

Abstract
Objective
Epilepsy develops in 70 to 90% of children with tuberous sclerosis complex (TSC) and is often resistant to medication. Recently, the concept of preventive antiepileptic treatment to modify the natural history of epilepsy has been proposed. EPISTOP was a clinical trial designed to compare preventive versus conventional antiepileptic treatment in TSC infants.
Methods
In this multicenter study, 94 infants with TSC without seizure history were followed with monthly video electroencephalography (EEG), and received vigabatrin either as conventional antiepileptic treatment, started after the first electrographic or clinical seizure, or preventively when epileptiform EEG activity before seizures was detected. At 6 sites, subjects were randomly allocated to treatment in a 1:1 ratio in a randomized controlled trial (RCT). At 4 sites, treatment allocation was fixed; this was denoted an open‐label trial (OLT). Subjects were followed until 2 years of age. The primary endpoint was the time to first clinical seizure.
Results
In 54 subjects, epileptiform EEG abnormalities were identified before seizures. Twenty‐seven were included in the RCT and 27 in the OLT. The time to the first clinical seizure was significantly longer with preventive than conventional treatment [RCT: 364 days (95% confidence interval [CI] = 223–535) vs 124 days (95% CI = 33–149); OLT: 426 days (95% CI = 258–628) vs 106 days (95% CI = 11–149)]. At 24 months, our pooled analysis showed preventive treatment reduced the risk of clinical seizures (odds ratio [OR] = 0.21, p = 0.032), drug‐resistant epilepsy (OR = 0.23, p = 0.022), and infantile spasms (OR = 0, p < 0.001). No adverse events related to preventive treatment were noted.
Interpretation
Preventive treatment with vigabatrin was safe and modified the natural history of seizures in TSC, reducing the risk and severity of epilepsy. ANN NEUROL 2021;89:304–314
Tuberous sclerosis complex (TSC) is a multisystem disease caused by inactivating mutations in either TSC1 or TSC2. 1 Tumors form in many organs in TSC, including the brain, heart, skin, kidneys, and lungs. TSC is increasingly diagnosed prenatally, because TSC‐related cardiac rhabdomyomas are commonly observed on routine fetal ultrasound. 2
TSC is a major cause of severe and drug‐resistant epilepsy, with focal seizures and infantile spasms occurring in about 80% of TSC infants. 3 , 4 Neurodevelopmental comorbidities including intellectual disability and autism are also common in TSC. 3 , 4 Current guidelines recommend antiepileptic treatment after 2 unprovoked clinical seizures or after 1 seizure in patients at high risk (>60%) of recurrent seizures. 5 Although immediate antiepileptic treatment after seizure onset in children with TSC decreases the risk of neurodevelopmental complications, 50 to 60% of children develop intellectual disability. 6 , 7
In the majority of individuals with TSC, clinical seizures (CS) are preceded by asymptomatic, epileptiform activity (EA) on electroencephalography (EEG), followed by asymptomatic electrographic seizures (ES). 7 , 8 , 9 , 10 Efforts to disrupt or prevent progression of epileptogenesis to clinical epilepsy are current areas of epilepsy research. 7 , 10 , 11 Recently, the World Health Organization together with the International Bureau for Epilepsy highlighted an unmet need for epilepsy prevention research. 12
In 2011, a pilot open‐label study among 14 infants with TSC, using video‐EEG monitoring, showed that starting antiepileptic treatment when EA or ES were first detected improved epilepsy‐related outcomes at the age of 2 years compared to a historical control group for which treatment was begun after clinical seizures. 7 This led to recommendations that all infants with TSC should be monitored with video‐EEG. 13
In this controlled multicenter study, we compared the safety and efficacy of preventive antiepileptic treatment with vigabatrin, introduced at the first detection of EA on video‐EEG, with conventional antiepileptic treatment begun after the onset of seizures, either clinical or electrographic.
Patients and Methods
Study Design
This study was part of the EPISTOP project (Long‐Term, Prospective Study Evaluating Clinical and Molecular Biomarkers of Epileptogenesis in a Genetic Model of Epilepsy–Tuberous Sclerosis Complex, NCT02098759). It was carried out from March 2014 to October 2018 at 9 sites in Europe and 1 site in Australia. The study was approved by local ethics committees at all study sites, and caregivers of all participants signed informed consent before enrollment. It adhered to the International Conference on Harmonization Guidelines for Good Clinical Practice and the Declaration of Helsinki. The study was registered with the ClinicalTrials.gov database, number NCT02098759.
We had intended to perform a randomized controlled trial (RCT) at all 10 enrollment sites. However, the RCT was not approved by ethics boards at 4 sites, and hence subjects at those sites were enrolled in a parallel open‐label trial (OLT), with treatment according to local clinical practice: preventive treatment at 2 sites and conventional treatment at 2 others.
In both the RCT and OLT, the criteria for preventive and/or conventional treatment and EA diagnosis as well as the EEG scoring system were identical.
Participants
Subjects were children aged ≤4 months with definite TSC diagnosis according to consensus criteria, 14 who had no prior CS and no ES on baseline video‐EEG. Exclusion criteria were lack of definite TSC diagnosis, prior epileptic seizure, prior antiepileptic treatment, and any condition considered by the investigator to hinder participation in the study.
Randomization and Blinding
In the RCT, computerized randomization was performed centrally using variably sized permuted blocks stratified for study sites. The local EEG reader sent EEG scores to the central reader, and the central reader randomly allocated eligible patients to either preventive or conventional treatment. The treating physicians and subjects' caregivers were blinded to EEG data, and they did not know whether the treatment was randomly allocated after EA or started conventionally because of seizures captured on video‐EEG.
Neuropsychologists performing neuropsychological tests and electrophysiologists assessing EEGs were blinded to treatment allocation and clinical history of the patients.
Procedures
From enrollment to study completion at the age of 24 months, subjects were seen every 4 weeks for children aged ≤6 months, every 6 weeks for children aged >6 to 12 months, and every 8 weeks for children aged >12 months, as recommended by standard guidelines. 13 , 15
Conventional treatment was given after the onset of either ES or CS. Preventive treatment was given when EA was found on EEG prior to seizures. EA was defined as unifocal discharges during >10% of the recording time; multifocal discharges (involving ≥2 areas of the brain); or widespread/generalized epileptiform activity, including hypsarrhythmia. Electrographic seizures were defined as ictal EEG activity with no clinical correlate on video.
At each visit, we performed video‐EEGs and gathered data on the occurrence and frequency of clinical seizures captured by the subjects' caregivers in seizure diaries. Sleep and awake video‐EEGs were recorded for ≥1 hour and were assessed for EA, ES, and CS. Because local interpretation led to immediate treatment intervention in some subjects, subjects with discordant EEG interpretation by the local and central EEG readers were excluded from further analysis (n = 6). The patients also had neurodevelopmental testing with the Bayley Scales of Infant Development, 3rd edition (BSID‐III) and the Autistic Diagnostic Observation Schedule, 2nd edition (ADOS‐2).
Both preventive and conventional treatments consisted of vigabatrin, given at 100 to 150mg/kg/day in both the OLT and RCT. In patients with EA randomized to vigabatrin who developed seizures, and in those who received vigabatrin as conventional treatment and seizure control was not achieved, additional antiepileptic drugs were prescribed according to the judgment of the treating physician. Infants who never developed EA or seizures by the age of 2 years did not receive any antiepileptic treatment. At the age of 2 years, preventive vigabatrin was tapered off in patients who did not develop seizures.
Outcome Measures
The primary endpoint was the time from birth to the first CS. Secondary endpoints were assessed at age 2 years, and included the proportion of patients with CS, drug‐resistant epilepsy (defined as failure of 2 trials of antiepileptic drugs, either as monotherapies or in combination, to achieve sustained seizure freedom), 16 history of infantile spasms or hypsarrhythmia on EEG, any EEG abnormalities at any time, autistic features (according to ADOS‐2) and neurodevelopmental delay (BSID‐III cognitive score < 70). The percentage of days with seizures calculated from patients' diaries was also a secondary endpoint.
The safety of patients in the study was monitored by an independent ethics board supported by a biostatistician. Adverse events were recorded during each follow‐up visit.
Statistical Analysis
The study was designed to have 80% power to detect a 50% improvement in the primary study endpoint, assuming that 70% of patients would have EA before the onset of seizures and 100 would be enrolled. 8 The protocol specified that the study would be stopped early if interim analyses after 70% enrollment showed a benefit in the primary endpoint (p ≤ 0.01).
Seizure‐free survival in preventive and conventional treatment groups was analyzed using Kaplan–Meier curves. Log‐rank test was used to analyze the primary endpoint among all patients who received at least 1 dose of treatment. Median times to events together with 95% confidence intervals (CIs) were calculated using bootstrap resampling for right‐censored data with the Kaplan–Meier method. In both study arms, differences in the baseline characteristics of the study groups and frequency of secondary endpoints between patients who completed the study on either preventive or conventional treatment were reported as odds ratios (ORs) and 95% CIs using Fisher exact test.
In the RCT, we compared patients who were randomly assigned to either preventive or conventional treatment. In the OLT, we compared patients who received at least 1 dose of either preventive treatment or conventional treatment. In both the RCT and the OLT, subjects who developed CS or ES before EA were not eligible for randomization and thus were excluded from analysis of the primary and secondary outcomes. However, they were followed until the age of 24 months. Pooled analyses of patients who received preventive and conventional treatment in both RCT and OLT were also performed.
Statistical significance was set at p < 0.05, 2‐tailed for both the primary and secondary endpoints. Analyses were performed using R (v3.5.2), with the packages tidyverse, survival, and survminer.
Results
Patients
One hundred one patients with a provisional diagnosis of TSC were screened for eligibility, 94 were included in the study (Fig 1). Twenty‐two patients had either CS or ES before EA was seen on EEG, 7 never had EA or seizures, and 4 dropped out before EA was found. In 7 patients, the central and local analyses of EEG were discordant, and they were excluded from further analysis. Thus, 54 patients were eligible for preventive treatment. Twenty‐seven subjects were included in the RCT and 27 in the OLT. In the RCT, 13 were randomly allocated to preventive treatment, and 14 to conventional treatment, of whom 1 dropped out. Thus, 26 children completed the RCT. In the conventional treatment arm, all patients received vigabatrin after CS; none had ES occurring before CS.
FIGURE 1.
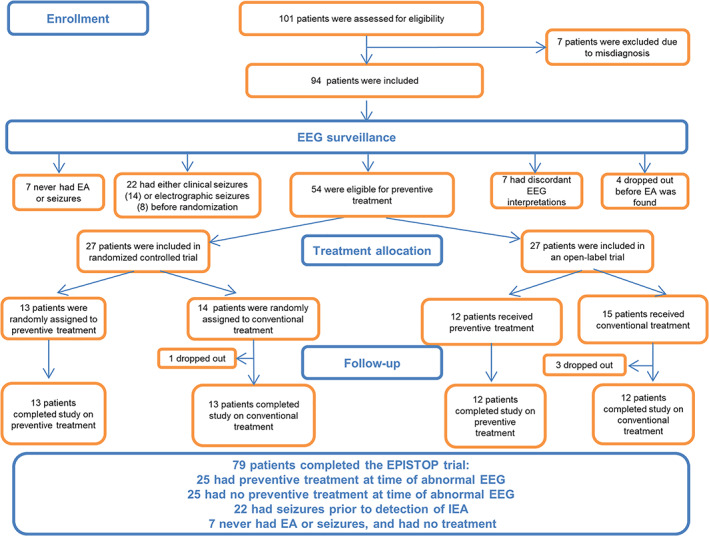
EPISTOP subject flow chart. EEG = electroencephalogram, EA = epileptiform activity.
In the OLT, 12 and 15 patients received preventive and conventional treatment, respectively, with 3 dropping out from the conventional arm. Thus, 12 patients completed the study on preventive treatment, and 12 patients on conventional treatment in the OLT (see Fig 1). All patients in the conventional treatment arm received vigabatrin after CS; in 3 of them, ES were observed prior to but on the same day as CS. Baseline clinical characteristics, including TSC1 or TSC2 mutation, brain magnetic resonance imaging findings, and gestational age did not differ significantly between the patients who received preventive or conventional treatment in either the RCT or OLT (Table 1).
TABLE 1.
Baseline Characteristics of Patients in Different Treatment Groups
| Characteristic | Randomized Controlled Trial | Open‐Label Trial | ||
|---|---|---|---|---|
| Preventive Treatment, n = 13 | Conventional Treatment, n = 14 | Preventive Treatment, n = 12 | Conventional Treatment, n = 15 | |
| Age at enrollment, days | ||||
| Mean ± SD | 36.1 ± 23.3 | 33.9 ± 27.6 | 40.2 ± 26.6 | 43.0 ± 26.6 |
| Median (IQR) | 36 (19–49) | 26.5 (16–35) | 33 (23–58) | 32 (19–68) |
| Sex, n (%) | ||||
| Female | 10 (77%) | 6 (43%) | 4 (33%) | 5 (33%) |
| Male | 3 (23%) | 8 (57%) | 8 (67%) | 10 (67%) |
| Mutation, n (%) | ||||
| TSC1 | 1 (8%) | 2 (14%) | 2 (17%) | 5 (33%) |
| TSC2 | 12 (92%) | 12 (86%) | 10 (83%) | 10 (67%) |
| No mutation identified | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Brain MRI findings, n (%) | ||||
| Subependymal nodules | 12 (92%) | 14 (100%) | 12 (100%) | 13 (87%) |
| SEGAs | 1 (8%) | 1 (7%) | 2 (17%) | 0 (0%) |
| Radial migration lines | 10 (77%) | 12 (86%) | 10 (83%) | 10 (67%) |
| Tubers | 11 (85%) | 14 (100%) | 10 (83%) | 11 (73%) |
| Median tuber volume as proportion of total brain volume (range) | 2.896 (0.000–14.43) | 2.687 (0.115–11.039 | 2.813 (1.122–7.748) | 0.585 (0.000–6.669) |
Includes dropouts after treatment allocation and excludes patients who never had epileptiform activity/seizures and were not treated. No statistically significant differences between preventive and conventional groups and between randomized and open‐label trials were found.
IQR = interquartile range; MRI = magnetic resonance imaging; SD = standard deviation; SEGA = subependymal giant cell astrocytoma; TSC1 = tuberous sclerosis complex gene 1; TSC2 = tuberous sclerosis complex gene 2.
The median dosages of vigabatrin were similar in all groups: 133mg/kg/day in the preventive treatment group in the RCT (range = 35–156mg/kg/day), 139mg/kg/day in the conventional treatment group in the RCT (range = 83–171mg/kg/day), 136mg/kg/day in the preventive treatment group in the OLT (range = 50–178mg/kg/day), and 136mg/kg/day in the conventional treatment group in the OLT (range = 43–167mg/kg/day). The median dose was calculated for the period from the day of treatment onset to the end of the study.
Primary Outcome
In the RCT, the median time from birth to the first clinical seizure was about 4 times longer with preventive treatment than with conventional treatment (614 days, 95% CI = 364–infinity vs 151 days, 95% CI = 99–215; for patients who developed seizures: 364 days, 95% CI = 223–535 vs 124 days, 95% CI = 33–149; Table 2). In the OLT, this time was also about 4 times longer with preventive versus conventional treatment (602 days, 95% CI = 463–infinity vs 124 days, 95% CI = 78–242; for patients who developed seizures: 426 days, 95% CI = 258–628 vs 106 days, 95% CI = 11–149; see Table 2 ). In a pooled analysis of both the RCT and the OLT, the median seizure onset was day 614 (95% CI = 474–infinity) in infants who received preventive treatment and day 124 (95% CI = 114–200) in children treated conventionally (for patients who developed seizures: day 390, 95% CI = 275–550 for patients treated preventively vs day 118, 95% CI = 44–139 for standard treatment path; see Table 2).
TABLE 2.
Epilepsy Outcomes in the Randomized Controlled Trial and Open‐Label Trial Patients
| Outcome | Randomized Controlled Trial | Open‐Label Trial | Pooled Analysis | |||
|---|---|---|---|---|---|---|
| PTx (95% CI) | CTx (95% CI) | PTx (95% CI) | CTx (95% CI) | PTx (95% CI) | CTx (95% CI) | |
| Median time to clinical seizures, days a | 614 (364–Inf) | 124 (118–215) | 602 (463–Inf) | 124 (78–242) | 614 (474–Inf) | 124 (114–200) |
| Median time to first clinical or electrographic seizure, days a | 505 (230–Inf) | 124 (99–215) | 587 (390–Inf) | 124 (78–242) | 587 (397–Inf) | 124 (114–200) |
| Median time from EA to first seizure, b days a | 496 (274–Inf) | 103 (56–158) | 561 (389–Inf) | 36 (21–124) | 561 (435–Inf) | 61 (41–124) |
| Median age at treatment onset, days a | 77 (42–110) | 131 (102–222) | 116 (91–147) | 124 (81–249) | 96 (77–118) | 131 (102–201) |
| Median age at treatment onset for patients who received treatment, days c | 75 (44–112) | 142 (40–160) | 124 (97–160) | 111 (13–146) | 9996 (54–115) | 125 (51–149) |
| Median proportion of days with seizures c | 12% (8.9–33.3) | 54.3% (47.6–91.7) | 6.4% (5.14–14.8) | 37.6% (1.54–60.3) | 8.0% (6.65–15.8) | 43.5% (5.47–46.4) |
| Median BSID‐III Cognitive Scale c | 71.6 (60–90) | 69.7 (52.5–72.5) | 84.2 (75–100) | 80.8 (65–110) | 77.1 (64.5–82) | 73 (64.5–82) |
Medians and confidence intervals were obtained by using bootstrap technique for right‐censored data with Kaplan–Meier method.
Either electrographic seizure or clinical seizure, whichever was detected first.
Medians and basic 2‐sided nonparametric confidence intervals were obtained by bootstrap method.
BSID‐III = Bayley Scales of Infant Development, 3rd edition; CI = confidence interval; CTx = conventional treatment; EA = epileptiform activity; Inf = infinity; PTx = preventive treatment.
In the RCT and OLT, patients in the preventive treatment group were about 3 times more likely to remain free of clinical seizures over the study period than those in the conventional treatment group (46% vs 15% in RCT, 50% vs 17% in OLT; log‐rank p = 0.011; Fig 2).
FIGURE 2.
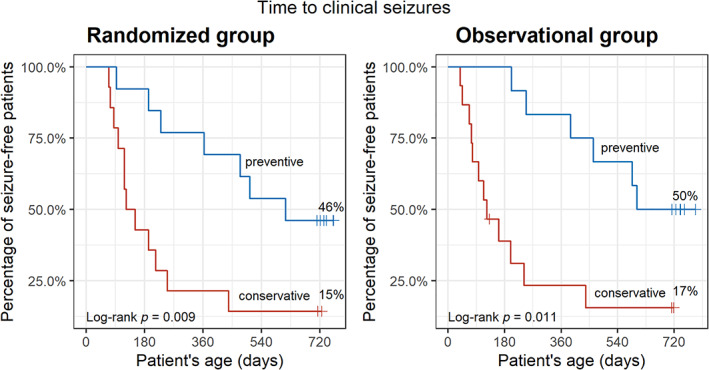
Time (in days) to clinical epileptic seizures in EPISTOP patients receiving preventive or conventional antiepileptic treatment in the randomized controlled trial and open‐label trial. Probability values are from the log‐rank test.
Key Secondary Outcomes
The median proportion of days with seizures until age 2 years was significantly lower in those receiving preventive treatment compared to conventional treatment in the RCT (17% vs 62%, p = 0.022), in the OLT (7% vs 35%, p = 0.030; see Table 2), and in the pooled analysis (8% vs 39%, p = 0.002; see Table 2). At the age of 2 years, the frequency of drug‐resistant epilepsy was significantly lower in patients receiving preventive treatment than conventional treatment in the RCT (31% vs 77%, OR = 0.15, 95% CI = 0.02–0.98, p = 0.047), trended to significance in the OLT (23% vs 50%, OR = 0.35, 95% CI = 0.04–2.45, p = 0.40), and was significant in the pooled analysis (28% vs 64%, OR = 0.23, 95% CI = 0.06–0.83, p = 0.025; Tables 3 and 4).
TABLE 3.
Secondary Endpoints at 24 Months of Life
| Number (%) of Patients With | Randomized Controlled Trial | Open‐Label Trial | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PTx | CTx | Estimated Difference (95% CI) | Estimated Odds Ratio (95% CI) | p a | PTx | CTx | Estimated Difference (95% CI) | Estimated Odds Ratio (95% CI) | p a | |
| Clinical seizures | 7 (54) | 11 (85) | −30.77 (−64.22, 2.68) | 0.23 (0.02, 1.74) | 0.202 | 6 (50) | 10 (77) | −33.33 (−68.62, 1.95) | 0.21 (0.02, 1.72) | 0.193 |
| Drug‐resistant epilepsy | 4 (31) | 10 (77) | −46.15 (−80.12, −12.18) | 0.15 (0.02, 0.98) | 0.047 | 3 (25) | 6 (50) | −25 (−62.42, 12.42) | 0.35 (0.04, 2.45) | 0.400 |
| Infantile spasms | 0 | 4 (31) | −30.77 (−55.86, −5.68) | 0 (0, 1.36) | 0.096 | 0 | 6 (50) | −46.15 (−73.25, −19.05) | 0 (0, 0.7) | 0.015 |
| Abnormal EEG | 11 (85) | 11 (85) | 0 (−27.74, 27.74) | 1 (0.06, 16.14) | 1.000 | 6 (50) | 8 (67) | 16.67 (−22.21, 55.55) | 1.94 (0.29, 14.3) | 0.680 |
| Hypsarrhythmia | 1 (8) | 1 (8) | 0 (−20.49, 20.49) | 1 (0.01, 84.9) | 1.000 | 1 (8) | 2 (15) | −7.05 (−32.14, 18.03) | 0.51 (0.01, 11.25) | 1.000 |
| Autism b | 4 (33) | 2 (16) | 16.67 (−17.33, 50.67) | 2.41 (0.26, 33.18) | 0.640 | 3 (25) | 2 (17) | 10 (−27.7, 47.7) | 1.67 (0.14, 25.6) | 1.000 |
| Neurodevelopmental delay c | 4 (33) | 6 (50) | −16.67 (−55.55, 22.21) | 0.51 (0.07, 3.4) | 0.680 | 2 (17) | 3 (25) | −10 (−47.7, 27.7) | 0.6 (0.04, 6.94) | 1.000 |
Fisher test.
High risk of autism according to Autistic Diagnostic Observation Schedule, 2nd edition score.
Bayley Scales of Infant Development, 3rd edition cognitive score < 70.
CI = confidence interval; CTx = conventional treatment; EEG = electroencephalogram; PTx = preventive treatment.
TABLE 4.
Secondary Endpoints at 24 Months in Pooled Analysis of the Randomized and Observational Arms
| Number (%) of Patients With | PTx | CTx | Patients Excluded from Analyses a | Patients without EA and Seizures | Estimated Difference, PTx–CTx (95% CI) | Estimated Odds Ratio, PTx:CTx (95% CI) | p b |
|---|---|---|---|---|---|---|---|
| Clinical seizures | 13 (52) | 21 (84) | 16 (73) | — | −32 (−56.29, −7.71) | 0.21 (0.04, 0.9) | 0.032 |
| Drug‐resistant epilepsy | 7 (28) | 16 (64) | 13 (59) | — | −36 (−61.76, −10.24) | 0.23 (0.06, 0.83) | 0.025 |
| Infantile spasms | 0 | 10 (40) | 3 (14) | — | −40 (−59.2, −20.8) | 0 (0, 0.33) | 0.001 |
| Abnormal EEG | 17 (68) | 19 (76) | 16 (73) | — | 8 (−16.79, 32.79) | 1.48 (0.36, 6.34) | 0.754 |
| Hypsarrhythmia | 2 (8) | 3 (12) | 1 (5) | — | −4 (−20.59, 12.59) | 0.64 (0.05, 6.19) | 1.000 |
| Autism c | 7 (32) | 4 (18) | 10 (50) | 0 | −13.64 (−38.91, 11.63) | 2.06 (0.43, 11.58) | 0. 488 |
| Neurodevelopmental delay d | 6 (25) | 9 (41) | 10 (48) | 0 | −13.64 (−41.36, 14.08) | 0.55 (0.12, 2.27) | 0.526 |
Due to presentation of clinical or electrographic seizures before interictal epileptiform activity (n = 15 and n = 7 in the randomized clinical trial and open‐label trial, respectively).
Fisher test, PTx and CTx compared.
High risk of autism according to Autistic Diagnostic Observation Schedule, 2nd edition score.
Bayley Scales of Infant Development, 3rd edition cognitive score < 70.
CI = confidence interval; CTx = conventional treatment; EA, epileptiform activity; EEG = electroencephalogram; PTx = preventive treatment.
In both the RCT and OLT, none of the patients who received preventive treatment developed infantile spasms, compared to 4 of 13 (31%) patients on conventional treatment in the RCT and 6 of 12 (50%) patients in the OLT (see Table 3). This difference was statistically significant both in the OLT and in the pooled analysis (p = 0.015 and p < 0.001, respectively; see Tables 3 and 4).
No EPISTOP subject who completed the study had severe intellectual disability (cognitive developmental quotient < 50) at the age of 2 years (BSID‐III). The median scores on the BSID‐III Cognitive Scale in children aged 2 years receiving preventive and conventional treatment were similar (ie, 76 and 72.5, respectively; see Table 4). The incidence of neurodevelopmental delay by the age of 2 years was lower in the preventive (33% in the RCT and 20% in the OLT) than in the conventional treatment group (50% in the RCT and 30% in the OLT), but the difference was not significant (see Table 3). The overall risk of autistic features (defined as ADOS‐2 > 12) in the EPISTOP cohort was low (20%), and there was no significant difference according to preventive versus conventional treatment (see Tables 3 and 4).
Data from patients excluded from the RCT or the OLT due to the presentation of CS or ES before EA (15 and 7, respectively) are presented in Table 4.
Adverse Events
Adverse events were typical for the antiepileptic drugs used and the age of the patients. There were no adverse events related to preventive treatment. There was 1 death, which was due to cardiac arrest during epilepsy surgery in a patient who had received conventional treatment. Otherwise, subjects receiving preventive and conventional treatment had similar rates of adverse events (Table 5).
TABLE 5.
Adverse Events
| Event | Events, n (events per patient) | p, Preventive vs Conventional | ||||
|---|---|---|---|---|---|---|
| Preventive Treatment, n = 25 | Conventional Treatment, n = 29 | Patient with no EA and No Seizures, n = 7 | Patients with Seizures prior to EA, n = 22 | Patients with Discordant EEG Analysis or Who Dropped Out before Treatment Allocation, n = 11 | ||
| All adverse events | 20 (0.80) | 36 (1.24) | 2 (0.29) | 40 (1.82) | 5 (0.45) | 0.153 |
| Serious adverse events | 11 (0.44) | 23 (0.79) | 0 | 37 (1.68) | 1 (0.09) | 0.132 |
| All serious adverse events | ||||||
| Death | 0 | 1 (0.03) a | 0 | 0 | 0 | 1 |
| Status epilepticus | 2 (0.08) | 7 (0.24) | 0 | 20 (0.9) | 1 (0.09) | 0.12 |
| Febrile seizures | 2 (0.08) | 5 (0.17) | 0 | 2 (0.09) | 0 | 0.22 |
| Surgery due to epilepsy | 1 (0.04) | 1 (0.03) | 0 | 11 (0.5) | 0 | 1 |
| Upper respiratory tract infection | 2 (0.08) | 4 (0.14) | 1 (0.14) | 3 (0.14) | 0 | 0.503 |
| Pneumonia | 2 (0.08) | 2 (0.07) | 0 | 1 (0.04) | 0 | 0.878 |
| Renal failure | 0 | 1 (0.03) | 0 | 0 | 0 | 1 |
| Surgery due to kidney tumors | 1 (0.04) | 1 (0.03) | 0 | 0 | 0 | 1 |
| Surgery due to cardiac tumors | 1 (0.04) | 1 (0.03) | 0 | 0 | 0 | 1 |
| Nonserious adverse events reported twice or more | ||||||
| Upper respiratory tract infection | 2 (0.08) | 2 (0.07) | 0 | 2 (0.09) | 2 (0.18) | 0.878 |
| Cardiac arrhythmia | 3 (0.12) | 4 (0.14) | 1 (0.14) | 2 (0.09) | 1 (0.09) | 0.846 |
| Feeding problems | 1 (0.04) | 1 (0.03) | 0 | 1 (0.04) | 0 | 1 |
| Anemia | 0 | 1 (0.03) | 0 | 0 | 1 (0.09) | 1 |
| Constipation | 0 | 2 (0.07) | 0 | 1 (0.04) | 0 | 0.494 |
| Influenza | 1 (0.04) | 2 (0.07) | 0 | 0 | 0 | 0.646 |
| Skin hyperkeratosis | 1 (0.04) | 1 (0.03) | 0 | 0 | 0 | 1 |
| Growth of SEGA | 1 (0.04) | 0 | 0 | 1 (0.04) | 0 | 0.463 |
Analysis includes the patients who dropped out from the study (n = 8) and patients with discordant EEG assessments (n = 7). Patients receiving preventive treatment were pooled from RCT and OLT. Patients receiving conventional treatment were pooled from RCT and OLT.
Due to cardiac arrest during epilepsy surgery.
EA = epileptiform activity; EEG = electroencephalogram; OLT = open‐label trial; RCT = randomized controlled trial; SEGA = subependymal giant cell astrocytoma.
Discussion
Despite great progress in the management of epilepsy and an increasing number of antiepileptic drugs, one‐third of patients are refractory to available medications, and many more suffer epilepsy‐related comorbidities such as intellectual disability and autism. 17 There is also increasing preclinical evidence that epilepsy can be prevented or even cured in animal models of acquired 18 or genetic epilepsy. 19 , 20 However, previous clinical trials aimed at preventing epilepsy after brain injury or stroke have failed because of the low risk of epilepsy in the studied populations and lack of reliable biomarkers. 21 In this prospective, multicenter study, we showed that preventive antiepileptic treatment with vigabatrin at the onset of EA seen on EEG delayed the time to the onset of clinical and electrographic seizures, reduced the risk of epileptic seizures, and decreased the severity of epilepsy in infants with TSC. Our findings support the use of serial video‐EEG monitoring beginning at the time of diagnosis of TSC in infants and the immediate initiation of antiepileptic treatment with vigabatrin at the onset of EA.
Our study included 2 interventions: frequent video‐EEG monitoring from the time of diagnosis of TSC in all patients, and preventive antiepileptic treatment at the onset of EA if this occurred before seizures. Serial video‐EEGs enabled early diagnosis of electrographic and subtle clinical seizures, for which caregiver identification can be difficult, and initiation of conventional antiepileptic treatment earlier than in historic cohorts of infants with TSC. It also allowed our preventive treatment to be initiated at the onset of EA.
Currently, epilepsy is diagnosed after seizures. 5 Until recently, conventional practice was to inform parents of infants with TSC about the risk of seizures and ask them to seek referral to a neurologist when seizures occurred. Because the first seizures in TSC are often subtle, and frequently missed by caregivers, this results in significant diagnostic and therapeutic delay.
A previous report indicated that TSC children who received antiepileptic treatment later than 1 week after the first clinical seizure all had intellectual disability, and 76% had drug‐resistant epilepsy, whereas of those in whom antiepileptic treatment was introduced immediately after CS onset, 61% had intellectual disability and 35% had drug‐resistant seizures. 6 , 7 In EPISTOP, preventive treatment was associated with a >2‐fold reduction in the risk of drug‐resistant epilepsy compared to conventional treatment (28% vs 64% in the RCT; Table 4). Neurodevelopmental delay at age 2 years was seen in only 33% of those on preventive treatment and 50% of those on conventional treatment. Furthermore, none of the patients in EPISTOP had severe learning disability (cognitive developmental quotient < 50) at age 2 years. However, the improvement in intellectual disability in the preventive versus conventional treatment group was not significant. This may be due to the small number of patients studied, follow‐up to only age 2 years, and/or the benefit of intensive clinical care and seizure monitoring for those on conventional treatment. It cannot be excluded, however, that the complex processes leading to epilepsy might contribute to neuropsychiatric disorders in TSC prior to the onset of treatment triggered by the epileptiform EEG. 22 All patients without epilepsy and epileptiform EEG had normal intellectual function, supporting the concept that epilepsy is the main contributor to intellectual disability in TSC. 23 , 24
There is evidence that developmental delay in TSC is associated with an earlier age at seizure onset. 25 , 26 , 27 Thus, delaying the onset of epileptic seizures with preventive treatment may reduce intellectual disability overall in TSC. Long‐term follow‐up of our 2011 open‐label study showed that improved epilepsy and neurodevelopmental outcomes were maintained into school age. 28
Infantile spasms are seen in 50 to 70% of children with TSC, and are associated with both drug‐resistance and intellectual disability. 4 , 29 , 30 Importantly, in EPISTOP, none of the children who received preventive treatment developed infantile spasms throughout the 2‐year course of the study, in contrast to 10 of 25 (40%) receiving conventional treatment.
Our study had limitations. Patient recruitment is not easy, because a TSC diagnosis is usually not anticipated, and parents face numerous issues at this moment in their lives. In addition, the intensive visit schedule and monitoring were difficult for many subjects, contributing to a relatively high dropout rate of 10%. Monthly video‐EEG monitoring was not sufficient to identify EA prior to subclinical or clinical seizures in 22 patients (23%). Moreover, EA is not perfect as a predictor of epileptogenesis in TSC, as it was reported recently that presence of EA had a 77% positive predictive value for development of seizures. 10 In EPISTOP, 2 subjects in the OLT had no evidence of clinical or electrographic seizures at age 2 years, but experienced their first clinical seizure before age 3 years. Furthermore, a preventive strategy is not possible in patients who present with clinical seizures as newborns, which occurs in about 5% of patients with TSC. 31 Lastly, interpretation of EEG, especially in young infants, can be difficult and is often associated with poor inter‐rater reliability. 32 , 33 In the EPISTOP trial, all EEGs were read both locally and centrally by 3 independent EEG readers. There were no discrepancies between them in terms of the recognition of electrographic or clinical seizures, but in 7 cases (7.4%) there was no consensus on the diagnosis of epileptiform activity. Thus, methods more sensitive than EEG to identify epileptogenesis are needed. In selected populations of patients with an extremely high risk of epilepsy such as TSC, antiseizure interventions without an abnormal EEG should be considered.
On the other hand, the intervention we used did not require any additional health resource use, given that EEG monitoring is recommended in all TSC infants. 13 The only difference from conventional care is to initiate vigabatrin treatment at the onset of EA on EEG.
In conclusion, this EPISTOP study has shown that it may be possible to change the natural history of severe infantile epilepsy through early intervention with antiepileptic therapy. Such treatment, introduced at the onset of epileptiform activity detected by serial video‐EEG and before the onset of seizures, delayed the onset of epilepsy, reduced the risk of seizures, and prevented infantile spasms in infants with TSC.
Author Contributions
K.K., D.J.K., and S.J. contributed to the conception and design of the study. All authors contributed to the acquisition and analysis of data. K.K., D.J.K., K.R., K.W., K.Sa., J.G.‐W., and S.J. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
TABLE S1. EPISTOP Investigators Who Participated in This Study and Their Institutional Affiliations
Acknowledgments
All authors were supported by the 7th Framework Program of the European Commission within the Large‐Scale Integrating Project EPISTOP (Proposal No. 602391–2; www.epistop.eu). Additionally, K.K., K.Si., J.G.‐W., J.B., K.Sa., D.D.‐P., and S.J. were supported by the Polish Ministerial funds for science (years 2014–2018). K.K., P.K., R.N., J.B., K.Sa., D.D.‐P., R.M., B.B., J.d.R., and L.L. are members of the European Reference Network EpiCare.
Members of the EPISTOP Investigators are available as an online supplementary file (Table S1).
Contributor Information
Katarzyna Kotulska, Email: k.kotulska@ipczd.pl.
the EPISTOP Investigators:
J. Anink, E. Aronica, B. Benova, A. Benvenuto, M. Blazejczyk, A. Bongaerts, J. Borkowska, D. Breuillard, D. Chmielewski, P. Curatolo, M. Dabrowska, D. Domańska‐Pakieła, M. Feucht, K. Giannikou, J. Głowacka‐Walas, L. Hamieh, A. Harȩza, Ch. Hertzberg, H. Hulshof, F. Huschner, A. Iyer, A. Jansen, F. Jansen, B. Janssen, J. Jaworski, S. Jùźwiak, M. Kaczorowska‐Frontczak, K. Kotulska, P. Krsek, D. Kwiatkowski, L. Lagae, K. Lehmann, A. Leusman, N. Maćkowiak, J. Mills, R. Moavero, A. Muelebner, R. Nabbout, J. de Ridder, K. Riney, K. Sadowski, S. Samueli, C. Scheldeman, T. Scholl, A. Sciuto, K. Sijko, M. Słowińska, A. Tempes, J. van Scheppingen, B. Verhelle, J. Vervisch, M. Urbańska, B. Weschke, and K. Wojdan
References
- 1. Salussolia CL, Klonowska K, Kwiatkowski DJ, Sahin M. Genetic etiologies, diagnosis, and treatment of tuberous sclerosis complex. Annu Rev Genomics Hum Genet 2019;31:217–240. [DOI] [PubMed] [Google Scholar]
- 2. Dragoumi P, O'Callaghan F, Zafeiriou DI. Diagnosis of tuberous sclerosis complex in the fetus. Eur J Paediatr Neurol 2018;22:1027–1034. [DOI] [PubMed] [Google Scholar]
- 3. Overwater IE, Bindels‐de Heus K, Rietman AB, et al. Epilepsy in children with tuberous sclerosis complex: chance of remission and response to antiepileptic drugs. Epilepsia 2015;56:1239–1245. [DOI] [PubMed] [Google Scholar]
- 4. Nabbout R, Belousova E, Benedik MP, et al. Epilepsy in tuberous sclerosis complex: findings from the TOSCA study. Epilepsia Open 2019;4:73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE Official Report: a practical clinical definition of epilepsy. Epilepsia 2014;55:475–482. [DOI] [PubMed] [Google Scholar]
- 6. Cusmai R, Moavero R, Bombardieri R, et al. Long‐term neurological outcome in children with early‐onset epilepsy associated with tuberous sclerosis. Epilepsy Behav 2011;22:735–739. [DOI] [PubMed] [Google Scholar]
- 7. Jóźwiak S, Kotulska K, Domańska‐Pakieła D, et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol 2011;15:424–431. [DOI] [PubMed] [Google Scholar]
- 8. de Groen A‐EC, Bolton J, Bergin AM, et al. The evolution of subclinical seizures in children with tuberous sclerosis complex. J Child Neurol 2019;34:770–777. [DOI] [PubMed] [Google Scholar]
- 9. Domańska‐Pakieła D, Kaczorowska M, Jurkiewicz E, et al. EEG abnormalities preceding the epilepsy onset in tuberous sclerosis complex patients—prospective study of 5 patients. Eur J Paediatr Neurol 2014;18:458–468. [DOI] [PubMed] [Google Scholar]
- 10. Wu JY, Goyal M, Peters JM, et al. Scalp EEG spikes predict impending epilepsy in TSC infants: a longitudinal observational study. Epilepsia 2019;60:2428–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roach ES, Kwiatkowski DJ. Seizures in tuberous sclerosis complex: hitting the target. Lancet 2016;388:2062–2064. [DOI] [PubMed] [Google Scholar]
- 12. World Health Organization . Epilepsy: a public health imperative. 2019. Available at: https://www.who.int/mental_health/neurology/epilepsy/report_2019/en/. Last accessed June 9, 2020.
- 13. Curatolo P, Nabbout R, Lagae L, et al. Management of epilepsy associated with tuberous sclerosis complex: updated clinical recommendations. Eur J Paediatr Neurol 2018;22:738–748. [DOI] [PubMed] [Google Scholar]
- 14. Northrup H, Krueger DA, Northrup H, et al. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013;49:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Curatolo P, Jóźwiak S, Nabbout R, TSC Consensus Meeting for SEGA and Epilepsy Management . Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol 2012;16:582–586. [DOI] [PubMed] [Google Scholar]
- 16. Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2009;51:1069–1077. [DOI] [PubMed] [Google Scholar]
- 17. Reilly C, Atkinson P, Das KB, et al. Neurobehavioral comorbidities in children with active epilepsy: a population‐based study. Pediatrics 2014;133:586–593. [DOI] [PubMed] [Google Scholar]
- 18. Pitkänen A, Lukasiuk K. Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol 2011;10:173–186. [DOI] [PubMed] [Google Scholar]
- 19. Marguet SL, Le‐Schulte VTQ, Merseburg A, et al. Treatment during a vulnerable developmental period rescues a genetic epilepsy. Nat Med 2015;21:1436–1444. [DOI] [PubMed] [Google Scholar]
- 20. Zeng L‐H, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol 2008;63:444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thompson K, Pohlmann‐Eden B, Campbell LA, Abel H. Pharmacological treatments for preventing epilepsy following traumatic head injury. Cochrane Database Syst Rev 2015;8:CD009900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moavero R, Benvenuto A, Emberti Gialloreti L, et al. Early clinical predictors of autism spectrum disorder in infants with tuberous sclerosis complex: results from the EPISTOP study. J Clin Med 2019;8:788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet 2001;68:64–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y‐Y, Pang L‐Y, Ma S‐F, et al. Epilepsy may be the major risk factor of mental retardation in children with tuberous sclerosis: a retrospective cohort study. Epilepsy Behav 2017;77:13–18. [DOI] [PubMed] [Google Scholar]
- 25. Capal JK, Bernardino‐Cuesta B, Horn PS, et al. Influence of seizures on early development in tuberous sclerosis complex. Epilepsy Behav 2017;70:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yates JR, MacLean C, Higgins JNP, et al. The Tuberous Sclerosis 2000 Study: presentation, initial assessments and implications for diagnosis and management. Arch Dis Child 2011;96:1020–1025. [DOI] [PubMed] [Google Scholar]
- 27. Jozwiak S, Goodman M, Lamm SH. Poor mental development in patients with tuberous sclerosis complex: clinical risk factors. Arch Neurol 1998;55:379–384. [DOI] [PubMed] [Google Scholar]
- 28. Jozwiak S, Słowińska M, Borkowska J, et al. Preventive antiepileptic treatment in tuberous sclerosis complex: long‐term, prospective trial. Pediatr Neurol 2019;101:18–25. [DOI] [PubMed] [Google Scholar]
- 29. Chu‐Shore CJ, Major P, Camposano S, et al. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 2010;51:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Curatolo P, Seri S, Verdecchia M, Bombardieri R. Infantile spasms in tuberous sclerosis complex. Brain Dev 2001;23:502–507. [DOI] [PubMed] [Google Scholar]
- 31. Kotulska K, Jurkiewicz E, Domańska‐Pakieła D, et al. Epilepsy in newborns with tuberous sclerosis complex. Eur J Paediatr Neurol 2014;18:714–721. [DOI] [PubMed] [Google Scholar]
- 32. Grant AC, Abdel‐Baki SG, Weedon J, et al. EEG interpretation reliability and interpreter confidence: a large single‐center study. Epilepsy Behav 2014;32:102–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jing J, Herlopian A, Karakis I, et al. Interrater reliability of experts in identifying interictal epileptiform discharges in electroencephalograms. JAMA Neurol 2019;77:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1. EPISTOP Investigators Who Participated in This Study and Their Institutional Affiliations