

Abstract
The chemical reduction of a π‐expanded polycyclic framework comprising a cyclooctatetraene moiety, octaphenyltetrabenzocyclooctatetraene, with lithium metal readily affords the corresponding tetra‐anion instead of the expected aromatic dianion. As revealed by X‐ray crystallography, the highly contorted tetra‐anion is stabilized by coordination of two internally bound Li+, while two external cations remain solvent separated. The variable‐temperature 7Li NMR spectra in THF confirm the presence of three types of Li+ ions and clearly differentiate internal binding, consistent with the crystal structure. Density‐functional theory calculations suggest that the formation of the highly charged tetra‐reduced carbanion is stabilized through Li+ coordination under the applied experimental conditions.
Keywords: polycycles, density-functional calculations, reduction, structure elucidation, X-ray diffraction
A highly contorted π‐expanded polycyclic tetra‐anion comprising a cyclooctatetraene moiety is readily formed upon reduction of the neutral precursor with lithium metal. The highly charged carbanion crystallizes with four lithium counterions, two of which stabilize the contorted structure through internal coordination. Density‐functional theory calculations rationalize the unexpected formation of the tetra‐anion and provide insights into its electronic properties.
Cyclooctatetraene (COT), an 8‐membered representative of the family of [n]annulenes,[ 1 , 2 ] was initially synthesized back in 1911. [3] Due to its analogous stoichiometric composition to 6‐membered π‐system benzene, COT was originally expected to feature aromaticity, which was, however, contradicted by its divergent chemical behavior. In 1947, X‐ray diffraction analysis of COT revealed a boat‐like conformation of the 8‐membered ring with alternating bond lengths and D 2d symmetry. [4] While the eight π‐electrons in COT formally suggest its antiaromaticity, the scaffolds avoid such destabilization through deviation from planarity, preventing electronic communication between the individual p‐orbitals.
According to Hückel's rule planar, π‐conjugated cycles with 10 π‐electrons are expected to be aromatic, which motivated experiments towards aromatization of COT by two‐electron reduction. In 1960, COT was successfully reduced to the corresponding COT2− dianion upon treatment with potassium metal in THF. The reduction resulted in a concomitant downfield shift of the 1H NMR signal, suggesting the occurrence of a diamagnetic ring current in the doubly‐reduced species. [5] Consequently, planarization of the 8‐membered scaffold was proposed and later confirmed through X‐ray crystallographic analysis of a dianion derived from 1,3,5,7‐tetramethyl‐substituted COT derivative.[ 6 , 7 ] Although further reduction beyond dianions have been observed for larger annulenes,[ 8 , 9 ] tetraanionic COT4− remains a theoretical consideration. [10]
The possibility to render polycyclic aromatic hydrocarbons (PAHs) electron acceptors through incorporation of suitable nonbenzenoid moieties is particularly attractive and has been successfully demonstrated for cyclopentannulated systems [11] with corannulene and related geodesic PAHs being the most paradigmatic examples. [12] Such PAHs form stable carbanions upon reduction [13] and can act as n‐type semiconductors when appropriately functionalized.[ 11c , 14 ] However, related efforts involving PAHs with embedded 8‐membered COT rings remain comparably scarce.[ 2d , 15 ] For example, π‐expanded COT derivatives such as tetrabenzocyclooctatetraene (TBCOT)[ 16 , 17 ] and octaphenyltetrabenzocyclooctatetraene (OPTBCOT; 1) [18] have been reported and studied (Scheme 1). The latter was obtained in a Diels–Alder cycloaddition between 1,2,3,4‐tetraphenylcyclopenta‐1,3‐dienone and 5,6,11,12‐tetradehydrodibenzo[a,e]cyclooctene and subjected to oxidative cyclodehydrogenation under different conditions. [18] Furthermore, the saddle‐shaped COT moiety is an important structural element for the construction of negatively curved carbon‐rich compounds and carbon allotropes.[ 19 , 20 ] Recently, a unique COT‐containing molecular precursor towards negatively curved nanocarbon has been reported, [21] along with a remarkable contorted chiral PAH with three embedded COT moieties having the shape of a monkey saddle. [22] However, the multi‐electron accepting properties of π‐expanded COT‐containing PAHs have not yet been investigated.
Scheme 1.
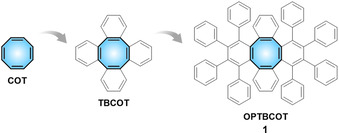
Consecutive π‐expansion of COT.
The crystal structure of OPTBCOT (C72H48, 1) was first reported back in 1997. [18] This PAH was crystallized as C72H48⋅CS2 (1‐CS2) with one interstitial CS2 molecule being fully wrapped by the peripheral phenyl rings of 1 (see Figure S12 in the Supporting Information). Using slow evaporation of THF solution of 1, colorless plate‐shaped crystals were obtained in this work (Yield: ≈93 %) and confirmed by X‐ray diffraction to be C72H48⋅THF (1‐THF) (Figure 1). [24] To exclude the effect of solvents, the gas phase sublimation of 1 in vacuo at 300 °C was also used for crystal growth affording a new non‐solvated (NS) polymorph (Yield: ≈84 %), 1‐NS (Figure S13).
Figure 1.
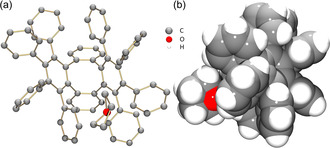
Crystal structure of 1‐THF. [23] a) Front view (ball‐and‐stick model, H‐atoms are omitted). b) Side view (space‐filling model).
In the solid‐state structure of 1‐THF, the OPTBCOT molecules of the same conformation are stacked into a 1D column held by C−H⋅⋅⋅π (2.682(5)–2.762(5) Å) and π⋅⋅⋅π (3.128(5) Å) interactions between the peripheral phenyl rings (Figure 2 a). The 1D columns are further assembled into a 3D network through C−H⋅⋅⋅π interactions, with the distances ranging from 2.777(5) Å to 2.836(5) Å (Figure 2 b). Similar solid‐state structure is observed in 1‐NS (Figure S18), although the packing is slightly tighter with the volume per C‐atom reduced to 17.6 Å3 from 18.2 Å3 in 1‐THF.
Figure 2.
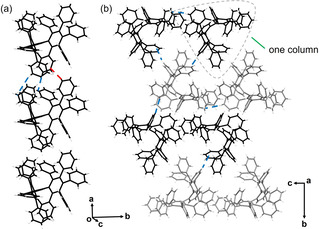
Solid‐state structure of 1‐THF: a) 1D column and b) 3D network, capped‐stick models. Two conformations are shown in gray and black. THF molecules are omitted. C−H⋅⋅⋅π and π⋅⋅⋅π interactions are shown in blue and red, respectively.
The conformational flexibility of 1 can be illustrated by evaluation of the tetraphenylene core changes in three different polymorphs. In 1‐CS2, the linear solvent molecule perfectly fits in the internal cavity of 1 (Figure S12), allowing the host to be symmetric. In contrast, the internal void space in 1‐THF is filled by the twisted peripheral phenyl rings (Figure 1), thus increasing the asymmetry of the tetraphenylene core. Compared to 1‐CS2, the central 8‐membered ring in 1‐THF becomes asymmetrically distorted, as can be seen from the increase of torsion angles at the positions β, γ, ζ, and η by about 13.7° (Table S4). Similar distortion is also observed in 1‐NS (Figure S13) with the average torsion angles at the above positions increased by 13.4°. The dihedral angles of the peripheral phenyl rings remain unchanged, but the dihedral angles I/R and K/R in 1‐THF and 1‐NS are increased by 10.4° and 9.2° (Table S3), illustrating added twist of the tetraphenylene core in comparison with 1‐CS2. The observed structural variations realized under different crystallization conditions illustrate a sufficient core flexibility of 1.
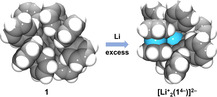
To gain insight into the electron accepting properties of OPTBCOT comprising a COT moiety embedded into a large contorted polycyclic framework, we investigated chemical reduction of 1 with lithium metal in THF. The reaction mixture was stirred at room temperature for 24 hours, and the product was successfully isolated in the form of bulk single crystals using slow diffusion of hexanes into the THF solution (Yield: ≈75 %, see the Supporting Information for more details). The X‐ray diffraction analysis confirmed the formation of [{Li+(THF)4}2{Li+ 2(1 4−)}] (2), which was crystallized with two interstitial THF molecules as 2⋅2 THF. There are two independent molecules in the unit cell (Figure S19), but since their geometric parameters are very close, only one is discussed below (see the Supporting Information for average values).
In the crystal structure of 2 (Figure 3), there are two internally coordinated Li+ ions with the Li‐Li separation of 3.351(11) Å. Specifically, Li1 is sandwiched by two peripheral phenyl rings of 1 4− in an asymmetric η 6‐fashion, with the Li‐C distances ranging over 2.261(9)–2.584(9) Å (Figure 4). Li2 is inserted deeply into the internal cavity formed by the highly twisted core of 1 4− (Figure 3 b). Eight Li2‐C distances fall into the range of 2.303(9)–2.544(9) Å with four additional contacts being much longer (3.095(9)–3.150(9) Å). The short Li‐C contacts in 2 are close to those reported for internal Li+ ions in the sandwich formed by tetra‐reduced corannulene, [Li+ 5(C20H10 4−)]3−. [13] In 2, two additional ions, Li3 and Li4, are capped by four THF molecules each and remain solvent‐separated from the [Li+ 2(1 4−)]2− core, with all Li‐OTHF distances (1.914(8)–1.958(8) and 1.894(8)–1.943(8) Å, respectively) being close to those previously reported.[ 13a , 24 ]
Figure 3.
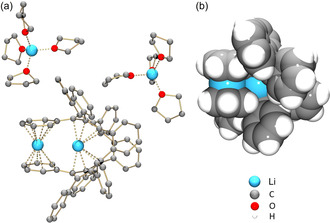
a) Crystal structure of 2, [23] ball‐and‐stick model. H‐atoms are omitted. b) Side view of [Li+ 2(1 4−)]2−, space‐filling model.
Figure 4.
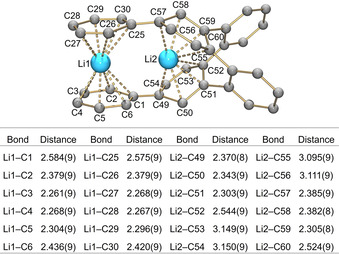
Metal coordination in [Li+ 2(1 4−)]2−, along with a table of Li‐C distances. Noncoordinated phenyl rings are omitted.
The geometry of the 1 4− anion has been analyzed by comparing the dihedral and torsion angles with those in neutral parent, 1‐THF, as both were crystallized from THF solution. Unlike 1‐THF, the central pocket in 1 4− is occupied by the rings A and E that are forced to rotate and fold in order to sandwich two internal Li+ ions (Figure 5). This is accompanied by a significant asymmetric distortion of the tetraphenylene core reflected by the change of the characteristic torsion angle between the rings A and E measured at 26.5° (vs. 138.8° in 1‐THF). Compared to 1‐THF, an increasing distortion is observed at positions β and ζ in the central 8‐membered ring of 1 4− (Table S4), with the average value increased by 6.9°.
Figure 5.
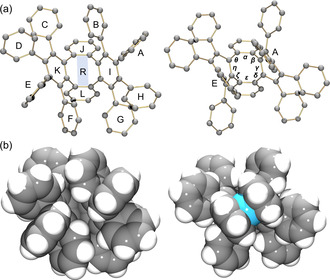
The cores of 1‐THF and [Li+ 2(1 4−)]2− in 2. a) Ball‐and‐stick models with rings and torsion angles labeled. b) Space‐filling models.
The solution behavior of 2 was investigated using variable temperature 7Li NMR spectroscopy. At room temperature (Figure S5), the only 7Li NMR resonance signal at −0.64 ppm corresponds to the solvated [Li+(THF)n] cations. Upon cooling, this peak slightly upshifts and two new signals appear in the low temperature 7Li NMR spectra (Figure S5). The 7Li resonance signal at −1.88 ppm (Figure 6) can be assigned to the sandwiched Li+ ion (Li1) while the signal at −5.08 ppm represents the deeply embedded Li+ ion (Li2). Importantly, the integration ratio of these three peaks of 2:1:1 is fully consistent with the three types of lithium ions present in the crystal structure. Remarkably, the internal lithium ions having different coordination environment can be clearly differentiated based on the characteristic 7Li NMR signal shifts. We have previously seen the tendency for the 7Li NMR signals to exhibit high‐negative values for deeply embedded Li+ ions in sandwich‐type assemblies formed by the tetra‐reduced corannulene.[ 13a , 13c , 25 ] In addition, 1H NMR spectroscopy illustrates that the 1 4− anion can be reversibly re‐oxidized back to the neutral state by exposure to air (Figure S9).
Figure 6.
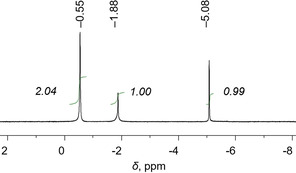
7Li NMR spectrum of 2 in THF‐d 8 at −80 °C.
The somewhat surprising formation of [Li+ 2(1 4−)]2− instead of the expected dianionic species can be rationalized by looking at the energetics of the equilibrium between these two forms, which was calculated on the DFT level of theory; solvent effects were taken into account by a conductor‐like continuum model (see the Supporting Information for details). According to our computations, the dianion 1 2− is mainly coordinated by one Li+ cation in solution. Therefore, we consider the equilibrium (neglecting the concentrations of lower/higher coordinated species):
| (1) |
The calculated free energy change ΔG° of this reaction is 0.6 kcal mol−1, i.e., the equilibrium constant K of this reaction is in the order of 1, and the actual concentration of the tetra‐reduced species will crucially depend on the concentration of the neutral parent 1. The concentration of 1, however, will be very low under the reducing reaction conditions (excess of Li metal), which “shifts” the equilibrium towards the tetra‐anion. The tetra‐anion (and dianion) enjoy significant stabilization through the coordinating Li+ cations and the solvent: both the dianion 1 2− and the tetraanion 1 4− cannot exist in vacuum as indicated by the calculated positive orbital energies of these species in vacuum (i.e, not all electrons are bound, Table S7).
Regarding the electronic properties of 1 and [Li+ 2(1 4−)]2−, it is informative to consider the nuclear independent chemical shifts (NICS) near the COT core. [26] We find clearly positive NICS values near the geometric center of the COT core of 1 indicating anti‐aromatic behavior (Figure S20). In case of [Li+ 2(1 4−)]2−, the NICS values near the COT core are of small magnitude (Figure S21) which points to a non‐aromatic character. Furthermore, natural population analysis reveals that the negative charge of 1 4− is delocalized over the [Li+ 2(1 4−)]2− scaffold with only a small fraction located at the COT core (−0.55 e) (Figures S22, S23).
In this work, the chemical reduction of OPTBCOT (1) with lithium metal has been investigated for the first time. In contrast to parent COT, four electrons are readily accepted by the π‐expanded and contorted polycyclic framework of 1. The resulting tetra‐anion, 1 4−, has been crystallized with four lithium counterions and was found to exhibit a highly nonplanar conformation, as revealed by X‐ray crystallography. This conformation is stabilized by coordination of two internally bound lithium cations, while two external cations remain solvent‐separated from the [Li+ 2(1 4−)]2− core. Notably, the low‐temperature 7Li NMR data in solution revealed characteristic signal shifts for three types of lithium ions observed in the crystal structure. Interestingly, the expected dianionic aromatic species with the planarized COT moiety has not been formed, under the experimental conditions used. The rationalization of this unexpected finding is provided by density functional theory calculations, suggesting that the formation of the tetra‐anion may be favored under the experimental reaction conditions.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial and instrumentational support of this work from the National Science Foundation (CHE‐2003411 and MRI‐1726724) is acknowledged (M.A.P.). NSF's ChemMatCARS Sector 15 is principally supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE‐1834750. The use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE‐AC02‐06CH11357. The generous funding by the Deutsche Forschungsgemeinschaft (DFG)—Project number 182849149—SFB 953 and project number 401247651—KI 1662/3‐1 is acknowledged (M.K.). Open access funding enabled and organized by Projekt DEAL.
Z. Zhou, Y. Zhu, Z. Wei, J. Bergner, C. Neiß, S. Doloczki, A. Görling, M. Kivala, M. A. Petrukhina, Angew. Chem. Int. Ed. 2021, 60, 3510.
Contributor Information
Prof. Dr. Milan Kivala, Email: milan.kivala@oci.uni-heidelberg.de.
Prof. Dr. Marina A. Petrukhina, Email: mpetrukhina@albany.edu.
References
- 1.
- 1a. Sondheimer F., Acc. Chem. Res. 1972, 5, 81–91; [Google Scholar]
- 1b. Huang N. Z., Sondheimer F., Acc. Chem. Res. 1982, 15, 96–102. [Google Scholar]
- 2.
- 2a. Kennedy R. D., Lloyd D., McNab H., J. Chem. Soc. Perkin Trans. 1 2002, 1601–1621; [Google Scholar]
- 2b. Marsella M. J., Acc. Chem. Res. 2002, 35, 944–951; [DOI] [PubMed] [Google Scholar]
- 2c. Spitler E. L., C. A. Johnson II , Haley M. M., Chem. Rev. 2006, 106, 5344–5386; [DOI] [PubMed] [Google Scholar]
- 2d. Stevenson C. D., Acc. Chem. Res. 2007, 40, 703–711; [DOI] [PubMed] [Google Scholar]
- 2e. Vogel E., Angew. Chem. Int. Ed. 2011, 50, 4278–4287; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4366–4375; [Google Scholar]
- 2f. Nishiuchi T., Iyoda M., Chem. Rec. 2015, 15, 329–346. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Willstätter R., Waser E., Ber. Dtsch. Chem. Ges. 1911, 44, 3423–3445; [Google Scholar]
- 3b. Willstätter R., Heidelberger M., Ber. Dtsch. Chem. Ges. 1913, 46, 517–527. [Google Scholar]
- 4.
- 4a. Kaufman H. S., Fankuchen I., Mark H., J. Chem. Phys. 1947, 6, 414–415; [Google Scholar]
- 4b. Kaufman H. S., Fankuchen I., Mark H., Nature 1948, 161, 165. [Google Scholar]
- 5.
- 5a. Katz T. J., J. Am. Chem. Soc. 1960, 82, 3784–3785; [Google Scholar]
- 5b. Katz T. J., J. Am. Chem. Soc. 1960, 82, 3785–3786. [Google Scholar]
- 6. Katz T. J., Strauss H. L., J. Chem. Phys. 1960, 32, 1873–1875. [Google Scholar]
- 7. Goldberg S. Z., Raymond K. N., Harmon C. A., Templeton D. H., J. Am. Chem. Soc. 1974, 96, 1348–1351. [Google Scholar]
- 8. Müllen K., Huber W., Meul T., Nakagawa M., Iyoda M., J. Am. Chem. Soc. 1982, 104, 5403–5411. [Google Scholar]
- 9. Kuwajima S., Soos Z. G., J. Am. Chem. Soc. 1987, 109, 107–113. [Google Scholar]
- 10. Monajjemi M., J. Struct. Chem. 2019, 60, 1361–1374. [Google Scholar]
- 11.
- 11a. Chase D. T., Fix A. G., Rose B. D., Weber C. D., Nobusue S., Stockwell C. E., Zakharov L. N., Lonergan M. C., Haley M. M., Angew. Chem. Int. Ed. 2011, 50, 11103–11106; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11299–11302; [Google Scholar]
- 11b. Bheemireddy S. R., Ubaldo P. C., Rose P. W., Finke A. D., Zhuang J., Wang L., Plunkett K. N., Angew. Chem. Int. Ed. 2015, 54, 15762–15766; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15988–15992; [Google Scholar]
- 11c. Wood J. D., Jellison J. L., Finke A. D., Wang L., Plunkett K. N., J. Am. Chem. Soc. 2012, 134, 15783–15789. [DOI] [PubMed] [Google Scholar]
- 12. Tsefrikas V. M., Scott L. T., Chem. Rev. 2006, 106, 4868–4884. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Zabula A. V., Filatov A. S., Spisak S. N., Rogachev A. Yu., Petrukhina M. A., Science 2011, 333, 1008–1011; [DOI] [PubMed] [Google Scholar]
- 13b. Zabula A. V., Spisak S. N., Filatov A. S., Petrukhina M. A., Angew. Chem. Int. Ed. 2012, 51, 12194–12198; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12360–12364; [Google Scholar]
- 13c. Filatov A. S., Zabula A. V., Spisak S. N., Rogachev A. Yu., Petrukhina M. A., Angew. Chem. Int. Ed. 2014, 53, 140–145; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 144–149; [Google Scholar]
- 13d. Zabula A. V., Spisak S. N., Filatov A. S., Rogachev A. Yu., Clérac R., Petrukhina M. A., Chem. Sci. 2016, 7, 1954–1961; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13e. Zabula A. V., Spisak S. N., Filatov A. S., Rogachev A. Yu., Petrukhina M. A., Acc. Chem. Res. 2018, 51, 1541–1549. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Bheemireddy S. R., Hautzinger M. P., Li T., Lee B., Plunkett K. N., J. Am. Chem. Soc. 2017, 139, 5801–5807; [DOI] [PubMed] [Google Scholar]
- 14b. Zeidell A. M., Jennings L., Fredrickson C. K., Ai Q., Dressler J. J., Zakharov L. N., Risko C., Haley M. M., Jurchescu O. D., Chem. Mater. 2019, 31, 6962–6970. [Google Scholar]
- 15. Kojima H., Bard A. J., Wong H. N. C., Sondheimer F., J. Am. Chem. Soc. 1976, 98, 5560–5565. [Google Scholar]
- 16. Hisaki I., Sonoda M., Tobe Y., Eur. J. Org. Chem. 2006, 833–847. [Google Scholar]
- 17.
- 17a. Hau C.-K., Chui S. S.-Y., Lu W., Che C.-M., Cheng P.-S., Mak T. C. W., Miao Q., Wong H. N. C., Chem. Sci. 2011, 2, 1068–1075; [Google Scholar]
- 17b. Wender P. A., Lesser A. B., Sirois L. E., Angew. Chem. Int. Ed. 2012, 51, 2736–2740; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2790–2794; [Google Scholar]
- 17c. Nishinaga T., Ohmae T., Aita K., Takase M., Iyoda M., Arai T., Kunugi Y., Chem. Commun. 2013, 49, 5354–5356. [DOI] [PubMed] [Google Scholar]
- 18. Müller M., Iyer V. S., Kübel C., Enkelmann V., Müllen K., Angew. Chem. Int. Ed. Engl. 1997, 36, 1607–1610; [Google Scholar]; Angew. Chem. 1997, 109, 1679–1682. [Google Scholar]
- 19. Rajca A., Safronov A., Rajca S., Shoemaker R., Angew. Chem. Int. Ed. Engl. 1997, 36, 488–491; [Google Scholar]; Angew. Chem. 1997, 109, 504–507. [Google Scholar]
- 20. Mackay A. L., Terrones H., Nature 1991, 352, 762.1881433 [Google Scholar]
- 21. Chen H., Miao Q., ChemPlusChem 2019, 84, 627–629. [DOI] [PubMed] [Google Scholar]
- 22. Kirschbaum T., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2020, 59, 270–274; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 276–280. [Google Scholar]
- 23. 2034668 (1-THF), 2034669 (1-NS), and 2034670 (2) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
- 24. Sumner N. J., Spisak S. N., Filatov A. S., Rogachev A. Yu., Zabula A. V., Petrukhina M. A., Organometallics 2014, 33, 2874–2878. [Google Scholar]
- 25. Filatov A. S., Spisak S. N., Zabula A. V., McNeely J., Rogachev A. Yu., Petrukhina M. A., Chem. Sci. 2015, 6, 1959–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schleyer P. v. R., Maerker C., Dransfeld A., Jiao H., van Eikema Hommes N. J. R., J. Am. Chem. Soc. 1996, 118, 6317–6318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary