

Abstract
Two N‐methylpyridinium compounds and analogous N‐protonated salts of 2‐ and 2,7‐substituted 4‐pyridyl‐pyrene compounds were synthesised and their crystal structures, photophysical properties both in solution and in the solid state, electrochemical and spectroelectrochemical properties were studied. Upon methylation or protonation, the emission maxima are significantly bathochromically shifted compared to the neutral compounds, although the absorption maxima remain almost unchanged. As a result, the cationic compounds show very large apparent Stokes shifts of up to 7200 cm−1. The N‐methylpyridinium compounds have a single reduction at ca. −1.5 V vs. Fc/Fc+ in MeCN. While the reduction process was reversible for the 2,7‐disubstituted compound, it was irreversible for the mono‐substituted one. Experimental findings are complemented by DFT and TD‐DFT calculations. Furthermore, the N‐methylpyridinium compounds show strong interactions with calf thymus (ct)‐DNA, presumably by intercalation, which paves the way for further applications of these multi‐functional compounds as potential DNA‐bioactive agents.
Keywords: chromophores, luminescent, pyrenes, pyridinium, viologens
Multi‐functional pyrene‐viologens: The synthesis, molecular and electronic structures, photophysical, electrochemical, and spectroelectrochemical properties of 2‐ and 2,7‐substituted para‐N‐methylpyridinium pyrene compounds are described along with detailed theoretical calculations. These compounds show strong interactions with calf thymus (ct)‐DNA, presumably by intercalation.
Introduction
Pyrene, a polycyclic aromatic hydrocarbon (PAH), is one of the most widely investigated fluorophores, as it possesses some unique properties, such as intense blue emission, a high fluorescence quantum yield, a long‐lived singlet state, and propensity for excimer and exciplex formation. [1] In addition to its rich photophysical properties, pyrene is chemically stable and can enhance intermolecular charge mobility. [1] Many pyrene derivatives have, therefore, been developed for a wide range of applications in modern scientific fields which include organic optoelectronic devices, such as organic light emitting diodes (OLEDs), organic field‐effect transistors (OFETs), and organic photovoltaic cells (OPVs). [2] The fluorescence of pyrene derivatives has been employed to investigate interactions in macromolecules and lipids,[ 3 , 4 ] and also in probes for signalling of biomolecules, such as oligonucleotides and nucleic acids (e.g., DNA). [5] In addition, their long fluorescence lifetimes and the possibility of excimer formation enable the exploration of advanced applications, such as the detection of DNA/RNA interactions both as a single label, [6] as well as excimer‐forming pairs or as multi‐pyrene probes. [7] The flat aromatic structure of pyrene derivatives facilitates its stacking with nucleobases and consequently intercalation into DNA/RNA. [8] However, intercalative binding to DNA/RNA has limited selectivity and, therefore, some interesting applications of those pyrene derivatives rely on their interactions with DNA or RNA grooves [9] or in combination with their turn‐on and ‐off excimers. [10] Pyrene derivatives have further been employed for applications including the determination of cellular oxygen concentrations or reactive oxygen species (ROS) in biological systems, [11] and the detection of various metal ions in solutions. [12] In addition, as the fluorescence of pyrene‐based compounds can be influenced by external stimuli, this has been exploited for various applications such as sensing temperature, [13] pressure, [14] pH, [15] and detection of small molecules. [16] The ability of pyrene to exhibit strong π⋅⋅⋅π interactions makes it a suitable probe to examine arene‐perfluoroarene interactions and associated electrostatic quadrupole‐quadrupole interactions, even in dilute solutions, e.g., with hexafluorobenzene and octafluoronaphthalene. [17] Furthermore, metal organic frameworks (MOFs) [18] and covalent organic frameworks (COFs) [19] have been reported with linkers containing a pyrene moiety.
Although pyrene is a symmetric molecule, its 10 peripheral reactive sites (C−H bonds) can be divided into three chemically inequivalent classes, depending on different electronic and steric features that correspond to different chemical reactivities.[ 20 , 21 ] The 1‐, 3‐, 6‐, and 8‐positions of pyrene, having maximum contributions of the highest occupied molecular orbital (HOMO), are favourable for electrophilic aromatic substitution. [22] Thus, the π‐orbitals of substituents at these positions participate very efficiently with pyrene's HOMO and LUMO. The 2‐ and 7‐positions of pyrene lie on a nodal plane in both the HOMO and LUMO, but are the least hindered sites. Therefore, sterically controlled reactions such as Ir‐catalysed direct borylation, are favourable at these sites. [23] Moreover, the substituents at the 2‐ or 2,7‐positions do not interact with the HOMO/LUMO, but can interact strongly with the HOMO‐1 and LUMO+1 of pyrene that have non‐zero contributions at these positions.[ 21 , 24 , 25 , 26 ] Consequently, the photophysical properties of pyrene derivatives with substituents at the 2‐ or 2,7‐ positions differ significantly from those with substituents at the 1‐, 3‐, 6‐, and 8‐positions. This is termed the “site‐effect”.[ 20 , 24 ] It has also been demonstrated by us and others that judicious incorporation of a combination of strong π‐donors/acceptors at the 2‐ or 2,7‐positions of pyrene switches the order of its HOMO/HOMO‐1 and LUMO/LUMO+1, respectively, which in turn greatly influences the photophysical and electrochemical properties.[ 21 , 25 , 26 ] The localised double bond (C=C) character at the 4‐, 5‐, 9‐, and 10‐positions can be exploited to derivatise pyrene at its K‐region.[ 27 , 28 ] Thus, various compounds with substituents at the 4‐, 5‐, 9‐, and 10‐positions with interesting photophysical and electrochemical properties have recently been reported.[ 20 , 29 , 30 ]
Methylviologens, or viologens in general, are di‐N,N’‐functionalised 4,4′‐bipyridyl salts, which have been extensively investigated in the past, and have attracted more attention recently.[ 31 , 32 ] The possibility of having three stable redox states, efficient electron accepting capabilities (oxidizing agent), and tunability of the substituents at the nitrogen make them an important class of compounds. The most common viologen compound, N,N’‐dimethyl‐4,4′‐dipyridyl dichloride (MV2+), commonly known as „paraquat“, can undergo two reversible reductions, forming a radical‐cation (MVC+) and a neutral species (MV). Consequently, the colour changes from colourless (MV2+) to blue‐violet (MVC+), and finally to yellow‐brown (MV) during its redox processes. [31] Such compounds have wide applications in electrochromic[ 32 , 33 ] and photochromic materials. [34] Moreover, viologen‐based materials have been explored over the years for their application in molecular shuttles and switches or machines. [35] Recently, viologen‐based compounds have gained much importance for their use in organic „green batteries“. [36] It should be noted that paraquat was one of the most widely used herbicides for decades; however, it has been banned in several countries due to its acute toxicity.[ 37 , 38 ]
Depending on the substituents attached at the nitrogen atoms, and the counteranions, the colour of viologen compounds can change dramatically. Various counteranions have been utilised, halides being the most common and, recently, non‐coordinating anions such as triflate and hexafluorophosphate have been employed. The counteranions also play an important role in the stability and solubility of viologens. [39] Significant efforts have been devoted to fine tune the electronic and photophysical properties of viologens by introducing further functionalisation such as additional bridging or incorporating a hetero‐atom in the molecule. A few examples are sulfide‐bridged viologens, [40] phosphole‐bridged “phosphaviologen”, [41] germanium‐bridged viologen, [42] thiophene‐based, [43] and π‐extended viologen compounds. [44]
Recently, we have reported the syntheses and photophysical properties of several mono‐ and bis‐pyridyl‐pyrene compounds substituted at various positions of pyrene. [20] It has been demonstrated that the protonation of pyridyl‐pyrene compounds shifts the emission bathochromically. [45] The protonation of other fluorophores containing a conjugated pyridyl moiety has also been demonstrated to exhibit bathochromically shifted emissions. [46] Thus, polyaromatic hydrocarbon (PAH)‐based viologen compounds or compounds containing pyridinium moieties have attracted attention recently due to their interesting photophysical properties which have various potential applications. [47] Konishi and co‐workers reported a pyrene‐based viologen compound where the extended π‐conjugation was elongated by incorporating an ethenyl group between the pyrene and pyridinium moieties. This acceptor‐π‐acceptor type dye exhibited an emission in the ‘tissue transparent window’ (650–1100 nm) and a two‐photon absorption band around 1000 nm with a large cross‐section, and thus performed better than analogous PAH dyes (e.g., with naphthalene and anthracene) in three‐dimensional (3D) imaging of mitochondria in living cells. [48] The photophysical properties of analogous 1,6‐bis(N‐methyl‐3’‐pyridinium)pyrene, 1,6‐bis(N‐methyl‐4’‐pyridinium)pyrene and 1,3,6,8‐tetrakis(N‐methyl‐4’‐pyridinium)pyrene have been reported along with their energy transfer processes on a clay surface in aqueous media. [49] However, detailed investigation of the photophysical properties along with electrochemical and spectroelectrochemical studies, and the interactions with biorelevant targets (e.g., DNA) of pyridinium‐pyrene compounds have not been documented so far. Herein, we report the structures, photophysical and electrochemical properties of mono‐ and bis‐(N‐methylpyridinium)‐pyrene and analogous protonated compounds (Scheme 1), as well as preliminary studies on DNA binding of cationic compounds 1M and 2M.
Scheme 1.
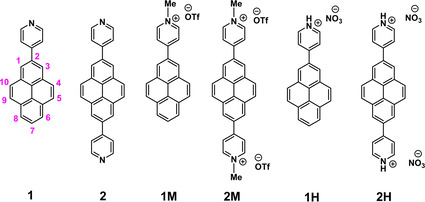
Compounds discussed in this study. Atom numbering of pyrene is also shown (left).
Results and Discussion
Synthesis
The neutral compounds 1 and 2 were synthesised via the Suzuki–Miyaura coupling reaction of 2‐(Bpin)pyrene and 2,7‐bis(Bpin)pyrene, respectively, with 4‐bromopyridine, as we have reported previously; [20] however, [Pd(dppf)Cl2] was used as the catalyst precursor instead of [Pd(PPh3)4]. The methylation reactions yielding compounds 1M and 2M were carried out with MeOTf in dry toluene under an argon atmosphere, and single crystals of 1M and 2M were obtained by slow evaporation of concentrated solutions of the compounds in MeCN. The salts 1H and 2H were obtained by reacting 1 and 2, respectively, with HNO3 in MeCN, and their single crystals were obtained by slow evaporation from MeCN and DMF solutions, respectively. All compounds were characterised by various spectroscopic methods. In the 1H NMR spectrum, the signals for the protons of both the pyridyl and pyrene moieties were found to be down‐field shifted, as expected, upon protonation or methylation. For example, the signals for the pyridyl protons appear at 8.75 and 8.57 ppm for 2M (Figure S7, Supporting Information), and at 9.04 and 8.57 ppm for 2H (Figure S11, Supporting Information), and the signals for the pyrene protons appear at 8.85 and 8.40 ppm for 2M, and at 9.0 and 8.43 for 2 H, which are significantly down‐field shifted compared to the signals observed for 2 at 8.79 and 7.81 ppm (pyridyl protons), and 8.45 and 8.19 ppm (pyrene protons) (Figure S3, Supporting Information). Similar down‐field shifts were also observed for the signals of 1M and 1H compared to that of 1 (Figures S1, S5, S9, Supporting Information).
Description of the crystal structures
The crystal structures of compounds 1M, 2M, 1H and 2H were analysed by single‐crystal X‐ray diffraction. Among these, only the structure of 1M is non‐centrosymmetric, and it was solved in the monoclinic space group P21. The unit cell of 1M contains two symmetry‐independent cations and two symmetry‐independent anions. The structures of the two cations differ mainly in the torsion angles between the planes of the pyridinium cations and the pyrene moieties, which are 18.2° and 22.7°, respectively (Figure 1 (a)). The cations stack in a parallel head‐to‐tail (anti) fashion and show π‐stacking (cation‐π) interactions between the pyridinium cation and pyrene moieties (interplanar separations (d) are in the range 3.34–3.41 Å). In addition, C−H⋅⋅⋅π interactions between these moieties with the methyl‐H atom are observed. The triflate anions exert various weak interactions including C−H⋅⋅⋅O and C−H⋅⋅⋅F interactions with the cations (Figure S13, Supporting Information).
Figure 1.
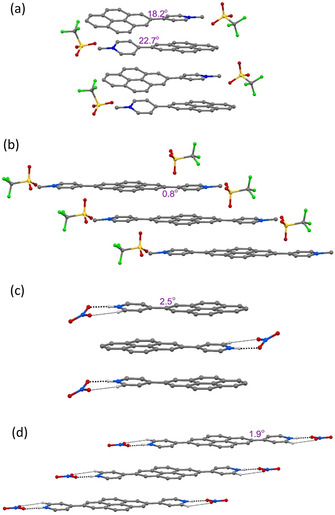
Molecular packing of (a) 1M, (b) 2M, (c) 1H, and (d) 2H in the solid state at 100 K. Only selected hydrogen atoms are shown for clarity. The numbers indicate the torsion angles between pyridyl and pyrene planes.
The salt 1H crystallizes in the monoclinic space group P21/c and, in contrast to 1M, only one symmetry‐independent cation (1‐H+) and nitrate ion are present in the unit cell. However, the molecular packing in 1H is similar to that in 1M, with the cations displaying parallel stacking in a head‐to‐tail fashion. The cation‐π interactions between the pyridinium cation and pyrene moieties (d=3.30–3.43 Å) are in a similar range to those in 1M. However, in contrast to 1M, the cation remains almost planar in 1H, as the torsion angle between pyridinium and pyrene moieties is only 2.5°. N−H⋅⋅⋅O and weak C−H⋅⋅⋅O hydrogen bonding interactions are observed between the nitrate anion and the pyridinium cation (Figure 1 (c)).
The crystal structure of 2M was refined in the monoclinic, centrosymmetric space group C2/m. The cation has 2/m (C 2h) symmetry, while the triflate anion is disordered and has mirror‐symmetry (Cs). The dications arrange in infinite, offset parallel stacks with strong π‐stacking (cation‐π) interactions between the pyridinium cation and pyrene moieties (d=3.35–3.37 Å) (Figure 1 (b)). Similar to the monocation 1H, the dication of 2M is almost planar with a torsion angle between the pyridinium and pyrene planes of 0.8°. The triflate anions interact with the dication via various weak C−F⋅⋅⋅π and C−H⋅⋅⋅O interactions (Figure S15, Supporting Information).
The salt 2H crystallizes in the monoclinic space group P21/c and the dication has inversion symmetry. The dications form offset parallel stacks with π‐stacking interactions between the pyridinium cation and pyrene moieties (d=3.37 and 3.39 Å) (Figure 1 (d)), similar to the arrangement in 2M. As in 1H and 2M, the molecule is almost planar, as the torsion angle between the pyridinium and pyrene planes is 1.9°. The nitrate ions are involved in N‐H⋅⋅⋅O and weak C−H⋅⋅⋅O hydrogen bonding interactions with the dication (Figure S23, Supporting Information).
In summary, the mono‐ or dications are almost planar in all compounds in the solid state (torsion angles: 0.8–2.5°), except for 1M (torsion angles: 18.2 and 22.7°). It is important to note that the observed torsion angles in the corresponding neutral compounds 1 (38.3°) and 2 (12.1° for α‐form and 38.6° for β‐form) [20] are larger than those in the cationic compounds, which are strongly diminished by protonation or methylation (except for 1M). The smaller torsion angles may also be a result of the large degree of π‐stacking interactions in the near‐planar cationic compounds. These show strong π‐stacking interactions between pyridinium and pyrene moieties as a result of protonation, which determine the parallel head‐to‐tail stacking in 1M and 1H and the offset parallel stacking in 2M and 2H. A similar π‐stacking interaction between pyridyl and pyrene moieties is not observed in the neutral compounds 1 and 2, in which π‐stacking is only present between the pyrene moieties (1) or both the pyrene and the pyridyl moieties (2). [20] In 1M, 1H, 2M and 2H, cation stacks weakly interact with the anions, which are located in between the stacks. A Hirshfeld surface analysis reveals the area and relative strengths of the intermolecular interactions and is presented in the Supporting Information (Figures S14 (1M), S18 (2M), S21 (1H), and S24 (2H)). The powder diffraction patterns of all samples were measured in order to examine the phase purity of the bulk samples. The observed powder diffraction patterns of all compounds agree very well with the respective simulated ones (Figures S25 and S26 in the Supporting Information). Small systematic shifts of the reflection positions can only be observed in 1M and 2M, which may be due to the sample preparation.
Photophysical properties
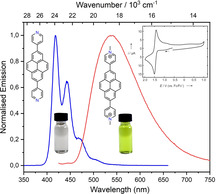
Detailed photophysical properties of all compounds are depicted in Figure 2. Absorption and emission were measured for 1M in THF, MeOH, MeCN, DMSO, and water, for 2M in MeOH, MeCN, DMSO, and water, as well as in the solid state for both compounds. For 1H and 2H, the absorption and emission spectra in MeCN are presented. The key photophysical data are summarised in Table 1. The neutral compounds 1 and 2 were measured in MeCN and in the solid state for comparison with the cations.
Figure 2.
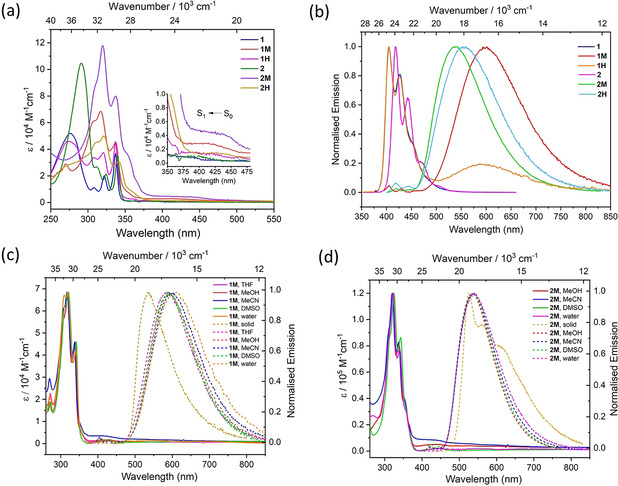
(a) Absorption, and (b) emission spectra of 1, 2, 1M, 2M, 1H, and 2H in MeCN (λ ex=337 nm); (c) absorption (solid lines), and emission (dashed lines) spectra of 1M in THF, MeOH, MeCN, DMSO, water and in the solid state; and (d) absorption (solid lines) and emission (dashed lines) spectra of 2M in MeOH, MeCN, DMSO, water and in the solid state. Very weak emission for 1M at 405–430 nm and for 2M at 420–440 nm might be due to the presence of traces of neutral compounds 1 and 2, respectively. All measurements were carried out in deoxygenated solvents.
Table 1.
Photophysical data for 1, 2, 1M, 2M, 1H, and 2H both in solution and in the solid state.
|
Compound |
Solvent or Solid |
λ abs [nm] |
ϵ [m −1 cm−1] |
λ em [nm] |
Apparent Stokes shift[a] [cm−1] |
Φ |
τ [ns] |
τ avg [b] [ns] |
τ 0 [c] [ns] |
k r [106 s−1] |
k nr [106 s−1] |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
1 |
MeCN |
362 337 322 308 |
1100 37 000 21 000 |
451 427 405 |
2900 |
0.22 |
36.1 |
– |
164 |
6.1 |
22 |
|
|
solid |
– |
– |
452 430 |
|
0.05 |
3.4 (32.6) 8.9 (37.6) 23.5 (29.8) |
6.9 |
|
|
|
|
1M |
THF |
418 337 319 |
|
586 |
6800 |
0.08 |
16 |
– |
200 |
5 |
58 |
|
|
MeOH |
418 337 318 |
|
593 |
7000 |
0.05 |
12.6 |
– |
252 |
4 |
75 |
|
|
MeCN |
418 337 317 |
2900 45 000 68 500 |
599 |
7200 |
0.10 |
25 |
– |
250 |
4 |
36 |
|
|
DMSO |
416 342 321 |
|
592 |
7100 |
0.11 |
22.6 |
– |
205 |
5 |
39 |
|
|
water |
425 340 318 309 |
|
608 |
7080 |
0.01 |
3 |
– |
300 |
3.3 |
330 |
|
|
solid |
– |
– |
535 |
– |
0.08 |
18.6 (60.7) 59.6 (39.3) |
25.5 |
|
|
|
|
1H |
MeCN |
421 337 319 |
[d] |
590 426 404 |
6800
|
– |
– |
– |
– |
– |
– |
|
|
solid |
– |
– |
545 |
|
0.05 |
8.4 (69.6) 23.2 (30.4) |
10.3 |
|
|
|
|
2 |
MeCN |
363 339 324 312 |
1400 35 000 |
469 442 418 |
3600 |
0.41 |
56.6 |
– |
138 |
7.2 |
10 |
|
|
solid |
– |
– |
482 453 |
– |
0.02 |
1.3 (24.8) 5.6 (34.3) 13.8 (40.9) |
3.5 |
|
|
|
|
2M |
MeOH |
438 337 320 |
|
536 |
4100 |
0.09 |
15 |
– |
167 |
6 |
61 |
|
|
MeCN |
437 337 320 |
4300 80 000 12 0000 |
538 |
4300 |
0.19 |
27 |
– |
142 |
7 |
30 |
|
|
DMSO |
440 342 325 |
|
537 |
4100 |
0.22 |
22.2 |
– |
101 |
10 |
35 |
|
|
water |
443 339 321 |
|
540 |
4050 |
0.12 |
15.5 |
– |
129 |
7.7 |
57 |
|
|
solid |
– |
– |
612 560 525 |
– |
0.03 |
3.3 (79.5) 13.4 (20.5)
|
3.9 |
|
|
|
|
2H |
MeCN |
428 341 321 |
1530 30 500 50 000 |
555 |
5300 |
0.07 |
15.6 |
– |
223 |
6.4 |
60 |
|
|
solid |
– |
– |
642 |
– |
0.0 |
1.2 (69) 4.8 (19.9) 19.9 (11.1) |
4 |
– |
– |
– |
[a] The Stokes shift is defined as the energy difference between the 0‐0 transitions of the absorption and the emission. However, as the S1←S0 transitions in substituted pyrene derivatives are usually broad with an undefined vibrational pattern, the band maxima of the absorption and emission have been used to determine the Stokes shift; consequently, we use the term „apparent Stokes shift“. [b] For multi‐exponential decays, the experimental average lifetime is given by τ avg=∑τ n B n/ ∑B n, where B n are pre‐exponential factors of the respective lifetime τ n. [c] The pure radiative lifetime, τ 0=τ/Φ; k r=1/τ 0; k nr=(1−Φ)/τ. [d] Partially dissociates in solution, see text.
The absorption spectra of 1 and 2 in MeCN are very similar and demonstrate „pyrene‐like“ absorptions in that the S1←S0 absorption (370–420 nm) is very weak with extinction coefficients ϵ below 1400 m −1 cm−1 (Figure 2 (a)). In general, the S1←S0 absorption of pyrene and 2‐ and 2,7‐substituted pyrene derivatives is attributed to a short‐axis‐polarised and transition‐dipole‐forbidden transition. [24] The S2←S0 transition was observed at 337 nm with vibrational progressions of 1382 and 1411 cm−1 for 1, and at 339 nm with vibrational progressions of 1365 and 1187 cm−1 for 2, which are much more intense (ϵ=37 000 and 35 000 m −1 cm−1) than the S1←S0 absorption, as also observed for unsubstituted pyrene (λ abs=334 nm for S2←S0 absorption, ϵ=55 000 m −1 cm−1). [50] The S1←S0 absorption of 2 is slightly bathochromically shifted compared to 1, by 175 cm−1. The absorption spectra of the cationic compounds 1M and 2M in MeCN are also very similar to that of pyrene, but show a stronger absorbance compared to 1 and 2 (Figure 2 (a)). Thus, their S1←S0 absorption is still a weak band with ϵ=3000–4300 m −1 cm−1; however, this band is bathochromically shifted and broad, covering ca. 100 nm (375–475 nm). The corresponding S2←S0 transitions of both 1M and 2M are less influenced as they are also observed at 337 nm in MeCN with vibrational progressions of 1872 and 1576 cm−1, respectively. The absorption of 1H appears to be the combination of the absorbance of 1 and 1M. All of the absorption bands in 1 and 1M are present in 1H with proportionate intensity. It seems that 1H undergoes dissociation in MeCN solution which is even more evident in the emission spectrum (Figure 2 (b)). The absorption of 2H resembles that of 2M in that its S1←S0 absorption (λ abs=428 nm, ϵ=1530 m −1 cm−1) is also transition‐dipole forbidden and broad. However, this band in 2M is further bathochromically shifted and more allowed compared to that in 2H. The S2←S0 transition of 2H occurs at 341 nm (ϵ=30 500 m −1 cm−1) with a vibrational progression of 1827 cm−1.
Furthermore, the absorption spectra of 1M and 2M were measured in various solvents to investigate possible solvent effects (see Table 1 and Figure 2 (c and d)). A very small shift (from 337 nm to 340 nm) was observed for 2M in DMSO, which shows a bathochromic shift of only 430 cm−1 compared to its absorption in MeOH or MeCN. Thus, no significant solvatochromism was observed in the absorption spectra, which was further confirmed by theoretical calculations (vide infra).
Although these compounds exhibited similar absorption characteristics, their emission characteristics are quite different and display interesting features. The emission of the neutral compounds 1 and 2 are pyrene‐like, which agrees with their absorption properties. Thus, the emission of 1 at λ em=405 nm in MeCN shows a vibrational progression of 1250 cm−1. The emission of 2 at λ em=418 nm is slightly bathochromically shifted compared to that of 1 and also possesses a vibrational progression of 1300 cm−1. On the other hand, the emissions of the cationic compounds 1M and 2M are different than that of pyrene. Their emission consists of a single broad band, which is strongly bathochromically shifted compared to 1 and 2, respectively. The emission of 1M occurs at λ em=599 nm in MeCN with an apparent Stokes shift of 7200 cm−1, which is the most bathochromically shifted in this series. It can be noted that this apparent Stokes shift is very large for 2‐ or 2,7‐substituted pyrenes,[ 20 , 21 , 24 ] which indicates a large change of the dipole moment in the excited state. Considering the energy of maximum emission intensity of 1 (λ em=405 nm) and 1M (λ em=599 nm), the emission of 1M is bathochromically shifted by 8000 cm−1, which is a large effect that is introduced by one methyl group. The emission of 2M occurs at λ em=538 nm with an apparent Stokes shift of 4300 cm−1. Considering the energy of maximum emission intensity of 2 (λ em=418 nm) and 2M, (λ em=538 nm), the emission of 2M is bathochromically shifted by 5300 cm−1. Interestingly, the emission of 1M is even more bathochromically shifted than 2M, which is an effect of a larger stabilization of the excited state in 1M due to the larger dipole moment compared to 2M, which has a symmetrical charge distribution. It can be noted that the emission of 2M is hypsochromically shifted compared to analogous 1,6‐bis(N‐methyl‐3’‐pyridinium)pyrene dichloride (λ em=591 nm) [49a] and bathochromically shifted compared to [1,3,6,8‐tetrakis(N‐methyl‐4‐pyridinium)pyrene][PF6]4 (λ em=485 nm). [49b]
To investigate the possible charge‐transfer (CT) nature in derivatives 1M and 2M, we conducted emission measurements in various solvents (see Table 1 and Figures 2 (c) and (d)). Indeed, the solvatochromic effect is very small. Very little change of λ em with the polarity of the solvent was observed for 1M. The difference of λ em between THF (586 nm) and water (608 nm) is 22 nm, which is only 617 cm−1. However, it can be concluded that 1M has greater CT nature than 2M, which is also in line with the lower extinction coefficients of the first transition. Interestingly, the solid‐state emission of 1M (λ em(solid)=535 nm) is considerably hypsochromically shifted compared to its solution emission (Figure 2(c)). On the other hand, the solid‐state emission of 2M (λ em(solid)=612 nm, 560 nm, 525 nm) is comparable to its solution‐phase features (Figure 2(d)).
The emission of 1H possesses two bands, one band at λ em=405 nm that shows a fine structure, which is identical to compound 1, and also a broad band at 590 nm, which is similar to that of 1M. Thus, it is evident that 1H undergoes dissociation and maintains an equilibrium between the neutral and protonated forms. The emission of 2H occurs at λ em=555 nm with an apparent Stokes shift of 5300 cm−1. Considering the energy of maximum emission intensity of 2 (λ em=418 nm) and 2M (λ em=538 nm), the emission of 2H is bathochromically shifted by 5900 cm−1 compared to that of 2, and by 570 cm−1 compared to that of 2M. Furthermore, we again observe that the mono‐substituted derivative 1H has stronger CT character than its disubstituted analogue 2H, as its emission is bathochromically shifted by 1070 cm−1.
The lifetime and quantum yields were measured in deoxygenated solutions. Monoexponential decays were obtained for the neutral and cationic compounds, and it is found that the neutral compounds possess a longer‐lived excited state compared to their cationic analogues (see Table 1). Furthermore, the non‐radiative decay rates of the latter ones are significantly increased (k nr=5.8–6.1×107 s−1), which is a result of their larger apparent Stokes shifts, fully in line with the energy gap law. [51] Similarly, the quantum yields of the neutral compounds are much higher compared to their cationic analogues. All of the compounds possess very low quantum yields in the solid state, which might be due to the absence of an effective suppression of the nonradiative decay, in the presence of strong π‐stacking interactions. This would also explain their shorter lifetimes in the solid state.
We found in our previous work that the solid state emissions of 1 and 2 strongly depend on crystallinity and particle size. [20] In addition, two polymorphic phases of 2 (α and β) have also been reported. While 2β displays a large π‐π overlap area, these interactions are much weaker in 2α. It is feasible to assume that the solid‐state emission is strongly influenced by the local environment and by the proportion of this local environment to the overall sample (Figures S29 and S30 in the Supporting Information). The overall solid‐state emission spectrum is therefore caused by an overlap of the emission spectra of the different polymorphs. However, these differences disappear when the samples are dissolved in solution.
Electrochemistry
Cyclic voltammetry studies were performed on the neutral and methylated compounds to investigate their electrochemical properties. The voltammograms were measured at a scan rate of 250 mV s−1 with 0.1 m [nBu4N][PF6] relative to the Fc/Fc+ couple. The first scan and then ten consecutive potential sweeps were measured for 1 in CH2Cl2 as presented in Figure 3 (a). For the very first scan, the formal potentials are E pa=+0.87 and +1.02 V, in the cathodic direction; however, no reduction event was observed. The peak currents for the redox processes increased steadily with each successive scan. This was an indication that some chemical reaction (e.g., dimerisation or polymerisation) might take place and there was deposition on the electrode surface. It should be noted that electrochemical polymerisation of pyrene compounds has been reported previously. [52] Electropolymerisation via anodic oxidation of unprotected bis(triarylamine)s leading to polymers with benzidine units is also known. [53] In the case of 2, ten consecutive potential sweeps are shown in Figure 3 (b). The formal potential E pa=+1.30 V was observed in the anodic direction; no reduction event was observed on the first scan. However, a reduction event can be observed only from the second scan. The reduction potential for the successive scans shifts to more negative values with a steady increase in the peak current (E pc=−1.60 V after 10 scans). This also indicates some chemical reaction or deposition on the electrode surface, as was observed for 1.[ 52 , 53 ] On the other hand, 1M shows an irreversible reduction (Figure 3 (c)), which takes place at E pc=−1.53 V, and the corresponding oxidation process completes at −0.94 V. In the case of 2M, a reversible reduction was observed with E 1/2=−1.47 V, which can be attributed either to a two‐electron reduction or two very closely spaced consecutive one‐electron reductions leading to the formation of the neutral species. No further reduction processes for either 1M or 2M were observed within the accessible potential window for the solvent (even in MeCN). The observed formal potentials for these two compounds are comparable; however, only 2M shows a reversible reduction. It can be noted that these observed reduction potentials for 1M and 2M occur at more negative values compared to the first one‐electron reduction of paraquat (MV2+), which occurs at −1.076 V vs. Fc+/Fc couple (the second one‐electron reduction of paraquat occurs at −1.51 V vs. Fc+/Fc couple in MeCN/[Et4N][PF6]).[ 31c , 31d , 41a , 54 ] Moreover, it can be noted that the extended methyl viologen 1,4‐bis(4'‐N‐methylpyridinium)benzene with [BF4]− counteranions, in which the pyridinium groups are separated by a phenylene moiety, showed a single two‐electron reduction signal at −0.91 V vs. SCE (i.e., −1.29 V vs. Fc+/Fc couple) [54] with 0.1 m MeCN/[Et4N][ClO4]. [31e] Another π‐extended viologen, i.e., 4,4’‐bis(4‐pyridinium‐n‐octyl)biphenyl with [PF6]− counteranions, showed one reversible two‐electron reduction at −1.48 V vs. Fc/Fc+ in THF, in a range comparable to that of 2M. [44a] It can be further noted that the reduction of 2M occurs at a much lower potential compared to that of our previously reported 2,7‐bis(BMes2)pyrene, an analogous acceptor‐pyrene‐acceptor system, which exhibits two reversible reduction events at −2.17 and −2.45 V vs. Fc/Fc+ in THF, indicating that the N‐methylpyridinium group is a stronger acceptor than ‐BMes2. [25b]
Figure 3.
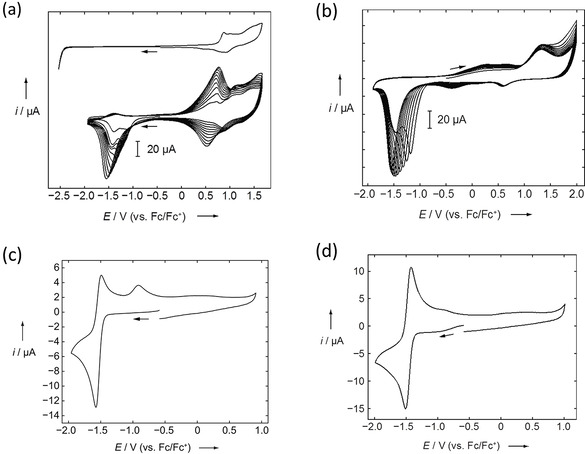
Cyclic voltammograms measured at 250 mV s−1 with 0.1 m [nBu4N][PF6] relative to the Fc/Fc+ couple: (a) the first scan (top) and ten consecutive potential sweeps (bottom) of 1 in CH2Cl2; (b) ten consecutive potential sweeps of 2 in CH2Cl2; (c) 1M in MeCN; (d) 2M in MeCN.
Spectroelectrochemistry
UV/Vis/NIR spectroelectrochemical measurements were carried out in CH2Cl2 for the neutral compounds 1 and 2, and in MeCN for the methylated compounds 1M and 2M, with 0.1 m [nBu4N][PF6] as the supporting electrolyte. Since reversible voltammograms were not observed for 1, 2 , and 1M in the cyclic voltammetry, we have presented their spectra in the Supporting Information (Figures S31–S33). Upon stepwise reduction of 2M, no absorption in the NIR region was observed; however, some changes in their UV/Vis absorption patterns were noticed (Figure 4). The absorption of reduced‐2M is very broad in the UV region with an extended tail in the visible region. The weaker broad peak at 400–450 nm increased, while the peak at 320 nm decreased upon reduction, and an intense peak at 295 nm appeared. Such an observation is attributed to the fact that 2M undergoes either a two‐electron reduction or two very closely spaced consecutive one‐electron reductions, leading to the formation of the neutral species, without detecting the radical monocation.
Figure 4.
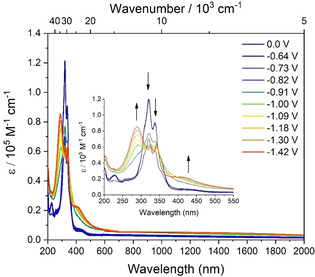
Spectroelectrochemical measurements of the stepwise reduction process of 2M with 0.1 m [nBu4N][PF6] in MeCN.
Theoretical calculations
Density functional theory (DFT) and time‐dependent density functional theory (TD‐DFT) calculations were carried out to rationalise the observed spectroscopic and electrochemical properties. The structures of these compounds were optimised at the PBE0/LANL2DZP level in THF including GD3BJ empirical dispersion (see Theoretical Computations in the Experimental Section). Selected experimentally determined and theoretically optimised structural parameters are listed in Tables S2 and S3 (Supporting Information). Overall, the optimised structural parameters for 1, 2, 1M, 2M, 1H, and 2H match well with the crystallographically determined parameters, except for the torsion angles for 1H and 2H, which deviate (ca. 25–30°) probably due to crystal packing effects in the solid state. Despite rather strong π‐stacking interactions observed between molecules in the cationic compounds (vide supra), periodic calculations on compound 2M, for example, (see Theoretical Computations in the Experimental Section) seem to indicate that this stacking hardly alters the electronic properties of each molecule. Indeed, the density of states (DOS) of 2M (Figure S34 in the Supporting Information) shows sharp peaks indicating weak electronic interactions between the dications in the solid.
HOMO–LUMO energy gaps are substantially larger for compounds 1 and 2 than for the cationic species (4.11 eV vs. ca. 3.15 eV, respectively), mainly due to some stabilization of the LUMO in the case of the latter ones. This suggests that the cationic compounds should absorb light at lower energies, as observed experimentally. This is supported by the maximum absorption and emission wavelengths which were also calculated at the TD‐DFT PBE0‐GD3BJ/LANL2DZP level in both MeCN and THF. No solvatochromic effect was observed in the calculations (see the comparison in Table S5 in the Supporting Information), which is in line with our experimental results. The main computed absorption wavelengths obtained in MeCN are given in Table 2 and compared with the experimentally observed values (see Table S5, in the Supporting Information, for values computed in THF). The corresponding simulated UV/Vis absorption spectra are shown in Figure S35 (Supporting Information). It can be noted that the calculated results agree better for the neutral compounds, with only a small deviation of a few nm, than for the methylated compounds, for which computed values are somewhat bathochromically shifted (ca. 850–4500 cm−1). Good agreement is also observed between computed and observed emission bands, although some blue shift is seen for the former ones with respect to the latter ones, especially for 1M in MeCN (Table S6, Supporting Information). The predominant frontier molecular orbitals which are involved in the main electronic transitions are reported in Table 2. Our calculations show that the pyrene‐like nature of the transitions is generally maintained. In compounds 1 and 2, pyrene's S1←S0 transition mostly involves a mixture of HOMO→LUMO+1 and HOMO‐1→LUMO electronic transitions and is transition dipole forbidden, and therefore has a very small oscillator strength, in agreement with the very small extinction coefficients observed experimentally (Table 2). In the cationic derivatives, the S1←S0 transitions remain forbidden as well with weak oscillator strengths, which is in agreement with the small extinction coefficients ϵ that we observed experimentally (1500–4300 m −1 cm−1).
Table 2.
Absorption wavelengths (λ calc, nm), oscillator strengths (f), and main electronic transitions calculated at the PBE0‐GD3BJ/LANL2DZP level in MeCN for compounds 1, 2, 1M, 2M, 1 H, and 2 H. Corresponding excited state numbers (Sn) are given in brackets. Experimental wavelengths (λ exp, nm) and their extinction coefficients (ϵ, M‐1 cm‐1) measured in MeCN are given for comparison.
|
Compound |
λ exp
|
ϵ [m −1 cm−1] |
λ calc |
f (Sn) |
Main electronic transitions [weight %][a] |
|---|---|---|---|---|---|
|
1 |
362 337 276 |
1100 37 000 52 000 |
355 338 276 |
0.013 (S1) 0.331 (S2) 1.572 (S5) |
H→L+1 (77 %); H‐1→L (21 %) H→L (90 %) H‐1→L+1 (91 %) |
|
1M |
418 337 269 |
2900 45 000 29 000 |
462 353 329 270 |
0.017 (S1) 0.031 (S2) 1.444 (S3) 0.142 (S8) |
H→L (98 %) H‐1→L (58 %); H→L+1 (40 %) H→L+1 (53 %); H‐1→L (41 %) H→L+3 (36 %); H‐1→L+1 (26 %); H‐1→L+2 (20 %); H‐3→L (13 %) |
|
1H |
421 337 274 |
[b] |
469 354 328 269 |
0.016 (S1) 0.034 (S2) 1.303 (S3) 0.146 (S8) |
H→L (98 %) H‐1→L (61 %); H→L+1 (37 %) H→L+1 (55 %); H‐1→L (37 %) H→L+3 (34 %); H‐1→L+1 (26 %); H‐3→L (20 %); H‐1→L+2 (14 %) |
|
2 |
363 339 291 |
1400 35 000 10 4000 |
371 342 296 |
0.017 (S1) 0.190 (S2) 2.553 (S5) |
H→L+1 (79 %); H‐1→L (20 %) H→L (83 %); H‐1→L+1 (15 %) H‐1→L+1 (83 %); H→L (14 %) |
|
2M |
437 337 320 |
4300 80 000 12 0000 |
468 399 362 332 |
0.027 (S1) 0.000 (S2) 0.436 (S3) 2.126 (S4) |
H→L (97 %) H→L+1 (99 %) H‐1→L (77 %); H→L+2 (22 %) H→L+2 (73 %); H‐1→L (21 %) |
|
2H |
428 341 321 |
1530 30 500 50 000 |
469 401 359 328 |
0.027 (S1) 0.000 (S2) 0.426 (S3) 1.887 (S4) |
H→L (97 %) H→L+1 (99 %) H‐1→L (80 %); H→L+2 (19 %) H→L+2 (75 %); H‐1→L (19 %) |
[a] H=HOMO and L=LUMO. [b] Partially dissociates in solution, see text.
As noted above, substituents at the 2‐ and 2,7‐positions do not contribute to pyrene's HOMO or LUMO.[ 21 , 24 , 25a , 25b , 26a ] However, they can have a profound effect on pyrene's HOMO‐1 and LUMO+1. Indeed, orbitals of the pyridine moiety in the neutral compounds 1 and 2 mix with pyrene's HOMO‐1/LUMO+1, although not very efficiently. Consequently, the influence on the first transition is very small for 1 and 2 and, therefore, remains of HOMO‐1→LUMO and HOMO→LUMO+1 character. However, protonation (compounds 1H/2H) or methylation (1M/2M) of the nitrogen, leads to a drastic stabilization of the LUMO+1, below the pyrene‐like LUMO (Figure 5). Hence, unlike pyrene, the LUMO of the cationic derivatives 1H, 2H, 1M, and 2M does not have a nodal plane through the long axis of the molecule, as observed previously with ‐BMes2 as the acceptor substituent.[ 21 , 24 , 25b ] This explains the stronger bathochromic shift of the first transition and strongly altered emission in these cationic compounds compared to 1 and 2. Our TD‐DFT calculations also show that the S1←S0 transitions in 1H, 2H, 1M, and 2M no longer involve a mixture of HOMO‐1→LUMO and HOMO→LUMO+1 electronic transitions as in pyrene or the neutral compounds 1 and 2, but are nearly pure HOMO→LUMO electronic transitions. The CT character of the excitations is nicely illustrated by the difference density plots between the S1 and S0 states shown in Table 3. The transferred charges (q) for 1H (q CT=0.862 e) and 1M (q CT=0.878 e) are higher than for their disubstituted (symmetrical) analogues 2H (q CT=0.743 e) and 2M (q CT =0.761 e), respectively. This is also fully in line with our observations which indicate that the S1←S0 transitions of 1H and 1M have smaller extinction coefficients, and their emissions are further bathochromically shifted compared to those of 2H and 2M, respectively, and that 1M shows some degree of solvatochromism compared to non‐solvatochromic 2M (Tables 1 and S5, Supporting Information).
Figure 5.
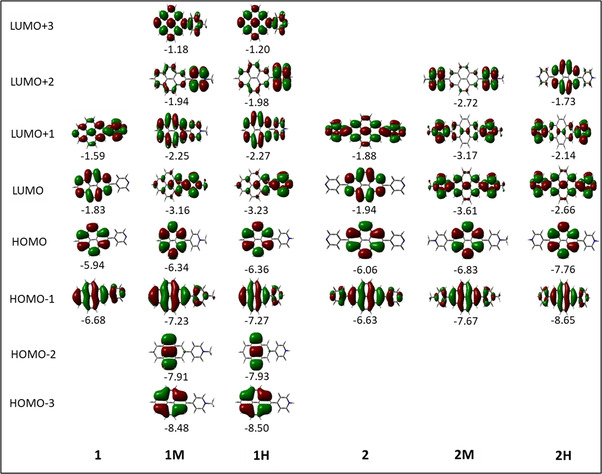
Representations of the frontier molecular orbitals and their energies (eV) calculated at the PBE0‐GD3BJ level in MeCN (isocontour value: ±0.03 au), involved in various electronic transitions.
Table 3.
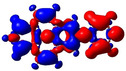
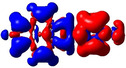
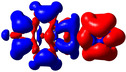
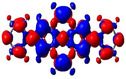
Density‐difference plots between relaxed excited state S1 and ground state S0 (with S1 geometry) (red=increase, blue=decrease of electron density) calculated at the PBE0‐GD3BJ/LANL2DZP level. Isocontour value: 0.03 au. q CT (e) and d CT (Å) correspond to the transferred charge and the distance between the positive (blue cross) and the negative (red cross) centroids of charge, respectively.
|
Compd |
q CT |
d CT |
|
ρ S0(r)−ρ S1(r) |
|---|---|---|---|---|
|
1 |
0.516 |
3.63 |
|
|
|
1M |
0.878 |
4.551 |
|
|
|
1H |
0.862 |
4.482 |
|
|
|
2 |
0.479 |
0.001 |
|
|
|
2M |
0.761 |
0.004 |
|
|
|
2H |
0.743 |
0.002 |
|
|
Interactions with ds‐DNA
As many pyrene derivatives showed intriguing biorelevant interactions, acting as probes for various DNA/RNA sequences[ 6 , 7 , 8 , 9 , 10 ] and intracellular mitochondrial recognition, [48] we performed preliminary studies on interactions of permanently charged N‐methylpyridinium derivatives 1M and 2M with the most commonly used ds‐DNA representative—calf thymus (ct)‐DNA. This DNA is characterised by a typical B‐helical secondary structure and almost equivalent number of GC‐(48 %) and AT‐(52 %) base‐pairs, thus representing a good target for both intercalative and minor groove binding mode. For the purpose of the study under biorelevant conditions, we prepared stock solutions of 1M and 2M in DMSO (1 mm), stored them at +4 oC, and aliquots were used for dilution in buffer (sodium cacodylate buffer, I=50 mm, pH 7.0) prior to each measurement. The stability of the stock solutions was confirmed by checking the UV/Vis absorption or emission spectra of the freshly prepared solutions in buffer. The UV/Vis absorption properties in the buffer (Figure S37, Supporting Information) revealed very similar shape, as noted for other organic solvents (Figure 2). However, the change in ratio of absorbances at wavelengths 316 nm/306 nm (1M) and 336 nm/319 nm (2M) at higher concentrations (>5×10−6 m) suggests intermolecular aggregation (Figure S37, Supporting Information). Therefore, all further experiments were performed at concentrations <5×10−6 m.
As a preliminary screening of the interactions of our compounds with ds‐DNA, we performed thermal denaturation experiments. It is well known that, upon heating, the ds‐helices of polynucleotides dissociate into two single stranded polynucleotides at a well‐defined temperature (T m value). Non‐covalent binding of small molecules to ds‐polynucleotides usually increases the thermal stability of the ds‐helices, thus resulting in an increased T m value, and this increase (ΔT m) can (in corroboration with other methods) be related to the various binding modes. [55] For example, if pyrene compounds intercalate into the ds‐DNA, ΔT m>2–5 °C can be expected, whereas the binding of pyrenes within the polynucleotide groove should have a negligible stabilising effect. [56] We observed that the addition of 1M monocation caused moderate stabilisation of the ct‐DNA against thermal denaturation (ΔTm=2 °C for r[1M]/[ctDNA]=0.2), whereas the dication 2M yielded strong stabilisation (ΔTm>16 °C for r[2M]/[ctDNA]=0.2), and such stabilisation is indicative of an intercalative binding mode.
The titration of 1M or 2M with ct‐DNA resulted in pronounced changes of their UV/Vis spectra (Figure 6(a) and Figures S40 and S41, Supporting Information), characterised by strong hypochromic and bathochromic effects typical of aromatic stacking interactions. Different responses were observed for 1M and 2M in the fluorimetric titrations with the ct‐DNA. For example, the emission of 1M was primarily quenched up to ratio r[1M]/[ctDNA]=0.3 and, upon further additions of ct‐DNA, its emission intensity increased (Figure S42, Supporting Information). The quenching of emission occurred at an excess of 1M over the ct‐DNA binding sites (r[1M]/[ctDNA] >0.3) and, therefore, it was attributed to aggregation of 1M along the ct‐DNA. The increased emission intensity at r[1M]/[ctDNA] ≪0.3 indicates binding of single 1M molecules to separate ct‐DNA binding sites. For the larger dicationic 2M, the addition of ct‐DNA caused only quenching of emission (Figure 6 (b) and Figure S43, Supporting Information), which can be attributed mainly to the binding of single 2M molecules to separate ct‐DNA binding sites. Analysis of the titration data by non‐linear fitting to Scatchard equation [57] gave binding constants of logKs=5.5 (for 1M, data fitted only for r[1M]/[ctDNA]≪0.3) and logKs=8.8 (for 2M), respectively, for Scatchard ratio n[bound dye]/[ctDNA]=0.3. The difference in binding constants matched well with the observed difference in thermal denaturation (ΔTm) values. Thus, it can be inferred that the dicationic compound 2M interacts more strongly with the ct‐DNA compared to the monocationic compound 1M.
Figure 6.
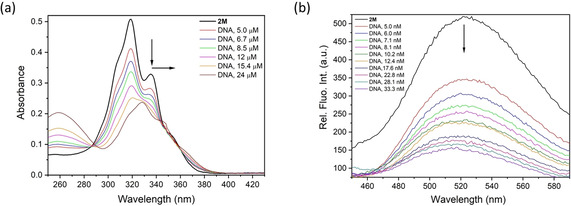
Fluorimetric titration of 2M with ct‐DNA at pH 7.0, sodium cacodylate buffer, I=0.05 m: a) changes in UV/Vis absorption of 2M (c=5×10−6 m); and b) changes in relative fluorescence intensity of 2M (c=3×10−8 m).
In order to obtain additional insight into the structural properties of small molecule/polynucleotide complexes, we have chosen the circular dichroism (CD) spectroscopic method, which is highly sensitive to conformational changes in the secondary structure of polynucleotides. In addition, achiral small molecules, such as 1M or 2M, can acquire an induced CD spectrum (ICD) upon binding to polynucleotides, which can give useful information about the modes of interaction. [58] Indeed, CD spectra (Figure 7) revealed weak negative ICD bands at λ=300–350 nm range for both, 1M and 2M. As pyrene absorbs in this range, such ICD bands are characteristic of the intercalative binding mode, which agrees well with the bathochromic and hypochromic changes in UV/Vis titrations, high binding constants and thermal denaturation results.
Figure 7.
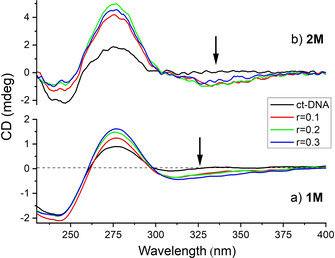
CD titration of ct‐DNA (c=2×10−5 m) with: a) 1M; and b) 2M; at molar ratios r=[compound]/[polynucleotide] and at pH 7.0, buffer sodium cacodylate, I=0.05 M.
Conclusions
We have presented structural, photophysical, and electrochemical studies on 2‐ and 2,7‐substituted pyridyl‐pyrenes and their N‐methylated compounds and analogous protonated salts, which are acceptor‐π and acceptor‐π‐acceptor systems, respectively. Their structure determination by X‐ray crystallography revealed that the torsion angle between the planes of pyrene and pyridyl moieties tends to become very small upon protonation or methylation, leading to almost planar structures, which agree well with the calculated optimised structures obtained at the PBE0‐GD3BJ/LANL2DZP level of theory. Although the absorption spectra show rather small changes upon protonation or methylation, their emission spectra were found to display large bathochromic shifts. The magnitude of such shifts is larger for the dipolar molecule 1M than for the symmetric molecule 2M. Thus, upon methylation of 1 (λ em=405 nm) and 2 (λ em=418 nm) to 1M (λ em=599 nm) and 2M (λ em=538 nm), respectively, their corresponding emission maxima shifted bathochromically by 8000 cm−1 and 5300 cm−1, respectively, in MeCN. Moreover, dipolar 1M shows an apparent Stokes shift of 7200 cm−1, which is very large for 2‐ and 2,7‐substituted pyrenes, indicating a rather large change of the dipole moment in the excited state. Symmetric, dicationic 2M shows an apparent Stokes shift of 4300 cm−1, which is smaller than that of 1M, due to its lack of a dipole moment. Compound 1M showed an irreversible reduction at −1.53 V, while 2M showed a reversible reduction at −1.47 V vs. Fc+/Fc couple in MeCN. These observed formal potentials are in accordance with other similar π‐conjugated extended viologens reported in the literature. However, the reduction of 2M occurs at a lower potential compared to 2,7‐bis(BMes2)pyrene, indicating that the N‐methylpyridinium group is a stronger acceptor than ‐BMes2. The reduction of 2M can be attributed either to a two‐electron reduction or to two closely spaced consecutive one‐electron reductions, leading to the formation of the neutral species, without producing the intermediate radical‐cation, which was further established by investigating the step‐wise reduction processes by spectroelectrochemical measurements. Our experimental observations are further confirmed by theoretical calculations performed at both the PBE0‐GD3BJ and CAM‐B3LYP levels of theory. The HOMO of both the neutral and methylated/protonated compounds remain rather unaffected. However, upon methylation or protonation, the LUMOs of the neutral compounds are destabilised, while the LUMO+1 orbitals are greatly stabilised and become the LUMOs. Thus, the new LUMOs of the methylated/protonated compounds do not have a nodal plane passing through the long axis of the molecule. Furthermore, preliminary studies reveal that 1M and 2M interact strongly with ct‐DNA by intercalation into the double helix, which makes them very interesting compounds for further studies of their biological activity. Detailed investigation of these interactions and their significance in biological systems are ongoing. Our research findings are expected to have significant impact on further investigations on the photophysics and electrochemistry of other PAH‐derived π‐conjugated viologen compounds and their potential applications in biological systems.
Experimental Section
General
The catalyst precursor [Ir(OMe)(COD)]2 was prepared according to a literature procedure [59] and B2pin2 was a gift from AllyChem Co. Ltd. The starting materials 2‐(Bpin)pyrene and 2,7‐bis(Bpin)pyrene were synthesised by following methods developed by Marder and co‐workers. [23c] The syntheses of 1, 2, 1M, and 2M were carried out using standard Schlenk or glovebox (Innovative Technology Inc.) techniques under an argon atmosphere. All of the compounds are, in general, stable, and can be handled in air. All solvents used for reactions were HPLC grade, dried using an Innovative Technology Inc. Solvent Purification System, and were further deoxygenated and stored under argon. All other chemicals were purchased from common commercial sources and were used without further purification.
1H and 13C{1H} solution NMR spectroscopic data were obtained at ambient temperature using a Bruker Avance 300 NMR spectrometer, operating at 300 MHz for 1H and 75 MHz for 13C{1H}. Chemical shifts (δ) were referenced to solvent residual peaks. High resolution mass spectrometry (HRMS) was performed with a Thermo Fisher Scientific Exactive Plus Orbitrap MS System with either an Atmospheric Sample Analysis Probe (ASAP) or Atmospheric Pressure Chemical Ionization (APCI) or an electrospray ionization (ESI) probe. Elemental analyses were performed on an Elementar vario MICRO cube elemental analyser.
Syntheses
Syntheses of compounds 1 and 2 were described previously. [20]
Synthesis of 1M: 1 (140 mg, 0.5 mmol) was stirred with methyl triflate (80 μl, 120 mg, 0.73 mmol) in dry toluene (10 mL) for 24 h under an argon atmosphere. The yellow precipitate obtained was isolated by filtration, washed with dry toluene and dried. Yield: 200 mg (90 %). Needle like single crystals were obtained by slowly evaporating an MeCN solution. 1 H NMR (300 MHz, CD3CN): δ=8.60 (d, J=7 Hz, 2 H), 8.59 (s, 2 H), 8.38 (d, J=7 Hz, 2 H), 8.30 (d, J=8 Hz, 2 H), 8.20 (AB multiplet, 4 H), 8.13 (t, J=8 Hz, 1 H), 4.27 ppm (s, 3 H). 13 C{1H} NMR (75 MHz, CD3CN): δ=157.1, 146.1, 132.7, 132.5, 131.6, 129.8, 128.5, 128.4, 127, 126.7, 126.1, 124.8, 124.6, 124.3, 120.1, 48.4 ppm. HRMS (ESI+): M/Z found: 294.1266; calculated M/Z for [M‐(OTf)]+ (C22H16N+)=294.1277. Elem. Anal. Calcd. (%) for C23H16NO3F3S: C 62.30, H 3.64, N 3.16, S 7.23; found: C 62.40, H 3.55, N 3.13, S 7.28.
Synthesis of 2M: 2 (180 mg, 0.5 mmol) was stirred with methyl triflate (180 μl, 270 mg, 1.64 mmol) in dry toluene (15 mL) over 24 h in an argon atmosphere. The yellow precipitate obtained was isolated by filtration, washed with dry toluene and dried under vacuum. Yield: 290 mg (85 %). Needle like single crystals were obtained by slowly evaporating a solution in MeCN. 1 H NMR (300 MHz, CD3CN): δ=8.85 (s, 4 H), 8.75 (d, J=7 Hz, 4 H), 8.57 (d, J=7 Hz, 4 H), 8.40 (s, 4 H), 4.36 ppm (s, 6 H). 13 C{1H} NMR (75 MHz, CD3CN): δ=133.5, 133.4, 129.9, 126.7, 126.2, 125.9, 123.5, 120.9, 48.7 ppm. HRMS (ESI+): M/Z found: 193.0883; calculated M/Z for [M‐2(OTf)]2+ (C28H22N2 2+)=193.0886. Elem. Anal. Calcd. (%) for C30H22N2O6F6S2: C 52.63, H 3.24, N 4.09, S 9.37: found: C 52.62, H 3.28, N 4.16, S 9.12.
Synthesis of 1H: 1 (140 mg, 0.5 mmol) was placed in MeCN (20 mL) and a few drops of nitric acid were added. The mixture was heated to 60 °C until all compounds dissolved. The clear solution was allowed to evaporate slowly. Needle shaped single crystals were obtained after a few days. Yield: 150 mg (88 %). 1 H NMR (300 MHz, d 4‐MeOD): δ=8.94 (m, 2 H), 8.81 (s, 2 H), 8.69 (m, 2 H), 8.32 (d, J=7 Hz, 2 H), 8.27 (AB multiplet, 4 H), 8.13 ppm (t, J=8 Hz, 1 H). HRMS (APCI+): M/Z found: 280.1116; calculated M/Z for [M‐(NO3)]+ (C21H14N+)=280.1121. Elem. Anal. Calcd. (%) for C21H14N2O3: C 73.68, H 4.12, N 8.18; found: C 73.53, H 4.13, N 8.12.
Synthesis of 2H: 2 (180 mg, 0.5 mmol) was placed in MeCN (20 mL) and a few drops of nitric acid were added. The mixture was stirred with heating at 60 °C for 24 h and then cooled to room temperature. The orange precipitate obtained was isolated by filtration. Yield: 210 mg (87 %). Needle shaped single crystals were obtained by recrystallization from DMF. 1 H NMR (300 MHz, [D6]DMSO): δ=9.04 (d, J=7 Hz, 4 H), 8.99 (s, 4 H), 8.58 (d, J=7 Hz, 4 H), 8.43 ppm (s, 4 H). HRMS (ESI+): M/Z found: 179.0722; calculated M/Z for [M‐2(NO3)]2+ (C26H18N2 2+)=179.0730. Elem. Anal. Calcd. (%) for C26H18N4O6: C 64.73, H 3.76, N 11.61; found: C 63.94, H 3.69, N 10.92.
Single crystal X‐ray diffraction
Single crystals suitable for X‐ray diffraction analysis were selected, coated in perfluoropolyether oil, and mounted on MiTeGen sample holders. Diffraction data were collected on a Bruker X8‐Apex II 4‐circle diffractometer with a CCD area detector using Mo‐Kα radiation monochromated by graphite or multi‐layer focusing mirrors. Diffraction data were collected at 100 K. The mounted crystals were cooled by a stream of cold nitrogen using Oxford Cryostreams or Bruker Kryoflex II low‐temperature devices. The frames were processed and corrected for Lorentz‐polarisation and absorption effects by employing the Bruker software packages. The structures were solved using the intrinsic phasing method (SHELXT). [60] All non‐hydrogen atoms were refined in anisotropic approximation, with hydrogen atoms ‘riding’ in idealised positions, by full‐matrix least squares against F 2 on all data, using SHELXL [60] software. Hirshfeld surfaces were calculated and analysed using the Crystal Explorer program (version 17.5). [61] Various structural information was extracted and graphics were produced using Mercury software. [62] Crystal data and experimental details are listed in Table S1 in the Supporting Information. Deposition numbers 2016793 (1H), 2016794 (1M), 2016795 (2H), and 2016796 (2M) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
General photophysical measurements
All measurements were performed in standard quartz cuvettes (1 cm × 1 cm cross‐section) fitted with screw caps. UV/Visible absorption spectra were recorded using an Agilent 8453 diode array UV/Vis spectrophotometer. Emission and excitation spectra were recorded using an Edinburgh Instruments FLSP920 spectrometer equipped with a cooled red PMT detector from Hamamatsu (R13456‐P), and with double monochromators, operating in right‐angle geometry mode. A 450 W continuous xenon arc lamp (Xe900) was used as the excitation source and was focused on the sample. All solutions used in photophysical measurements had concentrations lower than 5×10−6 m to eliminate the chances of excimer formation during fluorescence measurements.
Fluorescence quantum yield measurements
The fluorescence quantum yields were measured using a calibrated integrating sphere (150 mm inner diameter) from Edinburgh Instruments combined with the above FLSP920 spectrometer. The longest‐wavelength absorption maximum was chosen as the excitation wavelength, unless otherwise stated. The corresponding blank solvent was used as a reference for solution‐phase measurements and BaSO4 was used as reference for solid state measurements.
Fluorescence lifetime measurements
Lifetimes were measured using a 315.8 nm emitting picosecond pulsed diode laser as the light source in the time‐correlated single‐photon counting (TCSPC) mode using the Edinburgh Instruments FLS920 spectrometer. Emission decay was detected with a 3 nm emission‐slit band‐width. A 5000 kHz (every 200 ns) pulse was generated. The time range was set to 200 ns, and 1024 channels were used. Instrument response functions (IRFs) were measured from the scatter of an aqueous suspension of Ludox at the same excitation wavelength. A reconvolution‐fit was performed using Software F900 version 7.2.1. The quality of the decay fits was assessed by the calculated values of the reduced χ2 and Durbin‐Watson parameters, and visual inspection of the weighted and autocorrelated residuals.
Electrochemistry
Cyclic voltammetry measurements were carried out using a Gamry Instruments Reference 600 potentiostat. A standard arrangement of a three‐electrode cell configuration was set up using a platinum disk working electrode, a platinum wire counter electrode, and a silver wire, separated by a Vycor tip, as the reference electrode. Formal redox potentials are referenced to the ferrocene/ferrocenium ([Cp2Fe]+/0) redox couple by using ferrocene as an internal standard. Tetra‐n‐butylammonium hexafluorophosphate ([nBu4N][PF6]) was employed as the supporting electrolyte. Compensation for resistive losses (iR drop) was employed for all measurements.
Spectroelectrochemical measurements
Spectroelectrochemical experiments were carried out using an Agilent Cary 5000 spectrometer in combination with a designed sample compartment consisting of a cylindrical PTFE cell with an Infrasil® wedge window with an angle of 0.5°. An adjustable three‐in‐one electrode (6 mm platinum disc as working electrode, 1 mm platinum as counter electrode, and a pseudo reference electrode) was used. The potentials were adjusted with a Gamry 600 potentiostat. All experiments were measured at room temperature under an argon atmosphere.
Theoretical computations
All DFT calculations were carried out using the Gaussian09‐D01 suite of programs. [63] The geometries of all the compounds were optimised without symmetry constraints using the PBE0 functional[ 64 , 65 ] including Grimme's dispersion correction with Becke‐Johnson damping (GD3BJ),[ 66 , 67 ] and the LANL2DZ basis set augmented with polarisation functions for all atoms, except hydrogen (xyz coordinates of the optimised geometries are given in Table S4, Supporting Information). The nature of the stationary points after optimisation was checked by calculations of the harmonic vibrational frequencies to ensure that genuine minima were obtained. Time‐dependent density functional theory (TD‐DFT) calculations were performed at the same level of theory using the previously optimised geometries. For comparison, computations were also carried out using the CAM‐B3LYP functional [68] (Tables S5 and S6, Supporting Information). A better agreement with the experimentally observed values was obtained with the PBE0 functional. Solvent effects were taken into account by means of the polarisable continuum model (PCM). [69] The molecular orbitals and theoretical absorption spectra were drawn using the GaussView program. [70]
Periodic DFT calculations were performed on compound 2M using the VASP code. [71] The exchange‐correlation interaction was described within the generalised gradient approximation in the parametrisation of the Perdew‐Burke‐Ernzerhof functional. [64] Projector‐augmented wave potentials were used for all atoms. [72] Calculations were performed with a cut‐off energy of 530 eV. The electronic wave functions were sampled on dense densities in the irreducible BZ using the Monkhorst–Pack method. [73] Geometry optimisation including cell parameters and atomic positions were carried out without any symmetry constraints (see 2M‐sol‐opt.mol file in the Supporting Information).
Study of interactions with DNA
Stock solutions of 1M and 2M were prepared in DMSO (1 mm) and were diluted prior to use, whereby the DMSO content was always maintained at <0.1 %. The UV/Vis spectra were recorded on a Varian Cary 100 Bio spectrophotometer, fluorescence spectra were recorded on an Agilent Eclipse fluorimeter, and CD spectra were recorded on a JASCO J815 spectropolarimeter at 25.0 °C using appropriate quartz cuvettes of 1 cm path‐length. Calf thymus (ct)‐DNA was purchased from Aldrich, dissolved in sodium cacodylate buffer, I=0.05 m, pH 7.0, additionally sonicated and filtered through a 0.45 μm filter to obtain uniform rod‐like fragments of the B‐helical DNA. [74] Polynucleotide concentration was determined spectroscopically, by measuring the absorbance at 260 nm for molar extinction coefficient 6600 m −1 cm−1, whereby the concentration per nucleobase was obtained (traditionally referred as the concentration of phosphates).
For the study of interactions with DNA, the aqueous solutions of the compounds were buffered to pH 7.0 (sodium cacodylate buffer, I=0.05 m). Spectrophotometric titrations were performed by adding portions of DNA solution into the solution of the compound under investigation and CD experiments were performed by adding portions of stock solution of our compounds into the solution of DNA. In all titration experiments, dilution effects were taken into account.
Thermal melting curves for ds‐DNA and its complexes with 1M and 2M were determined as previously described, [55] by following the absorption change at 260 nm as a function of temperature. The absorbance of the ligands was subtracted from every curve and the absorbance scale was normalised. The T m values, the midpoints of the transition curves, were determined from the maximum of the first derivative and were checked graphically by the tangent method.[ 55 , 75 ] The ΔT m values were calculated by subtracting T m values of the free nucleic acid from that of the complex. Every ΔT m value reported was the average of at least two measurements. The error in ΔT m can be ±0.5 °C.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful for generous financial support by the Bavarian State Ministry of Science, Research, and the Arts for the Collaborative Research Network “Solar Technologies go Hybrid”, the DFG (GRK 2112), DAAD, the Julius‐Maximilians Universität Würzburg, and the Croatian Science Foundation grant No. IP‐2018‐01‐5475. G.K.K. thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship, and Michael Moos for his assistance with the spectroelectrochemical measurements. Open access funding enabled and organized by Projekt DEAL.
G. K. Kole, J. Merz, A. Amar, B. Fontaine, A. Boucekkine, J. Nitsch, S. Lorenzen, A. Friedrich, I. Krummenacher, M. Košćak, H. Braunschweig, I. Piantanida, J.-F. Halet, K. Müller-Buschbaum, T. B. Marder, Chem. Eur. J. 2021, 27, 2837.
Contributor Information
Prof. Dr. Ivo Piantanida, Email: Ivo.Piantanida@irb.hr.
Prof. Dr. Jean‐François Halet, Email: jean-francois.halet@univ-rennes1.fr.
Prof. Dr. Klaus Müller‐Buschbaum, Email: Klaus.Mueller-Buschbaum@anorg.chemie.uni-giessen.de.
Prof. Dr. Todd B. Marder, Email: todd.marder@uni-wuerzburg.de.
References
- 1. Figueira-Duarte T. M., Müllen K., Chem. Rev. 2011, 111, 7260–7314. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Kalyanasundaram K., Thomas J. K., J. Am. Chem. Soc. 1977, 99, 2039–2044; [Google Scholar]
- 2b. Mateo-Alonso A., Chem. Soc. Rev. 2014, 43, 6311–6324; [DOI] [PubMed] [Google Scholar]
- 2c. Jia W.-L., McCormick T., Liu Q.-D., Fukutani H., Motala M., Wang R.-Y., Tao Y., Wang S., J. Mater. Chem. 2004, 14, 3344–3350; [Google Scholar]
- 2d. Islam M. M., Hu Z., Wang Q., Redshaw C., Feng X., Mater. Chem. Front. 2019, 3, 762–781; [Google Scholar]
- 2e. Gong Y., Zhan X., Li Q., Li Z., Sci. China Chem. 2016, 59, 1623–1631; [Google Scholar]
- 2f. Ju H., Wang K., Zhang J., Geng H., Liu Z., Zhang G., Zhao Y., Zhang D., Chem. Mater. 2017, 29, 3580–3588; [Google Scholar]
- 2g. Liu M., Gong X., Zheng C., Gao D., Asian J. Org. Chem. 2019, 8, 722–730; [Google Scholar]
- 2h. Zhang S., Qiao X., Chen Y., Wang Y., Edkins R. M., Liu Z., Li H., Fang Q., Org. Lett. 2014, 16, 342–345; [DOI] [PubMed] [Google Scholar]
- 2i. Chercka D., Yoo S.-J., Baumgarten M., Kim J.-J., Müllen K., J. Mater. Chem. C 2014, 2, 9083–9086; [Google Scholar]
- 2j. Jung H., Kang S., Lee H., Yu Y.-J., Ho Jeong J., Song J., Jeon Y., Park J., ACS Appl. Mater. Interfaces 2018, 10, 30022–30028; [DOI] [PubMed] [Google Scholar]
- 2k. De Silva T. P. D., Youm S. G., Tamas G. G., Yang B., Wang C.-H., Fronczek F. R., Sahasrabudhe G., Sterling S., Quarels R. D., Chhotaray P. K., Nesterov E. E., Warner I. M., ACS Omega 2019, 4, 16867–16877; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2l. Jayabharathi J., Panimozhi S., Thanikachalam V., Sci. Rep. 2019, 9, 17555; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2m. Oniwa K., Kikuchi H., Shimotani H., Ikeda S., Asao N., Yamamoto Y., Tanigaki K., Jin T., Chem. Commun. 2016, 52, 4800–4803. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Winnik F. M., Chem. Rev. 1993, 93, 587–614; [Google Scholar]
- 3b. Winnik F. M., Winnik M. A., Tazuke S., J. Phys. Chem. 1987, 91, 594–597; [Google Scholar]
- 3c. Duhamel J., Langmuir 2012, 28, 6527–6538; [DOI] [PubMed] [Google Scholar]
- 3d. Zhang T., Taylor S. D., Palmer M., Duhamel J., Biophys. J. 2016, 111, 1267–1277; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Fowler M., Duhamel J., Qiu X. P., Korchagina E., Winnik F. M., J. Polym. Sci. Part B 2018, 56, 308–318; [Google Scholar]
- 3f. Kim D., Amos R., Gauthier M., Duhamel J., Langmuir 2018, 34, 8611–8621. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Sahoo D., Narayanaswami V., Kay C. M., Ryan R. O., Biochemistry 2000, 39, 6594–6601; [DOI] [PubMed] [Google Scholar]
- 4b. Somerharju P., Chem. Phys. Lipids 2002, 116, 57–74; [DOI] [PubMed] [Google Scholar]
- 4c. Zahid N. I., Ji L., Khyasudeen M. F., Friedrich A., Hashim R., Marder T. B., Abou-Zied O. K., Langmuir 2019, 35, 9584–9592. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Østergaard M. E., Hrdlicka P. J., Chem. Soc. Rev. 2011, 40, 5771–5788; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Wu C., Wang C., Yan L., Yang C. J., J. Biomed. Nanotechnol. 2009, 5, 495–504. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Yamana K., Zako H., Asazuma K., Iwase R., Nakano H., Murakami A., Angew. Chem. Int. Ed. 2001, 40, 1104–1106; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 1138–1140; [Google Scholar]
- 6b. Korshun V. A., Stetsenko D. A., Gait M. J., J. Chem. Soc. Perkin Trans. 1 2002, 1092–1104. [Google Scholar]
- 7.
- 7a. Kostenko E., Dobrikov M., Pyshnyi D., Petyuk V., Komarova N., Vlassov V., Zenkova M., Nucleic Acids Res. 2001, 29, 3611–3620; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Mahara A., Iwase R., Sakamoto T., Yamana K., Yamaoka T., Murakami A., Angew. Chem. Int. Ed. 2002, 41, 3648–3650; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 3800–3802; [Google Scholar]
- 7c. Astakhova I. V., Lindegaard D., Korshun V. A., Wengel J., Chem. Commun. 2010, 46, 8362–8364. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Christensen U. B., Pedersen E. B., Nucleic Acids Res. 2002, 30, 4918–4925; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Filichev V. V., Pedersen E. B., Org. Biomol. Chem. 2003, 1, 100–103. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Gröger K., Baretić D., Piantanida I., Marjanović M., Kralj M., Grabar M., Tomić S., Schmuck C., Org. Biomol. Chem. 2011, 9, 198–209; [DOI] [PubMed] [Google Scholar]
- 9b. Astakhova I. V., Malakhov A. D., Stepanova I. A., Ustinov A. V., Bondarev S. L., Paramonov A. S., Korshun V. A., Bioconjugate Chem. 2007, 18, 1972–1980. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Hernandez-Folgado L., Schmuck C., Tomić S., Piantanida I., Bioorg. Med. Chem. Lett. 2008, 18, 2977–2981; [DOI] [PubMed] [Google Scholar]
- 10b. Hernandez-Folgado L., Baretić D., Piantanida I., Marjanović M., Kralj M., Rehm T., Schmuck C., Chem. Eur. J. 2010, 16, 3036–3056; [DOI] [PubMed] [Google Scholar]
- 10c. Wu J., Zou Y., Li C., Sicking W., Piantanida I., Yi T., Schmuck C., J. Am. Chem. Soc. 2012, 134, 1958–1961; [DOI] [PubMed] [Google Scholar]
- 10d. Ma F., Liu W.-J., Qianyi Z., Zhang C.-Y., Chem. Commun. 2017, 53, 10596–10599. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Vaughn W. M., Weber G., Biochemistry 1970, 9, 464–473; [DOI] [PubMed] [Google Scholar]
- 11b. Oter O., Ribou A.-C., J. Fluoresc. 2009, 19, 389–397. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Bodenant B., Fages F., Delville M.-H., J. Am. Chem. Soc. 1998, 120, 7511–7519; [Google Scholar]
- 12b. Valeur B., Coord. Chem. Rev. 2000, 205, 3–40; [Google Scholar]
- 12c. Yang J.-S., Lin C.-S., Hwang C.-Y., Org. Lett. 2001, 3, 889–892; [DOI] [PubMed] [Google Scholar]
- 12d. Yang R.-H., Chan W.-H., Lee A. W. M., Xia P.-F., Zhang H.-K., Li K.’A., J. Am. Chem. Soc. 2003, 125, 2884–2885; [DOI] [PubMed] [Google Scholar]
- 12e. Kumari N., Dey N., Jha S., Bhattacharya S., ACS Appl. Mater. Interfaces 2013, 5, 2438–2445. [DOI] [PubMed] [Google Scholar]
- 13. Birks J. B., Lumb M. D., Munro I. H., Proc. R. Soc. London Ser. A 1964, 280, 289–297. [Google Scholar]
- 14. Templer R. H., Castle S. J., Curran A. R., Rumbles G., Klug D. R., Faraday Discuss. 1999, 111, 41–53. [DOI] [PubMed] [Google Scholar]
- 15. Pokhrel M. R., Bossmann S. H., J. Phys. Chem. B 2000, 104, 2215–2223. [Google Scholar]
- 16.
- 16a. Ueno A., Suzuki I., Osa T., Anal. Chem. 1990, 62, 2461–2466; [Google Scholar]
- 16b. Ikeda H., Nakamura M., Ise N., Oguma N., Nakamura A., Ikeda T., Toda F., Ueno A., J. Am. Chem. Soc. 1996, 118, 10980–10988; [Google Scholar]
- 16c. Fujiwara Y., Amao Y., Sens. Actuators B 2003, 89, 58–61; [Google Scholar]
- 16d. Aoyagi T., Ikeda H., Ueno A., Bull. Chem. Soc. Jpn. 2001, 74, 157–164. [Google Scholar]
- 17.
- 17a. Iwasaki T., Murakami S., Takeda Y., Tohnai N., Kambe N., Chem. Asian J. 2020, 15, 1349–1354; [DOI] [PubMed] [Google Scholar]
- 17b. Perry M., Carra C., Chrétien M. N., Scaiano J. C., J. Phys. Chem. A 2007, 111, 4884–4889; [DOI] [PubMed] [Google Scholar]
- 17c. Collings J. C., Roscoe K. P., Thomas R. Ll., Batsanov A. S., Stimson L. M., Howard J. A. K., Marder T. B., New J. Chem. 2001, 25, 1410–1417; [Google Scholar]
- 17d. Collings J. C., Roscoe K. P., Robins E. G., Batsanov A. S., Stimson L. M., Howard J. A. K., Clark S. J., Marder T. B., New J. Chem. 2002, 26, 1740–1746; [Google Scholar]
- 17e. Kilbinger A. F. M., Grubbs R. H., Angew. Chem. Int. Ed. 2002, 41, 1563–1566; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1633–1636. [Google Scholar]
- 18.
- 18a. Eddaoudi M., Kim J., Rosi N., Vodak D., Wachter J., O'Keeffe M., Yaghi O. M., Science 2002, 295, 469–472; [DOI] [PubMed] [Google Scholar]
- 18b. Rosi N. L., Kim J., Eddaoudi M., Chen B., O'Keeffe M., Yaghi O. M., J. Am. Chem. Soc. 2005, 127, 1504–1518; [DOI] [PubMed] [Google Scholar]
- 18c. Williams C. A., Blake A. J., Wilson C., Hubberstey P., Schröder M., Cryst. Growth Des. 2008, 8, 911–922. [Google Scholar]
- 19.
- 19a. Sick T., Rotter J. M., Reuter S., Kandambeth S., Bach N. N., Döblinger M., Merz J., Clark T., Marder T. B., Bein T., Medina D. D., J. Am. Chem. Soc. 2019, 141, 12570–12581; [DOI] [PubMed] [Google Scholar]
- 19b. Ghosh S., Tsutsui Y., Suzuki K., Kaji H., Honjo K., Uemura T., Seki S., Mol. Syst. Des. Eng. 2019, 4, 325–331. [Google Scholar]
- 20. Lu Q., Kole G. K., Friedrich A., Müller-Buschbaum K., Liu Z., Yu X., Marder T. B., J. Org. Chem. 2020, 85, 4256–4266. [DOI] [PubMed] [Google Scholar]
- 21. Merz J., Fink J., Friedrich A., Krummenacher I., Al Mamari H. H., Lorenzen S., Haehnel M., Eichhorn A., Moos M., Holzapfel M., Braunschweig H., Lambert C., Steffen A., Ji L., Marder T. B., Chem. Eur. J. 2017, 23, 13164–13180. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Vollmann H., Becker H., Corell M., Streeck H., Eur. J. Org. Chem. 1937, 1–159; [Google Scholar]
- 22b. Feng X., Hu J.-Y., Redshaw C., Yamato T., Chem. Eur. J. 2016, 22, 11898–11916. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Coventry D. N., Batsanov A. S., Goeta A. E., Howard J. A. K., Marder T. B., Perutz R. N., Chem. Commun. 2005, 2172–2174; [DOI] [PubMed] [Google Scholar]
- 23b. Mkhalid I. A. I., Barnard J. H., Marder T. B., Murphy J. M., Hartwig J. F., Chem. Rev. 2010, 110, 890–931; [DOI] [PubMed] [Google Scholar]
- 23c. Crawford A. G., Liu Z., Mkhalid I. A. I., Thibault M.-H., Schwarz N., Alcaraz G., Steffen A., Collings J. C., Batsanov A. S., Howard J. A. K., Marder T. B., Chem. Eur. J. 2012, 18, 5022–5035. [DOI] [PubMed] [Google Scholar]
- 24. Crawford A. G., Dwyer A. D., Liu Z., Steffen A., Beeby A., Pålsson L.-O., Tozer D. J., Marder T. B., J. Am. Chem. Soc. 2011, 133, 13349–13362. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Ji L., Lorbach A., Edkins R. M., Marder T. B., J. Org. Chem. 2015, 80, 5658–5665; [DOI] [PubMed] [Google Scholar]
- 25b. Ji L., Edkins R. M., Lorbach A., Krummenacher I., Brückner C., Eichhorn A., Braunschweig H., Engels B., Low P. J., Marder T. B., J. Am. Chem. Soc. 2015, 137, 6750–6753; [DOI] [PubMed] [Google Scholar]
- 25c. Merz J., Dietz M., Vonhausen Y., Wöber F., Friedrich A., Sieh D., Krummenacher I., Braunschweig H., Moos M., Holzapfel M., Lambert C., Marder T. B., Chem. Eur. J. 2020, 26, 438–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Kurata R., Ito A., Gon M., Tanaka K., Chujo Y., J. Org. Chem. 2017, 82, 5111–5121; [DOI] [PubMed] [Google Scholar]
- 26b. Kurata R., Tanaka K., Ito A., J. Org. Chem. 2016, 81, 137–145. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Moriarty R. M., Dansette P., Jerina D. M., Tetrahedron Lett. 1975, 16, 2557–2560; [Google Scholar]
- 27b. Zöphel L., Enkelmann V., Müllen K., Org. Lett. 2013, 15, 804–807; [DOI] [PubMed] [Google Scholar]
- 27c. Zöphel L., Beckmann D., Enkelmann V., Chercka D., Rieger R., Müllen K., Chem. Commun. 2011, 47, 6960–6962. [DOI] [PubMed] [Google Scholar]
- 28. Mochida K., Kawasumi K., Segawa Y., Itami K., J. Am. Chem. Soc. 2011, 133, 10716–10719. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Liu Z., Wang Y., Chen Y., Liu J., Fang Q., Kleeberg C., Marder T. B., J. Org. Chem. 2012, 77, 7124–7128; [DOI] [PubMed] [Google Scholar]
- 29b. Ji L., Krummenacher I., Friedrich A., Lorbach A., Haehnel M., Edkins K., Braunschweig H., Marder T. B., J. Org. Chem. 2018, 83, 3599–3606. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Hu J.-Y., Era M., Elsegood M. R. J., Yamato T., Eur. J. Org. Chem. 2010, 72–79; [Google Scholar]
- 30b. Sasaki S., Suzuki S., Igawa K., Morokuma K., Konishi G.-i., J. Org. Chem. 2017, 82, 6865–6873. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Weidel H., Russo M., Monatsh. Chem. 1882, 3, 850–885; [Google Scholar]
- 31b. Michaelis L., Hill E. S., J. Gen. Physiol. 1933, 16, 859–873; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31c. Bird C. L., Kuhn A. T., Chem. Soc. Rev. 1981, 10, 49–82; [Google Scholar]
- 31d. Bockman T. M., Kochi J. K., J. Org. Chem. 1990, 55, 4127–4135; [Google Scholar]
- 31e. Takahashi K., Nihira T., Akiyama K., Ikegami Y., Fukuyoc E., J. Chem. Soc. Chem. Commun. 1992, 620–622; [Google Scholar]
- 31f. Happ J. W., Ferguson J. A., Whitten D. G., J. Org. Chem. 1972, 37, 1485–1491. [Google Scholar]
- 32.
- 32a. Striepe L., Baumgartner T., Chem. Eur. J. 2017, 23, 16924–16940; [DOI] [PubMed] [Google Scholar]
- 32b. Ding J., Zheng C., Wang L., Lu C., Zhang B., Chen Y., Li M., Zhai G., Zhuang X., J. Mater. Chem. A 2019, 7, 23337–23360; [Google Scholar]
- 32c. Madasamy K., Velayutham D., Suryanarayanan V., Kathiresan M., Ho K.-C., J. Mater. Chem. C 2019, 7, 4622–4637; [Google Scholar]
- 32d. Woodward A. N., Kolesar J. M., Hall S. R., Saleh N.-A., Jones D. S., Walter M. G., J. Am. Chem. Soc. 2017, 139, 8467–8473; [DOI] [PubMed] [Google Scholar]
- 32e. Kim J.-W., Myoung J.-M., Adv. Funct. Mater. 2019, 29, 1808911; [Google Scholar]
- 32f. Kakibe T., Ohno H., J. Mater. Chem. 2009, 19, 4960–4964; [Google Scholar]
- 32g. Kao S.-Y., Kawahara Y., Nakatsuji S.i., Ho K.-C., J. Mater. Chem. C 2015, 3, 3266–3272. [Google Scholar]
- 33.
- 33a. Corr D., Rao S. N., Stobie N., Kinsella M., US Patent, 2009, US 7,567,371 B2;
- 33b. Archambeau S., Biver C., Berit-Debat F., Aiken S., Gabbutt C. D., Heron B. M., Roadbent T. D., European Patent Application, 2017, EP 3 115 433 A1;
- 33c. Kline W. M., Lorenzini R. G., Sotzing G. A., Color. Technol. 2014, 130, 73–80; [Google Scholar]
- 33d. Schoot C. J., Ponjee J. J., van Dam H. T., van Doorn R. A., Bolwijn P. T., Appl. Phys. Lett. 1973, 23, 64–65; [Google Scholar]
- 33e. Mortimer R. J., Rosseinsky D. R., Monk P. M. S., Electrochromic Materials and Devices, Wiley-VCH, Weinheim, 2015. [Google Scholar]
- 34.
- 34a. Nanasawa M., Miwa M., Hirai M., Kuwabara T., J. Org. Chem. 2000, 65, 593–595; [DOI] [PubMed] [Google Scholar]
- 34b. Zhang X., Clennan E. L., Arulsamy N., Weber R., Weber J., J. Org. Chem. 2016, 81, 5474–5486; [DOI] [PubMed] [Google Scholar]
- 34c. Kan W.-Q., Wen S.-Z., He Y.-C., Xu C.-Y., Inorg. Chem. 2017, 56, 14926–14935. [DOI] [PubMed] [Google Scholar]
- 35.
- 35a. Anelli P. L., Spencer N., Stoddart J. F., J. Am. Chem. Soc. 1991, 113, 5131–5133; [DOI] [PubMed] [Google Scholar]
- 35b. Benniston A. C., Harriman A., Angew. Chem. Int. Ed. Engl. 1993, 32, 1459–1461; [Google Scholar]; Angew. Chem. 1993, 105, 1553–1555; [Google Scholar]
- 35c. Bissell R. A., Cordova E., Kaifer A. E., Stoddart J. F., Nature 1994, 369, 133–137; [Google Scholar]
- 35d. Cheng C., McGonigal P. R., Schneebeli S. T., Li H., Vermeulen N. A., Ke C., Stoddart J. F., Nat. Nanotechnol. 2015, 10, 547–553; [DOI] [PubMed] [Google Scholar]
- 35e. Kahlfuss C., Métay E., Duclos M.-C., Lemaire M., Milet A., Saint-Aman E., Bucher C., Chem. Eur. J. 2015, 21, 2090–2106. [DOI] [PubMed] [Google Scholar]
- 36.
- 36a. Janoschka T., Martin N., Martin U., Friebe C., Morgenstern S., Hiller H., Hager M. D., Schubert U. S., Nature 2015, 527, 78–83; [DOI] [PubMed] [Google Scholar]
- 36b. Liu T., Wie X., Nie Z., Sprenkle V., Wang W., Adv. Energy Mater. 2016, 6, 1501449; [Google Scholar]
- 36c. Debruler C., Hu B., Moss J., Luo J., Liu T. L., ACS Energy Lett. 2018, 3, 663–668; [Google Scholar]
- 36d. Luo J., Hu B., Debruler C., Liu T. L., Angew. Chem. Int. Ed. 2018, 57, 231–235; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 237–241. [Google Scholar]
- 37.
- 37a. Summers L. A., The Bipyridinium Herbicides, 1980, Academic Press, London; [Google Scholar]
- 37b. Fischer H., Summers L. A., Tetrahedron 1976, 32, 615–618; [Google Scholar]
- 37c. Rockley J. E., Summers L. A., Aust. J. Chem. 1980, 33, 1397–1400. [Google Scholar]
- 38.
- 38a. Revkin A. C., Sci. Dig. 1983, 91, 36–38;10298869 [Google Scholar]
- 38b. Kamel F., Science 2013, 341, 722–723; [DOI] [PubMed] [Google Scholar]
- 38c. Tsai W.-T., Toxicol. Environ. Chem. 2013, 95, 197–206; [Google Scholar]
- 38d. Dinis-Oliveira R. J., Duarte J. A., Sánchez-Navarro A., Remião F., Bastos M. L., Carvalho F., Crit. Rev. Toxicol. 2008, 38, 13–71. [DOI] [PubMed] [Google Scholar]
- 39.
- 39a. Monk P. M. S., The viologens: physicochemical properties, synthesis, and applications of the salts of 4,4′-bipyridine, Wiley, Chichester, 1998; [Google Scholar]
- 39b. W. W. Porter III , Vaid T. P., J. Org. Chem. 2005, 70, 5028–5035; [DOI] [PubMed] [Google Scholar]
- 39c. Petersson J., Hammarström L., J. Phys. Chem. B 2015, 119, 7531–7540; [DOI] [PubMed] [Google Scholar]
- 39d. Li N., Liu Y. Y., Li Y., Zhuang J. B., Cui R. R., Gong Q., Zhao N., Tang B. Z., ACS Appl. Mater. Interfaces 2018, 10, 24249–24257; [DOI] [PubMed] [Google Scholar]
- 39e. Wang R.-T., Lee G.-H., Lai C. K., J. Mater. Chem. C 2018, 6, 9430–9444. [Google Scholar]
- 40. Benniston A. C., Hagon J., He X., Yang S., Harrington R. W., Org. Lett. 2012, 14, 506–509. [DOI] [PubMed] [Google Scholar]
- 41.
- 41a. Durben S., Baumgartner T., Angew. Chem. Int. Ed. 2011, 50, 7948–7952; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8096–8100; [Google Scholar]
- 41b. Stolar M., Borau-Garcia J., Toonen M., Baumgartner T., J. Am. Chem. Soc. 2015, 137, 3366–3371; [DOI] [PubMed] [Google Scholar]
- 41c. Greulich T. W., Yamaguchi E., Doerenkamp C., Lebbesmeyer M., Daniliuc C. G., Fukazawa A., Eckert H., Yamaguchi S., Studer A., Chem. Eur. J. 2017, 23, 6029–6033. [DOI] [PubMed] [Google Scholar]
- 42. Murakami K., Ohshita J., Inagi S., Tomita I., Electrochemistry 2015, 83, 605–608. [Google Scholar]
- 43.
- 43a. Alberto M. E., De Simone B. C., Cospito S., Imbardelli D., Veltri L., Chidichimo G., Russo N., Chem. Phys. Lett. 2012, 552, 141–145; [Google Scholar]
- 43b. Beneduci A., Cospito S., Crispini A., Gabriele B., Nicoletta F. P., Veltria L., Chidichimo G., J. Mater. Chem. C 2013, 1, 2233–2240; [Google Scholar]
- 43c. Hünig S., Langels A., Schmittel M., Wenner H., Perepichka I. F., Peters K., Eur. J. Org. Chem. 2001, 1393–1399. [Google Scholar]
- 44.
- 44a. W. W. Porter III , Vaid T. P., Rheingold A. L., J. Am. Chem. Soc. 2005, 127, 16559–16566; [DOI] [PubMed] [Google Scholar]
- 44b. Arrhenius T. S., Blanchard-Desce M., Dvolaitzky M., Lehn J.-M., Malthete J., Proc. Natl. Acad. Sci. USA 1986, 83, 5355–5359; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44c. Su Y.-S., Chen C.-F., Org. Lett. 2010, 12, 1888–1891. [DOI] [PubMed] [Google Scholar]
- 45.
- 45a. Giovanella U., Cariati E., Lucenti E., Pasini M., Galeotti F., Botta C., ChemPhysChem 2017, 18, 2157–2161; [DOI] [PubMed] [Google Scholar]
- 45b. Cariati E., Botta C., Danelli S. G., Forni A., Giaretta A., Giovanella U., Lucenti E., Marinotto D., Righetto S., Ugo R., Chem. Commun. 2014, 50, 14225–14228. [DOI] [PubMed] [Google Scholar]
- 46.
- 46a. Monkman A. P., Pålsson L.-O., Higgins R. W. T., Wang C., Bryce M. R., Batsanov A. S., Howard J. A. K., J. Am. Chem. Soc. 2002, 124, 6049–6055; [DOI] [PubMed] [Google Scholar]
- 46b. Fasina T. M., Collings J. C., Lydon D. P., Albesa-Jove D., Batsanov A. S., Howard J. A. K., Nguyen P., Bruce M., Scott A. J., Clegg W., Watt S. W., Viney C., Marder T. B., J. Mater. Chem. 2004, 14, 2395–2404; [Google Scholar]
- 46c. Chen X., Shen X. Y., Guan E., Liu Y., Qin A., Sun J. Z., Tang B. Z., Chem. Commun. 2013, 49, 1503–1505; [DOI] [PubMed] [Google Scholar]
- 46d. Chen S., Zhao M., Su J., Zhang Q., Tian X., Li S., Zhou H., Wu J., Tian Y., Dyes Pigm. 2017, 136, 807–816. [Google Scholar]
- 47.
- 47a. Kawamata J., Suzuki Y., Moritomo H., Fujiki S., PCT Int. Appl. 2016, WO 2016143335 A1 20160915 and; Kawamata J., Suzuki Y., Moritomo H., Fujiki S., Jpn. Kokai Tokkyo Koho, 2016, JP2016166154 A 20160915;
- 47b. Shukla A. D., Strawser D., Lucassen A. C. B., Freeman D., Cohen H., Jose D. A., Das A., Evmenenko G., Dutta P., van der Boom M. E., J. Phys. Chem. B 2004, 108, 17505–17511; [Google Scholar]
- 47c. Ding Z., Tian M., Guo L., Liu Z.-q., Yu X., Sens. Actuators B 2018, 276, 331–339; [Google Scholar]
- 47d. Hirsch T., Port H., Wolf H. C., Miehlich B., Effenberger F., J. Phys. Chem. B 1997, 101, 4525–4535; [Google Scholar]
- 47e. Shi Y., Liu J., Li M., Zheng J., Xu C., Electrochim. Acta 2018, 285, 415–423; [Google Scholar]
- 47f. Ge B.-D., Wei Q., Sun A.-H., Lin C.-Y., Duan X.-F., Li J.-H., Wang G.-M., Chem. Asian J. 2019, 14, 2086–2090; [DOI] [PubMed] [Google Scholar]
- 47g. Fudickar W., Linker T., Angew. Chem. Int. Ed. 2018, 57, 12971–12975; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13153–13157; [Google Scholar]
- 47h. Zhang R., Niu G., Li X., Guo L., Zhang H., Yang R., Chen Y., Yu X., Tang B. Z., Chem. Sci. 2019, 10, 1994–2000; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47i. Li D., Hu W., Wang J., Zhang Q., Cao X.-M., Ma X., Tian H., Chem. Sci. 2018, 9, 5709–5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.
- 48a. Niko Y., Moritomo H., Sugihara H., Suzuki Y., Kawamata J., Konishi G.-i., J. Mater. Chem. B 2015, 3, 184–190; [DOI] [PubMed] [Google Scholar]
- 48b. Abeywickrama C. S., Wijesinghe K. J., Stahelin R. V., Pang Y., Chem. Commun. 2017, 53, 5886–5889. [DOI] [PubMed] [Google Scholar]
- 49.
- 49a. Hagiwara S., Ishida Y., Miasui D., Shimada T., Takagi S., Clay Sci. 2013, 17, 7–10; [Google Scholar]
- 49b. Hagiwara S., Ishida Y., Masui D., Shimada T., Takagi S., Tetrahedron Lett. 2012, 53, 5800–5802; [Google Scholar]
- 49c. Sato K., Matsubara K., Hagiwara S., Saito K., Yagi M., Takagi S., Yui T., Langmuir 2015, 31, 27–31. [DOI] [PubMed] [Google Scholar]
- 50. Michl J., Thulstrup E. W., Spectroscopy with Polarized Light, VCH, Weinheim, 1986, p 368. [Google Scholar]
- 51.
- 51a. Englman R., Jortner J., Mol. Phys. 1970, 18, 145–164; [Google Scholar]
- 51b. Freed K. F., Jortner J., J. Chem. Phys. 1970, 52, 6272–6291; [Google Scholar]
- 51c. Bixon M., Jortner J., Cortes J., Heitele H., Michel-Beyerle M. E., J. Phys. Chem. 1994, 98, 7289–7299. [Google Scholar]
- 52. Bachman J. C., Kavian R., Graham D. J., Kim D. Y., Noda S., Nocera D. G., Shao-Horn Y., Lee S. W., Nat. Commun. 2015, 6, 7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.
- 53a. Heckmann A., Lambert C., Angew. Chem. Int. Ed. 2012, 51, 326–392; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 334–404; [Google Scholar]
- 53b. Lambert C., Nöll G., Synth. Met. 2003, 139, 57–62. [Google Scholar]
- 54.
- 54a. Connelly N. G., Geiger W. E., Chem. Rev. 1996, 96, 877–910; [DOI] [PubMed] [Google Scholar]
- 54b. Pavlishchuka V. V., Addison A. W., Inorg. Chim. Acta 2000, 298, 97–102. [Google Scholar]
- 55. Mergny J.-L., Lacroix L., Oligonucleotides 2003, 13, 515–537. [DOI] [PubMed] [Google Scholar]
- 56. Radić-Stojković M., Piotrowski P., Schmuck C., Piantanida I., Org. Biomol. Chem. 2015, 13, 1629–1633. [DOI] [PubMed] [Google Scholar]
- 57.
- 57a. Scatchard G., Ann. NY Acad. Sci. 1949, 51, 660–672; [Google Scholar]
- 57b. McGhee J. D., Hippel P. H. V., J. Mol. Biol. 1974, 86, 469–489. [DOI] [PubMed] [Google Scholar]
- 58.
- 58a. Eriksson M., Nordén B. in Methods in Enzymology, Vol. 340 (Eds.: Chaires J. B., Waring M. J.), Academic Press, San Diego: 2001, pp. 68–98; [DOI] [PubMed] [Google Scholar]
- 58b. Šmidlehner T., Piantanida I., Pescitelli G., Beilstein J. Org. Chem. 2018, 14, 84–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Uson R., Oro L. A., Cabeza J. A., Bryndza H. E., Stepro M. P. in Inorg. Synth., Vol. 23, (Ed.: Kirschner S.), New York, 1985, pp. 126–130. [Google Scholar]
- 60.
- 60a. Sheldrick G. M., Acta Crystallogr. Sect. A 2008, 64, 112–122; [DOI] [PubMed] [Google Scholar]
- 60b. Sheldrick G. M., Acta Crystallogr. Sect. A 2015, 71, 3–8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60c. Hübschle C. B., Sheldrick G. M., Dittrich B., J. Appl. Crystallogr. 2011, 44, 1281–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka, M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017.
- 62. Macrae C. F., Bruno I. J., Chisholm J. A., Edgington P. R., McCabe P., Pidcock E., Rodriguez-Monge L., Taylor R., van de Streek J., Wood P. A., J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar]
- 63.Gaussian 09, Revision D. 01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian Inc., Pittsburgh, PA, 2009.
- 64.
- 64a. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865–3868; [DOI] [PubMed] [Google Scholar]
- 64b. Perdew J. P., Ernzerhof M., Burke K. J., Chem. Phys. 1996, 105, 9982–9985. [Google Scholar]
- 65. Adamo C., Barone V., J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar]
- 66. Grimme S., Antony J., Ehrlich S., Krieg H. J., Chem. Phys. 2010, 132, 154104–154123. [DOI] [PubMed] [Google Scholar]
- 67. Grimme S., Ehrlich S., Goerigk L. J., Comput. Chem. 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- 68. Yanai T., Tew D. P., Handy N. C., Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar]
- 69. Tomasi J., Mennucci B., Cammi R., Chem. Rev. 2005, 105, 2999–3093. [DOI] [PubMed] [Google Scholar]
- 70.R. Dennington, T. Keith, J. Millam, GaussView, Version5; Semichem Inc.: Shawnee Mission, KS, 2009.
- 71. Kresse G., Furthmüller J., Phys. Rev. B 1996, 54, 11169–11186. http://www.vasp.at. [DOI] [PubMed] [Google Scholar]
- 72. Blöchl P. E., Phys. Rev. B 1994, 50, 17953–17979. [DOI] [PubMed] [Google Scholar]
- 73. Monkhorst H. J., Pack J. D., Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar]
- 74. Chaires J. B., Dattagupta N., Crothers D. M., Biochemistry 1982, 21, 3933–3940. [DOI] [PubMed] [Google Scholar]
- 75. Stolić I., Mišković K., Magdaleno A., Silber A. M., Piantanida I., Bajić M., Glavaš-Obrovac L., Bioorg. Med. Chem. 2009, 17, 2544–2554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary