

Abstract
The role of somatic copy number alterations (SCNAs) that occur in colorectal tumors is poorly understood. SCNAs are correlated with corresponding gene expression changes that may contribute to neoplastic progression. Thus, we examined SCNAs and the expression of messenger RNAs (mRNAs) located at corresponding loci in colorectal neoplasia, a progression model of human neoplasm. We used 42 colorectal neoplastic samples, including adenomas, intramucosal cancers (IMC) and invasive colorectal cancers (CRC) that were microsatellite stable (MSS) using a genome‐wide SNP array and gene expression array (first cohort). In addition, validation analyses were examined (37 colorectal neoplasias). None of the mRNAs with a corresponding SCNA was found in the adenomas. However, three mRNAs, including ARFGEF2 at 20q13.13, N4BP2L2 at 13q13.1 and OLFM4 at 13q14.3 with a copy number (CN) gain at the corresponding locus were upregulated in IMCs of the first cohort. Moreover, upregulated expression of ARFGEF2 and OLFM4 was upregulated in the validation analysis. Finally, 28 mRNAs with gains of corresponding loci were pooled in invasive CRC of the first cohort. The mRNAs, including ACSS2 (20q11.22), DDX27 (20q13.13), MAPRE1 (20q11.21), OSBPL2 (20q11.22) and PHF20 (20q11.22‐q11.23) with CN gains of the corresponding loci were identified in 28 mRNAs. Four of these mRNAs (DDX27, MAPRE1, OSBPL2 and PHF20) were upregulated in the invasive CRC in the validation analysis. We conclude that specific 13q and 22q CN gains with gene expression changes in the corresponding loci may play an important role in IMC cells' progression into invasive CRC.
Keywords: array‐based analysis, colorectal adenoma, colorectal cancer, genome‐wide study, messenger RNA, somatic copy number alteration
Abbreviations
- CN
copy number
- CN‐LOH
copy neutral loss of heterozygosity
- CRC
colorectal cancer
- mRNA
messenger RNA
- MSI or MIN
microsatellite instability
- MSS
microsatellite stable
- SCNA
somatic copy number alteration
1. INTRODUCTION
Colorectal cancer (CRC) is one of the most common malignant neoplasms throughout the world. 1 Recent advances in detection and chemotherapeutic/biological agent‐based therapies have dramatically increased the survival rate of CRC patients. 2 Such advances in clinical medicine are based on noteworthy progress in basic cancer research, particularly molecular mechanism in colorectal pathogenesis. Despite such advances, CRC remains an uncontrollable disease.
Cancer cells are characterized by cytogenetic alterations that can be used to define specific disease entities and their prognostic markers. 3 , 4 Previous studies showed that there are two molecular CRC subtypes, including chromosomal (CIN, or microsatellite stable MSS) and those with microsatellite instabilities (MIN; MSI). CIN is observed in 80% to 90% of sporadic CRCs; this alteration is closely associated with an accelerated rate of gains or losses of whole or partial portions of chromosomes that are detected as somatic copy number alterations (SCNA) that are identified using an array‐based analysis. 4 , 5 , 6 , 7 MIN is estimated to occur in approximately 10% to 20% of sporadic CRCs. An alternative molecular pathway of sporadic CRC is characterized by instability in DNA microsatellite sequences. 4 , 5 , 6 , 7
Comprehensive CINs in CRC have been investigated by many researchers using either comparative genomic hybridization (CGH) or array‐based SNP arrays (SNP array). 8 , 9 , 10 These approaches have contributed to the detection of many chromosomal aberrations, including gains and losses, helping to evaluate complex disease progression. 11 SNP arrays may be suitable for gathering more detailed information regarding the possible underlying mechanism of chromosomal abnormalities and to evaluate genetic characteristics. In recent studies, it has been used in place of CGH arrays. 11 , 12 Accumulated chromosomal alterations involving key regulatory genes are essential genetic events in the initiation and progression of colorectal tumorigenesis. 11 , 12 , 13 Many alterations, such as SCNA can disrupt or change proper gene function. 14 Although there is wide agreement that chromosomal aberrations may involve genes that possess important function in tumor development, 14 , 15 , 16 such an assumption may not be necessarily true in chromosomal aberrations occurring in human neoplasia. Traditional theory suggests that tumor suppressor genes are inactivated by a loss of another chromosomal locus and that oncogenes are enhanced by gene amplification. 17 Thus, SCNA may be assumed to be accompanied by corresponding genetic alteration in colorectal tumors. 18 However, whether such functional abnormality is always apparent remains unknown, given that whether a change in SCNA does induce actual expression has not been identified in previous studies. 18 , 19 This issue might be further complicated by the finding that many chromosomal aberrations span large chromosomal regions that involve multiple genes. 18 , 19
We set out to determine whether SCNAs occurring in tumor cells were involved with the altered expression of genes in affected chromosomal regions. To approach this problem, we focused on colorectal tumors, including adenoma, intramucosal cancer (IMC), and invasive CRC with an MSS phenotype that evolves through a multistep accumulation of molecular alterations, including genetic and epigenetic abnormalities.
2. MATERIALS AND METHODS
2.1. Patients
This study used 42 tumor samples and normal colonic mucosa that were obtained from 42 enrolled patients with 15 colorectal adenomas (including tubular and tubulovillous adenomas), eight patients with IMC and 19 patients with CRC that invaded into the muscular layer without metastasis. Adenoma and IMC were diagnosed according to the modified World Health Organization (WHO) 2019 criteria. 20 In addition, the adenomas that we examined included low (LGA) and high‐grade adenomas (HGA). Clinicopathological findings were recorded according to the General Rules for Management of the Japanese Colorectal Cancer Association. 21 Finally, 37 colorectal tumors including 15 adenomas, eight IMCs and 14 invasive CRCs were examined for validation analysis (second cohort). The clinicopathological findings are shown in Table 1.
TABLE 1.
Clinicopathological findings of the colonic neoplasms in sampled cohorts
| Cohort 1 (microarray) (%) | Cohort 2 (validation test) (%) | |
|---|---|---|
| Sex | ||
| Male | 29 (69) | 25 (71.4) |
| Female | 13 (31) | 12 (28.6) |
| Age, median (range) | 67 (43‐81) | 66 (45‐85) |
| Location | ||
| C/A/T/D/S/R | 2/8/6/3/11/12 | 4/6/3/2/15/12 |
| Histological type | ||
| Conventional adenoma | 15 (35.7) | 15 (40.5) |
| High grade | 3 (20) | 4 (16.7) |
| Low grade | 12 (80) | 11 (73.3) |
| Intramural carcinoma | 8 (19) | 8 (21.6) |
| Colorectal cancer with MSS phenotype | 19 (45.2) | 14 (37.8) |
| MDA | 18 (94.7) | 13 (92.9) |
| MUC | 1 (5.3) | 1 (7.1) |
| Stage | ||
| II | 10 (52.6) | 9 (64.3) |
| III | 9 (47.4) | 5 (35.7) |
Abbreviations: A, ascending colon; C, cecum; D, descending colon; MDA, moderately differentiated adenocarcinoma; MUC, mucinous adenocarcinoma; R, rectum; S, sigmoid colon; T, transverse colon.
This study was approved by the local ethics committee of Iwate Medical University (approval number MH2020‐066), and all patients provided informed consent.
2.2. Crypt isolation method
Crypt isolation from the tumors and normal mucosa (distal site of the colon for surgical specimens) was performed as described elsewhere. 22 Tumor glands (adenomatous and intramucosal carcinomatous glands) were obtained from suspected target lesions as determined by magnification, whereas cancer glands in invasive CRC were taken from the invasive front. Briefly, fresh tissues were minced with a razor into small pieces and incubated at 37°C for 30 minutes in calcium‐ and magnesium‐free Hanks' balanced salt solution (CMF) containing 30 mM EDTA. The isolated crypts were immediately fixed in 70% ethanol and stored at 4°C until used for DNA/RNA extractions. The fixed isolated crypts were observed under a dissecting microscope (SZ60; Olympus, Tokyo, Japan). Some isolated crypts were routinely processed for histopathological analysis to confirm the histological nature of the isolated glands. Contamination, such as interstitial cells, was not evident in any of the 42 samples used for array‐based analysis and the 37 samples used for the validation test.
2.3. DNA extraction
For each patient, DNA was extracted from isolated tumors and normal glands using classical phenol‐chloroform extraction.
2.4. Analysis of MSI
The MSI status was determined using a consensus panel of five reference microsatellite markers (BAT25, BAT26, D2S123, D3S546 and D17S250) using a previously described method. 23 When no marker was altered, the tumors were defined as MSS. When only one marker was altered, the tumors were defined as low MSI. When two or more markers were altered, the tumors were defined as high MSI.
3. RNA EXTRACTION
Isolation of total RNA from cancer cells was performed using RNeasy Mini kits (Qiagen, Valencia, CA) in accord with the manufacturer's instructions. The nucleic acid concentration was determined using a Nanodrop1000 spectrophotometer (Thermo Fisher Scientific; Waltham, MA), and the RNA purity was verified using 1.5% denaturing agarose gels.
3.1. SNP array analysis
The Cytoscan HD (Affymetrix, UK) platform was used in the present study. This array contains more than 1.9 million non‐polymorphic markers and over 740 000 SNP markers with an average intragenic marker spacing of 880 bps and intergenic marker spacing of 1737 bps. These platforms are composed of microarrays containing non‐polymorphic probes for CNVs (copy number variations) from coding and noncoding regions of the human genome as well as polymorphic SNP probes. All procedures were performed as instructed by the manufacturer. The hybridized slides bearing DNA marked with biotin, were analyzed with a GeneChip Scanner 3000 7G (Affymetrix) and Chromosome Analysis Suite (ChAS) Software (Affymetrix). Detailed information for definition of abnormalities was previously described. 24
3.2. Classification of copy number alteration
In the present study, we classified SCNAs into five subtypes, including gain, loss of heterozygosity (LOH), copy neutral LOH (CN‐LOH), mosaic and mixed types, as previously reported. 24 Whereas LOH was considered a gross chromosomal change that results in loss of the entire gene and the surrounding region, gain was defined as a gross chromosomal change that was caused by a gain of the entire gene and the surrounding region. CN‐LOH was defined as an occurrence of LOH in the absence of the allelic loss (copy number [CN] ≧ 2). A mosaic pattern was defined as a mixture of normal and abnormal cells with SCNAs. Finally, a mixed pattern was a mixture of >2 SCNA patterns within one locus, such as LOH and LOH mosaic, or gain and LOH or gain and gain mosaic.
In the present study, a mosaic LOH was considered to be a LOH, whereas mosaic gain was regarded as a gain. In addition, a mixed pattern was classified into dominant patterns, such as LOH>gain, LOH; gain>LOH, gain.
4. CLARIOM S HUMAN ARRAY AND GENE EXPRESSION ANALYSIS
For each array experiment, 500 ng of total RNA was used for labeling using the Clariom S Human Array (Thermo Fisher Scientific). This array contains 21 453 messenger RNAs (mRNAs). Probe labeling, chip hybridization and scanning were performed according to the manufacturer's instructions. A Probe Set (gene‐exon) was considered expressed if ≥50% of the samples were detected above background (DABG) values below the DABG threshold (DABG <0.05). The array data were generated using the Transcriptome Analysis Console (TAC version 4.0.1.36) and analyzed with the Affymetrix Chromosome Analysis Suite v.4.1 (ChAS) (Affymetrix Inc.,).
To validate microarray results, quantitative reverse transcription polymerase chain reaction (qRT‐PCR) was performed. One microgram of total RNA was reverse transcribed to first‐strand cDNA with the Qiagen cDNA Synthesis Kit (Qiagen), and this cDNA was subsequently used as a template for quantitative PCR with gene‐specific primers. The ubiquitous β‐actin gene served as a control for constitutive gene expression. qRT‐PCR reactions were performed using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific) on a CFX96 Touch Real‐Time PCR Detection System (Bio‐Rad, Hercules, CA). Relative expression levels (2−ΔCt) were calculated according to the Livak and Schmittgen method. 25 Expression levels of each gene were compared with the expression level of actin.
4.1. Statistical analysis
The differences between SCNAs with an upregulated or downregulated corresponding gene expression level (including gain and non‐gain) were assessed with a Fisher's exact test. In addition, the differences in the frequencies of genotype (ex ARFGEF2/CN gain) between adenoma, IMC and invasive CRC were also assessed with a Fisher's exact test with an adjusted Bonferroni correction. If statistically significant differences among the three lesion types were detected, comparisons between two groups were performed using a Fisher's exact test. Differences in SCNA number including gain, LOH and CN‐LOH among the groups were evaluated using the Kruskal‐Wallis H test in Stat Mate‐III. A P‐value <.05 was accepted as significant. If statistical differences among the three lesion types were detected, comparisons between two groups were performed using the same method (Kruskal‐Wallis H test). Final statistical significance was determined after two group comparison. As a result, a P‐value <.05 was accepted as significant.
4.2. Workflow (experimental design)
The workflow used in the present study is summarized in Figure 1.
FIGURE 1.
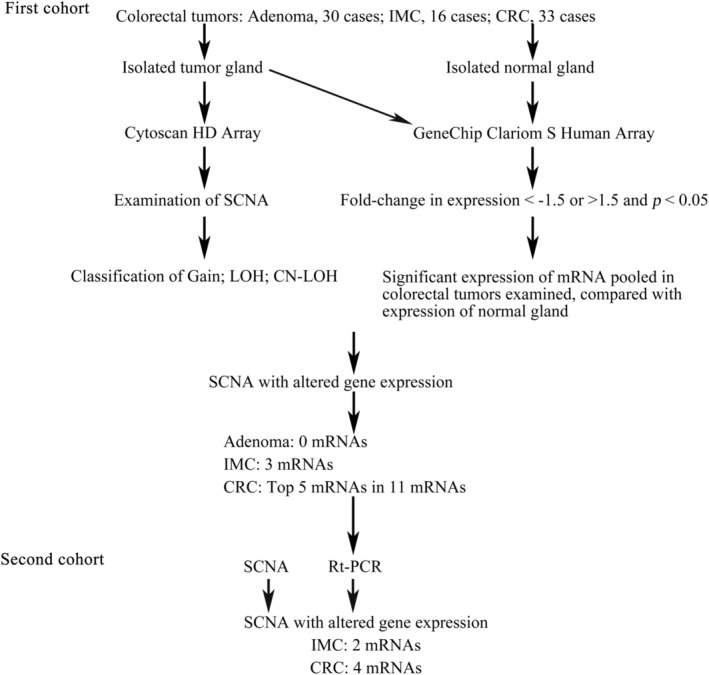
Workflow of experimental design. First, somatic copy number alteration (SCNA) was examined using a Cytoscan HD. Second, messenger RNAs (mRNA) expression was investigated according to the following criteria: fold‐change in expression (<−1.5 or >1.5) and P < .05 using a Clariom S Human Array, compared with that of normal gland. Third, SCNAs with altered gene expression were pooled in adenoma, intramucosal cancers (IMC) and colorectal cancer (CRC). Finally, the association of selected mRNAs with corresponding gene expression was validated
5. RESULTS
5.1. Microsatellite analysis of colorectal adenoma, IMC and invasive CRC
All tumors were classified into an MSS phenotype according to criteria previously reported. 23
5.2. mRNA expression profiling in colorectal tumors
We sought to identify potential mRNA biomarkers in 42 colorectal neoplasias, including 15 adenomas, eight IMCs and 19 invasive CRCs. We used global mRNA expression profiling in the analysis of the 42 colorectal neoplasias and compared them with normal gland samples as follows. First, we examined the difference in the expression levels of mRNA between isolated normal and neoplastic glands using a t test (P value) with an adjusted Benjamini‐Hochberg method/False discovery rate (FDR) correction. Second, such mRNAs were selected and grouped. As a result, we identified 31 differentially (upregulated) mRNAs in adenomas (Table 2). Using a similar method, three upregulated and five downregulated mRNAs were grouped in Table 3. Third, 29 mRNAs were groped in invasive CRC (14 upregulated and 15 downregulated; Table 5). Finally, individual data pooled in colorectal adenoma, IMC and invasive CRC are shown in Table S1 ( t test with FDR correction).
TABLE 2.
Associated expression of mRNA with copy number alteration in conventional adenoma
| Official symbol | Location | Up‐regulated case (n = 15) (%) | Pattern of copy number alteration (n = 15) (%) | P‐value | |
|---|---|---|---|---|---|
| Gain | Non‐gain | ||||
| MIR3654 | 7q33 | 10 (66.7) | 4 (26.7) | 11 (73.3) | .5604 |
| OLFM4 | 13q14.3 | 7 (46.7) | 3 (20) | 12 (80) | .0769 |
| GNAS | 20q13.32 | 9 (60) | 2 (13.3) | 13 (86.7) | 1.000 |
| RPS21 | 20q13.33 | 8 (53.3) | 2 (13.3) | 13 (86.7) | 1.000 |
| RPL41 | 12q13.2 | 10 (66.7) | 1 (6.7) | 14 (93.3) | 1.000 |
| CEACAM5 | 19q13.2 | 9 (60) | 1 (6.7) | 14 (93.3) | 1.000 |
| PIGR | 1q32.1 | 11 (73.3) | 0 (0) | 15 (100) | 1.000 |
| TMSB10 | 2p11.2 | 8 (53.3) | 0 (0) | 15 (100) | 1.000 |
| SPTBN1 | 2p16.2 | 8 (53.3) | 0 (0) | 15 (100) | 1.000 |
| EPCAM | 2p21 | 8 (53.3) | 0 (0) | 15 (100) | 1.000 |
| ITM2C | 2q37.1 | 8 (53.3) | 0 (0) | 15 (100) | 1.000 |
| CAST | 5q15 | 12 (80) | 0 (0) | 15 (100) | 1.000 |
| SLC12A2 | 5q23.3 | 7 (46.7) | 0 (0) | 15 (100) | 1.000 |
| RACK1 | 5q35.3 | 8 (53.3) | 0 (0) | 15 (100) | 1.000 |
| SNORD95 | 5q35.3 | 8 (53.3) | 0 (0) | 15 (100) | 1.000 |
| SNORD96A | 5q35.3 | 8 (53.3) | 0 (0) | 15 (100) | 1.000 |
| H4C3 | 6p22.2 | 9 (60) | 0 (0) | 15 (100) | 1.000 |
| EEF1A1 | 6q13 | 11 (73.3) | 0 (0) | 15 (100) | 1.000 |
| EEF1G | 11q12.3 | 10 (66.7) | 0 (0) | 15 (100) | 1.000 |
| RPS3 | 11q13.4 | 10 (66.7) | 0 (0) | 15 (100) | 1.000 |
| SNORD15A | 11q13.4 | 10 (66.7) | 0 (0) | 15 (100) | 1.000 |
| HSPA8 | 11q24.1 | 13 (86.7) | 0 (0) | 15 (100) | 1.000 |
| SNORD14C | 11q24.1 | 13 (86.7) | 0 (0) | 15 (100) | 1.000 |
| SNORD14D | 11q24.1 | 13 (86.7) | 0 (0) | 15 (100) | 1.000 |
| RPS28 | 19p13.2 | 10 (66.7) | 0 (0) | 15 (100) | 1.000 |
| CSTB | 21q22.3 | 8 (53.3) | 0 (0) | 15 (100) | 1.000 |
| RPL3 | 22q13.1 | 12 (80) | 0 (0) | 15 (100) | 1.000 |
| SNORD43 | 22q13.1 | 12 (80) | 0 (0) | 15 (100) | 1.000 |
| SNORD83B | 22q13.1 | 12 (80) | 0 (0) | 15 (100) | 1.000 |
| MT‐CO1 | MT | 9 (60) | 0 (0) | 15 (100) | 1.000 |
| MT‐CO2 | MT | 8 (53.3) | 0 (0) | 15 (100) | 1.000 |
Abbreviations: CN‐LOH, copy neutral loss of heterozygosity; LOH, loss of heterozygosity.
TABLE 3.
Associated increased expression level of mRNA with copy number alteration in intramucosal cancer
| Official symbol | Location | Up‐regulated case (n = 8) (%) | Pattern of copy number alteration (n = 8) (%) | P‐value | |
|---|---|---|---|---|---|
| Gain | Non‐gain | ||||
| N4BP2L2 | 13q13.1 | 6 (75) | 6 (75) | 2 (25) | .0357 |
| OLFM4 | 13q14.3 | 6 (75) | 6 (75) | 2 (25) | .0357 |
| ARFGEF2 | 20q13.13 | 6 (75) | 6 (75) | 2 (25) | .0357 |
Abbreviations: CN‐LOH, copy neutral loss of heterozygosity; LOH, loss of heterozygosity.
5.3. SCNAs in conventional adenoma, IMC and CRC
In CRC, the mean total number of chromosomal aberrations per patient was 516, with an average of 315 gains (range: 30‐644), 136 LOHs (range: 4‐480) and 66 copy‐neutral LOHs (range: 12‐185). In conventional adenoma, the mean total number of chromosomal aberrations per patient was 75, with an average of 35 gains (range: 1‐103), 18 LOHs (range: 10‐33) and 22 copy‐neutral LOHs (range: 8‐50). Finally, in IMC, the mean total number of chromosomal aberrations per patient was 337, with an average of 246 gains (range: 14‐641), 68 LOHs (rang: 29‐181) and 23 copy‐neutral LOHs (range: 4‐43). There were significant differences in the median values in the total numbers of SCNAs, gains and LOHs between adenoma and IMC or invasive CRC (P < .01). Statistically significant differences in the median number of CN‐LOH between invasive CRC and adenoma or IMC were found (Figure 2).
FIGURE 2.
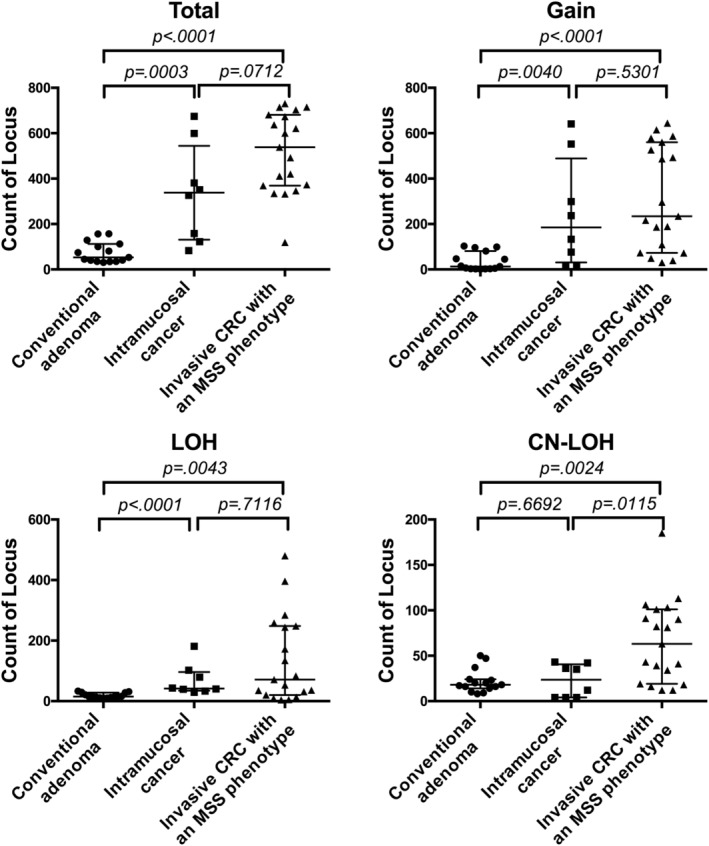
Total number of somatic copy number alterations (SCNAs), gains, loss of heterozygosity (LOHs) and copy neutral loss of heterozygosity (CN‐LOHs) in colorectal adenoma, intramucosal cancers (IMC) and colorectal cancer (CRC)
Regions of SCNAs, including gain, LOH and copy neutral LOH detected in more than 30% of cases were pooled in adenoma, IMC and invasive CRC. These SCNAs are listed in Table S2. In addition, the ideograms of adenoma, IMC and invasive CRC are depicted in Figure S1. Finally, a systematic analysis of CN gains and losses and altered regulation of corresponding mRNAs is shown in Table S3.
5.4. Integrated genome and transcriptome analysis
To identify the correlation of SCNA status with corresponding gene expression, we utilized a statistical approach. First, each mRNA and corresponding chromosomal location (eg, DDX27 located at 20q13.13) was explored in our three categories (colorectal adenoma, IMC and invasive CRC). Second, we examined the association of the corresponding chromosomal location with SCNA, including gain and non‐gain in adenoma, IMC and invasive CRC (eg, SCNA at 20q13.13 was a CN gain). In addition, we determined whether the SCNA pattern was correlated with a change in the corresponding gene expression level (upregulated or downregulated) using a Fisher's exact test (Tables 2, 3, 4, 5, 6). Finally, we showed that different patterns of SCNAs could be demonstrated, using CN levels and gene expression levels (Figure 3).
TABLE 4.
Associated decreased expression level of mRNA with copy number alteration in intramucosal cancer
| Official symbol | Location | Down‐regulated case (n = 8) (%) | Pattern of copy number alteration (n = 9) (%) | P‐value | |
|---|---|---|---|---|---|
| LOH | Non‐LOH (gain) | ||||
| FABP1 | 2p11.2 | 5 (62.5) | 0 (0) | 8 (100) | 1.000 |
| GSKIP | 14q32.2 | 4 (50) | 2 (25) | 6 (75) | .4286 |
| SELENOH | 11q12.1 | 3 (37.5) | 0 (0) | 8 (100) | 1.000 |
| TMX2 | 11q12.1 | 3 (37.5) | 1 (12.5) | 7 (87.5) | 1.000 |
| XKRY2 | Yq11.222 | 3 (37.5) | 1 (12.5) | 7 (87.5) | .375 |
Abbreviations: CN‐LOH, Copy neutral loss of heterozygosity; LOH, loss of heterozygosity.
TABLE 5.
Associated increased expression level of mRNA with copy number alteration in CRC
| Official symbol | Location | Up‐regulated case (n = 19) (%) | Pattern of copy number alteration (n = 19) (%) | P‐value | |
|---|---|---|---|---|---|
| Gain | Non‐gain | ||||
| DDX27 | 20q13.13 | 18 (94.7) | 18 (94.7) | 1 (5.3) | .0326 |
| MAPRE1 | 20q11.21 | 15 (79) | 17 (89.5) | 2 (10.5) | .0351 |
| ACSS2 | 20q11.22 | 15 (79) | 17 (89.5) | 2 (10.5) | .0351 |
| PHF20 | 20q11.22‐q11.23 | 16 (84.2) | 17 (89.5) | 2 (10.5) | .0175 |
| OSBPL2 | 20q13.33 | 15 (79) | 17 (89.5) | 2 (10.5) | .0351 |
| DHX35 | 20q11.23‐q12 | 10 (52.6) | 17 (89.5) | 2 (10.5) | 1.000 |
| SEMG1 | 20q13.12 | 9 (47.4) | 17 (89.5) | 2 (10.5) | .4737 |
| ZFP64 | 20q13.2 | 12 (63.2) | 17 (89.5) | 2 (10.5) | .1228 |
| CAB39L | 13q14.2 | 9 (47.4) | 16 (84.2) | 3 (15.8) | .2105 |
| DACH1 | 13q21.33 | 12 (63.2) | 16 (84.2) | 3 (15.8) | .5232 |
| TPX2 | 20q11.21 | 12 (63.2) | 16 (84.2) | 3 (15.8) | .5232 |
| DEFB126 | 20p13 | 12 (63.2) | 12 (63.2) | 7 (36.8) | .3261 |
| ODF1 | 8q22.3 | 11 (57.9) | 11 (57.9) | 8 (42.1) | .1809 |
| VIRMA | 8q22.1 | 11 (57.9) | 0 (0) | 19 (100) | 1.000 |
Abbreviations: CN‐LOH, Copy neutral loss of heterozygosity; LOH, loss of heterozygosity.
TABLE 6.
Associated decreased expression level of mRNA with copy number alteration in CRC
| Official symbol | Location | Down‐regulated case (n = 19) (%) | Pattern of copy number alteration (n = 19) (%) | P‐value | |
|---|---|---|---|---|---|
| LOH | Non‐LOH | ||||
| CNDP2 | 18q22.3 | 11 (57.9) | 12 (63.2) | 7 (36.8) | .0063 |
| TSPAN1 | 1p34.1 | 15 (79) | 6 (31.6) | 13 (68.4) | 1.000 |
| SH3BGRL3 | 1p36.11 | 8 (42.1) | 6 (31.6) | 13 (68.4) | 1.000 |
| B2M | 15q21.1 | 11 (57.9) | 6 (31.6) | 13 (68.4) | .3189 |
| PIGR | 1q32.1 | 16 (84.2) | 3 (15.8) | 16 (84.2) | 1.000 |
| MS4A12 | 11q12.2 | 12 (63.2) | 3 (15.8) | 16 (84.2) | 1.000 |
| MUC2 | 11p15.5 | 16 (84.2) | 2 (10.5) | 17 (89.5) | .2982 |
| SLC26A3 | 7q22.3‐q31.1 | 13 (68.4) | 1 (5.3) | 18 (94.7) | .3158 |
| FABP1 | 2p11.2 | 17 (89.5) | 1 (5.3) | 18 (94.7) | .1053 |
| CEACAM7 | 19q13.2 | 16 (84.2) | 1 (5.3) | 18 (94.7) | 1.000 |
| LGALS4 | 19q13.2 | 14 (73.7) | 1 (5.3) | 18 (94.7) | 1.000 |
| ITM2C | 2q37.1 | 18 (94.7) | 1 (5.3) | 18 (94.7) | 1.000 |
| CD63 | 12q13.2 | 11 (57.9) | 1 (5.3) | 18 (94.7) | .4211 |
| CA1 | 8q21.2 | 19 (100) | 0 (0) | 19 (100) | 1.000 |
| ZG16 | 16p11.2 | 16 (84.2) | 0 (0) | 19 (100) | 1.000 |
Abbreviations: CN‐LOH, Copy neutral loss of heterozygosity; LOH, loss of heterozygosity.
FIGURE 3.
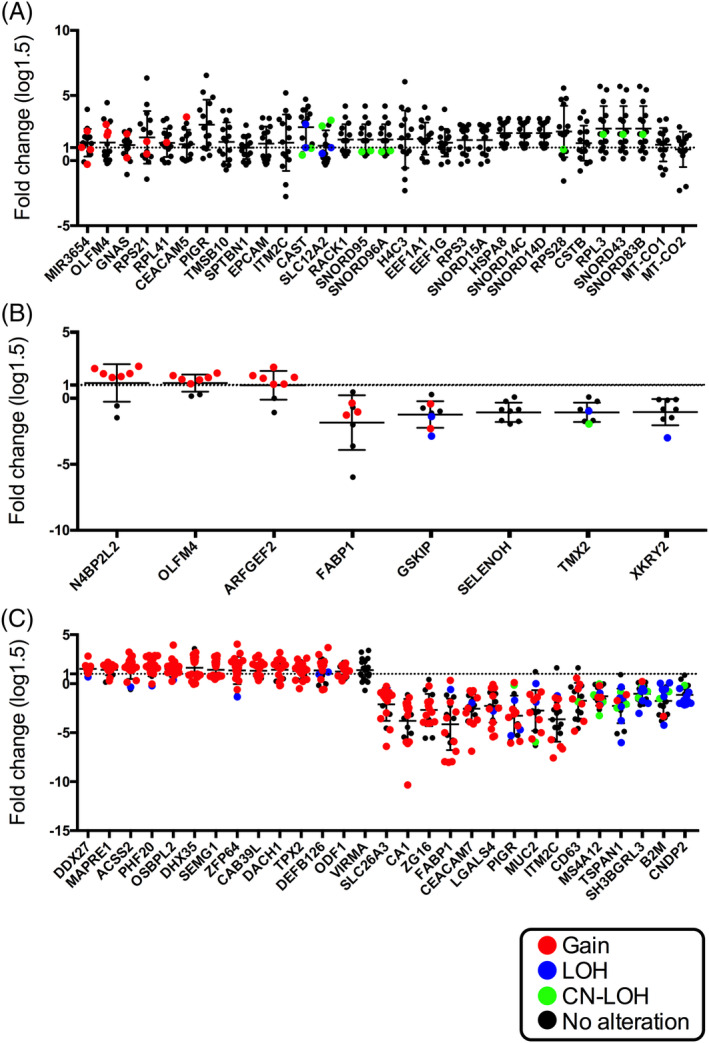
Expression levels of messenger RNAs (mRNAs) pooled in array‐based tests in colorectal adenoma, intramucosal cancer and colorectal cancer. Different patterns of somatic copy number alteration (SCNAs) are marked by different colors connecting copy number levels and gene expression levels. Red, gain; Blue, loss of heterozygosity (LOH), green, copy neutral LOH and black, no alteration
Analysis of the SCNA data and mRNA expression data revealed that none of the SCNAs had a correspondingly expressed gene in adenoma (Table 2) (Fisher's exact test). In IMC, we observed eight SCNAs (gains) with altered expression of genes, and three were correlated with upregulated expression of genes (Fisher's exact test). They included the following: ARFGEF2 (assay ID: Hs01573093_m1, ADP Ribosylation Factor Guanine Nucleotide Exchange Factor 2 at 20q13.13), N4BP2L2 (assay ID: Hs00263840_m1, neural precursor cell expressed developmentally down‐regulated protein 4 binding protein 2 Like 2 at 13q1.1) and OLFM4 (olfactomedin; assay ID: Hs00197437_m1 at 13q14.3) (Table 3). Next, we assessed the correlation between SCNA and the corresponding expression of mRNA in invasive CRC (Fisher's exact test). As a result, six mRNAs were identified out of 29 mRNAs in which a statistical change in expression level was found in invasive CRC, compared with that of normal gland (Fisher's exact test). They included the following six genes: ACSS2 (assay ID: Hs01120914_m1; Acyl‐coenzyme A synthetase short‐chain family member 2, 20q11.22); DDX27 (assay ID: Hs00916726_g1; DEAD‐Box Helicase 27, 20q13.13); MAPRE1 (assay ID: Hs01121100_g1; Microtubule Associated Protein RP/EB Family Member 1, 20q11.21); OSBPL2 (assay ID: Hs00375881_m1; oxysterol binding protein like 2, 20q11.22); PHF20 (PHD (assay ID: Hs00916778_m1; Plant Homeodomain Finger Protein 20, 20q11.22‐q11.23) (Table 5); and CNDP2 (Cytosolic non‐specific dipeptidase 2) However, expression of CNDP2 could not be examined due to the limited availability of RNA from isolated neoplastic glands. To summarize, SCNA gains correlated with gene expression in array‐based data.
We defined CN gain with corresponding gene expression, including ARFGEF2, N4BP2L2, OLFM, ACSS2, DDX27, MARRE1, OSBPL2 and PHF204 as a specific genotype (eg, termed upregulated ARFGEF2/CN gain, etc.). There were significant differences in the frequencies of the specific genotypes among the three lesions (adenoma, IMC and invasive CRC) using a Fisher's exact test analysis with Bonferroni correction. We found significant differences in the frequencies of three specific genotypes, including upregulated ARFGEF2/CN gain, upregulated N4BP2L2/CN gain and upregulated OLFM4/CN gain in comparisons of adenoma vs IMC, adenoma vs invasive CRC and IMC vs invasive CRC. In addition, the frequencies of upregulated ACSS2/CN gain, DDX27/CN gain, MARRE1/CN gain, OSBPL2/CN gain and PHF20/CN gain genotypes were statistically higher in invasive CRC than in adenoma or IMC using the same statistical method. The results are depicted in Table 7.
TABLE 7.
Comparison of specific mRNAs with corresponding SCNA gains between conventional adenoma, intramucosal cancer and CRC in array‐based data
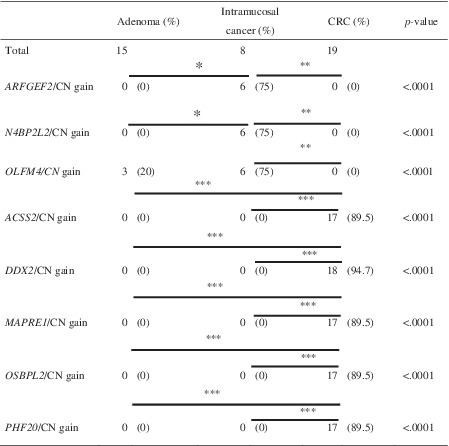
|
Abbreviations: CN, copy number; CRC, colorectal cancer; SCNA, somatic copy number alteration.
*Bonferroni corrected P‐value = .0009; **Bonferroni corrected P‐value = .0003; ***Bonferroni corrected P‐value <.0001.
None of the SCNAs had a correspondingly expressed gene in adenomas in the second cohort. In the validation cohort for IMC, the median values of the expression levels of ARFGEF2 and OLFM4 with CN gains of the corresponding loci (20q13.13 and 13q14.3, respectively) were upregulated. However, there was no association between the expression level of N4BPP2L2 and CN gain of 13q13.1 (Figure 4A). In addition, the median values of 4 mRNAs (DDX27 (20q13.13), MAPRE1 (20q11.21), OSBPL2 (20q11.22) and PHF20 (20q11.22‐q11.23) were upregulated in invasive CRC. However, upregulation of ACSS2 (20q11.22)) was not observed in invasive CRC (Figure 4B). Finally, SCNAs of IMC and invasive CRC used for the second cohort (validation cohort) were examined using the same method used in the first cohort. SCNAs of 20q13.13 and 13q14.3 with expression of corresponding genes including ARFGEF2 and OLFM4 were found in the second cohort (using a one‐sided t test). In addition, SCNAs of 20q13.13, 20q11.21, 20q11.22 and 21q11.22‐q11.23, which included expression of genes DDX27, MAPRE1, OSBPL2 and PHF20, were observed in the second cohort (using a one‐sided t test) (Table S4).
FIGURE 4.
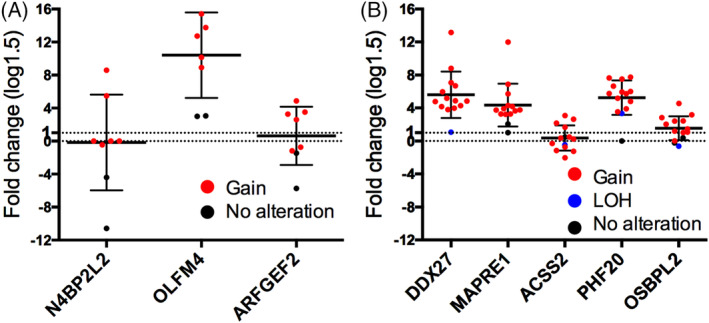
Expression levels of validated messenger RNAs (mRNAs) in intramucosal cancers (IMC) and invasive colorectal cancer. Different patterns of somatic copy number alteration (SCNAs) are marked by different colors connecting copy number levels and gene expression levels. A, Expression levels of mRNAs for ARFGF2, N4BP2L2 and OLFM4 in IMC. B, Expression level of messenger RNAs of ACSS2, DDX27, MAPRE1, OSBPL2 and PHF20 in colorectal cancer with a microsatellite stable phenotype. Red, gain; blue and black, no alteration
Taken together, two genotypes (upregulated ARFGEF2/CN gain and OLFM4/CN gain) were retained in IMC. Moreover, four genotypes, including upregulated DDX27/CN gain, MAPRE1/CN gain, OSBPL2/CN gain and PHF20/CN gain were sustained in invasive CRC (Table S4).
6. DISCUSSION
Our crypt isolation method enabled us to obtain pure tumor tissues and normal glands. 22 , 26 We used an array‐based method that is necessary to evaluate comprehensive genome‐wide alterations of a given tumor. 11 , 12 Therefore, crypt isolation could certify the origin of DNA or RNA extracted from epithelial tumor cells. This finding is important for the identification of the association between SCNA and expression of mRNA using human tumor tissue that is composed of both tumor cells and interstitial cells. However, different handling of different tissue specimens might also explain the differences in mRNA expression profiles. Moreover, the expression profile of mRNA may depend on sampling sites and differences between organs. Thus, the properties of isolated tumor glands could be influenced by such differences. In our experience, however, the crypt isolation method could be simply and reproducibly performed in any organ and sample site.
The most common changes in CRC are gains at 7p, 7q, 13q, 20p, 20q, Xp and Xq and losses at 8p, 17p, 18p and 18q. 14 Gains in chr8, chr10 and 21q were found more frequently in primary tumors, and gains in chr14, chr3p and chr18q were related to metastatic tumors. 14 , 27 Another study identified deletions from chr1p, 17p and 18q and gains in chr7 and 13q in metastatic CRC. Whereas those associations deserve further validation in larger populations, 28 those findings are consistent with our data. What our study emphasizes in particular is that SCNA by itself does not play a critical role in colorectal carcinogenesis. Rather, a specific genotype (eg, a CN gain with high expression of the corresponding gene) may contribute to neoplastic progression of colorectal neoplasia. This finding is consistent with the hypothesis that SCNA‐affected genes are closely associated with signaling pathways, pathological features, and therapeutic targets in CRC. 14 This understanding may facilitate the development of more effective molecular approaches and the evaluation of colorectal carcinogenesis when it is viewed as a heterogeneous disease.
Recent studies revealed that colorectal adenoma has a low level of SCNAs, compared with carcinomas. 11 , 12 By contrast, not only advanced adenomas but also early adenomas have SCNAs comparable to that of carcinomas as shown in a previous report. 29 Whether such SCNAs are correlated with the expression of corresponding mRNAs has remained unclear. However, we did not find the correlated expression of mRNAs with corresponding SCNAs. This finding is interesting because the expression of mRNA does not depend on SCNA in adenoma, suggesting that mechanisms other than SCNA, such as epigenetic alteration or unknown enhancers, play a major role in changing the expression of mRNA in adenoma cells. Collectively, the data suggest that SCNAs without functional expression of mRNA cannot progress from adenomatous cells into carcinomatous cells.
With regard to the IMCs that we examined here (interpreted as high‐grade colorectal adenomas in the West), it would be beneficial to identify them early in progression 11 , 12 because they are intermediates between adenomas and invasive CRCs. In the present study, the number of mRNAs that showed altered (upregulated/downregulated) expression of mRNA compared with those in normal crypts was lower in IMC than in adenoma. However, upregulated expression of mRNAs with CN gain, that is, ARFGEF2 (20q13.13) and OLFM4 (13q14.3) was commonly found in both the first and second cohorts. This finding suggests that those genes located at 20q13.13 and 13q14.3, respectively, are upregulated by CN gains in IMCs and may play important roles in the development of IMC. ARFGEF2, which is involved in Golgi transport, is closely associated with intracellular vesicular trafficking and is involved in the activation of ARFs by accelerating the replacement of bound GDP with GTP. 29 , 30 , 31 In addition, it may be responsible for guanine‐nucleotide exchange activity and also inhibition of brefeldin A, which inhibits protein transport from the endoplasmic reticulum to the golgi complex. 31 However, the genetic role of this gene in the development of CRC remains unknown. In addition, OLFM4 is selectively expressed in inflamed colonic epithelium and is an anti‐apoptotic factor that promotes tumor growth and is an extracellular matrix glycoprotein that facilitates cell adhesion. 32 , 33 High levels of expression of OLFM4 are found in CRC, suggesting that OLFM4 is a candidate diagnostic marker of CRC. 34 , 35 Moreover, this protein is a stem cell marker and potential biomarker of early CRC that involves IMC, supporting our finding. 32 , 33 We suggest that the current finding is novel in that the two genotypes, including upregulated ARFGEF2/CN gain and upregulated OLFM4/CN gain, may be responsible for the adenoma to cancer progression.
Genes with causal roles in cancers are often found in amplified genomic regions, whereas tumor suppressors may be embedded in deleted genomic segments. 14 , 36 , 37 In many cases, high‐level amplification or homozygous deletion may be necessary for the onset of neoplastic growth or invasion due to the requirement for activation of oncogenes or inactivation of tumor suppressor genes. 14 In the present study, we selected five mRNAs that were correlated with gains of their corresponding genes as identified in both first and second (validation) cohorts. Among them, four mRNAs including DDX27 (20q13.13), MAPRE1 (20q11.21), OSBPL2 (20q11.22) and PHF20 (20q11.22‐q11.23) were ultimately upregulated in invasive CRC. DDX27, which is associated with stem cell activity, was previously reported to be upregulated in CRC. 38 , 39 This is an interesting finding in that stem cell activity may play an important role in carcinogenesis. A recent study has shown that DDX27 promotes CRC growth and metastasis. 39 Furthermore, high expression of DDX27 predicts a poorer prognosis of CRC patients. 38 Upregulation of MAPRE1, which was associated with a gain (20q11.21), was found in gastric cancer. 40 , 41 Furthermore, overexpression of MAPRE1 was also observed in CRC. 42 Therefore, this molecule may be useful as a differential marker of colorectal adenoma from CRC. In addition, high expression of OSBPL2 was associated with a gain at this gene locus. OSBPL2 is associated with the p53/SREBF2/NFYA signal pathway. 43 OSBPL2 might be induced via this pathway and serve as a cancer‐induced molecule. Finally, expression of PHF20 which is associated with the p53 signal pathway has not been reported in CRC, but might be associated with its development. We suggest that four specific genotypes, including DDX27/CN gain, MAPRE1/CN gain, OSBPL2/CN gain and PHF20/CN gain, may be major molecular events in the progression to CRC. 43
There are some limitations to this study. First, heterogeneous expression of mRNAs might be essential to identify the roles of those that are highly expressed in CRC. 44 In the present study, we used a single sample from the invasive front of CRC. Therefore, heterogeneous expression of mRNAs could not be examined. The same would apply to heterogeneous occurrence of SCNA within the same tumor. Heterogeneity of expression of mRNAs and occurrence of SCNAs will be investigated in the near future. Second, the number of study participants may have been too small to identify the association of mRNA expression levels with SCNAs. Larger studies, such as that made possible by the Cancer Genome Alas (TCGA) and consensus molecular subtype (CMS) have already been performed in CRC. 8 , 9 , 10 However, we believe that the results of the present study contain new data. 8 , 9 , 10 For example, crypt isolation is a useful method to obtain target cells for molecular analysis and that method was utilized here as it permits the purification of neoplastic cells. In contrast, that approach was not utilized in TCGA. This difference is essential in understanding the differences between our data and those in TCGA. Moreover, early colorectal lesions, including colorectal adenoma and intramucosal cancer were target lesions in the present study. This point is crucial in explaining the unique aspects of the present study. Thus, we suggest that the current study provides new insight into the evaluation of colorectal carcinogenesis based on the “adenoma‐carcinoma sequence.” Finally, we could not make a meaningful association between SCNA and corresponding altered gene expression levels between LGA and HGA or HGA and IMC due to the small number of HGA cases. Larger studies will be needed to identify that association.
In conclusion, our results suggest that SCNAs with altered expression of corresponding cancer‐related genes could enhance the progression of cancer cells into a more pathologic state. This finding supports a hypothesis in which SCNAs with upregulated/downregulated expression of corresponding genes in cancer cells may be required for the progression of CRC. Here, specific genotypes that were closely associated with neoplastic progression occurring in IMC were different from those of invasive CRC. This finding suggests that clonal exchange might occur during neoplastic progression from intramucosal cancer to invasive CRC.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
Tamotsu Sugai, who is first author and the corresponding author, contributed to the preparation of the manuscript, including all aspects of the data collection and analysis. Mitsumasa Osakabe and Ryo Sugimoto performed all data collection and statistical analyses. Makoto Eizuka and Yoshihito Tanaka performed the collection of isolated tumors and normal glands. Naoki Yanagawa supported data preparation. Koki Otsuka, Akira Sasaki and Takayuki Matsumoto provided clinical support during the preparation of the manuscript. Hiromu Suzuki assisted the molecular analyses.
Supporting information
Figure S1 Ideogram of copy number alterations in 3 lesions: adenoma, IMC and invasive CRC in cohort 1. Chromosomes are ordered from 1 to 22. The colored horizontal lines represent the frequencies of gains, LOHs and CNLOHs. Lines on the left indicate losses (red, copy neutral LOH; gray, LOH), and those on the right (green) indicate gains.
Table S1 Table 1‐a Significant differential expression of each mRNA in conventional adenoma
Table S1‐b Significant differential expression of each mRNA in intramucosal cancer
Table S1‐c Significant differential expression of each mRNA in CRC
Table S2 Frequent CNA regions in conventional adenoma, intramucosal cancer and CRC with an MSS phenotype
Table S3‐a Association of expression of mRNA and copy number alteration in conventional adenoma in first cohort
Table S3‐b Association of expression of mRNA and copy number alteration in intramucosal cancer in first cohort
Table S3‐c Association of expression of mRNA and copy number alteration in CRC in first cohort
Table S4 Association of SCNAs with expression of corresponding genes in validation cohort.
ACKNOWLEDGMENTS
We gratefully acknowledge the technical assistance of Ms. E. Sugawara and Mrs. Ishikawa. We also thank the members of the Department of Molecular Diagnostic Pathology, Iwate Medical University, for its support.
Sugai T, Osakabe M, Sugimoto R, et al. A genome‐wide study of the relationship between chromosomal abnormalities and gene expression in colorectal tumors. Genes Chromosomes Cancer. 2021;60:250–262. 10.1002/gcc.22924
Tamotsu Sugai and Mitsumasa Osakabe contributed equally to this study.
Funding information Iwate Medical University
DATA AVAILABILITY STATEMENT
The data that support the findings of our study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. 2019;14(2):89‐103. 10.5114/pg.2018.81072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xie YH, Chen YX, Fang JY. Comprehensive review of target therapy for colorectal cancer. Signal Transduct Target Ther. 2020;5(1):22 10.1038/s41392-020-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ribeiro IP, Melo JB, Carreira IM. Cytogenetic and cytogenomics evaluation in cancer. Int J Mol Sci. 2019;20(19):4711 10.3390/ijms20194711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138(6):2059‐2072. 10.1053/j.gastro.2009.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386(6625):623‐627. 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 6. Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010;138(6):2088‐2100. 10.1053/j.gastro.2009.12.066. [DOI] [PubMed] [Google Scholar]
- 7. Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn. 2008;10(1):13‐27. 10.2353/jmoldx.2008.070082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330‐337. 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21(11):1350‐1356. 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sadanandam A, Lyssiotis CA, Homicsko K, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19(5):619‐625. 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sugai T, Eizuka M, Habano W, et al. Comprehensive molecular analysis based on somatic copy number alterations in intramucosal colorectal neoplasias and early invasive colorectal cancers. Oncotarget. 2018;9(33):22895‐22906. 10.18632/oncotarget.25112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eizuka M, Sugai T, Habano W, et al. Molecular alterations in colorectal adenomas and intramucosal adenocarcinomas defined by high‐density single‐nucleotide polymorphism arrays. J Gastroenterol. 2017;52(11):1158‐1168. 10.1007/s00535-017-1317-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takahashi Y, Sugai T, Habano W, et al. Molecular differences in the microsatellite stable phenotype between left‐sided and right‐sided colorectal cancer. Int J Cancer. 2016;139(11):2493‐2501. 10.1002/ijc.30377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang H, Liang L, Fang JY, Xu J. Somatic gene copy number alterations in colorectal cancer: new quest for cancer drivers and biomarkers. Oncogene. 2016;35(16):2011‐2019. 10.1038/onc.2015.304. [DOI] [PubMed] [Google Scholar]
- 15. Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444‐454. 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10(8):551‐564. 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal tumor development. N Engl J Med. 1988;319(9):525‐532. 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 18. Tsafrir D, Bacolod M, Selvanayagam ZT, et al. Relationship of gene expression and chromosomal abnormalities in colorectal cancer. Cancer Res. 2006;66(4):2129‐2137. 10.1158/0008-5472.CAN-05-2569. [DOI] [PubMed] [Google Scholar]
- 19. Berg M, Agesen TH, Thiis‐Evensen E, et al. Distinct high resolution genome profiles of early onset and late onset colorectal cancer integrated with gene expression data identify candidate susceptibility loci. Mol Cancer. 2010;9:100 10.1186/1476-4598-9-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hamilton SR, Sekine S. The 2019 WHO Classification of Tumours of the Digestive System. Lyon: World Health Organization; 2019:170‐173. [Google Scholar]
- 21. Japanese Society for Cancer of the Colon and Rectum . Japanese Classification of Colorectal Carcinoma. 2nd ed. Tokyo: Kanehara Co.; 2009:30‐63. [Google Scholar]
- 22. Habano W, Sugai T, Nakamura S, Yoshida T. A novel method for gene analysis of colorectal carcinomas using a crypt isolation technique. Lab Invest. 1996;74(5):933‐940. [PubMed] [Google Scholar]
- 23. Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248‐5257. [PubMed] [Google Scholar]
- 24. Tsuyukubo T, Ishida K, Osakabe M, et al. Comprehensive analysis of somatic copy number alterations in clear cell renal cell carcinoma. Mol Carcinog. 2020;59(4):412‐424. 10.1002/mc.23164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐delta delta C[T]) method. Methods. 2001;25:402‐408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26. Sugai T, Eizuka M, Takahashi Y, et al. Molecular subtypes of colorectal cancers determined by PCR‐based analysis. Cancer Sci. 2017;108(3):427‐434. 10.1111/cas.13164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xie T, Cho YB, Wang K, et al. Patterns of somatic alterations between matched primary and metastatic colorectal tumors characterized by whole‐genome sequencing. Genomics. 2014;104:234‐241. [DOI] [PubMed] [Google Scholar]
- 28. Gonzalez‐Gonzalez M, Fontanillo C, Abad MM, et al. Identification of a characteristic copy number alteration profile by high‐resolution single nucleotide polymorphism arrays associated with metastatic sporadic colorectal cancer. Cancer. 2014;120:1948‐1959. [DOI] [PubMed] [Google Scholar]
- 29. Carvalho B, Postma C, Mongera S, et al. Multiple putative oncogenes at the chromosome 20q amplicon contribute to colorectal adenoma to carcinoma progression. Gut. 2009;58(1):79‐89. 10.1136/gut.2007.143065. [DOI] [PubMed] [Google Scholar]
- 30. Yilmaz S, Gokben S, Serdaroglu G, et al. The expanding phenotypic spectrum of ARFGEF2 gene mutation: cardiomyopathy and movement disorder. Brain Dev. 2016;38(1):124‐127. 10.1016/j.braindev.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 31. Sheen VL, Ganesh VS, Topcu M, et al. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet. 2004;36(1):69‐76. 10.1038/ng1276. [DOI] [PubMed] [Google Scholar]
- 32. Liu W, Li H, Hong SH, Piszczek GP, Chen W, Rodgers GP. Olfactomedin 4 deletion induces colon adenocarcinoma in ApcMin/+ mice. Oncogene. 2016;35(40):5237‐5247. 10.1038/onc.2016.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van der Flier LG, Haegebarth A, Stange DE, van de Wetering M, Clevers H. OLFM4 is a robust marker for stem cells in human intestine and marks a subset of colorectal cancer cells. Gastroenterology. 2009;137(1):15‐17. 10.1053/j.gastro.2009.05.035. [DOI] [PubMed] [Google Scholar]
- 34. Jang BG, Kim HS, Kim KJ, Rhee YY, Kim WH, Kang GH. Distribution of intestinal stem cell markers in colorectal precancerous lesions. Histopathology. 2016;68(4):567‐577. 10.1111/his.12787. [DOI] [PubMed] [Google Scholar]
- 35. Huang MY, Wang HM, Chang HJ, Hsiao CP, Wang JY, Lin SR. Overexpression of S100B, TM4SF4, and OLFM4 genes is correlated with liver metastasis in Taiwanese colorectal cancer patients. DNA Cell Biol. 2012;31(1):43‐49. 10.1089/dna.2011.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haan JC, Labots M, Rausch C, et al. Citeb genomic landscape of metastatic colorectal cancer. Nat Commun. 2014;5:5457 10.1038/ncomms6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xie T, Ario G, Lamb JR, et al. A comprehensive characterization of genome‐wide copy number aberrations in colorectal cancer reveals novel oncogenes and patterns of alterations. PLoS One. 2012;7:e42001 10.1371/journal.pone.0042001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang C, Li D, Bai Y, et al. DEAD‐box helicase 27 plays a tumor‐promoter role by regulating the stem cell‐like activity of human colorectal cancer cells. Onco Targets Ther. 2018;12:233‐241. 10.2147/OTT.S190814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tang J, Chen H, Wong CC, et al. DEAD‐box helicase 27 promotes colorectal cancer growth and metastasis and predicts poor survival in CRC patients. Oncogene. 2018;37(22):3006‐3021. 10.1038/s41388-018-0196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Feng Y, Shi C, Wang D, Wang X, Chen Z. Integrated analysis of dna copy number changes and gene expression identifies key genes in gastric cancer. Comput Biol. 2020;27(6):877‐887. 10.1089/cmb.2019.0149. [DOI] [PubMed] [Google Scholar]
- 41. Kim K, Lee HC, Park JL, et al. Epigenetic regulation of microRNA‐10b and targeting of oncogenic MAPRE1 in gastric cancer. Epigenetics. 2011;6(6):740‐751. 10.4161/epi.6.6.15874. [DOI] [PubMed] [Google Scholar]
- 42. Taguchi A, Rho JH, Yan Q, et al. MAPRE1 as a plasma biomarker for early‐stage colorectal cancer and adenomas. Cancer Prev Res. 2015;8(11):1112‐1119. 10.1158/1940-6207.CAPR-15-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Q, Lin C, Zhang C, et al. 25‐hydroxycholesterol down‐regulates oxysterol binding protein like 2 (OSBPL2) via the p53/SREBF2/NFYA signaling pathway. J Steroid Biochem Mol Biol. 2019;187:17‐26. 10.1016/j.jsbmb.2018.10.018. [DOI] [PubMed] [Google Scholar]
- 44. Bastiaenen VP, Klaver CEL, van der Heijden MCS, et al. A mouse model for peritoneal metastases of colorectal origin recapitulates patient heterogeneity. Lab Invest. 2020;100(11):1465‐1474. 10.1038/s41374-020-0448-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Ideogram of copy number alterations in 3 lesions: adenoma, IMC and invasive CRC in cohort 1. Chromosomes are ordered from 1 to 22. The colored horizontal lines represent the frequencies of gains, LOHs and CNLOHs. Lines on the left indicate losses (red, copy neutral LOH; gray, LOH), and those on the right (green) indicate gains.
Table S1 Table 1‐a Significant differential expression of each mRNA in conventional adenoma
Table S1‐b Significant differential expression of each mRNA in intramucosal cancer
Table S1‐c Significant differential expression of each mRNA in CRC
Table S2 Frequent CNA regions in conventional adenoma, intramucosal cancer and CRC with an MSS phenotype
Table S3‐a Association of expression of mRNA and copy number alteration in conventional adenoma in first cohort
Table S3‐b Association of expression of mRNA and copy number alteration in intramucosal cancer in first cohort
Table S3‐c Association of expression of mRNA and copy number alteration in CRC in first cohort
Table S4 Association of SCNAs with expression of corresponding genes in validation cohort.
Data Availability Statement
The data that support the findings of our study are available from the corresponding author upon reasonable request.