

Abstract
Background and Aims
Up‐regulation of the E2F‐dependent transcriptional network has been identified in nearly every human malignancy and is an important driver of tumorigenesis. Two members of the E2F family, E2F7 and E2F8, are potent repressors of E2F‐dependent transcription. They are atypical in that they do not bind to dimerization partner proteins and are not controlled by retinoblastoma protein. The physiological relevance of E2F7 and E2F8 remains incompletely understood, largely because tools to manipulate their activity in vivo have been lacking.
Approach and Results
Here, we generated transgenic mice with doxycycline‐controlled transcriptional activation of E2f7 and E2f8 and induced their expression during postnatal development, in adulthood, and in the context of cancer. Systemic induction of E2f7 and, to lesser extent, E2f8 transgenes in juvenile mice impaired cell proliferation, caused replication stress, DNA damage, and apoptosis, and inhibited animal growth. In adult mice, however, E2F7 and E2F8 induction was well tolerated, yet profoundly interfered with DNA replication, DNA integrity, and cell proliferation in diethylnitrosamine‐induced liver tumors.
Conclusion
Collectively, our findings demonstrate that atypical E2Fs can override cell‐cycle entry and progression governed by other E2F family members and suggest that this property can be exploited to inhibit proliferation of neoplastic hepatocytes when growth and development have subsided during adulthood.
Abbreviations
- BrdU
5‐bromo‐2′‐deoxyuridine
- Cdc6
cell division cycle 6
- cDNA
complementary DNA
- CHK1
checkpoint kinase 1
- CMV
cytomegalovirus
- DEN
diethylnitrosamine
- EGFP
enhanced green fluorescent protein
- FACS
fluorescence‐activated cell sorting
- γ‐H2AX
gamma H2A.X variant histone
- HCC
hepatocellular carcinoma
- M2‐rtTA
M2 reverse tetracycline transactivator
- pRb
retinoblastoma protein
- Rad51
Rad51 recombinase
- RPE
retinal pigment epithelium
- Tg
transgenic
The cell cycle consists of a sequence of tightly controlled and ordered events, which together ensure proper genetic inheritance. Central among these events is the oscillatory activity of the E2F transcription program. Individual E2F family members bind to hundreds of target genes, many of which are essential for DNA replication and repair. Hence, entry into S phase depends on activation of this program.( 1 , 2 ) Once S phase has started, E2F transcription becomes silenced again. E2F7 and E2F8 are the repressing family members that redundantly mediate this downswing during late S and G2 phases.( 3 , 4 , 5 , 6 , 7 ) They are referred to as atypical E2Fs because they possess unique structural features compared to the other E2Fs. They cannot bind to dimerization partner proteins and instead act as homodimers or heterodimers.( 6 , 8 ) Furthermore, their activity is not controlled by retinoblastoma protein (pRb). This is important, because RB is inactivated, mutated, or lost in a wide array of human cancers.( 9 , 10 ) Manipulation of E2F7/8 can uncouple E2F target gene transcription from pRb activity.( 11 ) The resulting unrestrained E2F transcription is thought to contribute to cancer progression in a variety of cancer types, including hepatocellular carcinoma (HCC). Accordingly, we observed that conditional deletion of E2f7 and E2f8 in mice caused up‐regulation of many E2F target genes, resulting in spontaneous liver cancer and accelerated skin cancer progression.( 12 , 13 ) These results indicate that atypical E2Fs are capable of controlling proliferation under stress conditions. For example, DNA damage can trigger a p53‐dependent up‐regulation of E2F7 to block cell‐cycle progression.( 11 , 14 ) These data led us to hypothesize that tipping the balance between activator and repressor E2Fs in favor of repressors might be a potent strategy to inhibit tumor proliferation. Notwithstanding these tumor‐suppressing effects, in vitro studies aimed at overexpressing atypical E2Fs have yielded conflicting results. A number of studies have indicated that E2F8 overexpression can promote proliferation of lung, breast, and liver cancer cell lines.( 15 , 16 , 17 ) Other work demonstrated that overexpression of E2F7 and E2F8 is a potent inhibitor of proliferation in HeLa, MEF, and U2OS cells.( 1 , 3 , 5 , 8 , 18 )
To resolve these issues, we employed an in vivo genetic approach to study the consequences of inducible E2F target gene repression on proliferation of normal and cancer cells. We created doxycycline‐inducible E2f7/8‐transgenic (Tg) mice and demonstrate that transgene induction markedly inhibited tissue proliferation in juvenile mice by perturbing S‐phase progression. Adult mice tolerated the transgene induction better than juvenile mice. Moreover, induction of E2F7 and, to a lesser extent, E2F8 revealed a consistent reduction of liver tumor growth through repression of gene transcription.
Materials and Methods
Animal Experiments
Animal experiments were approved by the Utrecht University Animal Ethics Committee (approval no. AVD108002016626) and performed according to institutional and national guidelines. Doxycycline (2 g/kg) was administrated ad libitum in pellets to all experimental mice (Bio Services). All lines were generated by Prof. Dr. Jan van Deursen (Mayo Clinic) according to a described methodology( 34 ) and maintained on a mixed genetic background, 129/Sv × C57Bl/6 × Friend virus B‐type. Additional information is available in the Supporting Information.
Generation of Inducible Cell Lines and Cell Culture
Inducible cell lines were created by introducing consecutively the pLenti CMV TetR and our synthesized E2F7/8 containing plasmids into RPE‐hTert cells using a third‐generation lentiviral packaging system. In addition to E2F7/8, we used E2F7/8 complementary DNAs (cDNAs) with both DNA binding domains mutated as described.( 35 ) pLenti CMV TetR Blast (716‐1) and pLenti CMV/TO Puro DEST (670‐1) were a gift from Eric Campeau and Paul Kaufman (Addgene plasmids 17492, http://n2t.net/addgene:17492; RRID:Addgene_17492, and 17293, http://n2t.net/addgene:17293; RRID:Addgene_17293). The HeLa/TO E2F8DBDmut. cells were available in our lab from previous studies.( 1 ) Further information is available in the Supporting Information.
HeLa and retinal pigment epithelium (RPE) cells were maintained in Dulbecco’s modified Eagle medium supplemented with 10% Tet System–approved fetal bovine serum (Clontech; CL631106) and 1% penicillin‐streptomycin at 37°C, 5% CO2. Overexpression was induced by adding 0.2 μg/mL doxycycline (Sigma‐Aldrich) to the cell culture medium.
For the proliferation assay, HeLa/TO E2F7 cells were seeded at a density of 1 × 105 cells in a 60‐cm Petri dish. Doxycycline was refreshed every 2 days. Cells were harvested in duplicate daily and counted using a BioRad TC20 Automated Cell Counter.
Flow Cytometry and Fluorescence‐Activated Cell Sorting
For determination of DNA content, cells were trypsinized, washed twice with phosphate‐buffered saline (PBS), fixed with 70% ethanol, and stored at 4ºC. Nuclei suspensions from frozen liver tissue were obtained as described.( 36 ) Details can be found in the Supporting Information.
Fluorescence‐activated cell sorting (FACS) based on enhanced green fluorescent protein (EGFP) fluorescence was performed on a BD‐Influx. Cells were collected in cell culture medium and immediately plated at a density of 5 × 103 cells per well for the colony formation assay.
RNA Isolation, cDNA, and Quantitative Real‐Time PCR
RNA isolation, cDNA synthesis, and quantitative PCR were performed based on the manufacturers’ instructions: Qiagen (RNeasy Kits), Thermo Fisher Scientific (cDNA synthesis kits), and Bio‐Rad (SYBR Green Master Mix). Reactions were performed in duplicate, and the relative amount of cDNA was normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) using the ΔΔCt method. Quantitative real‐time PCR primer sequences are provided in Supporting Table S1.
Immunoblotting
Protein lysates were obtained with radio immunoprecipitation assay buffer (50 mM Tris‐HCl [pH 7.5], 1 mM ethylene diamine tetraacetic acid, 150 mM NaCl, 0.25% deoxycholic acid, 1% Nonidet‐P40), 1 mM NaF, and NaV3O4 and protease inhibitor cocktail (11873580001; Sigma‐Aldrich). After centrifugation at full speed (12g) for 10 minutes, supernatants were collected and we proceeded to a standard sodium dodecyl sulfate–polyacrylamide gel electrophoresis immunoblot. Immunoblot antibodies are shown in Supporting Table S3.
Immunohistochemistry and Histopathological Analysis
Tissues were embedded in paraffin and sectioned at 4 μm. Details on antibodies, procedures, and quantifications are outlined in the Supporting Information and Supporting Table S3. Liver tumors were classified as described.( 37 , 38 )
Fiber Analysis
Cells were pulse‐labeled with 25 μM chlorodeoxyuridine followed by 250 μM iododeoxyuridine for 20 minutes each. The rest of the protocol was performed as described.( 24 ) Pictures were taken with a Leica SPE‐II‐DMI4000 confocal microscope using a ×63 objective and the LAS‐AF, HCS basic module software. Track lengths and quantification of number of origins fired were manually analyzed with ImageJ software. Replication track lengths were calculated using the conversion factor 1 μm = 2.59 kb.( 39 )
Colony Formation Assays
HeLa cells were seeded after FACS in duplicate at low density (5 × 103/well in six‐well plates) from three different cell clones. Cells were incubated with or without doxycycline for 10 days. For fixation, medium was removed, and cells were washed twice with PBS and fixed with acetic acid/methanol 1:7 (vol/vol) for 5 minutes. Then, colonies were stained with 0.5% crystal violet for 2 hours. Pictures represent entire wells (34.8 mm diameter), taken with a Nikon camera. For quantification in Fig. 2F and Supporting Fig. S2H, pictures were loaded in Photoshop CS6. For each condition, 10 fields (300 × 300 pixels) were randomly selected. Positive colony staining was measured with the Magic Wand Tool, with the tolerance value set to 16. Relative intensity was defined as the ratio of positive purple‐staining area (pixels) over the total area of the field.
Fig. 2.
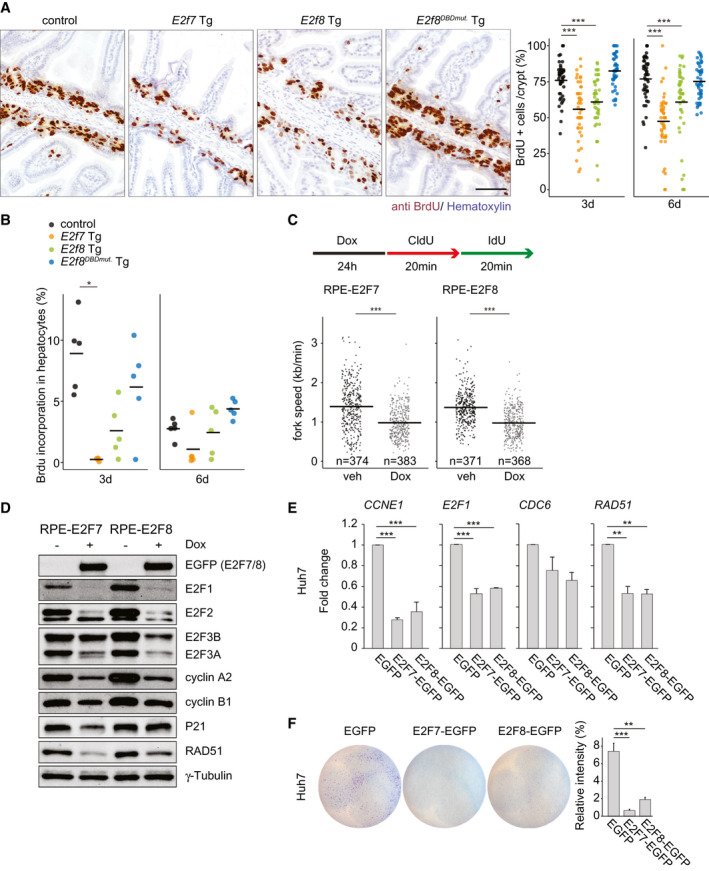
E2F7 and E2F8 expression inhibits ongoing DNA replication. (A) Immunohistochemistry using anti‐BrdU to label S‐phase cells in small intestines of Tg mice treated for 6 days with doxycycline. Scale bar, 50 μm. Dot plot shows quantification of BrdU‐positive area in five fields using the ×20 objective. Crossbars represent averages per genotype (n = 45 crypts/genotype, n = 3 mice/genotype). (B) Dot plot shows the percentage of BrdU‐positive hepatocyte nuclei per condition. Crossbars represent average values per condition (n = 5 mice). E2f7 Tg after 3‐day doxycycline (n = 4). (C) Experimental work flow and quantification of replication fork speed in the cell lines and conditions indicated. Data from two separate experiments are pooled. Only ongoing replication forks (red+green) were included in the quantification. Crossbars represent average values per condition. (D) Protein expression of cell cycle regulators in the indicated RPE inducible cell lines after 24 hours of doxycycline treatment. (E) Transcript levels of CDC6, E2F1, and RAD51 in Huh7 cells. EGFP‐positive cells were sorted 24 hours after transfections and directly collected for RNA analysis. Fold changes were adjusted to average of EGFP controls and GAPDH and RSP18 were used to normalize the expression. Data represent average ± SEM of at least two different experiments. (F) Clonogenic survival assay of EGFP FACS‐sorted Huh7 cells. Cells were transfected with the indicated plasmids for 24 hours before FACS‐sorting and replated for 72 hours. Histogram shows the quantification of relative intensity from each condition. Data represent average ± SEM (two independent experiments were performed in at least duplicate). Data information: In (A,B,E,F), *P < 0.05, **P < 0.01, ***P < 0.001 (Kruskal‐Wallis one‐way analysis of variance on ranks and Dunnett’s method for multiple comparisons versus control). In (C) ***P < 0.001 (Mann‐Whitney rank sum test). Abbreviations: CCNE1, cyclin E1; CldU, chlorodeoxyuridine; IdU, iododeoxyuridine; Dox, doxycycline; veh, vehicle.
Statistics
The number of independent experiments, the number of mice, and the type of statistical analysis for each figure are indicated in the legends. All statistical analyses were performed using SigmaPlot 13.0 software. Every cell culture experiment was repeated at least 2 times and with conditions in duplicate. Specific statistical tests for each data set are mentioned in the figure legends. Asterisks indicate where significant differences were seen. Where relevant for understanding the figure and individual comparisons, we indicate with “n.s.” that significance was not reached.
Results
E2F7/8 Overexpression Inhibits Proliferation In Vivo
To study the physiological consequences of atypical E2F repressor activation, we generated Tg mice with doxycycline‐inducible overexpression of E2F7 or E2F8. Gene constructs encoding EGFP‐tagged E2F7 or E2F8 (hereafter E2f7 Tg and E2f8 Tg) under the control of a tetracycline‐responsive element were inserted in the collagen type I alpha 1 chain locus. The M2 reverse tetracycline transactivator (M2‐rtTA) is expressed from the ROSAβgeo26 mouse locus, allowing doxycycline‐inducible expression of the transgene (Fig. 1A). A third Tg mouse carrying a mutated version of E2f8 containing mutations in both DNA‐binding domains (E2f8DBDmut. Tg) was created to validate that the effects of E2F8 overexpression were caused by transcriptional repression. Mice on doxycycline chow but only expressing M2‐rtTA served as negative controls.
Fig. 1.
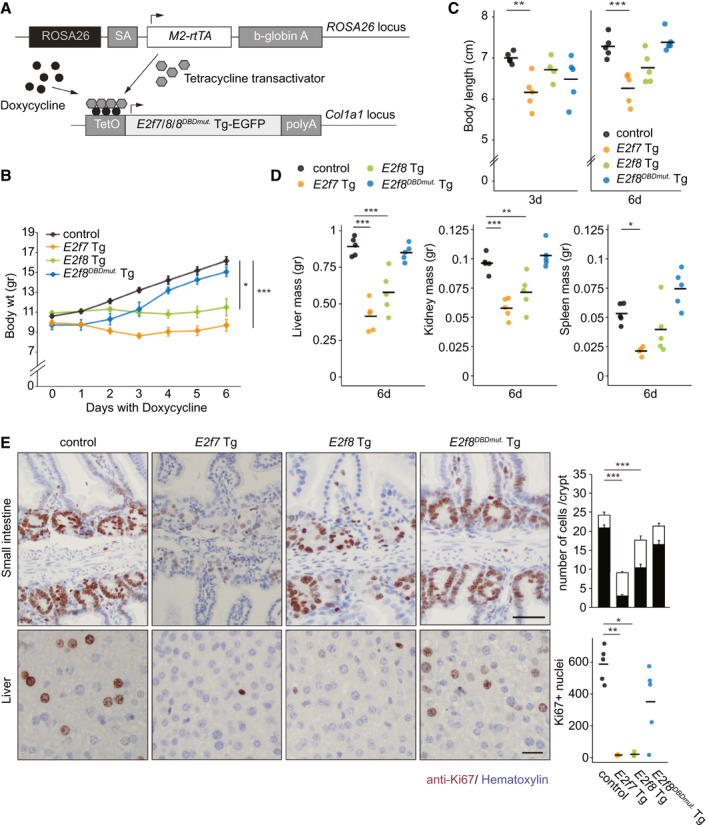
Inducible Tg E2F7 and E2F8 expression blocks proliferation in juvenile mice. (A) Schematic representation of the Tg model. (B) Growth curves of Tg mice. Error bars represents SEM (n = 10, days 0‐3; n = 5, dasy 4‐6 of doxycycline treatment). (C) Body lengths of juvenile mice after 3 and 6 days of transgene induction with doxycycline. Bars represent average (n = 5 mice). (D) Liver, kidney, and spleen mass of Tg mice after 6 days with doxycycline. Organs were weighed postfixation with 4% paraformaldehyde. Crossbars represent averages per genotype (n = 5 mice). E2f7 Tg spleen n = 4. (E) Representative pictures of Ki67 showing proliferating cells in small intestines (top) and livers (bottom) of Tg mice after 3 days of doxycycline treatment. Scale bars, 50 μm (top) and 20 μm (bottom). Small intestine quantification: total count and Ki67‐positive nuclei per crypt of small intestines. Error bars represent SEM (n = 45 crypts/genotype, n = 3 mice/genotype). Liver: Total Ki67‐positive nuclei in 10 fields (×40 objective). Crossbars represent averages per genotype (n = 5 mice). Data information: In (B‐E), *P < 0.05, **P < 0.01, ***P < 0.001 (Kruskal‐Wallis one‐way analysis of variance on ranks and Dunnett’s method for multiple comparisons versus control). Abbreviations: Col1a1, collagen type I alpha 1 chain; SA, splice acceptor.
Because expression of E2F target genes strongly correlates with proliferation rates, we anticipated the strongest impact of E2F7/8 overexpression in rapidly proliferating tissues. Therefore, we first induced the transgenes in juvenile mice immediately after weaning, when the proliferation rates are high in multiple tissues. To this end, we fed 21‐day‐old juvenile mice with doxycycline for 3 or 6 days.
Doxycycline caused a strong induction of E2f7 Tg and E2f8 Tg transcripts and proteins in the respective Tg mice (Supporting Fig. S1A‐C) and repressed key E2F target transcripts such as cell division cycle 6 (Cdc6), chromatin licensing and DNA replication factor 1 (Cdt1), and Rad51 recombinase (Rad51) (Supporting Fig. S1D). Immunohistochemistry on liver tissues revealed that transgenic E2F7 and E2F8 were nuclear proteins (Supporting Fig. S1E). After 6 days of transgene induction, E2f7 Tg and E2f8 Tg mice showed a clear growth reduction, measured by body weight and length, compared to control littermates and E2f8DBDmut. Tg mice (Fig. 1B,C). Furthermore, weights of multiple organs—including liver, kidney, spleen, and thymus—were markedly reduced in the E2f7 and, to a lesser extent, E2f8 Tg mice (Fig. 1D; Supporting Fig. S1F,G).
Immunohistochemical analysis of Ki67 confirmed a marked reduction of cycling cells in small intestinal crypts of E2f7 and E2f8 Tg mice, thus reducing the number of cells per crypt in each transgenic model (Fig. 1E; Supporting Fig. S1H). These phenotypes were more profound in E2f7 Tg mice than their E2f8 Tg counterparts. The development of E2f8DBDmut. Tg mice did not deviate from normal development of controls.
Collectively, the data demonstrate that E2F7 or E2F8 overexpression is sufficient to repress cell proliferation during postnatal development through a mechanism that involves DNA binding.
E2F7/8 Overabundance Inhibits DNA Replication In Vivo
Next, we examined whether the observed growth inhibitions were due to perturbations in DNA replication. To this end, mice treated with doxycycline for 3 days were injected with the thymidine analogue 5‐bromo‐2′‐deoxyuridine (BrdU) 2 hours prior to euthanasia. Indeed, the incorporation of BrdU was reduced in both small intestinal crypts and hepatocytes of juvenile E2f7 and E2f8 Tg mice (Fig. 2A,B; Supporting Fig. S2A). Reductions were most profound in E2f7 Tg mice and not observed in E2f8DBDmut. Tg mice. In complementary experiments, we evaluated S‐phase progression upon E2F7 and E2F8 overexpression on human RPE cells with doxycycline‐inducible expression of EGFP‐tagged E2F7 and E2F8 (Supporting Fig. S2B,C). DNA replication fork progression was visualized and quantified by performing a DNA fiber analysis.( 19 ) We found that the speed of replication fork progression and the percentage of new origins that fired were significantly reduced by transgene induction (Fig. 2C; Supporting Fig. S2D,E), confirming that overexpression of our E2F7 and E2F8 fusion proteins leads to replication stress. As expected, RPE cell lines expressing the DNA‐binding mutant versions of our E2F7 and E2F8 transgenes showed no signs of replication stress (Supporting Fig. S4A‐C).
In addition, expression of activator E2Fs, cyclin A2 and cyclin B1 was decreased in RPE cells upon induction of E2F7 and E2F8 (Fig. 2D). The DNA repair protein RAD51 and the cyclin‐dependent kinase (CDK) inhibitor P21, which are both known E2F7/8 target genes, were also strongly repressed in E2F7/8 expressing cells (Fig. 2D). Surprisingly, p21 levels were profoundly induced in HeLa cells after E2F7 induction, which could potentially be explained by defective checkpoint functions. Importantly, the inhibitory effect of E2F7 on proliferation was consistently observed independently of the genetic background (HeLa versus RPE) and p53/Rb status (Fig. 2D; Supporting Figs. S2F and S3A‐D). In addition, we observed repression of DNA replication and DNA repair transcripts as well as an inhibitory proliferative effect on EGFP‐sorted HCC‐derived cell lines with null and mutated p53, Hep3B and Huh7, respectively, upon transfection with E2F7‐EGFP and E2F8‐EGFP plasmids (Fig. 2E,F; Supporting Fig. S2G,H).
Together, these data show that overabundance of E2F7 and E2F8 causes DNA replication stress through transcriptional repression of multiple essential cell cycle genes.
E2F7/8 Overexpression Causes DNA Damage and Apoptosis
Because E2F7 and E2F8 repress multiple DNA repair genes, their overexpression would not only predispose cells to replication stress–induced double strand breaks but also inhibit the repair of such lesions.( 1 , 20 , 21 ) Indeed, E2f7 and E2f8 Tg mice showed a marked increase of gamma H2A.X variant histone (γ‐H2AX)–positive nuclei in intestines and livers after 3 and 6 days with doxycycline, indicating severe DNA damage in these tissues (Fig. 3A,B). On the other hand, E2f8DBDmut. Tg mice were unaffected. In addition, we observed increased rates of apoptosis in small intestinal crypts of E2f7 and E2f8 Tg mice, suggesting that the DNA damage was severe enough to be fatal (Fig. 3C; Supporting Fig. S5A). As a second tissue, we analyzed liver, but overall apoptosis rates were extremely low, precluding reliable assessment of differences between genotypes (Supporting Fig. S5B). Induction of E2F7 or E2F8 expression also caused severe DNA damage in RPE cells (Fig. 2E; Supporting Fig. S5C,D). Collectively, these data indicate that E2F7 and E2F8 overexpression creates replication stress–induced DNA damage.
Fig. 3.
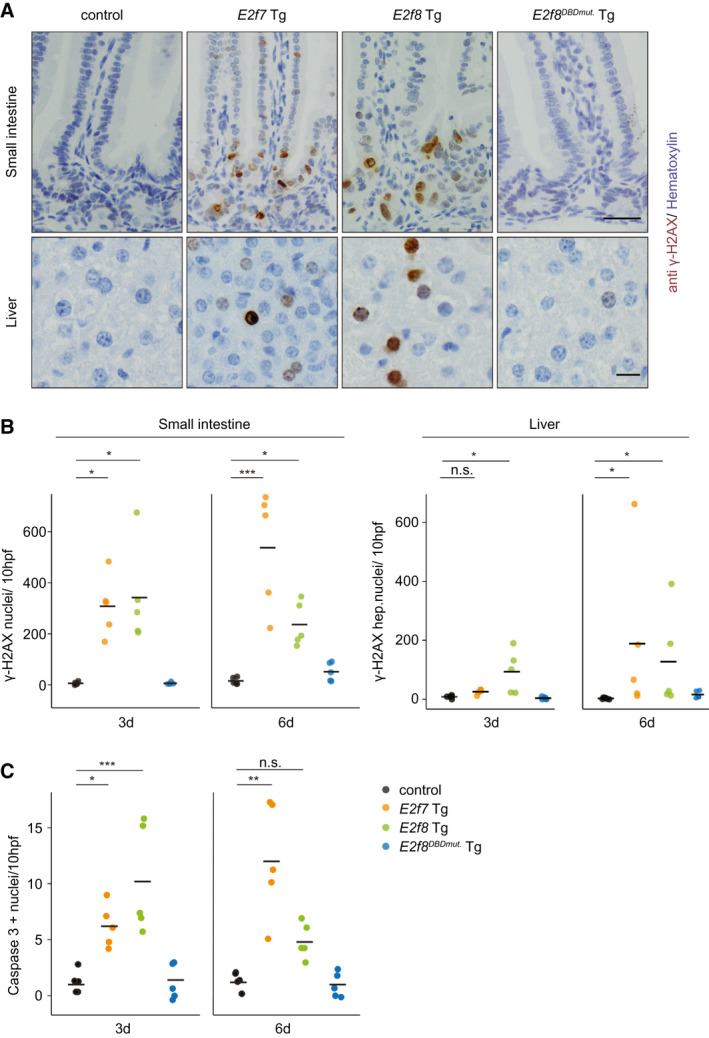
Replication stress induced by Tg E2F7/8 expression leads to DNA damage and apoptosis. (A) Representative pictures of γ‐H2AX immunohistochemistry of small intestines (top) and livers (bottom) of Tg mice after 6 days of doxycycline treatment. Scale bars, 50 μm (top) and 20 μm (bottom). (B) Quantification of total γ‐H2AX‐positive nuclei in 10 fields using ×40 objective of small intestine and liver sections after 3 and 6 days of doxycycline treatment. Crossbars represent average (n = 5 mice). (C) Quantification of total cleaved caspase 3–positive nuclei in 10 fields using ×40 objective of small intestine sections after 3 and 6 days of doxycycline treatment. Crossbars represent average (n = 5 mice). Data information: In (B,C), *P < 0.05, **P < 0.01, ***P < 0.001 (Kruskal‐Wallis one‐way analysis of variance on ranks and Dunnett’s method for multiple comparisons versus control). Abbreviations: hpf, high‐power field; n.s., not significant.
E2F7/8 Transgene Induction in Liver Tumors Causes Replication Stress and Inhibits Neoplastic Growth
To study the in vivo consequences of elevated E2F7/8 levels in cancer cells, we used our Tg mice to induce E2F7 and E2F8 expression in a liver cancer model. To this end, control, E2f7 Tg , E2f8 Tg, and E2f8DBDmut. Tg male mice were injected once with the liver‐specific carcinogen diethylnitrosamine (DEN) at postnatal day 14 (Fig. 4A). After 9 months, we confirmed that 20 randomly chosen mice had macroscopic tumors, although there was substantial variation in tumor burden between mice (Supporting Fig. S6A,B). Interestingly, 1 month of transgene induction neither affected body weights nor caused obvious structural or functional abnormalities in proliferative tissues such as small intestine (Supporting Fig. S6C,D). These findings suggest that mice tolerate E2F7 and E2F8 overexpression much better in adulthood than during postnatal development.
Fig. 4.
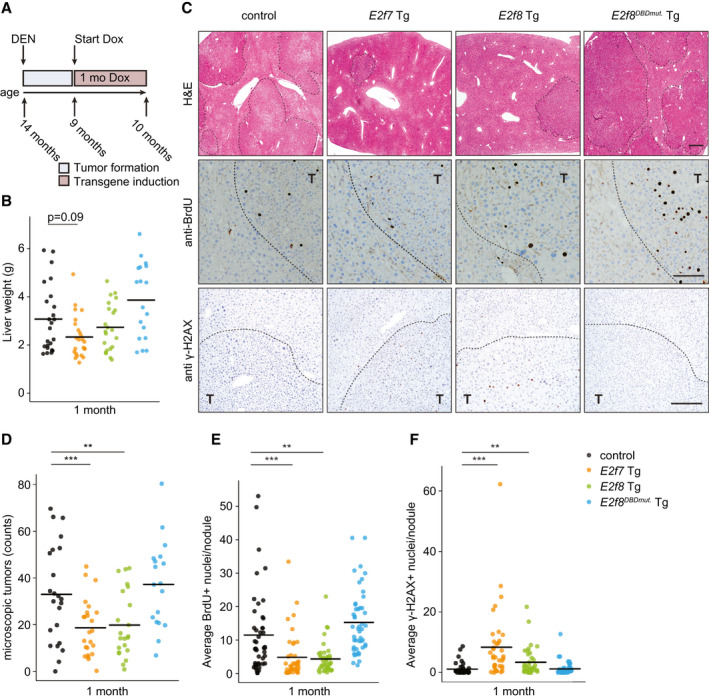
Proliferation of DEN‐induced liver tumors is inhibited by induction of transgenic E2F7 expression. (A) Schematic overview of the tumor experiment. (B) Gross liver weights after 1 month of doxycycline treatment. Crossbars indicate the average per genotype (control n = 24, E2f7 Tg n = 24, E2f8 Tg n = 22, E2f8DBDmut. Tg n = 18 mice). (C) Representative pictures of hematoxylin and eosin–stained (top), BrdU‐stained (middle), and γ‐H2AX‐stained (bottom) liver sections after 1 month of doxycycline treatment. Liver tumors are outlined with dashed lines. Scale bars, 500 μm (top), 100 μm (middle), and 50 μm (bottom). (D) Dot plot showing the numbers of microscopic liver tumors detected in hematoxylin and eosin–stained sections per mouse after 1 month of doxycycline. Crossbars represent average per genotype (control n = 23, E2f7 Tg n = 24, E2f8 Tg n = 22, E2f8DBDmut. Tg n = 18 mice). (E) Quantification of BrdU immunohistochemically stained liver sections from (C). Each dot represents the average of BrdU‐positive hepatocytes, counted in five fields (×40 objective), per nodule. At least three random tumor nodules were analyzed per mouse, n = 10 mice/genotype) (control n = 46, E2f7 Tg n = 40, E2f8 Tg n = 42, E2f8DBDmut. Tg n = 47 nodules). (F) Average number of γ‐H2AX‐positive hepatocyte nuclei per field (five fields, ×40 objective) per tumor nodule. At least three random tumor nodules were analyzed per animal, n = 10 mice/genotype. Crossbars represent average values per genotype (control n = 41, E2f7 Tg n = 38, E2f8 Tg n = 38, E2f8DBDmut. Tg n = 50 nodules). Data information: In (B,D‐F), *P < 0.05, **P < 0.01, ***P < 0.001 (Kruskal‐Wallis one‐way analysis of variance on ranks and Dunnett’s method for multiple comparisons versus control). Abbreviations: Dox, doxycycline; H&E, hematoxylin and eosin; T, tumor.
Analysis of gross liver weights and number of macroscopically visible tumors suggested that E2F7 overexpression reduced DEN‐induced tumor burden, although differences did not reach statistical significance due to the large variation among animals (Fig. 4B; Supporting Fig. S6E). E2f8 Tg mice showed a similar but weaker trend, whereas the mean tumor burden in E2f8DBDmut. Tg mice was nearly identical to that of control mice. For more in‐depth analysis, we performed detailed histological analysis on seven independent liver sections of each mouse in the study. This analysis revealed a highly significant decrease in the numbers of histologically detectable tumors in E2f7 and E2f8 Tg livers, as well as a reduction in the ratio between tumor and normal liver tissue (Fig. 4C,D; Supporting Fig. S6F). Importantly, tumor nodules of E2f7 and E2f8 Tg mice showed reduced BrdU incorporation compared to controls, indicative of reduced DNA replication (Fig. 4C,E), which was accompanied by an increase in DNA damage, as measured with γ‐H2AX staining (Fig. 4C,F).
Histological analysis revealed that the majority of the nodules in all genotypes are premalignant lesions, characterized as focal cellular alteration. HCCs were diagnosed in a very low percentage in all genotypes (Supporting Fig. S7A). Notably, BrdU incorporation was reduced in tumor nodules of E2f7 and E2f8 Tg mice independently of their grade of malignancy (Supporting Fig. S7B). These data are consistent with a model in which E2f7 and E2f8 overexpression inhibits tumorigenesis by blocking cell proliferation in DEN‐induced liver cancers. Additionally, in the absence of DEN treatment, adult E2f7 and E2f8 Tg mice that were long term–treated with doxycycline developed spontaneous liver tumor with decreased incidence compared to control animals (Supporting Fig. S7C).
Taken together, these data suggest that E2F7 and E2F8 can inhibit tumor growth by interfering with DNA replication, irrespective of the grade of malignancy. Importantly, adult mice were able to tolerate the induction of the transgenes with little effect on their quality of life.
Heterogeneity in Transgene Expression Underlies Variation in Liver Tumor Inhibition by E2F7
To determine whether the heterogeneity in tumor burden among E2F7 and E2F8 Tg mice might be due to differences in transgene expression, we immunolabeled tumor level sections of DEN‐treated mice for the EGFP tags of the Tg proteins. Surprisingly, we observed completely negative tumor nodules side by side with strongly positive nodules in E2f7 and, to a lesser extent, E2f8 Tg liver tumors (Fig. 5A; Supporting Fig. S8A). Analysis of EGFP‐positive versus EGFP‐negative tumor nodules did not show differences in vascularization. Thus, variation in accessibility to doxycycline is unlikely to explain the variation in transgene expression (Supporting Fig. S8B). The vast majority of the tumors from E2f8DBDmut. Tg mice presented high numbers of EGFP‐positive cells (Supporting Fig. S8A), indicating that E2F7 and E2F8 transgene expression was selected against in a subset of tumors. Thus, E2F7/8 transgene induction may be such a potent mechanism of inhibiting tumorigenesis that transgene silencing seems to be an important requirement for tumor growth (Fig. 5B).
Fig. 5.
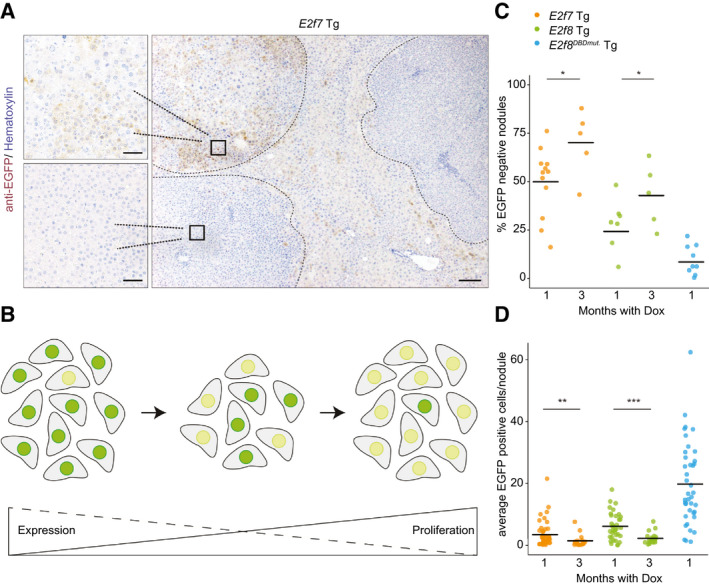
Antiproliferative effects of E2F7/8 overexpression cause the appearance of liver tumor nodules lacking transgene expression. (A) Representative anti‐EGFP immunohistochemistry pictures showing E2f7‐EGFP Tg expression in livers treated for 1 month with doxycycline. Scale bars, 200 μm (main), 50 μm (side pictures). Dashed lines delineate tumor nodules. (B) Proposed model of the observed and described variation in expression and proliferative capacity of cells overexpressing E2F7 both in cell culture system and in mice. Dark green and yellow nuclei represent high and low E2F7‐EGFP expression levels, respectively. (C) Percentage of immunohistochemically stained EGFP‐negative nodules relative to the total number of microscopic tumors in the indicated genotype. Bars represents average per genotype. *P < 0.05 (t test). (D) Dot plot represents the count of EGFP‐positive hepatocyte nuclei in five fields (×40 objective) per nodule. At least three random tumor nodules were analyzed per animal, n = at least 10 mice/genotype). **P < 0.01, ***P < 0.001 (Mann‐Whitney rank sum test).
To further investigate this phenomenon, we kept an additional cohort of DEN‐treated mice on prolonged transgene induction of 3 months with doxycycline (Supporting Fig. S9A). Interestingly, average liver weights of control, E2f7, and E2f8 Tg mice were similar and all higher after 3 months than after 1 month of transgene induction (Supporting Fig. S9B). Moreover, quantification of EGFP‐stained tissue sections revealed a marked increase in the percentages of completely EGFP‐negative nodules over time in E2f7 and E2f8 Tg mice (Fig. 5C). In contrast, E2f8DBDmut. Tg mice presented very low percentages of EGFP‐negative nodules. We also saw that the average number of EGFP‐positive cells per nodule clearly decreased in the period from 1 month to 3 months with doxycycline in the E2f7 and E2f8 Tg mice (Fig. 5D). Importantly, genotyping of 3 month–treated tumor samples showed the presence of the transgene locus, indicating that loss of the transgene locus is an unlikely scenario (Supporting Fig. S9C). However, despite comparable mRNA expression of the Tet activator in juvenile livers and liver tumors after 3 months of doxycycline, we observed a strong reduction of EGFP mRNA in the tumor samples (Supporting Fig. S9D). This indicates that transgene expression in tumor cells might be epigenetically silenced, which is a known phenomenon for tetracycline‐inducible cytomegalovirus (CMV) promoters.( 22 , 23 , 24 , 25 , 26 )
To further support this concept of E2F7/8‐mediated selection pressure, we then set out to mimic it in vitro. To test this, we cultured HeLa cell lines with stable doxycycline‐inducible expression of EGFP‐tagged E2F7. We previously demonstrated strong inhibition of proliferation and induction of apoptosis for up to 4 days,( 1 , 18 ) but now we induced E2F7 overexpression for up to 20 days. Quantitative PCR and flow‐cytometric analysis revealed a near‐complete loss of E2F7‐EGFP expressing cells within 12 days (Fig. 6A; Supporting Fig. S10A). Genotyping PCRs on these HeLa/Tet On cell lines demonstrated that the E2F7‐EGFP locus was still clearly detectable after 20 days of doxycycline. These data again suggest epigenetic silencing of the inducible E2F7‐EGFP (Supporting Fig. S10B). Accordingly, E2F7‐EGFP cell lines cultured for 20 days in doxycycline proliferated at a similar rate as noninduced cells, whereas acute induction caused a near‐complete inhibition of proliferation (Fig. 6B). Importantly, a control cell line carrying doxycycline‐inducible EGFP had very stable expression levels over the course of 20 days, suggesting again that the loss of expression in E2F7‐EGFP cell lines is due to the selection pressure caused by the antiproliferative effect of E2F7 (Fig. 6A; Supporting Fig. S10A).
Fig. 6.
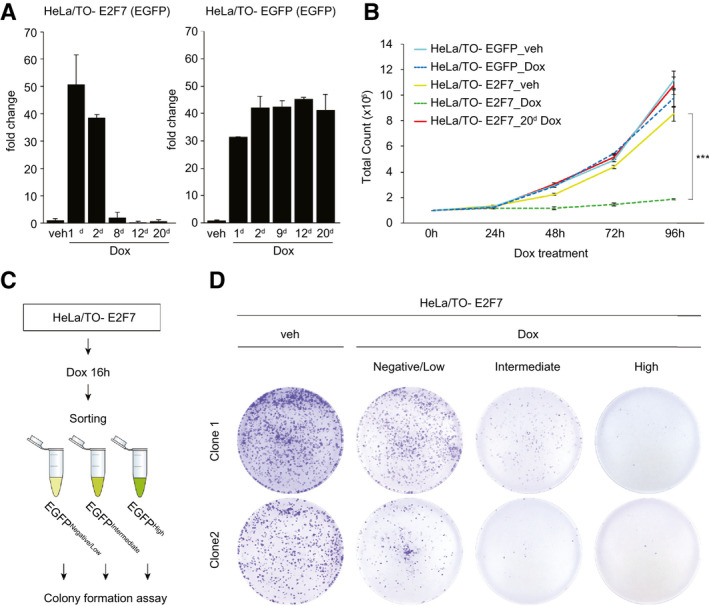
Selection pressure by inducible overexpression of E2F7 in vitro favors proliferation of cells with low levels of E2F7‐EGFP. (A) Quantitative PCR results showing EGFP transcript expression in inducible E2F7‐EGFP HeLa cells and EGFP after 1, 2, 8, 12, and 20 days of doxycycline administration. Two independent experiments. (B) Proliferation curves of HeLa cells with inducible E2F7‐EGFP and EGFP after doxycycline treatment. Colored lines represent average ± SD of two technical replicates for each time point. Representative curves of two independent experiments. (C) Experimental scheme of FACS experiment to determine E2F7‐EGFP overexpression levels on clonogenic capacity of HeLa cells. (D) Representative images of colony formation assay with cells sorted in (C) from two independent clones. Two independent experiments were performed in duplicate for three different cell clones of inducible E2F7‐EGFP HeLa cell lines. Abbreviations: Dox, doxycycline; veh, vehicle.
We then used FACS on inducible HeLa E2F7‐EGFP based on expression levels shortly after doxycycline induction (16 hours), and we analyzed the colony‐forming capacity of these cells. This assay revealed that HeLa cells with high expression of E2F7 cannot survive long term compared to negative or very low expressers and vehicle condition (Fig. 6C,D). Even when we removed the doxycycline after sorting and replating, high E2F7 expressing cells did not proliferate, indicating that they are irreversibly damaged after only 16 hours of E2F7 induction (Supporting Fig. S10C). The colony‐forming capacity of cells expressing E2F7DBDmut. was not affected, confirming that transcriptional repression is the mechanism of DNA damage induction (Supporting Fig. S10D).
Collectively, our data reveal that Tg expression of E2F7 has a strong dose‐dependent antiproliferative effect on cancer cells in vivo. This creates a strong growth advantage for cancer cells losing transgene expression.
Deregulation of E2F‐Dependent Transcription Is Associated With Disease Progression and Poor Survival
To determine the relevance of our findings to human HCC, we compared patients with HCC (The Cancer Genome Atlas Liver Hepatocellular Carcinoma) with high versus low expression of E2F7 and E2F8 target genes (Supporting Fig. S11A). We observed that deregulated expression of these target genes was strongly associated with poor survival. Interestingly, when we divided patients according to tumor stage (American Joint Committee on Cancer stages I‐III), we observed only a strong correlation in the advanced stages of HCC (II and III; Supporting Fig. S11B). Collectively, these results further support the notion that E2F‐dependent transcription contributes to cell proliferation and disease progression, resulting in poor clinical outcome in patients with HCC.
Discussion
Up‐regulation of E2F‐dependent transcription due to alterations in the CDK/RB/E2F pathway is an important characteristic of HCC. Our in vitro and in vivo studies demonstrate that boosting atypical E2F repressor activity can efficiently block deregulated E2F‐dependent transcription, leading to reduced proliferation of neoplastic hepatocytes. Mechanistically, we show that overexpression of E2F7 or E2F8 represses transcription of E2F target genes involved in DNA replication and DNA repair, resulting in DNA replication stress and DNA damage. Importantly, we show that ubiquitous induction of atypical E2F activity inhibits liver tumor growth without a major impact on the health status of adult mice. These findings could open a therapeutic avenue for patients with HCC through increasing the transcriptional repressor activity of atypical E2Fs.
The Tg mouse model presented here provides a powerful tool to inactivate E2F‐dependent transcription at any desired time and allowed us to evaluate this effect during mouse development and liver cancer progression. The notion that E2F7 overexpression virtually blocks postnatal development is consistent with previous work showing that combined loss of the activators E2f1 and E2f3a results in growth retardation, dysplasia of multiple tissues, and multiorgan failure.( 27 ) However, loss of activator E2Fs can either cause impaired S‐phase entry or S‐phase progression. Thus, in that model it is difficult to distinguish the relative importance of these two functions. In addition, E2F1‐3 can switch from activators to repressors, depending on tissue context and Rb status.( 28 ) The advantage of inducible E2F7/8 induction is that its effect is largely confined to S/G2‐phase progression: we previously demonstrated that both endogenous and exogenous E2F7 and E2F8 are highly efficiently degraded through anaphase promoting complex/cyclosome relying on cadherin 1 (APC/CCdh1) during G1.( 18 ) This means that the Tg E2f7/8 only accumulates once S phase has already begun. Therefore, the cell cycle defects in multiple organs in the juvenile E2f7/8 Tg mice described here now unequivocally show that unscheduled inactivation of E2F transcription during S phase strongly impairs cell cycle progression.
Inhibition of cell cycle progression by the E2f7 and E2f8 transgenes in normal cells as well as cancer cells clearly supports their role as tumor suppressors. Nevertheless, E2F7 and in particular E2F8 have also been suggested to have oncogenic effects. This idea is based on the notion that E2F7 and E2F8 are highly expressed in cancer and that their transcript levels positively correlate with poor prognosis.( 15 , 16 , 17 ) However, E2F7 and E2F8 are E2F target genes themselves, whose expression levels peak during S phase. Therefore, high E2F7/8 mRNA levels simply correlate with a high percentage of cycling cells, and hence poor prognosis. To understand the biological actions of atypical E2Fs on tumor growth, it should be taken into account that they are tightly regulated by multiple posttranslational mechanisms, including APC/CCdh1 and checkpoint kinase 1 (CHK1), indicating that mRNA levels do not necessarily reflect high activity of atypical E2Fs.( 18 , 21 ) A second notion is that some in vitro studies using cancer cell lines suggested that overexpression of E2F8 promoted tumor cell proliferation.( 15 , 16 , 17 , 30 ) However, we now demonstrate that E2F7/8 overexpression invokes a strong selection pressure on cells. Thus, generation of stable E2F7/8‐overexpressing cell lines could be problematic because it would favor the selection of severely adapted cell lines. Nevertheless, we cannot exclude that cell‐type context or concomitant mutations in cancer‐related genes uncover oncogenic functions of E2F7 or E2F8.
Our study shows that severe repression of E2F‐dependent transcription during S phase causes clear signs of DNA replication stress. Because replication stress is often seen in cancer cells, it is likely that tipping the balance between atypical repressors and activator E2Fs toward the former will have a particularly strong impact on the cell‐cycle progression of cancer cells under DNA damaging conditions. In this respect, it is interesting to note that CHK1 inhibits repressor E2F activity under conditions of replication stress to maintain a high level of E2F‐dependent transcription for boosting DNA repair and restarting DNA replication.( 20 , 31 ) As a consequence, the cell‐cycle arrest seen in CHK1‐depleted cells is rescued by additional loss of atypical E2Fs.( 21 ) Together with these insights, our data suggest that low levels of E2F‐dependent transcription sensitize cancer cells to replication stress. In further support of this notion, recent work showed that high levels of E2F‐dependent transcription, through loss of E2F7, promote resistance of cancer cells toward poly(adenosine diphosphate‐ribose) polymerase inhibitors and interstrand‐crosslinking drugs such as cisplatin and mitomycin C.( 32 , 33 )
In conclusion, the inducible Tg mouse model presented here strongly suggests that a basal level of E2F‐dependent transcription is essential for the proliferation of mammalian cells. In addition, we provide a strong rationale to combine the inhibition of E2F‐dependent transcription during S phase with DNA‐damaging reagents to achieve synergistic cancer‐killing effects. Future studies should explore whether the transcriptional repressor activity of atypical E2Fs can be boosted in patients with HCC, for example, by applying small molecules that specifically inhibit the degradation of E2F7/8.
Author Contributions
E.M., B.W., E.A.v.L., and R.Y. performed experiments. M.J.M.T. and S.C.v.E. provided technical assistance during mice necropsies and tissue processing. S.C.v.E. and M.H.K. performed immunohistochemical staining. L.B. performed the histopathological analysis of tumors. B.W., E.A.v.L., and R.Y. designed and tested the construct to develop the mouse models. J.v.D. generated the mice. All authors contributed to the revision of the manuscript. E.M., B.W., and A.d.B. conceived the study design and experimental approaches and data analysis. E.M., J.v.D., B.W., and A.d.B. wrote the manuscript.
Supporting information
Supplementary Material
Acknowledgment
We thank Wout Puijk and the rest of the animal caretakers for excellent care of our mice during experiments. Ger Arkesteijn (Faculty of Veterinary Medicine, Utrecht University, Utrecht, the Netherlands) and Reinier van der Linden (Hubrecht Institute, Utrecht, the Netherlands) for providing professional assistance with FACS. We also thank Rosan Heijboer for generation of the RPE inducible cell lines and Rachel Thomas for her contribution to the manuscript revision.
Supported by the Dutch Cancer Society (UU2013‐5777, 11941/2018‐2, to B.W. and A.d.B.), an Utrecht Life Sciences Infrastructure Grant (call 2014‐I), and ZonMW (91116011).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Westendorp B, Mokry M, Koerkamp G, Marian JA, Holstege FCP, Cuppen E, et al. E2F7 represses a network of oscillating cell cycle genes to control S‐phase progression. Nucleic Acids Res 2012;40:3511‐3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bertoli C, Skotheim J, de Bruin RAM. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 2013;14:518‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Di Stefano L, Jensen M, Helin K. E2F7, a novel E2F featuring DP‐independent repression of a subset of E2F‐regulated genes. EMBO J 2003;22:6289‐6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Bruin A, Maiti B, Jakoi L, Timmers C, Buerki R, Leone G. Identification and characterization of E2F7, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem 2003;278:42041‐42049. [DOI] [PubMed] [Google Scholar]
- 5. Maiti B, Li J, de Bruin A, Gordon F, Timmers C, Opavsky R, et al. Cloning and characterization of mouse E2F8, a novel mammalian E2F family member capable of blocking cellular proliferation. J Biol Chem 2005;280:18211‐18220. [DOI] [PubMed] [Google Scholar]
- 6. Logan N, Graham A, Zhao X, Fisher R, Maiti B, Leone G, et al. E2F‐8: an E2F family member with a similar organization of DNA‐binding domains to E2F‐7. Oncogene 2005;24:5000‐5004. [DOI] [PubMed] [Google Scholar]
- 7. Chen H, Tsai S, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 2009;9:785‐797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Logan N, Delavaine L, Graham A, Reilly C, Wilson J, Brummelkamp T, et al. E2F‐7: a distinctive E2F family member with an unusual organization of DNA‐binding domains. Oncogene 2004;23:5138‐5150. [DOI] [PubMed] [Google Scholar]
- 9. Nevins JR. The rb/E2F pathway and cancer. Hum Mol Genet 2001;10:699‐703. [DOI] [PubMed] [Google Scholar]
- 10. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995;81:323‐330. [DOI] [PubMed] [Google Scholar]
- 11. Aksoy O, Chicas A, Zeng T, Zhao Z, McCurrach M, Wang X, et al. The atypical E2F family member E2F7 couples the p53 and RB pathways during cellular senescence. Genes Dev 2012;26:1546‐1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thurlings I, Martínez López LM, Westendorp B, Zijp M, Kuiper R, Tooten P, et al. Synergistic functions of E2F7 and E2F8 are critical to suppress stress‐induced skin cancer. Oncogene 2017;36:829‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kent L, Rakijas J, Pandit S, Westendorp B, Chen H, Huntington J, et al. E2f8 mediates tumor suppression in postnatal liver development. J Clin Invest 2016;126:2955‐2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carvajal L, Hamard P, Tonnessen C, Manfredi J. E2F7, a novel target, is up‐regulated by p53 and mediates DNA damage‐dependent transcriptional repression. Genes Dev 2012;26:1533‐1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park S, Platt J, Lee J, López Giráldez F, Herbst R, Koo J. E2F8 as a novel therapeutic target for lung cancer. J Natl Cancer Inst 2015;107 10.1093/jnci/djv151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ye L, Guo L, He Z, Wang X, Lin C, Zhang X, et al. Upregulation of E2F8 promotes cell proliferation and tumorigenicity in breast cancer by modulating G1/S phase transition. Oncotarget 2016;7:23757‐23771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lv Y, Xiao J, Liu J, Xing F. E2F8 is a potential therapeutic target for hepatocellular carcinoma. J Cancer 2017;8:1205‐1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boekhout M, Yuan R, Wondergem A, Segeren H, van Liere E, Awol N, et al. Feedback regulation between atypical E2Fs and APC/CCdh1 coordinates cell cycle progression. EMBO Rep 2016;17:414‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tuduri S, Tourrière H, Pasero P. Defining replication origin efficiency using DNA fiber assays. Chromosome Res 2010;18:91‐102. [DOI] [PubMed] [Google Scholar]
- 20. Bertoli C, Herlihy A, Pennycook B, Kriston‐Vizi J, de Bruin RAM. Sustained E2F‐dependent transcription is a key mechanism to prevent replication‐stress‐induced DNA damage. Cell Rep 2016;15:1412‐1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuan R, Vos H, van Es R, Chen J, Burgering B, Westendorp B, et al. Chk1 and 14‐3‐3 proteins inhibit atypical E2Fs to prevent a permanent cell cycle arrest. EMBO J 2018;37:e97877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nuo MT, Yuan JL, Yang WL, Gao XY, He N, Liang H, et al. Promoter methylation and histone modifications affect the expression of the exogenous DsRed gene in transgenic goats. Genet Mol Res 2016;15 10.4238/gmr.15038560. [DOI] [PubMed] [Google Scholar]
- 23. Ngai SC, Rosli R, Al Abbar A, Abdullah S. DNA methylation and histone modifications are the molecular lock in lentivirally transduced hematopoietic progenitor cells. Biomed Res Int 2015;2015:346134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu Y, Lowy M, Elble R. Tet‐on lentiviral transductants lose inducibility when silenced for extended intervals in mammary epithelial cells. Metab Eng Commun 2016;3:64‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu P, Aller MI, Baron U, Cambridge S, Bausen M, Herb J, et al. Silencing and un‐silencing of tetracycline‐controlled genes in neurons. PLoS One 2007;2:e533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gödecke N, Zha L, Spencer S, Behme S, Riemer P, Rehli M, et al. Controlled re‐activation of epigenetically silenced tet promoter‐driven transgene expression by targeted demethylation. Nucleic Acids Res 2017;45:e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tsai S, Opavsky R, Sharma N, Wu L, Naidu S, Nolan E, et al. Mouse development with a single E2F activator. Nature 2008;454:1137‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chong J, Wenzel P, Sáenz Robles MT, Nair V, Ferrey A, Hagan J, et al. E2f1‐3 switch from activators in progenitor cells to repressors in differentiating cells. Nature 2009;462:930‐934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deng Q, Wang Q, Zong W, Zheng D, Wen Y, Wang K, et al. E2F8 contributes to human hepatocellular carcinoma via regulating cell proliferation. Cancer Res 2010;70:782‐791. [DOI] [PubMed] [Google Scholar]
- 30. Chang H, Song J, Wu J, Zhang Y. E2F transcription factor 8 promotes cell proliferation via CCND1/p21 in esophageal squamous cell carcinoma. Onco Targets Ther 2018;11:8165‐8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bertoli C, Klier S, McGowan C, Wittenberg C, de Bruin RAM. Chk1 inhibits E2F6 repressor function in response to replication stress to maintain cell‐cycle transcription. Curr Biol 2013;23:1629‐1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clements K, Thakar T, Nicolae C, Liang X, Wang H, Moldovan G. Loss of E2F7 confers resistance to poly‐ADP‐ribose polymerase (PARP) inhibitors in BRCA2‐deficient cells. Nucleic Acids Res 2018;46:8898‐8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mitxelena J, Apraiz A, Vallejo Rodríguez J, García Santisteban I, Fullaondo A, Alvarez Fernández M, et al. An E2F7‐dependent transcriptional program modulates DNA damage repair and genomic stability. Nucleic Acids Res 2018;46:4546‐1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nam H, van Deursen J. Cyclin B2 and p53 control proper timing of centrosome separation. Nat Cell Biol 2014;16:538‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li J, Ran C, Li E, Gordon F, Comstock G, Siddiqui H, et al. Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev Cell 2008;14:62‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pandit S, Westendorp B, Nantasanti S, van Liere E, Tooten PCJ, Cornelissen PWA, et al. E2F8 is essential for polyploidization in mammalian cells. Nat Cell Biol 2012;14:1181‐1191. [DOI] [PubMed] [Google Scholar]
- 37. Costa DA, Gil RM, Paula Santos N, Rocha A, Colaço A, Lopes C, et al. The N‐nitrosodiethylamine mouse model: sketching a timeline of evolution of chemically‐induced hepatic lesions. Anticancer Res 2014;34:7029‐7037. [PubMed] [Google Scholar]
- 38. Thoolen B, Maronpot R, Harada T, Nyska A, Rousseaux C, Nolte T, et al. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol Pathol 2010;38(7 Suppl.):5S‐81S. [DOI] [PubMed] [Google Scholar]
- 39. Benedict B, van Harn T, Dekker M, Hermsen S, Kucukosmanoglu A, Pieters W, et al. Loss of p53 suppresses replication‐stress‐induced DNA breakage in G1/S checkpoint deficient cells. Elife 2018;7; pii: e37868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 1998;140:1285‐1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material