

Abstract
Several genome wide association studies of colorectal cancer (CRC) have identified single nucleotide polymorphisms (SNPs) on chromosome 15q13.3 associated with CRC risk. To identify functional variant(s) underlying this association, we investigated SNPs in linkage disequilibrium with the risk‐associated SNP rs4779584 that overlapped regulatory regions/enhancer elements characterized in colon‐related tissues and cells. We identified several SNP‐containing regulatory regions that exhibited enhancer activity in vitro, including one SNP (rs1406389) that correlated with allele‐specific effects on enhancer activity. Deletion of either this enhancer or another enhancer that had previously been reported in this region correlated with decreased expression of GREM1 following CRISPR/Cas9 genome editing. That GREM1 is one target of these enhancers was further supported by an expression quantitative trait loci correlation between rs1406389 and GREM1 expression in the transverse but not sigmoid colon in the Genotype‐Tissue Expression dataset. Taken together, we conclude that the 15q13.3 region contains at least two functional variants that map to distinct enhancers and impact CRC risk through modulation of GREM1 expression.
Keywords: colorectal cancer, enhancers, genomics, GWAS, SNP, SNV
1. INTRODUCTION
Genome wide association studies (GWAS) were designed to help elucidate “missing heritability” and in doing so lead to the identification of novel genes involved in disease risk, including colorectal cancer (CRC). For example, a single nucleotide polymorphism (SNP) associated with CRC risk identified on 11q23.1 led to the identification of previously uncharacterized genes COLCA1 and COLCA2 (Biancolella et al., 2014; Peltekova et al., 2014). However, discovering the biological mechanism of SNPs underlying CRC risk remains a challenge because few risk‐associated variants are themselves functional/causal and most functional/causal variants map to regulatory regions such as enhancers that in turn regulate expression of critical target genes. The emerging picture is that many functional/causal SNPs associated with CRC risk map to enhancers and lead to altered gene expression (Chen et al., 2019; Corradin et al., 2014; Xia & Wei, 2019).
A high proportion of CRC GWAS loci contain members of the bone morphogenetic proteins (BMP)/transforming growth factor beta (TGFβ) signaling pathways, for example, TGFB1, SMAD7, BMP4, BMP2, and GREM1 (Broderick et al., 2007; COGENT Study et al., 2008; Huyghe et al., 2019; Jia et al., 2013; Kupfer et al., 2014; Law et al., 2019; Peters et al., 2012, 2013; Schmit et al., 2019; Tenesa et al., 2008; Tomlinson et al., 2008; Whiffin et al., 2014; Zhang et al., 2014; 2014). While the role of BMP/TGFβ signaling has long been implicated in CRC development, validating the role of genes within these signaling pathways as target genes of risk enhancers is challenging, regardless of the proximity to the associated variant. For example, the risk association at chromosome 18q21.1, intronic to SMAD7, involves multiple functional SNPs that can affect activity of an enhancer element that in turn correlates with altered SMAD7 transcription levels (Fortini et al., 2014).
Several GWAS have implicated SNP rs4779584 (NC_000015.10:g.32702555T>C) on chromosome 15q13.3 in risk of CRC (Jaeger et al., 2008; Middeldorp et al., 2009; Peters et al., 2012, 2013; Tanskanen et al., 2018; Whiffin et al., 2014), and studies have shown rs4779584 to be associated with earlier onset of CRC (Giráldez et al., 2012), serrated polyps (Hang et al., 2019), microsatellite stable CRC (Lubbe et al., 2012), and, perhaps surprisingly, reduced risk of death from CRC (Xing et al., 2011). The nearest gene mapping to rs4779584 is the BMP antagonist GREM1. Tomlinson et al. (2011) performed fine‐mapping to further explore the GWAS signal on 15q13.3 and identified a neighboring SNP, rs16969681 (NC_000015.10:g.32700910C>T; r 2 = .2, CEU), that was more highly associated with CRC than rs4779584, although it did not account for the total risk burden associated with rs4779584 (Tomlinson et al., 2011). The intergenic region surrounding rs4779584 is duplicated in four different known copy number variants associated with hereditary mixed polyposis syndrome (HMPS), implicating a regulatory element linked to GREM1 expression levels in the etiology of HMPS (Jaeger et al., 2012; McKenna et al., 2019; Rohlin et al., 2016; Venkatachalam et al., 2011). In the duplicated region, a candidate enhancer element containing both rs4779584 and rs16969681 was found to physically interact with the promoter of GREM1, and follow up studies determined that rs16969681 disrupted CDX2 and TCF7L2 transcription factor binding sites (Lewis et al., 2014). In a separate study using imputation and fine mapping, Whiffin et al. (2014) found the highest risk association with rs1406389 (NC_000015.10:g.32717277A>T), a SNP in high linkage disequilibrium (LD) with rs4779584 (Whiffin et al., 2013). These data suggest that there may be additional functional/causal variants within this region.
To identify novel functional variants, we characterized regulatory regions/enhancers across the chromosome 15q13.3 GWAS region. Incorporating ChIP‐seq profiles of colon tissues and cell lines, we identified several candidate enhancer elements that contained SNPs in LD with the risk associated SNP rs4779584. In addition to the enhancer element containing rs16969681, we identified a novel enhancer element, the activity of which was found to be modulated by rs1406389 in in vitro enhancer activity assays. To determine the gene target(s) of these enhancer elements, we used CRISPR/Cas9 genome editing to delete the enhancer regions, and examined the correlation between these SNPs and expression of nearby genes including GREM1 and FMN1. Deletion of either risk enhancer correlated with reduced GREM1 gene expression but not expression of FMN1. Taken together, these data suggest that the associated risk for CRC on 15q13.3 is due to functional/causal variants that map to at least two enhancer elements that in turn modulate GREM1 gene expression.
2. MATERIALS AND METHODS
2.1. Cell culture
HCT116, SW480, RKO, and SW948 CRC cell lines were obtained from the American Type Culture Collection (ATCC). HCT116, SW480, and SW948 cells were grown in McCoy's 5A (Mediatech) supplemented with 10% fetal bovine serum (Omega Scientific, Inc.), and 1% Penicillin/Streptomycin, and incubated at 37°C and 5% CO2. RKO cells were grown in DMEM (Mediatech) supplemented with 10% fetal bovine serum (Omega Scientific, Inc.), and 1% Penicillin/Streptomycin, and incubated at 37°C and 5% CO2.
2.2. Plasmids and luciferase assays
DNA fragments corresponding to the candidate enhancer regions were PCR amplified from normal human genomic DNA, and subcloned into a Sac II restriction enzyme site (in both directions) upstream of a thymidine kinase (TK) minimal promoter‐firefly‐luciferase vector (courtesy of Dr. G. A. Coetzee, Van Andel Research Institute) using CloneAmp HiFi PCR Premix and the In‐Fusion HD cloning kit (Clontech). See Table S1 for genomic coordinates and primer sequences. Fragments were cloned using the In‐Fusion HD cloning kit (Takara). Plasmid clones were sequenced by Sanger sequencing (Genewiz) to confirm the presence of the candidate variants and the absence of any PCR amplification‐induced mutations.
A region of Chr8q24 previously shown to have no activity in any of the cell lines served as the negative control, and a region of Chr8q24 that was previously shown to have enhancer activity in all of the cell lines served as the positive control. For enhancer assays SW480 (10 × 104 cells/well), HCT116 and RKO (ATCC) cells (6 × 104 cells/well) were seeded into 96‐well plates. Cells were cotransfected with reporter plasmids and constitutively active pRL‐TK Renilla luciferase plasmid (Promega) using Lipofectamine 2000 Reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. After 24 h cells were harvested and extracts were assayed for luciferase activity using the Dual‐ Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions, and measured using a Tecan Infinite F200Pro Microplate Reader. The ratio of normalized luminescence from the experimental sample to the negative control reporter was calculated for each sample, and defined as the relative luciferase activity. Luciferase activity was tested in a minimum of three independent clones for each allele. The data are presented as mean ±SD of four independent transfection experiments. Two‐side p values between alleles were calculated using Student's t test.
2.3. Linkage disequilibrium
Lists of SNPs in LD (r 2 >.2) were generated using rAggr (raggr.usc.edu) based on 1000 Genomes Project (ph 2) and HapMap Project genotype databases in the CEU population.
2.4. Expression quantitative trait loci (eQTL) analysis
Initial eQTL data were identified through the Genotype‐Tissue Expression (GTEx) portal on 07/18/2019 (v7 data release). Given the strength of our experimental data, targeted hypothesis and lack of multiple testing, we chose to use a nominally significant threshold (p = .05) to test for the effect of genetic variation at these two SNPs GREM1 expression. Additional eQTL data were identified through the GTEx portal on 8/10/2020 (v8 data release).
2.5. CRISPR/Cas9 genome editing
Upstream and downstream CRISPR gRNAs were designed flanking the two enhancer elements using http://crispr.mit.edu (chr15:32992304‐32994184, chr15:33008912‐33009921), and cloned into the GeneArt OFP reporter (Thermo Fisher Scientific) and pSpCas9(BB)‐2A‐GFP (AddGene). Vectors were cotransfected into SW948 using Lipofectamine 3000. Cells were sorted for green and orange coexpression using a MoFlo Astrios FACS and expanded for 3 weeks before DNA and RNA harvesting. Genomic DNA was purified using Qiagen Robot EZ1 Tissue protocol and enhancer deletion was confirmed with internal and external PCR amplifications. RNA was purified from three samples using Trizol following manufacturer's instructions. One microgram of RNA was used in the High Capacity RNA‐to‐cDNA kit and amplified with GREM1, FMN1, and GUSB (endogenous control) TaqMan assays (Thermo Fisher Scientific), in triplicate for each RNA preparation on an AB 7900HT RT‐PCR system. The qPCR was performed twice and analyzed with SDS v2.4, RQManager v1.2.1, Expression Suite v1.0.3 (Thermo Fisher Scientific).
3. RESULTS
3.1. Region analysis
Several GWAS have shown chromosome 15q13.3 to be associated with CRC risk (Peters et al., 2012, 2013; Schmit et al., 2019; Tomlinson et al., 2008; Whiffin et al., 2014). The region on chromosome 15, tagged by the SNP rs4779584 and defined using an r 2 cutoff of .2 in the CEU population, is 185 kb in length and includes 45 SNVs in LD with rs4779584 (Figure S1). Of those SNPs, 25 were in LD with an r 2 value greater than .5 across 31 kb (Figure 1). To identify candidate functional SNPs, we overlaid the SNPs in LD with rs4779584 with ChIP‐seq tracks of histone marks H3K4 monomethylation or H3K27 acetylation, histone marks commonly associated with enhancer elements. As in our previous studies (Biancolella et al., 2014; Fortini et al., 2014), we referenced ChIP‐seq data derived from normal colon crypts (Cohen et al., 2017) and CRC cell lines (Akhtar‐Zaidi et al., 2012). We identified six putative enhancer elements defined by histone ChIP‐seq peaks that contained at least one SNP in LD with SNP rs4779584 of r 2 more than .62 (CEU population; Figure 1).
Figure 1.
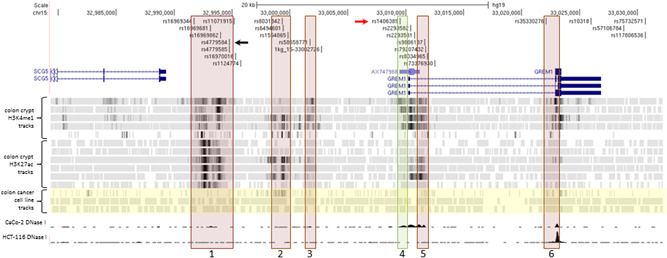
Region of chromosome 15 associated with CRC risk. A detailed view of the region between genes SCG5 and GREM1 showing putative enhancer elements containing SNPs in LD (r 2 >.2 CEU population) with rs4779584 (black arrow). Red boxes denote regions with enhancer activity in luciferase assays that is unaffected by haplotype of the encompassed SNPs, including the previously characterized region 2. Green box denotes allele‐specific enhancer region 4, including rs1406389 (red arrow). Histone ChIP‐seq tracks for H3K4me1 and H3K27ac modifications from individual normal colon crypts aligned below genes indicate potential enhancer elements (Cohen et al., 2017). Highlighted in yellow are ChIP‐seq tracks from CRC cell lines SW480 H3K4me1, HCT116 H3K4me1, and HCT116 H3K27ac. Bottom two tracks are CRC cell lines CaCo‐2 and HCT116 DNase 1 hypersensitivity tracks from the UW ENCODE group showing regions of open chromatin in their respective cell lines (Davis et al., 2018; The ENCODE Project Consortium, 2012). Figure S1 for the complete region defined by linkage disequilibrium (LD) block with lead SNP rs4779584, r 2 more than .2 CEU population. CRC, colorectal cancer; SNP, single nucleotide polymorphism
The majority of the six putative enhancers lie in the intergenic region between genes SCG5 and GREM1, while two putative enhancers are intronic of GREM1. Although the 3′ end of the gene FMN1 lies within the LD region, none of the SNPs in LD fall inside putative enhancers within the boundaries of the FMN1 gene. Importantly, the majority of the six putative enhancers were identified through analysis of ChIP‐seq data from normal colon crypt samples rather than CRC cell lines, highlighting the importance of using physiologically relevant normal tissue samples in addition to cancer cell lines for post‐GWAS functional follow up analyses.
3.2. Candidate enhancer region characterization
All six putative enhancers shown in Figure 1 were cloned into vectors (Table S1) and tested for enhancer activity in cell‐based luciferase assays. Vector constructs were then transfected into CRC cell lines RKO, HCT116, SW480, and SW948. All six regions demonstrated enhancer activity in SW948 in at least one orientation (Figure S2A). Additionally, at least one orientation was positive in one or more of the three other CRC cell lines. QuickChange mutagenesis was used to create constructs containing the common haplotypes of each of the SNPs encompassed in the enhancer fragments for haplotypes not captured during the cloning process. The region 4 enhancer that lies upstream of the GREM1 promoter contained one linked SNP, rs1406389. As shown in Figure 2, luciferase activity in the forward direction showed allele specific effects on enhancer activity, with the A allele fragment demonstrating higher activity than the T allele fragment in all of the cell lines tested (Figure 2).
Figure 2.
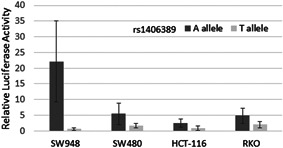
Allele‐specific enhancer activity. Enhancer depicted as region 4 in Figure 1 was cloned into a luciferase enhancer assay construct including SNP rs1406389 alleles A and T. The construct with the A allele demonstrated statistically significantly higher activity than the T allele (p values for SW948 = 5.77 × 10−6, SW480 = 1.12 × 10−3, HCT116 = 3.20 × 10−4, RKO = 1.58 × 10−6 were calculated using the two‐sided Student's t test). SNP, single nucleotide polymorphism
We found no statistically significant difference between the enhancer activities of the respective haplotype constructs for candidate enhancer regions 1, 2, 3, 5, and 6. Note that this also includes candidate enhancer region 1, which contains five SNPs including the SNP rs4779584 that had been characterized in detail in previous studies (Lewis et al., 2014). In this region, allele‐specific effects were assessed alternating the alleles of SNP rs16969681 alone or the two major haplotypes of rs16969344 (NC_000015.10:g.32699512C>G), rs16969681 (NC_000015.10:g.32700910C>T), rs16969862, rs4779584, and rs4779585 (NC_000015.10:g.32702576C>G) but no differences reached the threshold for statistical significance (Figure S2B).
3.3. CRISPR/Cas9 enhancer disruption
To provide additional evidence that GREM1 is a target of these enhancers, we used CRISPR/Cas9 genome editing of both the region 1 (containing rs16969681) and region 4 enhancers (containing rs1406389). The CRC line SW948 was chosen for these experiments as it expressed detectable levels of GREM1 using qPCR. Two guide RNA sequences were designed to cut at the boundaries of the enhancers, as depicted by the boxes in Figure 1 that had been tested in the luciferase assays (see Figure 2). Each gRNA sequence was cloned into a CRISPR vector containing the Cas9 gene and either a green or orange fluorescent protein marker gene. After transfection, SW948 cells were sorted for green and orange fluorescence and passaged as pools of transfected cells. These pools were assessed for deletion of the enhancer sequences by PCR with primers internal and external to the desired DNA break points. Both pools showed that a large fraction of cells contained disruption of the relevant sequence (Figure S3). RNA was purified from the same cellular pools and expression of the genes GREM1 and the nearby gene FMN1 were determined using TaqMan qPCR gene expression assays relative to cells grown and processed side by side with a mock CRISPR/Cas9 deletion. FMN1 was chosen as a control gene as it was not expected to be affected by deletion of either enhancer based on our eQTL analyses.
As shown in Figure 3, disruption of the region 1 enhancer resulted in a statistically significant reduction in GREM1 transcript levels, while not significantly altering FMN1 expression. This pattern was also seen with the disruption of the novel region 4 enhancer, although the reduction in GREM1 expression was not as great as that seen following deletion of the region 1 enhancer. Again, there was no significant change in FMN1 expression. It is important to note that the gene expression changes were detectable in a pooled population of cells containing a mix of deleted and intact enhancers.
Figure 3.
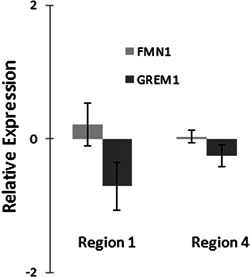
Gene expression changes due to genome editing of enhancers. Genomic regions corresponding to enhancer regions 1 and 4 were targeted for deletion in SW948 cells using CRISPR/Cas9 technology. Pools of transfected cells were analyzed using qPCR and Taqman gene expression assays for GREM1, FMN1, and GUSB (control). Targeting of both regions 1 and 4 enhancers resulted in a decrease in GREM1 expression levels, while FMN1 expression levels did not change significantly. qPCR, quantitative polymerase chain reaction
3.4. eQTL analysis in normal colon tissue
We next determined whether the candidate functional SNPs in region 1 (rs16969681) or region 4 enhancers (rs1406389) correlated with gene expression levels of nearby genes in healthy colon tissue. We examined the correlation between these SNPs and the expression of GREM1, FMN1, SCG5 in normal colon tissues in the GTEx dataset (v7 data release; GTEx Consortium et al., 2017). We found that rs1406389 was nominally significantly associated with GREM1 expression in transverse (p = 6.00E−03), but not sigmoid colon (Figure 4), with the T allele correlating with reduced expression of GREM1. No significant associations were seen between rs1406389 and FMN1 or SCG5 gene expression in either tissue (data not shown). This SNP was also found to be nominally significantly associated with GREM1 expression in other tissues in the gastrointestinal tract including the gastroesophageal junction of the esophagus (p = .05), and the terminal ileum of the small intestine (p = .03). Importantly, a recent meta‐analysis of this SNP indicates a highly significant, multitissue effect on GREM1 expression (RE2 p = 8.65 E−12), with transverse colon having the greatest m‐value (0.911), a measure of posterior probability for that SNP being an eQTL in that tissue (Han & Eskin, 2011). We found no evidence for a correlation between rs16969681 and gene expression of any of these genes in colon or any gastrointestinal‐related tissues. In the most recent GTEx data release (v8), there are no significant eQTL relationships with any SNPs and the GREM1 gene, including rs1406389 in either transverse or sigmoid colon. However, rs1406389 and rs4779584 exhibit strong eQTLs with GREM1 in liver (p values of 1.10E−15 and 2.70E−16, respectively; Table S2). Additional studies will be required to further elucidate the effects of these SNPs on the expression of GREM1 in normal colon tissues and colon tumors.
Figure 4.
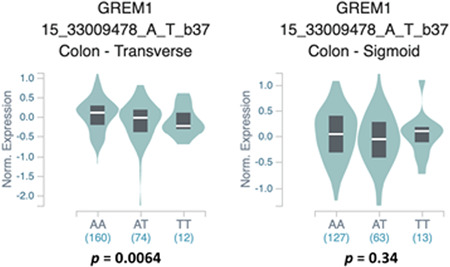
Violin plots showing the correlation between normalized GREM1 gene expression and rs1406389 genotype. The T allele was significantly correlated with reduced GREM1 expression in transverse colon but not sigmoid colon. Data were generated using the Single‐Tissue eQTLs function in the GTEx Data Portal. eQTL, expression quantitative trait loci; GTEx, Genotype‐Tissue Expression
4. DISCUSSION
Elucidation of the role of SNPs residing in enhancer elements on the control of GREM1 gene expression in colon cells is important for understanding the mechanism of CRC risk conferred by these single nucleotide germline variants. Colon epithelium cell identities are determined along the colon crypt axis through morphogen gradients of WNT (wingless‐type MMTV integration site family) and BMP/TGFβ proteins. These gradients are maintained with the help of BMP antagonists including GREMLIN1. In normal colon tissues, GREM1 is expressed in the myofibroblasts around the base of colon crypts, which in turn affects cells within the stem cell niche at the base of the crypt (Kosinski et al., 2007). This fact may complicate the analysis of eQTLs in colon tissue, since tissue preparation of samples may collect only the colon epithelial cells of the colon crypts. Expression of GREM1 in intestinal epithelia can cause cells outside of the crypt base to acquire stem‐like properties leading to tumorigenesis in mice (Davis et al., 2015). Studies have also linked GREM1 expression with epithelial to mesenchymal transition in CRC invasion fronts, and thus with CRC progression (Karagiannis et al., 2015).
Duplication of the region upstream of GREM1 leads to HMPS in humans, due to aberrant expression of GREM1 in epithelial tissues (Jaeger et al., 2012; McKenna et al., 2019; Rohlin et al., 2016; Venkatachalam et al., 2011). Here, we extend on previous findings to show that these duplications include multiple enhancers modulating the expression of the GREM1 gene. It is perhaps therefore unsurprising that dysregulation of GREM1 may play an important role in CRC and thus that this region was identified in CRC GWAS. Despite this, identifying the underlying mechanistic basis of risk within this genomic region has remained somewhat elusive and has revealed the genetic complexity of CRC risk.
Fine‐mapping studies have been helpful in identifying putative functional SNPs for a handful of GWAS risk loci for CRC. Whiffin, et al reported that the top SNP associated with CRC risk was rs1406389 (Whiffin et al., 2013). The lead SNP in the region, rs4779584 was also one of the top 10 associated SNPs in this study. Of the other eight SNPs with the ten lowest p values for association with CRC, we found six mapping to enhancer regions that we investigated in our study. While we did not see allele specific effects on enhancer activity in our cell‐based experiments for the majority of these SNPs, including SNPs within a previously reported CRC enhancer (Tomlinson et al., 2011), we cannot rule out that, in the context of the genome, they may have subtle effects on GREM1 expression, and thus perhaps belong to a physiologically relevant risk haplotype.
Indeed, while we observed no allele specific effects on enhancer activity that reached statistical significance for SNP rs16969681 alone or the two major haplotypes of rs16969344, rs16969681, rs16969862, rs4779584, and rs4779585 using in vitro enhancer activity assays, we did observe an effect on GREM1 expression following deletion of the entire enhancer containing this SNP, as well as the novel enhancer that we report here, following CRISPR/Cas9 genome editing. It is also interesting note that while rs4779584 and rs1406389 demonstrate strong eQTLs in liver tissue in the latest GTEx data release, the GREM1 region has not been associated with hepatocellular carcinoma in any published GWAS studies to date (Buniello et al., 2019). These data further emphasize the fact that multiple functional assays may be needed to identify functional/causal variants involved in risk of CRC and other genetic diseases. In future studies, it may be possible to leverage the differences in LD structure in populations in addition to the CEU cohort to refine the list of candidate SNPs by comparing the major haplotypes and the CRC risk conferred by the GREM1 region in those populations and thus help establish whether the risk signals are due to causal effects or merely correlation because of LD.
It is important to note that the allele of rs1406389 correlated with CRC risk by GWAS shows lower levels of GREM1 expression in colon tissues and enhancer activity in in vitro assays, while increased copy numbers of GREM1 enhancers are associated with HMPS. It is yet unclear how individual enhancer SNPs in the germline affect overall tissue expression levels over the course of development including in cancerous lesions. Using in silico predictions of transcription factor binding using the TRANSFAC database, candidate transcription factors that may preferentially bind the A allele over the T allele include glucocorticoid receptor (GRβ), FOXA1, and XBP1 (Farré et al., 2003; Messeguer et al., 2002).
Currently, GWAS findings still account for only a relatively small amount of heritability associated with CRC. This is due in part to the fact that there are likely many more risk associations left to discover. It also relates to the fact that few associated variants identified through GWAS are themselves functional. These data presented here, combined with our studies on CRC GWAS loci 11q23.1 and 18q21.1, reveal that a growing number of GWAS risk loci contain multiple functional/causal variants, implying that comprehensive analyses of GWAS regions needs to be performed to fully characterize the underlying mechanistic and biological impact of risk association and the cumulative effect of multiple risk enhancers will undoubtedly help improve our understanding of the biologic mechanisms underlying the total genetic risk.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
This work was supported by NIH/NCI grants CA143237 (GC) and CA204279 (GC). The Genotype‐Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript was obtained from: the GTEx Portal on 07/18/2019. The NHGRI‐EBI GWAS Catalog is funded by NHGRI Grant Number 2U41HG007823, and delivered by collaboration between the NHGRI, EMBL‐EBI, and NCBI. GWAS Catalog data was accessed for this manuscript on 08/09/2020. This study was approved by the Institutional Review Boards of the University of Southern California and the University of Virginia.
Fortini BK, Tring S, Devall MA, Ali MW, Plummer SJ, Casey G. SNPs associated with colorectal cancer at 15q13.3 affect risk enhancers that modulate GREM1 gene expression. Human Mutation. 2021;42:237–245. 10.1002/humu.24166
DATA AVAILABILITY STATEMENT
Data that support the findings of this study are available in the Gene Expression Omnibus, reference number GSE77737 (Cohen et al., 2017) and from the ENCODE Project (https://www.encodeproject.org/; Davis et al., 2018; The ENCODE Project Consortium, 2012), reference numbers ENCSR000ENM and ENCSR000EMI, accessed using the UCSC Genome Browser. All other data are contained within the paper.
REFERENCES
- Akhtar‐Zaidi, B. , Cowper‐Sal‐lari, R. , Corradin, O. , Saiakhova, A. , Bartels, C. F. , Balasubramanian, D. , Myeroff, L. , Lutterbaugh, J. , Jarrar, A. , Kalady, M. F. , Willis, J. , Moore, J. H. , Tesar, P. J. , Laframboise, T. , Markowitz, S. , Lupien, M. , & Scacheri, P. C. (2012). Epigenomic enhancer profiling defines a signature of colon cancer. Science (New York, N.Y.), 336(6082), 736–739. 10.1126/science.1217277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancolella, M. , Fortini, B. K. , Tring, S. , Plummer, S. J. , Mendoza‐Fandino, G. A. , Hartiala, J. , Hitchler, M. J. , Yan, C. , Schumacher, F. R. , Conti, D. V. , Edlund, C. K. , Noushmehr, H. , Coetzee, S. G. , Bresalier, R. S. , Ahnen, D. J. , Barry, E. L. , Berman, B. P. , Rice, J. C. , Coetzee, G. A. , & Casey, G. (2014). Identification and characterization of functional risk variants for colorectal cancer mapping to chromosome 11q23.1. Human Molecular Genetics, 23(8), 2198–2209. 10.1093/hmg/ddt584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick, P. , Carvajal‐Carmona, L. , Pittman, A. M. , Webb, E. , Howarth, K. , Rowan, A. , Lubbe, S. , Spain, S. , Sullivan, K. , Fielding, S. , Jaeger, E. , Vijayakrishnan, J. , Kemp, Z. , Gorman, M. , Chandler, I. , Papaemmanuil, E. , Penegar, S. , Wood, W. , Sellick, G. , … Houlston, R. S. (2007). A genome‐wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nature Genetics, 39(11), 1315–1317. 10.1038/ng.2007.18 [DOI] [PubMed] [Google Scholar]
- Buniello, A. , MacArthur, J. A. L. , Cerezo, M. , Harris, L. W. , Hayhurst, J. , Malangone, C. , McMahon, A. , Morales, J. , Mountjoy, E. , Sollis, E. , Suveges, D. , Vrousgou, O. , Whetzel, P. L. , Amode, R. , Guillen, J. A. , Riat, H. S. , Trevanion, S. J. , Hall, P. , Junkins, H. , … Parkinson, H. (2019). The NHGRI‐EBI GWAS Catalog of published genome‐wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Research, 47(D1), D1005–D1012. 10.1093/nar/gky1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. , Kichaev, G. , Bien, S. A. , MacDonald, J. W. , Wang, L. , Bammler, T. K. , Auer, P. , Pasaniuc, B. , & Lindström, S. (2019). Genetic associations of breast and prostate cancer are enriched for regulatory elements identified in disease‐related tissues. Human Genetics, 138(10), 1091–1104. 10.1007/s00439-019-02041-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- COGENT Study , Houlston, R. S. , Webb, E. , Broderick, P. , Pittman, A. M. , Di Bernardo, M. C. , Lubbe, S. , Chandler, I. , Vijayakrishnan, J. , Sullivan, K. , Penegar, S. , Colorectal Cancer Association Study Consortium, Carvajal‐Carmona, L. , Howarth, K. , Jaeger, E. , Spain, S. L. , Walther, A. , Barclay, E. , Martin, L. , & Dunlop, M. G. (2008). Meta‐analysis of genome‐wide association data identifies four new susceptibility loci for colorectal cancer. Nature Genetics, 40(12), 1426–1435. 10.1038/ng.262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, A. J. , Saiakhova, A. , Corradin, O. , Luppino, J. M. , Lovrenert, K. , Bartels, C. F. , Morrow, J. J. , Mack, S. C. , Dhillon, G. , Beard, L. , Myeroff, L. , Kalady, M. F. , Willis, J. , Bradner, J. E. , Keri, R. A. , Berger, N. A. , Pruett‐Miller, S. M. , Markowitz, S. D. , & Scacheri, P. C. (2017). Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nature Communications, 8(1), 1–13. 10.1038/ncomms14400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradin, O. , Saiakhova, A. , Akhtar‐Zaidi, B. , Myeroff, L. , Willis, J. , Cowper‐Sal lari, R. , Lupien, M. , Markowitz, S. , & Scacheri, P. C. (2014). Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Research, 24(1), 1–13. 10.1101/gr.164079.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, C. A. , Hitz, B. C. , Sloan, C. A. , Chan, E. T. , Davidson, J. M. , Gabdank, I. , Hilton, J. A. , Jain, K. , Baymuradov, U. K. , Narayanan, A. K. , Onate, K. C. , Graham, K. , Miyasato, S. R. , Dreszer, T. R. , Strattan, J. S. , Jolanki, O. , Tanaka, F. Y. , & Cherry, J. M. (2018). The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Research, 46(D1), D794–D801. 10.1093/nar/gkx1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, H. , Irshad, S. , Bansal, M. , Rafferty, H. , Boitsova, T. , Bardella, C. , Jaeger, E. , Lewis, A. , Freeman‐Mills, L. , Giner, F. C. , Rodenas‐Cuadrado, P. , Mallappa, S. , Clark, S. , Thomas, H. , Jeffery, R. , Poulsom, R. , Rodriguez‐Justo, M. , Novelli, M. , Chetty, R. , … Leedham, S. J. (2015). Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nature Medicine, 21(1), 62–70. 10.1038/nm.3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farré, D. , Roset, R. , Huerta, M. , Adsuara, J. E. , Roselló, L. , Albà, M. M. , & Messeguer, X. (2003). Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Research, 31(13), 3651–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortini, B. K. , Tring, S. , Plummer, S. J. , Edlund, C. K. , Moreno, V. , Bresalier, R. S. , Barry, E. L. , Church, T. R. , Figueiredo, J. C. , & Casey, G. (2014). Multiple functional risk variants in a SMAD7 enhancer implicate a colorectal cancer risk haplotype. PLoS One, 9(11), e111914 10.1371/journal.pone.0111914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giráldez, M. D. , López‐Dóriga, A. , Bujanda, L. , Abulí, A. , Bessa, X. , Fernández‐Rozadilla, C. , Muñoz, J. , Cuatrecasas, M. , Jover, R. , Xicola, R. M. , Llor, X. , Piqué, J. M. , Carracedo, A. , Ruiz‐Ponte, C. , Cosme, A. , Enríquez‐Navascués, J. M. , Moreno, V. , Andreu, M. , Castells, A. , & Castellví‐Bel, S. , Gastrointestinal Oncology Group of the Spanish Gastroenterological Association . (2012). Susceptibility genetic variants associated with early‐onset colorectal cancer. Carcinogenesis, 33(3), 613–619. 10.1093/carcin/bgs009 [DOI] [PubMed] [Google Scholar]
- GTEx Consortium Aguet, F. , Brown, A. , Castel, S. , et al. (2017). Genetic effects on gene expression across human tissues. Nature, 550(7675), 204–213. 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, B. , & Eskin, E. (2011). Random‐effects model aimed at discovering associations in meta‐analysis of genome‐wide association studies. The American Journal of Human Genetics, 88(5), 586–598. 10.1016/j.ajhg.2011.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang, D. , Joshi, A. D. , He, X. , Chan, A. T. , Jovani, M. , Gala, M. K. , Ogino, S. , Kraft, P. , Turman, C. , Peters, U. , Bien, S. A. , Lin, Y. , Hu, Z. , Shen, H. , Wu, K. , Giovannucci, E. L. , & Song, M. (2019). Colorectal cancer susceptibility variants and risk of conventional adenomas and serrated polyps: Results from three cohort studies. International Journal of Epidemiology, 49(1), 259–269. 10.1093/ije/dyz096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyghe, J. R. , Bien, S. A. , Harrison, T. A. , Kang, H. M. , Chen, S. , Schmit, S. L. , Conti, D. V. , Qu, C. , Jeon, J. , Edlund, C. K. , Greenside, P. , Wainberg, M. , Schumacher, F. R. , Smith, J. D. , Levine, D. M. , Nelson, S. C. , Sinnott‐Armstrong, N. A. , Albanes, D. , Alonso, M. H. , … Seminara, D. (2019). Discovery of common and rare genetic risk variants for colorectal cancer. Nature Genetics, 51(1), 76–87. 10.1038/s41588-018-0286-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger, E. , Leedham, S. , Lewis, A. , Segditsas, S. , Becker, M. , Cuadrado, P. R. , Davis, H. , Kaur, K. , Heinimann, K. , Howarth, K. , HMPS Collaboration East, J. , Taylor, J. , Thomas, H. , & Tomlinson, I. (2012). Hereditary mixed polyposis syndrome is caused by a 40‐kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nature Genetics, 44(6), 699–703. 10.1038/ng.2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger, E. , Webb, E. , Howarth, K. , Carvajal‐Carmona, L. , Rowan, A. , Broderick, P. , Walther, A. , Spain, S. , Pittman, A. , Kemp, Z. , Sullivan, K. , Heinimann, K. , Lubbe, S. , Domingo, E. , Barclay, E. , Martin, L. , Gorman, M. , Chandler, I. , Vijayakrishnan, J. , … Tomlinson, I. (2008). Common genetic variants at the CRAC1 (HMPS) locus on chromosome 15q13.3 influence colorectal cancer risk. Nature Genetics, 40(1), 26–28. 10.1038/ng.2007.41 [DOI] [PubMed] [Google Scholar]
- Jia, W.‐H. , Zhang, B. , Matsuo, K. , Shin, A. , Xiang, Y.‐B. , Jee, S. H. , Kim, D.‐H. , Ren, Z. , Cai, Q. , Long, J. , Shi, J. , Wen, W. , Yang, G. , Delahanty, R. J. , Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO)Colon Cancer Family Registry (CCFR) Ji, B.‐T. , Pan, Z.‐Z. , Matsuda, F. , … Zheng, W. (2013). Genome‐wide association analyses in East Asians identify new susceptibility loci for colorectal cancer. Nature Genetics, 45(2), 191–196. 10.1038/ng.2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis, G. S. , Musrap, N. , Saraon, P. , Treacy, A. , Schaeffer, D. F. , Kirsch, R. , Riddell, R. H. , & Diamandis, E. P. (2015). Bone morphogenetic protein antagonist gremlin‐1 regulates colon cancer progression. Biological Chemistry, 396(2), 163–183. 10.1515/hsz-2014-0221 [DOI] [PubMed] [Google Scholar]
- Kosinski, C. , Li, V. S. W. , Chan, A. S. Y. , Zhang, J. , Ho, C. , Tsui, W. Y. , Chan, T. L. , Mifflin, R. C. , Powell, D. W. , Yuen, S. T. , Leung, S. Y. , & Chen, X. (2007). Gene expression patterns of human colon tops and basal crypts and BMP antagonists as intestinal stem cell niche factors. Proceedings of the National Academy of Sciences, 104(39), 15418–15423. 10.1073/pnas.0707210104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupfer, S. S. , Skol, A. D. , Hong, E. , Ludvik, A. , Kittles, R. A. , Keku, T. O. , Sandler, R. S. , & Ellis, N. A. (2014). Shared and independent colorectal cancer risk alleles in TGFβ‐related genes in African and European Americans. Carcinogenesis, 35(9), 2025–2030. 10.1093/carcin/bgu088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law, P. J. , Timofeeva, M. , Fernandez‐Rozadilla, C. , Broderick, P. , Studd, J. , Fernandez‐Tajes, J. , Farrington, S. , Svinti, V. , Palles, C. , Orlando, G. , Sud, A. , Holroyd, A. , Penegar, S. , Theodoratou, E. , Vaughan‐Shaw, P. , Campbell, H. , Zgaga, L. , Hayward, C. , Campbell, A. , … Dunlop, M. G. (2019). Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nature Communications, 10(1), 1–15. 10.1038/s41467-019-09775-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, A. , Freeman‐Mills, L. , de la Calle‐Mustienes, E. , Giráldez‐Pérez, R. M. , Davis, H. , Jaeger, E. , Becker, M. , Hubner, N. C. , Nguyen, L. N. , Zeron‐Medina, J. , Bond, G. , Stunnenberg, H. G. , Carvajal, J. J. , Gomez‐Skarmeta, J. L. , Leedham, S. , & Tomlinson, I. (2014). A polymorphic enhancer near GREM1 influences bowel cancer risk through differential CDX2 and TCF7L2 binding. Cell Reports, 8(4), 983–990. 10.1016/j.celrep.2014.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubbe, S. J. , Whiffin, N. , Chandler, I. , Broderick, P. , & Houlston, R. S. (2012). Relationship between 16 susceptibility loci and colorectal cancer phenotype in 3146 patients. Carcinogenesis, 33(1), 108–112. 10.1093/carcin/bgr243 [DOI] [PubMed] [Google Scholar]
- McKenna, D. B. , Van Den Akker, J. , Zhou, A. Y. , Ryan, L. , Leon, A. , O'Connor, R. , Shah, P. D. , Rustgi, A. K. , & Katona, B. W. (2019). Identification of a novel GREM1 duplication in a patient with multiple colon polyps. Familial Cancer, 18(1), 63–66. 10.1007/s10689-018-0090-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messeguer, X. , Escudero, R. , Farré, D. , Núñez, O. , Martínez, J. , & Albà, M. M. (2002). PROMO: Detection of known transcription regulatory elements using species‐tailored searches. Bioinformatics, 18(2), 333–334. 10.1093/bioinformatics/18.2.333 [DOI] [PubMed] [Google Scholar]
- Middeldorp, A. , Jagmohan‐Changur, S. , van Eijk, R. , Tops, C. , Devilee, P. , Vasen, H. F. A. , Hes, F. J. , Houlston, R. , Tomlinson, I. , Houwing‐Duistermaat, J. J. , Wijnen, J. T. , Morreau, H. , & van Wezel, T. (2009). Enrichment of low penetrance susceptibility loci in a Dutch familial colorectal cancer cohort. Cancer Epidemiology, Biomarkers & Prevention, 18(11), 3062–3067. 10.1158/1055-9965.EPI-09-0601 [DOI] [PubMed] [Google Scholar]
- Peltekova, V. D. , Lemire, M. , Qazi, A. M. , Zaidi, S. H. E. , Trinh, Q. M. , Bielecki, R. , Rogers, M. , Hodgson, L. , Wang, M. , D'Souza, D. J. A. , Zandi, S. , Chong, T. , Kwan, J. Y. Y. , Kozak, K. , Borja, R. , Timms, L. , Rangrej, J. , Volar, M. , Chan‐Seng‐Yue, M. , … Hudson, T. J. (2014). Identification of genes expressed by immune cells of the colon that are regulated by colorectal cancer‐associated variants. International Journal of Cancer, 134(10), 2330–2341. 10.1002/ijc.28557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, U. , Hutter, C. M. , Hsu, L. , Schumacher, F. R. , Conti, D. V. , Carlson, C. S. , Edlund, C. K. , Haile, R. W. , Gallinger, S. , Zanke, B. W. , Lemire, M. , Rangrej, J. , Vijayaraghavan, R. , Chan, A. T. , Hazra, A. , Hunter, D. J. , Ma, J. , Fuchs, C. S. , Giovannucci, E. L. , … Casey, G. (2012). Meta‐analysis of new genome‐wide association studies of colorectal cancer risk. Human Genetics, 131(2), 217–234. 10.1007/s00439-011-1055-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, U. , Jiao, S. , Schumacher, F. R. , Hutter, C. M. , Aragaki, A. K. , Baron, J. A. , Berndt, S. I. , Bézieau, S. , Brenner, H. , Butterbach, K. , Caan, B. J. , Campbell, P. T. , Carlson, C. S. , Casey, G. , Chan, A. T. , Chang–Claude, J. , Chanock, S. J. , Chen, L. S. , Coetzee, G. A. , … Hsu, L. , Colon Cancer Family Registry and the Genetics and Epidemiology of Colorectal Cancer Consortium . (2013). Identification of genetic susceptibility loci for colorectal tumors in a genome‐wide meta‐analysis. Gastroenterology, 144(4), 799–807. 10.1053/j.gastro.2012.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlin, A. , Eiengard, F. , Lundstam, U. , Zagoras, T. , Nilsson, S. , Edsjo, A. , Pedersen, J. , Svensson, J. , Skullman, S. , Karlsson, B. G. , Bjork, J. , & Nordling, M. (2016). GREM1 and POLE variants in hereditary colorectal cancer syndromes. Genes, Chromosomes & Cancer, 55(1), 95–106. 10.1002/gcc.22314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmit, S. L. , Edlund, C. K. , Schumacher, F. R. , Gong, J. , Harrison, T. A. , Huyghe, J. R. , Qu, C. , Melas, M. , Van Den Berg, D. J. , Wang, H. , Tring, S. , Plummer, S. J. , Albanes, D. , Alonso, M. H. , Amos, C. I. , Anton, K. , Aragaki, A. K. , Arndt, V. , Barry, E. L. , … Taverna, D. (2019). Novel common genetic susceptibility loci for colorectal cancer. Journal of the National Cancer Institute, 111(2), 146–157. 10.1093/jnci/djy099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanskanen, T. , van den Berg, L. , Välimäki, N. , Aavikko, M. , Ness‐Jensen, E. , Hveem, K. , Wettergren, Y. , Bexe Lindskog, E. , Tõnisson, N. , Metspalu, A. , Silander, K. , Orlando, G. , Law, P. J. , Tuupanen, S. , Gylfe, A. E. , Hänninen, U. A. , Cajuso, T. , Kondelin, J. , Sarin, A.‐P. , … Aaltonen, L. A. (2018). Genome‐wide association study and meta‐analysis in Northern European populations replicate multiple colorectal cancer risk loci. International Journal of Cancer, 142(3), 540–546. 10.1002/ijc.31076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenesa, A. , Farrington, S. M. , Prendergast, J. G. D. , Porteous, M. E. , Walker, M. , Haq, N. , Barnetson, R. A. , Theodoratou, E. , Cetnarskyj, R. , Cartwright, N. , Semple, C. , Clark, A. J. , Reid, F. J. L. , Smith, L. A. , Kavoussanakis, K. , Koessler, T. , Pharoah, P. D. P. , Buch, S. , Schafmayer, C. , … Dunlop, M. G. (2008). Genome‐wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nature Genetics, 40(5), 631–637. 10.1038/ng.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium . (2012). An integrated encyclopedia of DNA elements in the human genome. Nature, 489(7414), 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson, I. P. M. , Carvajal‐Carmona, L. G. , Dobbins, S. E. , Tenesa, A. , Jones, A. M. , Howarth, K. , Palles, C. , Broderick, P. , Jaeger, E. E. M. , Farrington, S. , Lewis, A. , Prendergast, J. G. D. , Pittman, A. M. , Theodoratou, E. , Olver, B. , Walker, M. , Penegar, S. , Barclay, E. , Whiffin, N. , … Dunlop, M. G. (2011). Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part of the missing heritability of colorectal cancer. PLoS Genetics, 7(6), e1002105 10.1371/journal.pgen.1002105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson, I. P. , Webb, E. , Carvajal‐Carmona, L. , Broderick, P. , Howarth, K. , Pittman, A. M. , Spain, S. , Lubbe, S. , Walther, A. , Sullivan, K. , Jaeger, E. , Fielding, S. , Rowan, A. , Vijayakrishnan, J. , Domingo, E. , Chandler, I. , Kemp, Z. , Qureshi, M. , Farrington, S. M. , … Houlston, R. S. (2008). A genome‐wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nature Genetics, 40(5), 623–630. 10.1038/ng.111 [DOI] [PubMed] [Google Scholar]
- Venkatachalam, R. , Verwiel, E. T. P. , Kamping, E. J. , Hoenselaar, E. , Görgens, H. , Schackert, H. K. , van Krieken, J. H. J. M. , Ligtenberg, M. J. L. , Hoogerbrugge, N. , van Kessel, A. G. , & Kuiper, R. P. (2011). Identification of candidate predisposing copy number variants in familial and early‐onset colorectal cancer patients. International Journal of Cancer, 129(7), 1635–1642. 10.1002/ijc.25821 [DOI] [PubMed] [Google Scholar]
- Whiffin, N. , Dobbins, S. E. , Hosking, F. J. , Palles, C. , Tenesa, A. , Wang, Y. , Farrington, S. M. , Jones, A. M. , Broderick, P. , Campbell, H. , Newcomb, P. A. , Casey, G. , Conti, D. V. , Schumacher, F. , Gallinger, S. , Lindor, N. M. , Hopper, J. , Jenkins, M. , Dunlop, M. G. , … Houlston, R. S. (2013). Deciphering the genetic architecture of low‐penetrance susceptibility to colorectal cancer. Human Molecular Genetics, 22(24), 5075–5082. 10.1093/hmg/ddt357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiffin, N. , Hosking, F. J. , Farrington, S. M. , Palles, C. , Dobbins, S. E. , Zgaga, L. , Lloyd, A. , Kinnersley, B. , Gorman, M. , Tenesa, A. , Broderick, P. , Wang, Y. , Barclay, E. , Hayward, C. , Martin, L. , Buchanan, D. D. , Win, A. K. , Hopper, J. , Jenkins, M. , … Dunlop, M. G. (2014). Identification of susceptibility loci for colorectal cancer in a genome‐wide meta‐analysis. Human Molecular Genetics, 23(17), 4729–4737. 10.1093/hmg/ddu177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, J.‐H. , & Wei, G.‐H. (2019). Enhancer dysfunction in 3D genome and disease. Cells, 8(10), 1281 10.3390/cells8101281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing, J. , Myers, R. E. , He, X. , Qu, F. , Zhou, F. , Ma, X. , Hyslop, T. , Bao, G. , Wan, S. , Yang, H. , & Chen, Z. (2011). GWAS‐identified colorectal cancer susceptibility locus associates with disease prognosis. European Journal of Cancer, 47(11), 1699–1707. 10.1016/j.ejca.2011.02.004 [DOI] [PubMed] [Google Scholar]
- Zhang, B. , Jia, W.‐H. , Matsuda, K. , Kweon, S.‐S. , Matsuo, K. , Xiang, Y.‐B. , Shin, A. , Jee, S. H. , Kim, D.‐H. , Cai, Q. , Long, J. , Shi, J. , Wen, W. , Yang, G. , Zhang, Y. , Li, C. , Li, B. , Guo, Y. , Ren, Z. , … Zheng, W. (2014). Large‐scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nature Genetics, 46(6), 533–542. 10.1038/ng.2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B. , Jia, W.‐H. , Matsuo, K. , Shin, A. , Xiang, Y.‐B. , Matsuda, K. , Jee, S. H. , Kim, D.‐H. , Cheah, P. Y. , Ren, Z. , Cai, Q. , Long, J. , Shi, J. , Wen, W. , Yang, G. , Ji, B.‐T. , Pan, Z.‐Z. , Matsuda, F. , Gao, Y.‐T. , … Zheng, W. (2014). Genome‐wide association study identifies a new SMAD7 risk variant associated with colorectal cancer risk in East Asians. International Journal of Cancer, 135(4), 948–955. 10.1002/ijc.28733 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
Data that support the findings of this study are available in the Gene Expression Omnibus, reference number GSE77737 (Cohen et al., 2017) and from the ENCODE Project (https://www.encodeproject.org/; Davis et al., 2018; The ENCODE Project Consortium, 2012), reference numbers ENCSR000ENM and ENCSR000EMI, accessed using the UCSC Genome Browser. All other data are contained within the paper.