PubMed] [Google Scholar]
- 8b. Yokoi A., Kotake Y., Takahashi K., Kadowaki T., Matsumoto Y., Minoshima Y., Sugi N. H., Sagane K., Hamaguchi M., Iwata M., Mizui Y., FEBS J. 2011, 278, 4870–4880; [DOI] [PubMed] [Google Scholar]
- 8c. Lan L., Appelman C., Smith A. R., Yu J., Larsen S., Marquez R. T., Liu H., Wu X., Gao P., Roy A., Anbanandam A., Gowthaman R., Karanicolas J., De Guzman R. N., Rogers S., Aube J., Ji M., Cohen R. S., Neufeld K. L., Xu L., Mol. Oncol. 2015, 9, 1406–1420; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Martínez-Montiel N., Rosas-Murrieta N. H., Martínez-Montiel M., Gaspariano-Cholula M. P., Martínez-Contreras R. D., Biomed Res. Int. 2016, 3681094; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. Cretu C., Agrawal A. A., Cook A., Will C. L., Fekkes P., Smith P. G., Luhrmann R., Larsen N., Buonamici S., Pena V., Mol. Cell 2018, 70, 265–273.e268. [DOI] [PubMed] [Google Scholar]
- 9. Roos M., Pradere U., Ngondo R. P., Behera A., Allegrini S., Civenni G., Zagalak J. A., Marchand J.-R., Menzi M., Towbin H., Scheuermann J., Neri D., Caflisch A., Catapano C. V., Ciaudo C., Hall J., ACS Chem. Biol. 2016, 11, 2773–2781. [DOI] [PubMed] [Google Scholar]
- 10. D'Agostino V. G., Sighel D., Zucal C., Bonomo I., Micaelli M., Lolli G., Provenzani A., Quattrone A., Adami V., SLAS Discovery 2019, 24, 314–331. [DOI] [PubMed] [Google Scholar]
- 11. Roy A., Expert Opin. Drug Discovery 2019, 14, 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Beckmann B. M., Castello A., Medenbach J., Pfluegers Arch. 2016, 468, 1029–1040; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Jankowsky E., Harris M. E., Nat. Rev. Mol. Cell Biol. 2015, 16, 533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Moss E. G., Tang L., Dev. Biol. 2003, 258, 432–442; [DOI] [PubMed] [Google Scholar]
- 13b. Loughlin F. E., Gebert L. F. R., Towbin H., Brunschweiger A., Hall J., Allain F. H. T., Nat. Struct. Mol. Biol. 2012, 19, 84–89; [DOI] [PubMed] [Google Scholar]
- 13c. Nam Y., Chen C., Gregory R. I., Chou J. J., Sliz P., Cell 2011, 147, 1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wagner R. W., Matteucci M. D., Grant D., Huang T., Froehler B. C., Nat. Biotechnol. 1996, 14, 840–844. [DOI] [PubMed] [Google Scholar]
- 15. Obad S., dos Santos C. O., Petri A., Heidenblad M., Broom O., Ruse C., Fu C., Lindow M., Stenvang J., Straarup E. M., Hansen H. F., Koch T., Pappin D., Hannon G. J., Kauppinen S., Nat. Genet. 2011, 43, 371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eckstein F., Antisense Nucleic Acid Drug Dev. 2000, 10, 117–121. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Geary R. S., Norris D., Yu R., Bennett C. F., Adv. Drug Delivery Rev. 2015, 87, 46–51; [DOI] [PubMed] [Google Scholar]
- 17b. Geary R. S., Watanabe T. A., Truong L., Freier S., Lesnik E. A., Sioufi N. B., Sasmor H., Manoharan M., Levin A. A., J. Pharmacol. Exp. Ther. 2001, 296, 890–897; [PubMed] [Google Scholar]
- 17c. Crooke S. T., Vickers T. A., Liang X.-h., Nucleic Acids Res. 2020, 48, 5235–5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martin P., Helv. Chim. Acta 1995, 78, 486–504. [Google Scholar]
- 19.
- 19a. Baek M.-S., Yu R. Z., Gaus H., Grundy J. S., Geary R. S., Oligonucleotides 2010, 20, 309–316; [DOI] [PubMed] [Google Scholar]
- 19b. Yu R. Z., Geary R. S., Monteith D. K., Matson J., Truong L., Fitchett J., Levin A. A., J. Pharm. Sci. 2004, 93, 48–59. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Egli M., Manoharan M., Acc. Chem. Res. 2019, 52, 1036–1047; [DOI] [PubMed] [Google Scholar]
- 20b. Donner A. J., Bell T. A., Greenlee S., Graham M. J., Crooke R. M., Nucleic Acid Ther. 2018, 28, 297–306. [DOI] [PubMed] [Google Scholar]
- 21. Wang L., Nam Y., Lee A. K., Yu C., Roth K., Chen C., Ransey E. M., Sliz P., Cell Rep. 2017, 18, 2664–2675. [DOI] [PubMed] [Google Scholar]
- 22. Wang L., Rowe R. G., Jaimes A., Yu C., Nam Y., Pearson D. S., Zhang J., Xie X., Marion W., Heffron G. J., Daley G. Q., Sliz P., Cell Rep. 2018, 23, 3091–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Schapira M., Calabrese M. F., Bullock A. N., Crews C. M., Nat. Rev. Drug Discovery 2019,18, 949–963; [DOI] [PubMed] [Google Scholar]
- 23b. Collins, Wang H., Caldwell J. J., Chopra R., Biochem. J. 2017, 474, 1127–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Paiva S.-L., Crews C. M., Curr. Opin. Chem. Biol. 2019, 50, 111–119; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Zoppi V., Hughes S. J., Maniaci C., Testa A., Gmaschitz T., Wieshofer C., Koegl M., Riching K. M., Daniels D. L., Spallarossa A., Ciulli A., J. Med. Chem. 2019, 62, 699–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.
- 25a. Epstein A. C. R., Gleadle J. M., McNeill L. A., Hewitson K. S., O'Rourke J., Mole D. R., Mukherji M., Metzen E., Wilson M. I., Dhanda A., Tian Y.-M., Masson N., Hamilton D. L., Jaakkola P., Barstead R., Hodgkin J., Maxwell P. H., Pugh C. W., Schofield C. J., Ratcliffe P. J., Cell 2001, 107, 43–54; [DOI] [PubMed] [Google Scholar]
- 25b. Jaakkola P., Mole D. R., Tian Y.-M., Wilson M. I., Gielbert J., Gaskell S. J., von Kriegsheim A., Hebestreit H. F., Mukherji M., Schofield C. J., Maxwell P. H., Pugh C. W., Ratcliffe P. J., Science 2001, 292, 468–472. [DOI] [PubMed] [Google Scholar]
- 26. Corson T. W., Aberle N., Crews C. M., ACS Chem. Biol. 2008, 3, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Cantrill C., Chaturvedi P., Rynn C., Petrig Schaffland J., Walter I., Wittwer M. B., Drug Discovery Today 2020, 25, 969–982; [DOI] [PubMed] [Google Scholar]
- 27b. Gadd M. S., Testa A., Lucas X., Chan K.-H., Chen W., Lamont D. J., Zengerle M., Ciulli A., Nat. Chem. Biol. 2017, 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Bargagna-Mohan P., Baek S.-H., Lee H., Kim K., Mohan R., Bioorg. Med. Chem. Lett. 2005, 15, 2724–2727; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Sakamoto K. M., Kim K. B., Kumagai A., Mercurio F., Crews C. M., Deshaies R. J., Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28c. Schneekloth J. S., Fonseca F. N., Koldobskiy M., Mandal A., Deshaies R., Sakamoto K., Crews C. M., J. Am. Chem. Soc. 2004, 126, 3748–3754. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Miller C. M., Wan W. B., Seth P. P., Harris E. N., Nucleic Acid Ther. 2018, 28, 86–96; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29b. Miller C. M., Donner A. J., Blank E. E., Egger A. W., Kellar B. M., Østergaard M. E., Seth P. P., Harris E. N., Nucleic Acids Res. 2016, 44, 2782–2794; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29c. Juliano R. L., Ming X., Carver K., Laing B., Nucleic Acid Ther. 2014, 24, 101–113; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29d. Juliano R. L., Nucleic Acid Ther. 2018, 28, 166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Soifer H. S., Koch T., Lai J., Hansen B., Hoeg A., Oerum H., Stein C. A., Methods Mol. Biol. 2012, 815, 333–346. [DOI] [PubMed] [Google Scholar]
- 31. Crooke S. T., Baker B. F., Pham N. C., Hughes S. G., Kwoh T. J., Cai D., Tsimikas S., Geary R. S., Bhanot S., Nucleic Acid Ther. 2018, 28, 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Smith B. E., Wang S. L., Jaime-Figueroa S., Harbin A., Wang J., Hamman B. D., Crews C. M., Nat. Commun. 2019, 10, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Emmerich C. H., Cohen P., Biochem. Biophys. Res. Commun. 2015, 466, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang J., Ratanasirintrawoot S., Chandrasekaran S., Wu Z., Ficarro S. B., Yu C., Ross C. A., Cacchiarelli D., Xia Q., Seligson M., Shinoda G., Xie W., Cahan P., Wang L., Ng S.-C., Tintara S., Trapnell C., Onder T., Loh Y.-H., Mikkelsen T., Sliz P., Teitell M. A., Asara J. M., Marto J. A., Li H., Collins J. J., Daley G. Q., Cell Stem Cell 2016, 19, 66–80. [DOI] [PubMed] [Google Scholar]
- 35.
- 35a. Viswanathan S. R., Powers J. T., Einhorn W., Hoshida Y., Ng T. L., Toffanin S., O'Sullivan M., Lu J., Phillips L. A., Lockhart V. L., Shah S. P., Tanwar P. S., Mermel C. H., Beroukhim R., Azam M., Teixeira J., Meyerson M., Hughes T. P., Llovet J. M., Radich J., Mullighan C. G., Golub T. R., Sorensen P. H., Daley G. Q., Nat. Genet. 2009, 41, 843–848; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35b. Balzeau J., Menezes M. R., Cao S., Hagan J. P., Front. Genet. 2017, 10.3389/fgene.2017.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Towbin H., Wenter P., Guennewig B., Imig J., Zagalak J. A., Gerber A. P., Hall J., Nucleic Acids Res. 2013, 41, e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.
- 37a. Piskounova E., Polytarchou C., Thornton J. E., LaPierre R. J., Pothoulakis C., Hagan J. P., Iliopoulos D., Gregory R. I., Cell 2011, 147, 1066–1079; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37b. Balzer E., Moss E. G., RNA Biol. 2007, 4, 16–25. [DOI] [PubMed] [Google Scholar]
- 38. Maniaci C., Hughes S. J., Testa A., Chen W., Lamont D. J., Rocha S., Alessi D. R., Romeo R., Ciulli A., Nat. Commun. 2017, 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shibata H., Huynh D. P., Pulst S.-M., Hum. Mol. Genet. 2000, 9, 1303–1313. [DOI] [PubMed] [Google Scholar]
- 40.
- 40a. Hamada N., Ito H., Nishijo T., Iwamoto I., Morishita R., Tabata H., Momiyama T., Nagata K.-I., Sci. Rep. 2016, 6, 30805; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40b. Gehman L. T., Stoilov P., Maguire J., Damianov A., Lin C.-H., Shiue L., Ares M., Mody I., Black D. L., Nat. Genet. 2011, 43, 706–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.
- 41a. Jin Y., Suzuki H., Maegawa S., Endo H., Sugano S., Hashimoto K., Yasuda K., Inoue K., EMBO J. 2003, 22, 905–912; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41b. Zhang C., Zhang Z., Castle J., Sun S., Johnson J., Krainer A. R., Zhang M. Q., Genes Dev. 2008, 22, 2550–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cieniková Z., Damberger F. F., Hall J., Allain F. H. T., Maris C., J. Am. Chem. Soc. 2014, 136, 14536–14544. [DOI] [PubMed] [Google Scholar]
- 43. Malinowska A. L., Hall J., Advances in Nucleic Acid Therapeutics, The Royal Society of Chemistry, London, 2019, pp. 151–180. [Google Scholar]
- 44.
- 44a. Crinelli R., Bianchi M., Gentilini L., Magnani M., Nucleic Acids Res. 2002, 30, 2435–2443; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44b. An H.-J., Gwon M.-G., Gu H., Kim H.-J., Park K.-K., Kim J.-Y., Leem J., Youn S. W., Int. J. Mol. Sci. 2020, 21, 0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44c. Denichenko P., Mogilevsky M., Cléry A., Welte T., Biran J., Shimshon O., Barnabas G. D., Danan-Gotthold M., Kumar S., Yavin E., Levanon E. Y., Allain F. H., Geiger T., Levkowitz G., Karni R., Nat. Commun. 2019, 10, 1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.
- 45a. Turner J. J., Arzumanov A. A., Gait M. J., Nucleic Acids Res. 2005, 33, 27–42; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45b. Halloy F., Iyer P. S., Ćwiek P., Ghidini A., Barman-Aksözen J., Wildner-Verhey van Wijk N., Theocharides A. P. A., Minder E. I., Schneider-Yin X., Schümperli D., Hall J., Nucleic Acids Res. 2020, 48, 4658–4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary