

Abstract
Aims
Angiotensin II (AngII) is a potential contributor to the development of abdominal aortic aneurysm (AAA). In aortic vascular smooth muscle cells (VSMCs), exposure to AngII induces mitochondrial fission via dynamin-related protein 1 (Drp1). However, pathophysiological relevance of mitochondrial morphology in AngII-associated AAA remains unexplored. Here, we tested the hypothesis that mitochondrial fission is involved in the development of AAA.
Methods and results
Immunohistochemistry was performed on human AAA samples and revealed enhanced expression of Drp1. In C57BL6 mice treated with AngII plus β-aminopropionitrile, AAA tissue also showed an increase in Drp1 expression. A mitochondrial fission inhibitor, mdivi1, attenuated AAA size, associated aortic pathology, Drp1 protein induction, and mitochondrial fission but not hypertension in these mice. Moreover, western-blot analysis showed that induction of matrix metalloproteinase-2, which precedes the development of AAA, was blocked by mdivi1. Mdivi1 also reduced the development of AAA in apolipoprotein E-deficient mice infused with AngII. As with mdivi1, Drp1+/− mice treated with AngII plus β-aminopropionitrile showed a decrease in AAA compared to control Drp1+/+ mice. In abdominal aortic VSMCs, AngII induced phosphorylation of Drp1 and mitochondrial fission, the latter of which was attenuated with Drp1 silencing as well as mdivi1. AngII also induced vascular cell adhesion molecule-1 expression and enhanced leucocyte adhesion and mitochondrial oxygen consumption in smooth muscle cells, which were attenuated with mdivi1.
Conclusion
These data indicate that Drp1 and mitochondrial fission play salient roles in AAA development, which likely involves mitochondrial dysfunction and inflammatory activation of VSMCs.
Keywords: Abdominal aortic aneurysm, Mitochondria, Vascular smooth muscle cell, Senescence, Inflammation
Graphical Abstract
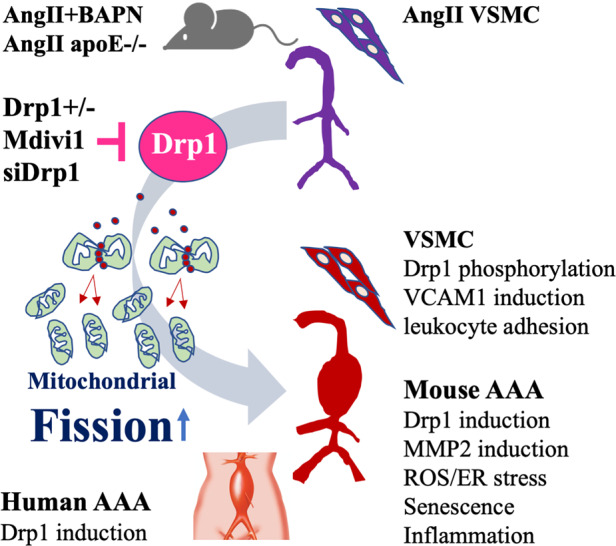
1. Introduction
Abdominal aortic aneurysm (AAA) is characterized as a localized dilation of the abdominal aorta that expands to >50% of the normal diameter with an increased risk of rupture. AAA is a significant cause of morbidity and mortality and is the 13th leading cause of death in USA. Aging, male gender, and smoking are the major risk factors developing AAA. At present, there is no pharmacological treatment to prevent AAA development and the mortality remains high (>65%) upon rupture.1,2 Increasingly, endovascular repair of AAA is utilized over open repair surgery, though, neither option is a cure for patients with AAA. However, greater than 50% of endovascular repair patients do not survive beyond 8 years.2 Therefore, there is an unmet clinical need to develop medical therapies or at the very least, a better understanding of AAA aetiology.
Human AAA pathology involves disruption of the medial vascular smooth muscle cell (VSMC) architecture, infiltration of inflammatory cells, and local activation of matrix metalloproteinases (MMPs).3 While the driving mechanism that integrates these events is still unclear, activation of the angiotensin II type-1 (AT1) receptor has been observed in animal models of AAA.4,5 Our laboratories have described a signalling pathway from AT1 receptor activation in VSMCs involving a disintegrin and metalloproteinase-17 (ADAM17), caveolin-1, and epidermal growth factor receptor (EGFR) in the formation and progression of AAA.6–8 However, other potential VSMC mechanisms may exist in the development of AAA and require further exploration.
Mitochondrial dysfunction has been consistently linked to cardiovascular disorders.9 Mitochondria are dynamic organelles which undergo fission and fusion events to maintain their homoeostasis.10 Importantly, a shift towards mitochondrial fission has been observed in variety of cardiovascular disease settings.11 Mitochondrial fission is mainly driven by the GTPase dynamin-related protein 1 (Drp1) which upon activation, translocates to the mitochondrion from the cytosol, associates with its receptors, and oligomerizes to encircle and divide the organelle.10,12 In cultured rat aortic VSMCs, exposure to angiotensin II (AngII) induces mitochondrial fission.13 Furthermore, AngII-induced mitochondrial fission, reactive oxygen species production, reduced membrane potential, and enhanced VSMC proliferation were prevented by the putative Drp1 inhibitor, mdivi1.13 In addition, Drp1 activation has been implicated in endothelial dysfunction,14 inflammation,15 and stress-induced senescence.16 However, pathophysiological relevance of Drp1 and mitochondrial dynamics in aneurysmal diseases have not been fully explored.
A widely utilized murine model of AAA infuses AngII in male apolipoprotein E (apoE) or LDL receptor −/− mice fed a high-fat diet. These models lead to acceleration of atherosclerosis together with high incidence of AAA and recapitulate many elements of human AAA pathology4 including premature vascular senescence.17 In a non-atherogenic model of AAA involving AngII infusion together with consumption of a lysyl oxidase inhibitor, β-aminopropionitrile (BAPN) in C57BL/6 mice on a standard diet,18 we have reported a key role for EGFR in development and rupture of AAA.6,7 This process likely involves AngII-induced EGFR transactivation and subsequent extracellular signal-regulated kinase (ERK) activation in VSMCs.5 Importantly, Drp1 phosphorylation by ERK is one of the key post-translational mechanisms by which Drp1 mediates mitochondrial fission.19 Accordingly, using these two murine models of AAA together with human AAA samples, we sought to test the hypothesis that enhanced mitochondrial fission via Drp1 is crucial to the development of AAA pathology.
2. Methods
An expanded methods section can be found in Supplementary material online, Appendix A. Key reagent and resources were described in Supplementary material online, Table S1.
2.1 Human subjects and specimens
Written informed consent was obtained from all subjects before participation and human surgical AAA specimens were utilized under approvals of Temple University (No. 21973) and Okayama University (No. 1810-030) Institutional Review Boards in accordance with the Helsinki Declaration. Formalin-fixed control aortas were obtained from Advanced Tissue Services and with assurances that the donors have no medical history or diagnosed disease. Attempts were made to have age-matched controls.
2.2 Animal protocols
All animal procedures were performed with prior approval of the Temple University Institutional Animal Care and Use Committee (No. 4625) and in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals. For all AngII infusion procedures, mice were anesthetized by isoflurane inhalation (induction dose: 3–5%, maintenance dose: 1–2%) and osmotic mini-pumps containing AngII were implanted subcutaneously into the dorsum of the mouse. About 8–10-week-old male C57BL/6J mice (stock 000664) and 10–12-week-old male apoE−/− (B6.129P2-Apoetm1Unc/J stock 002052) were obtained from the Jackson Laboratory. About 8–10-week-old male Drp1+/− mice (C57BL/6-129), generated as previously described20 were backcrossed three times with C57BL/6 mice. Animal genotypes were determined using a Phusion High Fidelity Polymerase kit (New England BioLabs, No. M0530L). To differentiate Drp1+/+ from Drp1+/− animals, the Eurofin Genomics primers D1 (5′-CACTGAGAGCTCTATATGTAGGC-3′), D3 (5′-ACCAAAGTAAGGAATAGCTGTTG-3′), and D5 (5′-GAGTACCTAAAGTGGACAAGAGGTCC-3′) were used. Littermate Drp1+/+ mice were used as controls. Standard laboratory diet and water were available ad libitum.
In our first AAA model, AngII was delivered at a rate of 1 μg/kg/min for 4 weeks via subcutaneous infusion (Alzet micro-osmotic mini pump 1004) in male C57BL/6 mice. In addition, mice were received BAPN at a dose of 0.625 mg/mL which was added to the drinking water during the first 2 weeks of AngII infusion (we identified this combination as an AAA model with ∼12% chance of rupture). Control C57BL/6 mice were sham-operated. In the treatment group, mdivi1 solubilized in 10 μL DMSO at 25 mg/kg or vehicle (10 μL DMSO) was administered with 90 μL corn oil by intraperitoneal (i.p.) injection, three times per week. For animals that died before the completion of the study, necropsy was performed when possible to define the causes of death. The presence of haematoma in thoracic or abdominal cavity indicated thoracic aorta or abdominal aorta rupture as a cause of death. In our second model, apoE−/− mice kept on a standard diet and infused with AngII (1 µg/kg/min) for 4 weeks along with the tri-weekly mdivi1 or vehicle as in the first model. Third, we used Drp1+/− mice to complement our pharmacological findings and examine the specificity to which Drp1 contributes to aneurysm development. About 8–10-week-old male Drp1+/− and control Drp1+/+ mice were infused with AngII (1 µg/kg/min) for 4 weeks and received BAPN in drinking water at a dose of 1 mg/mL as a means to enhance AAA developments, as demonstrated in our past work.18 After study, all animals were anaesthetized by isoflurane inhalation (2–5%) and then euthanized by exsanguination and bilateral thoracotomy.
2.3 Rat abdominal aortic VSMCs
Abdominal aortic VSMCs were isolated from 12-week-old male Sprague–Dawley rat (300–350 g Charles River Laboratories). VSMCs were pooled from two rats and renewed every 2 months. A total of 24 rats were used for the in vitro experiments. To obtain VSMCs, rats were anesthetized with 5% isoflurane and euthanized by exsanguination and bilateral thoracotomy. Abdominal aortas were then collected by cutting the aorta below the level of the diaphragm. VSMCs were isolated by explant method and cultured in DMEM and 10% FBS as previously described for thoracic VSMCs.21 VSMCs were used between passage 3–9 in all experiments.
2.4 Statistical analysis
Data are presented as mean ± SEM. Unless otherwise stated, comparisons were performed via unpaired Student’s t-test for two groups, or via one-way or two-way (only for internal diameter analysis in vivo) ANOVA with the post hoc Tukey (in vitro assays) and Bonferroni (in vivo assays) for multiple groups or log-rank (Mantel–Cox) test (Kaplan–Meier survival curves) using Prism software (GraphPad, San Diego, CA, USA). Differences were considered statistically significant at P < 0.05.
3. Results
3.1 Drp1 expression is enhanced in human AAA
To investigate potential relevance of the mitochondrial fission inducer Drp1 in human AAA, immunohistochemical analysis was performed on human AAA specimen and control aortas. While semiquantitative, enhanced Drp1 protein expression was suggested in all human AAA samples compared with control aortas. IgG served as antibody control for specificity of Drp1 labelling (Figure 1 and see Supplementary material online, Figure S1 and Table S2).
Figure 1.
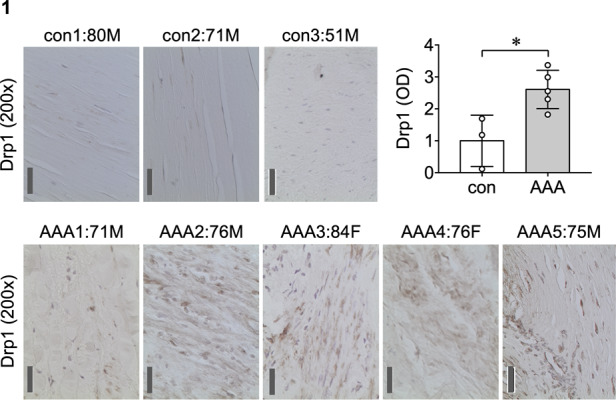
Drp1 expression was increased in human AAA samples. Immunohistochemical staining of Drp1 was demonstrated in human AAA samples and control abdominal aortas. Control abdominal aorta: con1 80 M (80-year-old male), con2 71 M, and con3 51 M. AAA1 71 M, AAA2 76 M, AAA3 84 F (84-year-old female), AAA4 76 F, and AAA5 75 M (see additional data with control IgG staining in Supplementary material online, Figure S1 and patient characteristics in Supplementary material online, Table S2). Scale bar; 50 μm. Data are expressed as mean±SEM. Statistical significance was determined by a two-tailed unpaired Student’s t-test. *P < 0.05 control n = 3 vs. AAA n = 5.
3.2 Drp1 inhibition protects against AAA development in mice
Next, we examined the effect of a mitochondrial fission inhibitor, mdivi122 in the AngII plus BAPN model of AAA (see Supplementary material online, Figure S2A). Mice treated with mdivi1 showed attenuation of AAA development assessed by measurement of external and internal diameters of the abdominal aortas as well as by histology (Figure 2A–C). Van Giesson staining further indicates disruption of elastic lamina structures in mouse AAA which was prevented with mdivi1 (see Supplementary material online, Figure S2B). In contrast, hypertension observed in mice treated with AngII plus BAPN was unaltered by mdivi1 (see Supplementary material online, Figure S2C). There was no difference observed in heart rate among the groups (see Supplementary material online, Figure S2D). In addition, mdiv1 completely protected against fatal aneurysm rupture which occurs at a rate of ∼12% with the current dose of BAPN (see Supplementary material online, Figure S2E).
Figure 2.

Mitochondrial fission inhibitor-1 mdivi1 decreased AAA formation induced by AngII plus BAPN. About 8–10-week-old C57BL/6J male mice were treated with AngII (1 μg/kg/min subcutaneous infusion) for 4 weeks with BAPN in drinking water (0.625 g/L) for the first 2 weeks (AII+B) with mdivi1 (mdiv) injections (25 mg/kg i.p. 3 times/week) or vehicle (vehi) injections (10 μL DMSO plus 90 μL corn oil i.p. 3 times/week) for 4 weeks. Control mice were sham-operated. (A) Representative photographs of the mouse aortas were demonstrated. Scale bar is 3 mm. Maximum external diameter/D of the aortas was determined in the aorta picture after aortic isolation and cleaning, sham n = 6, vehi AII+B n = 16, mdiv AII+B n = 12. Data are expressed as mean±SEM. Statistical significance was determined by one-way ANOVA with Bonferroni post hoc test. ****P < 0.0001. (B) Maximal aortic internal diameter/D was determined by echo analysis using high-resolution 2D imaging (B mode) with high-frequency ultrasound (VisualSonics Vevo2100) on Days 0 (0 W), 14 (2 W), 21 (3 W), and 28 (4 W) of the study, sham n = 5, vehi AII+B n = 12, mdiv AII+B n = 10. Data are expressed as mean±SEM. Statistical significance was determined by two-way ANOVA with Bonferroni post hoc test. *P < 0.05, ***P < 0.001, ****P < 0.0001. (C) Histological assessment was done by Masson’s Trichrome staining following euthanasia of animals at 28 days, dissection of abdominal aorta and paraffin embedding and sectioning (5 μm thick), n = 4. Scale bar; 200 μm (40×), 50 μm (200×).
In a second model of AAA, AngII was infused in apoE−/− mice fed a normal diet. Consistent with the AngII plus BAPN model, treatment of mdivi1 significantly reduced AAA development in this model (see Supplementary material online, Figure S3). In addition, no rupture was observed in any group from this cohort of mice.
Although mdivi1 is widely considered to be a Drp1 inhibitor, it may have off-target effects independent of Drp1.23,24 Thus, to obtain direct evidence for Drp1 in AAA development, we used Drp1 heterozygous (Drp1+/−) mice which are viable with no obvious phenotype in contrast to Drp1 null (Drp1−/−) mice which are embryonic lethal.20 While control Drp1+/+ mice treated with AngII plus BAPN developed AAA, there was partial, yet significant, reduction of AAA development in Drp1+/− mice (Figure 3). The incomplete prevention of AAA seen in Drp1+/− mice may be due to the activation of endogenous Drp1, which appears to maintain mitochondrial homoeostasis under basal conditions in Drp1+/− mice.25
Figure 3.

Drp1 heterozygous mice had attenuated AAA development in an AngII plus BAPN model. About 8–10-week-old Drp1 heterozygous mice (Drp1+/−) and control Drp1+/+ mice were treated with AngII infusion (1 μg/kg/min) for 4 weeks with BAPN in drinking water (0.625 g/L) for the first 2 weeks (AII+B) or sham-operated. (A) Maximum external diameter/D of the aortas was determined, Drp1+/+ sham n = 4, Drp1+/+ AII+B n = 6, Drp1+/− sham n = 4, Drp1+/− AII+B n = 5. Scale bar is 3 mm. Data are expressed as mean±SEM. Statistical significance was determined by one-way ANOVA with Bonferroni post hoc test. *P < 0.05, **P < 0.01. (B) Maximal aortic internal diameter/D was determined by echo analysis on Days 0 (0 W), 14 (2 W), 21 (3 W), and 28 (4 W) of the study, Drp1+/+ sham n = 4, Drp1+/+ AII+B n = 6, Drp1+/− sham n = 4, Drp1+/− AII+B n = 5. Data are expressed as mean ± SEM. Statistical significance was determined by two-way ANOVA with Bonferroni post hoc test. *P < 0.05, **P < 0.01, ****P < 0.0001. (C) Histological assessment was done by Masson’s Trichrome staining following euthanasia of animals at 28 days, dissection of abdominal aorta and paraffin embedding and sectioning (5 μm thick), n = 4. Scale bar; 200 μm (40×), 50 μm (200×).
To examine the regulation of Drp1 in AAA, Drp1 expression in mouse abdominal aortas was evaluated with immunohistochemistry. Similar to our observations in human AAA samples, AngII plus BAPN treatment increased Drp1 expression in abdominal aorta of C57BL6 mice, which was not observed with mdivi1 treatment (Figure 4A). Moreover, Drp1 protein expression in abdominal aorta was enhanced by AngII plus BAPN treatment in Drp1+/+ mice, which was attenuated in Drp1+/− mice (Figure 4B). In addition, slightly lower expression of vascular Drp1 was observed in sham-operated Drp1+/− mice. Limited silencing of Drp1 protein in Drp1+/− mice is a known issue of these mice as reported previously in the brain25 which could be due to compensation. Upon AAA induction, the compensation may no longer be effective to meet the demand for Drp1 induction associated with AAA.
Figure 4.

Drp1 expression was decreased in mdivi1-treated C57BL6 mice and Drp1 heterozygous mice following AngII plus BAPN administration. (A) Male C57BL/5 mice were treated as in Figure 2. sham n = 3, vehi AII+B n = 4, mdiv AII+B n = 3. (B) Mice were treated as in Figure 3. Drp1+/+ sham n = 3, Drp1+/+ AII+B n = 3, Drp1+/− sham n = 3, Drp1+/− AII+B n = 3. Representative immunostaining of Drp1 or isotype-matched IgG staining was shown. Scale bar is 50 μm. Data are expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA with Bonferroni post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001.
3.3 Protection of AAA by Drp1 inhibition is associated with reduced stress responses including inflammatory leucocyte infiltration and senescence
Local inflammation plays a critical role in AAA development.3 To evaluate inflammation associated with AAA, leucocyte infiltration in abdominal aortas was assessed by immunostaining of CD45. CD45 positive cell numbers were increased in mouse AAAs induced by AngII plus BAPN, which were attenuated with mdivi1 co-treatment (Figure 5A). CD45 positive cells were also reduced in aortas from Drp1+/− mice treated with AngII plus BAPN compared to the control mice (Figure 5B). To determine the type of immune cells that are present in mouse AAA, F4/80 staining was utilized to detect macrophages. F4/80 positive cells were detected in AAA, which was reduced with the mdivi1 co-treatment (see Supplementary material online, Figure S4). To investigate potential downstream mechanism(s) through which Drp1 contributes to aortic inflammation, oxidative stress and ER stress were assessed with 4-hydorxynonenal and KDEL staining, respectively. As we have reported,6–8 expression of these markers were enhanced in mice treated with AngII plus BAPN whereas administration of mdivi1 attenuated these responses (see Supplementary material online, Figure S5).
Figure 5.

CD45 positive cells were decreased in C57BL6 mice treated with mdivi1 or Drp1 heterozygous mice following AngII plus BAPN administration. (A) Mice were treated as in Figure 2. sham n = 4, vehi AII+B n = 4, mdiv AII+B n = 4. (B) Mice were treated as in Figure 3. Drp1+/+ sham n = 3, Drp1+/+ AII+B n = 3, Drp1+/− sham n = 3, Drp1+/− AII+B n = 3. Representative immunostaining of CD45 or isotype-matched IgG staining in suprarenal abdominal aortic tissue was shown. Scale bar is 50 μm. Data are expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA with Bonferroni post hoc test. *P < 0.05, **P < 0.01, and ***P < 0.001.
VSMC senescence has also been observed in AAA and represents a potential mechanism for inflammation and medial degeneration.17,26 We found increased β-galactosidase activity in AAA segments of mice treated with AngII and BAPN, which was attenuated with mdivi1 treatment (see Supplementary material online, Figure S6A). The senescent phenotype seen in the mouse AAA samples and its attenuation by mdivi1 were confirmed with detection of lipofuscin, a marker for age-related lysosomal dysfunction and senescence27 (see Supplementary material online, Figure S6B). Senescence cells also show enhanced expression of a cell cycle suppressor p16Ink4a.28 Here, we found a trend of enhanced p16Ink4a immunoreactivity in AAA, which was not observed in abdominal aortas co-treated with mdivi1 (see Supplementary material online, Figure S6C). Consistent with our mouse model, human AAA samples showed higher staining of lipofuscin and p16Ink4a compared to control aortas (see Supplementary material online, Figure S7).
To test if the improvements in the AAA pathophysiology by mdivi1 are indeed due to the inhibition of salient early signalling events, abdominal aortas were harvested at Day 7 of the AAA induction protocol, a time point before vessel wall expansion is observed. Indeed, mdivi1 treatment prevented the increase in protein expression of MMP-2, an early indicator of AAA pathology29 and a similar trend was observed with expression of vascular cell adhesion molecule-1 (VCAM-1) (see Supplementary material online, Figure S8A and B). We also confirmed that mdivi1 attenuated enhancement of the gelatinase (MMP-2 and MMP-9) activity in abdominal aortas in mice treated with AngII and BAPN for 7 days (see Supplementary material online, Figure S8C).
3.4 Drp1 mediates mitochondrial fission upon AngII exposure
It has been demonstrated that VSMCs play a critical role in formation of AAA in mouse models infused with AngII.6,17,30–32 To investigate the role of Drp1 and mitochondrial fission in promoting a VSMC phenotype associated with AAA, rat abdominal aortic VSMCs were stimulated with AngII. AngII induced mitochondrial fission, which was attenuated with Drp1 silencing (Figure 6A and B). Mdivi1 pre-treatment also attenuated AngII-induced mitochondrial fission in VSMCs (Figure 6C). To evaluate the effect of mdivi1 on mitochondrial morphology in vivo, medial layers of the mouse aortas were analysed by electron microscopy. Through morphometric analysis of mitochondrial ultrastructure, we observed decreased aspect ratio of mitochondria in medial layers of AAA tissues, which was attenuated by mdivi1, a finding consistent with prevention of mitochondrial fission (Figure 6D).
Figure 6.

AngII-induced mitochondrial fission via Drp1 activation. (A) Serum-starved rat abdominal aortic VSMCs infected with adenovirus encoding Drp1 siRNA (siDrp1) or control non-silencing siRNA (siCon) at 100 moi (multiplicity of infection) and the mitochondria-labelling adenovirus, mito-dsRed (10 moi) for 48 h were stimulated with 100 nM AngII (AII) for 2 h. (−) basal control. Mitochondrial fragmentation count (MFC) was calculated using ImageJ analysis. MFC; mitochondria count×100/total mitochondria area (pixcels) per cell was measured. Scale bar is 15 μm and the box is 4× zoomed. Data (mean ± SEM) were from two independent experiments with HPF images, n = 10. Statistical significance was determined by one-way ANOVA with Tukey post hoc test. *P < 0.05 and ****P < 0.0001. (B) Confirmation of Drp1 knockdown in siDrp or siCon adenovirus infected rat aortic abdominal VSMCs by immunoblotting. The adenoviruses (100 moi) were infected for 48 h. For quantification, densitometry analysis was performed and normalized with corresponding GAPDH density. Data are expressed as mean ± SEM. Statistical significance was determined by a two-tailed unpaired Student’s t-test. *P < 0.05 control siCon n = 4 vs. siDrp1 n = 4. (C) Serum-starved rat abdominal aortic VSMCs were pre-treated for 60 min with mdivi1 (mdiv, 20 μM) or its vehicle (0.1% DMSO final) then stimulated with 100 nM AngII (AII) for 2 h. (−) basal control. Mitochondria was stained with MitoTracker Red. MFC was measured. Data were from three independent experiments with HPF images, DMSO—n = 10, DMSO AII n = 15, mdiv—n = 11, and mdiv AII n = 10. Data are expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA with Tukey post hoc test. *P < 0.05. (D) Mice were treated as in Figure 2. Aortic mitochondrial image analysis with transmission electron microscopy. Dissected abdominal aortas from each treatment group (2 sham, 3 AII+B vehi, and 3 AII+B mdiv) were incubated in glutaraldehyde fixative and the mitochondria in the medial layer were imaged by transmission electron microscopy. Scale bar is 200 nm. Aspect ratio was calculated as long axis/short axis of mitochondria by ImageJ analysis. Counted mitochondria numbers in each group were sham n = 83, AII+B vehi n = 164, and 3 AII+B mdiv n = 159. Data are expressed as mean ± SEM. Statistical significance was determined by one-way ANOVA with Bonferroni post hoc test. **P < 0.01 and ****P < 0.0001.
Drp1 possesses two important phosphorylation sites, where Ser616 phosphorylation is stimulatory and Ser637 phosphorylation is inhibitory.33 Here, we evaluated Drp1 phosphorylation status in response to AngII by Phos-tag SDS–PAGE, which dissociates a phosphorylated protein which migrates slower.34 In addition, we tested whether ERK, as a kinase known to directly phosphorylate Drp1 at Ser616,19 played a role in Drp1 phosphorylation. VSMCs were pre-treated with an ERK inhibitor U0126 or its vehicle, then stimulated by AngII. We observed that AngII increased Drp1 upper band, which was attenuated by the ERK inhibitor (see Supplementary material online, Figure S9A). We confirmed that AngII stimulated Drp1 Ser616 phosphorylation as well as ERK Thr202/Tyr204 phosphorylation, which were attenuated by the ERK inhibitor (see Supplementary material online, Figure S9B). Thus, the ERK inhibitor prevented mitochondrial fission induced by AngII in VSMCs (see Supplementary material online, Figure S9C).
3.5 Mdivi1 attenuated inflammatory phenotype and enhanced oxygen consumption in abdominal aortic VSMCs stimulated with AngII
To examine whether mitochondrial fission mediates inflammatory responses in vitro, VSMCs were stimulated with AngII with or without pre-treatment of mdivi1. AngII-induced inflammatory responses assessed by VCAM-1 expression and THP-1 adhesion were prevented by the mdivi1 pre-treatment in abdominal aortic VSMCs (see Supplementary material online, Figure S10). Enhanced mitochondrial oxygen consumption of VSMCs has been implicated in senescence and senescence-associated inflammatory phenotype in VSMCs.35 To evaluate the potential functional alteration of mitochondria in response to AngII-induced mitochondrial fission, cellular oxygen consumption rate (OCR) was evaluated by Seahorse Extracellular Flux assay in VSMCs with or without treatment of mdivi1. AngII increased basal OCR, maximal respiration, and ATP-linked OCR, which were attenuated with mdivi1 (see Supplementary material online, Figure S11A and B). In line with the observation that mdivi1 did not suppress basal OCR, mdivi1 did not alter basal complex I activity (see Supplementary material online, Figure S11C). Taken together, these data suggest that mitochondrial respiratory stimulation via mitochondrial fission potentially contributes to the VSMC inflammatory phenotype induced by AngII.
4. Discussion
In the present study, we provide evidence that both mdivi1 and Drp1 heterozygous mice are effective in attenuating AAA development in two distinct murine models. Taken together with our findings in humans and abdominal aortic VSMCs, we speculate that the underlying mechanism involves up-regulation of Drp1-mediated mitochondrial fission which results in pro-inflammatory VSMC phenotype leading to AAA progression. Thus, Drp1 can be considered a potential therapeutic target against AAA development. These findings expand our understanding of the AAA pathophysiology by bringing into focus Drp1 and the mitochondria as key participatory elements in aneurysmal aortic diseases.
Alterations in mitochondrial dynamics, including those involving Drp1, have been implicated in several vascular pathophysiological processes such as endothelial dysfunction,14 constriction of mesenteric arteries,36 neointimal hyperplasia,37 calcification,38 and atherosclerosis.39 There is also accumulating evidence indicating that disruption of mitochondrial functions is associated with AAA in both mouse models and humans. These alterations include enhanced mitochondrial reactive oxygen species production,40–42 reduced mitochondrial biogenesis,42,43 and mitochondria-initiated apoptosis.44 Moreover, deletion of mitochondrial uncoupling protein-2 in apoE−/− mice enhanced AAA phenotype induced by AngII independent of hypertension.45 Our findings along with these studies support the potential contribution of mitochondrial dynamics in AAA aetiology.
In humans, hypertension is frequently associated with AAA and is a risk factor for rupture. However, there is limited evidence for hypertension as a potential risk factor for AAA incidence compared with smoking.2,46 A prior study showed that hydralazine did not prevent AAA in hypercholesterolaemic mice with AngII infusion while blood pressure was normalized.47 Several studies have also shown that the hypertension is not necessary for the AAA development in mice.48–50 In this study, we found that while AAA development was prevented by mdivi1, it did not alter hypertension in mice treated with AngII plus BAPN. These data are consistent with our past findings that signalling elements downstream of the AT1 receptors, namely caveolin-1,7 EGFR,8 and VSMC ADAM17,6 are involved in AngII plus BAPN model of AAA with no alteration of blood pressure. It is likely that these upstream signalling components regulate Drp1-dependent mitochondrial fission and AAA induced by AngII since each element has the ability to regulate ERK,5 which we found to be upstream of AngII-mediated Drp1 Ser616 phosphorylation and mitochondrial fission in VSMCs. In addition, mdivi1 has been reported to protect against cardiac hypertrophy and fibrosis without alteration of blood pressure in Dahl-salt sensitive rats.51 Interestingly, this protection was associated with prevention of Drp1 induction and oxidative stress, which were recapitulated in the aortic vasculature in the present study.
VSMCs are considered as one of the key cell types that contribute to AAA pathophysiology.3 Accordingly, accumulating data suggest several smooth muscle-specific processes that contribute to AAA development in mice.6,17,30–32 Moreover, several phenotypic modulations have also been reported in VSMCs which contribute to AAA pathology,52,53 including MMP secretion, senescence, susceptibilities to apoptosis, and pro-inflammatory responses. Our studies with mdivi1 show attenuation of MMP-2 induction in the aorta preceding AAA progression as well as VCAM-1 induction and leucocyte adhesion in VSMCs suggesting that mdivi1 attenuates the induction of VSMC dysfunctional phenotype promoted by AAA risk factors. Although VSMCs are highly implicated in our study, we are aware that additional experiments are needed to selectively narrow down our observations to a specific cell type such as with conditional knockdown animals. For instance, macrophage activation and shift to the M1 phenotype have been strongly implicated in AAA development with contributions to inflammation and ECM remodelling.54 Our data thus indicate that Drp1 and mitochondrial fission in macrophages may participate in AAA in this model. T cells have also been reported to contribute to AAA development55 and T-cell function appears to require Drp1.56 In contrast, B2 cells were known to suppress AAA in the mouse elastase model,57 whereas total B-cell depletion is protective in a mouse AngII model.58 Mouse AT1 receptors consist of AT1a and AT1b, both of which are expressed in the abdominal aorta. However, silencing of smooth muscle or endothelial AT1a59 as well as systemic deletion of AT1b60 did not alter AAA development in LDL receptor −/− mice infused with AngII. It is possible that there might be compensation by either receptors or the distinct cell type(s) to maintain the pathology. Thus, whether Drp1 and mitochondrial fission contribute to AAA progression via a mechanism requiring VSMCs, endothelial cells or a type of immune cells remains to be studied.
In addition to the well-studied role of VSMC oxidative stress in AAA,3 the role of ER stress in VSMCs has been increasingly investigated in human and murine models of AAA, which likely involves the AngII-EGFR transactivation cascade.6–8,42 It is also important to note that attenuation of protein misfolding by chemical chaperone protected against the formation of AAA in an AngII-dependent model.61 Consistent with these concepts, we found mdivi1 attenuated oxidative stress and ER stress in the AAA model.
We also demonstrated that mdivi1 treatment reduced markers for senescence in the AAA model and that the senescence marker, p16Ink4a, was enriched in human AAA samples. While senescent cells usually possess fused mitochondria,62 literature suggests that Drp1 and mitochondrial fission can mediate senescence in certain conditions.16,63 Our findings are in line with those reported in human VSMCs cultured from AAA lesions which were prone to replicative senescence26 and demonstrated telomere attrition.64 Overall, these findings support the view point that enhanced mitochondrial fission is part of a potential molecular mechanism by which enhanced renin–angiotensin system accelerates cellular stress and senescence in age-related aortic pathology.65
Using VSMCs from abdominal aorta, we observed that AngII-induced mitochondrial fission was associated with enhanced oxygen consumption, which was attenuated with mdivi1 treatment. Increase in oxygen consumption in thoracic VSMCs stimulated by AngII has been linked to oxidative stress.66 In addition, increased basal oxygen consumption was associated with mitochondrial fission in VSMCs from mouse thoracic aortas incubated with platelet-derived growth factor.37 However, further investigation is necessary to determine whether VSMC metabolic adaptation or maladaptation in response to mitochondrial fission has any contribution to AAA.
While a multitude of studies supports mdivi1 as a mitochondrial fission inhibitor,24 a recent study demonstrated that mdiv1 inhibited mitochondrial complex I activity in vitro at high concentrations (50–100 μM).23 Although we used a lower concentration (20 μM) which did not alter baseline complex I activity in vitro and confirmed AAA protection with Drp1 hetero-deficient mice, it is possible that complex I inhibition by mdivi1 may have contributed to the protective effects against AAA in vivo, which is a limitation of this study. However, we believe that this concern is offset somewhat given the similarity in experimental results from the Drp1+/− mice and the mdiv1 approaches. In addition, it has been reported that mdivi1 and genetic deficiency of Drp1 inhibit mitophagy.67 Thus, the potential involvement of mitophagy in AAA remains to be explored. Another limitation is a lack of translational studies such as to test the effect of mdivi1 on established or developing AAA in mice. Lastly, while human AAA data consistently support our mouse AAA findings, our data are limited to dissecting models of AAA since AngII and BAPN were utilized.68 Therefore, it is interesting to further test if our findings are applicable to other non-dissecting models of AAA.
In conclusion, pharmacological as well as genetic inhibition of Drp1 attenuated AAA development in mouse models. These data together with our human and in vitro findings suggest a potential role for mitochondrial fission in promotion of pro-inflammatory phenotype in abdominal aortic VSMCs leading to AAA development.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Authors’ contributions
H.A.C., S.C., K.J.P., K.O., and T.K. performed experiments and contributed to the discussion. E.T.C., S.K., H.A.U., and N.O. provided expertise in human AAA and contributed to the data interpretation and discussion. H.A.C., R.S., V.R., and S.E. conceived, designed, and co-ordinated the research plan and wrote/edited the paper.
Supplementary Material
Acknowledgement
We thank Dr Hiromi Sesaki for gifting us the Drp1+/− mice.
Conflict of interest: none declared.
Funding
This work was supported by the National Institutes of Health Grants (RO1HL128324 to S.E. and V.R., RO1HL133248 to S.E., RO1DK111042 to R.S. and S.E., RO1NS109382 to S.E., and F30HL146006 to H.A.C.).
Time for primary review: 18 days
Translational perspective
Mitochondrial fission/fusion regulation is critical to maintain mitochondrial homoeostasis. A shift towards mitochondrial fission is associated with a variety of cardiovascular diseases. A GTPase, Drp1 is known to mediate mitochondrial fission and we found evidence of Drp1 dysregulation in human and mouse abdominal aortic aneurysms (AAA). The results from the present study demonstrate that pharmacological as well as genetic inhibition of mitochondrial fission via Drp1 prevents AAA formation and vascular smooth muscle aneurysmal phenotype in mouse models of the disease. These data indicate that intervening in this pathway may have therapeutic potential for treating AAA.
References
- 1. Sakalihasan N, Limet R, Defawe OD. Abdominal aortic aneurysm. Lancet 2005;365:1577–1589. [DOI] [PubMed] [Google Scholar]
- 2. Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol 2011;8:92–102. [DOI] [PubMed] [Google Scholar]
- 3. Quintana RA, Taylor WR. Cellular mechanisms of aortic aneurysm formation. Circ Res 2019;124:607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu H, Rateri DL, Bruemmer D, Cassis LA, Daugherty A. Involvement of the renin-angiotensin system in abdominal and thoracic aortic aneurysms. Clin Sci (Lond) 2012;123:531–543. [DOI] [PubMed] [Google Scholar]
- 5. Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, Scalia R, Eguchi S. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev 2018;98:1627–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kawai T, Takayanagi T, Forrester SJ, Preston KJ, Obama T, Tsuji T, Kobayashi T, Boyer MJ, Cooper HA, Kwok HF, Hashimoto T, Scalia R, Rizzo V, Eguchi S. Vascular ADAM17 (a Disintegrin and Metalloproteinase Domain 17) is required for angiotensin II/beta-aminopropionitrile-induced abdominal aortic aneurysm. Hypertension 2017;70:959–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takayanagi T, Crawford KJ, Kobayashi T, Obama T, Tsuji T, Elliott KJ, Hashimoto T, Rizzo V, Eguchi S. Caveolin 1 is critical for abdominal aortic aneurysm formation induced by angiotensin II and inhibition of lysyl oxidase. Clin Sci (Lond) 2014;126:785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Obama T, Tsuji T, Kobayashi T, Fukuda Y, Takayanagi T, Taro Y, Kawai T, Forrester SJ, Elliott KJ, Choi E, Daugherty A, Rizzo V, Eguchi S. Epidermal growth factor receptor inhibitor protects against abdominal aortic aneurysm in a mouse model. Clin Sci (Lond) 2015;128:559–565. [DOI] [PubMed] [Google Scholar]
- 9. Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW II, Kitsis RN, Otsu K, Ping P, Rizzuto R, Sack MN, Wallace D, Youle RJ. Mitochondrial function, biology, and role in disease: a scientific statement from the American Heart Association. Circ Res 2016;118:1960–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 2012;46:265–287. [DOI] [PubMed] [Google Scholar]
- 11. Hall AR, Burke N, Dongworth RK, Hausenloy DJ. Mitochondrial fusion and fission proteins: novel therapeutic targets for combating cardiovascular disease. Br J Pharmacol 2014;171:1890–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sesaki H, Adachi Y, Kageyama Y, Itoh K, Iijima M. In vivo functions of Drp1: lessons learned from yeast genetics and mouse knockouts. Biochim Biophys Acta 2014;1842:1179–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lim S, Lee SY, Seo HH, Ham O, Lee C, Park JH, Lee J, Seung M, Yun I, Han SM, Lee S, Choi E, Hwang KC. Regulation of mitochondrial morphology by positive feedback interaction between PKCdelta and Drp1 in vascular smooth muscle cell. J Cell Biochem 2015;116:648–660. [DOI] [PubMed] [Google Scholar]
- 14. Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess MA, Levit A, Kim B, Hartman ML, Joseph L, Shirihai OS, Vita JA. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011;124:444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kang YJ, Bang BR, Han KH, Hong L, Shim EJ, Ma J, Lerner RA, Otsuka M. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat Commun 2015;6:8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim YM, Youn SW, Sudhahar V, Das A, Chandhri R, Cuervo Grajal H, Kweon J, Leanhart S, He L, Toth PT, Kitajewski J, Rehman J, Yoon Y, Cho J, Fukai T, Ushio-Fukai M. Redox regulation of mitochondrial fission protein Drp1 by protein disulfide isomerase limits endothelial senescence. Cell Rep 2018;23:3565–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen HZ, Wang F, Gao P, Pei JF, Liu Y, Xu TT, Tang X, Fu WY, Lu J, Yan YF, Wang XM, Han L, Zhang ZQ, Zhang R, Zou MH, Liu DP. Age-associated sirtuin 1 reduction in vascular smooth muscle links vascular senescence and inflammation to abdominal aortic aneurysm. Circ Res 2016;119:1076–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kanematsu Y, Kanematsu M, Kurihara C, Tsou TL, Nuki Y, Liang EI, Makino H, Hashimoto T. Pharmacologically induced thoracic and abdominal aortic aneurysms in mice. Hypertension 2010;55:1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell 2015;57:537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol 2009;186:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Forrester SJ, Elliott KJ, Kawai T, Obama T, Boyer MJ, Preston KJ, Yan Z, Eguchi S, Rizzo V. Caveolin-1 deletion prevents hypertensive vascular remodeling induced by angiotensin II. Hypertension 2017;69:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 2008;14:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, Francis TC, Kennedy NW, Picton LK, Kumar T, Uppuluri S, Miller AM, Itoh K, Karbowski M, Sesaki H, Hill RB, Polster BM. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell 2017;40:583–594.e586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smith G, Gallo G. To mdivi-1 or not to mdivi-1: is that the question? Dev Neurobiol 2017;77:1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manczak M, Sesaki H, Kageyama Y, Reddy PH. Dynamin-related protein 1 heterozygote knockout mice do not have synaptic and mitochondrial deficiencies. Biochim Biophys Acta 2012;1822:862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liao S, Curci JA, Kelley BJ, Sicard GA, Thompson RW. Accelerated replicative senescence of medial smooth muscle cells derived from abdominal aortic aneurysms compared to the adjacent inferior mesenteric artery. J Surg Res 2000;92:85–95. [DOI] [PubMed] [Google Scholar]
- 27. Georgakopoulou EA, Tsimaratou K, Evangelou K, Fernandez Marcos PJ, Zoumpourlis V, Trougakos IP, Kletsas D, Bartek J, Serrano M, Gorgoulis VG. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2012;5:37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van Deursen JM. The role of senescent cells in ageing. Nature 2014;509:439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maguire EM, Pearce SWA, Xiao R, Oo AY, Xiao Q. Matrix metalloproteinase in abdominal aortic aneurysm and aortic dissection. Pharmaceuticals 2019;12:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parastatidis I, Weiss D, Joseph G, Taylor WR. Overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 2013;33:2389–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Salmon M, Johnston WF, Woo A, Pope NH, Su G, Upchurch GR Jr, Owens GK, Ailawadi G. KLF4 regulates abdominal aortic aneurysm morphology and deletion attenuates aneurysm formation. Circulation 2013;128:S163–S174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Imanishi M, Chiba Y, Tomita N, Matsunaga S, Nakagawa T, Ueno M, Yamamoto K, Tamaki T, Tomita S. Hypoxia-inducible factor-1alpha in smooth muscle cells protects against aortic aneurysms—brief report. Arterioscler Thromb Vasc Biol 2016;36:2158–2162. [DOI] [PubMed] [Google Scholar]
- 33. Breitzig MT, Alleyn MD, Lockey RF, Kolliputi N. A mitochondrial delicacy: dynamin-related protein 1 and mitochondrial dynamics. Am J Physiol Cell Physiol 2018;315:C80–C90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kinoshita E, Kinoshita-Kikuta E, Koike T. Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat Protoc 2009;4:1513–1521. [DOI] [PubMed] [Google Scholar]
- 35. Gardner SE, Humphry M, Bennett MR, Clarke MC. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1alpha-dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol 2015;35:1963–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu MY, Jin J, Li SL, Yan J, Zhen CL, Gao JL, Zhang YH, Zhang YQ, Shen X, Zhang LS, Wei YY, Zhao Y, Wang CG, Bai YL, Dong DL. Mitochondrial fission of smooth muscle cells is involved in artery constriction. Hypertension 2016;68:1245–1254. [DOI] [PubMed] [Google Scholar]
- 37. Wang L, Yu T, Lee H, O’Brien DK, Sesaki H, Yoon Y. Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc Res 2015;106:272–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rogers MA, Maldonado N, Hutcheson JD, Goettsch C, Goto S, Yamada I, Faits T, Sesaki H, Aikawa M, Aikawa E. Dynamin-related protein 1 inhibition attenuates cardiovascular calcification in the presence of oxidative stress. Circ Res 2017;121:220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z, Zou MH. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes 2017;66:193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, Yoshimura K, Aoki H, Tsutsui H, Noda T, Sagara J, Taniguchi S, Takahashi M. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler Thromb Vasc Biol 2015;35:127–136. [DOI] [PubMed] [Google Scholar]
- 41. Li Q, Youn JY, Siu KL, Murugesan P, Zhang Y, Cai H. Knockout of dihydrofolate reductase in mice induces hypertension and abdominal aortic aneurysm via mitochondrial dysfunction. Redox Biol 2019;24:101185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Navas-Madroñal M, Rodriguez C, Kassan M, Fité J, Escudero JR, Cañes L, Martínez-González J, Camacho M, Galán M. Enhanced endoplasmic reticulum and mitochondrial stress in abdominal aortic aneurysm. Clin Sci (Lond) 2019;133:1421–1438. [DOI] [PubMed] [Google Scholar]
- 43. Gabrielson M, Vorkapic E, Folkesson M, Welander M, Matussek A, Dimberg J, Lanne T, Skogberg J, Wagsater D. Altered PPARgamma coactivator-1 alpha expression in abdominal aortic aneurysm: possible effects on mitochondrial biogenesis. J Vasc Res 2016;53:17–26. [DOI] [PubMed] [Google Scholar]
- 44. Sinha I, Sinha-Hikim AP, Hannawa KK, Henke PK, Eagleton MJ, Stanley JC, Upchurch GR Jr. Mitochondrial-dependent apoptosis in experimental rodent abdominal aortic aneurysms. Surgery 2005;138:806–811. [DOI] [PubMed] [Google Scholar]
- 45. Yan P, Chen K, Wang Q, Yang D, Li D, Yang Y. UCP-2 is involved in angiotensin-II-induced abdominal aortic aneurysm in apolipoprotein E-knockout mice. PLoS One 2017;12:e0179743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carino D, Sarac TP, Ziganshin BA, Elefteriades JA. Abdominal aortic aneurysm: evolving controversies and uncertainties. Int J Angiol 2018;27:58–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cassis LA, Gupte M, Thayer S, Zhang X, Charnigo R, Howatt DA, Rateri DL, Daugherty A. ANG II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol 2009;296:H1660–H1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Subramanian V, Uchida HA, Ijaz T, Moorleghen JJ, Howatt DA, Balakrishnan A. Calpain inhibition attenuates angiotensin II-induced abdominal aortic aneurysms and atherosclerosis in low-density lipoprotein receptor-deficient mice. J Cardiovasc Pharmacol 2012;59:66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen X, Rateri DL, Howatt DA, Balakrishnan A, Moorleghen JJ, Morris AJ, Charnigo R, Cassis LA, Daugherty A. Amlodipine reduces AngII-induced aortic aneurysms and atherosclerosis in hypercholesterolemic mice. PLoS One 2013;8:e81743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miao XN, Siu KL, Cai H. Nifedipine attenuation of abdominal aortic aneurysm in hypertensive and non-hypertensive mice: mechanisms and implications. J Mol Cell Cardiol 2015;87:152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hasan P, Saotome M, Ikoma T, Iguchi K, Kawasaki H, Iwashita T, Hayashi H, Maekawa Y. Mitochondrial fission protein, dynamin-related protein 1, contributes to the promotion of hypertensive cardiac hypertrophy and fibrosis in Dahl-salt sensitive rats. J Mol Cell Cardiol 2018;121:103–106. [DOI] [PubMed] [Google Scholar]
- 52. Ailawadi G, Moehle CW, Pei H, Walton SP, Yang Z, Kron IL, Lau CL, Owens GK. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg 2009;138:1392–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Airhart N, Brownstein BH, Cobb JP, Schierding W, Arif B, Ennis TL, Thompson RW, Curci JA. Smooth muscle cells from abdominal aortic aneurysms are unique and can independently and synergistically degrade insoluble elastin. J Vasc Surg 2014;60:1033–1041; discussion 1041–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Raffort J, Lareyre F, Clement M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol 2017;14:457–471. [DOI] [PubMed] [Google Scholar]
- 55. Zhou HF, Yan H, Cannon JL, Springer LE, Green JM, Pham CT. CD43-mediated IFN-gamma production by CD8+ T cells promotes abdominal aortic aneurysm in mice. J Immunol 2013;190:5078–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Simula L, Pacella I, Colamatteo A, Procaccini C, Cancila V, Bordi M, Tregnago C, Corrado M, Pigazzi M, Barnaba V, Tripodo C, Matarese G, Piconese S, Campello S. Drp1 controls effective T cell immune-surveillance by regulating T cell migration, proliferation, and cMyc-dependent metabolic reprogramming. Cell Rep 2018;25:3059–3073.e3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Meher AK, Johnston WF, Lu G, Pope NH, Bhamidipati CM, Harmon DB, Su G, Zhao Y, McNamara CA, Upchurch GR Jr, Ailawadi G. B2 cells suppress experimental abdominal aortic aneurysms. Am J Pathol 2014;184:3130–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schaheen B, Downs EA, Serbulea V, Almenara CC, Spinosa M, Su G, Zhao Y, Srikakulapu P, Butts C, McNamara CA, Leitinger N, Upchurch GR Jr, Meher AK, Ailawadi G. B-cell depletion promotes aortic infiltration of immunosuppressive cells and is protective of experimental aortic aneurysm. Arterioscler Thromb Vasc Biol 2016;36:2191–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rateri DL, Moorleghen JJ, Knight V, Balakrishnan A, Howatt DA, Cassis LA, Daugherty A. Depletion of endothelial or smooth muscle cell-specific angiotensin II type 1a receptors does not influence aortic aneurysms or atherosclerosis in LDL receptor deficient mice. PLoS One 2012;7:e51483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Poduri A, Owens AP, Howatt DA, Moorleghen JJ, Balakrishnan A, Cassis LA, Daugherty A. Regional variation in aortic AT1b receptor mRNA abundance is associated with contractility but unrelated to atherosclerosis and aortic aneurysms. PLoS One 2012;7:e48462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Qin Y, Wang Y, Liu O, Jia L, Fang W, Du J, Wei Y. Tauroursodeoxycholic acid attenuates angiotensin ii induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice by inhibiting endoplasmic reticulum stress. Eur J Vasc Endovasc Surg 2017;53:337–345. [DOI] [PubMed] [Google Scholar]
- 62. Ziegler DV, Wiley CD, Velarde MC. Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell 2015;14:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nishimura A, Shimauchi T, Tanaka T, Shimoda K, Toyama T, Kitajima N, Ishikawa T, Shindo N, Numaga-Tomita T, Yasuda S, Sato Y, Kuwahara K, Kumagai Y, Akaike T, Ide T, Ojida A, Mori Y, Nishida M. Hypoxia-induced interaction of filamin with Drp1 causes mitochondrial hyperfission-associated myocardial senescence. Sci Signal 2018;11:eaat5185. [DOI] [PubMed] [Google Scholar]
- 64. Cafueri G, Parodi F, Pistorio A, Bertolotto M, Ventura F, Gambini C, Bianco P, Dallegri F, Pistoia V, Pezzolo A, Palombo D. Endothelial and smooth muscle cells from abdominal aortic aneurysm have increased oxidative stress and telomere attrition. PLoS One 2012;7:e35312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cooper HA, Scalia R, Rizzo V, Eguchi S. Angiotensin II- and Alzheimer-type cardiovascular aging. Circ Res 2018;123:651–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kyaw M, Yoshizumi M, Tsuchiya K, Kirima K, Tamaki T. Antioxidants inhibit JNK and p38 MAPK activation but not ERK 1/2 activation by angiotensin II in rat aortic smooth muscle cells. Hypertens Res 2001;24:251–261. [DOI] [PubMed] [Google Scholar]
- 67. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 2015;116:264–278. [DOI] [PubMed] [Google Scholar]
- 68. Senemaud J, Caligiuri G, Etienne H, Delbosc S, Michel JB, Coscas R. Translational relevance and recent advances of animal models of abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol 2017;37:401–410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.