

Abstract
Normal cardiac contractile and relaxation functions are critically dependent on a continuous energy supply. Accordingly, metabolic perturbations and impaired mitochondrial bioenergetics with subsequent disruption of ATP production underpin a wide variety of cardiac diseases, including diabetic cardiomyopathy, dilated cardiomyopathy, hypertrophic cardiomyopathy, anthracycline cardiomyopathy, peripartum cardiomyopathy, and mitochondrial cardiomyopathies. Crucially, there are no specific treatments for preventing the onset or progression of these cardiomyopathies to heart failure, one of the leading causes of death and disability worldwide. Therefore, new treatments are needed to target the metabolic disturbances and impaired mitochondrial bioenergetics underlying these cardiomyopathies in order to improve health outcomes in these patients. However, investigation of the underlying mechanisms and the identification of novel therapeutic targets have been hampered by the lack of appropriate animal disease models. Furthermore, interspecies variation precludes the use of animal models for studying certain disorders, whereas patient-derived primary cell lines have limited lifespan and availability. Fortunately, the discovery of human-induced pluripotent stem cells has provided a promising tool for modelling cardiomyopathies via human heart tissue in a dish. In this review article, we highlight the use of patient-derived iPSCs for studying the pathogenesis underlying cardiomyopathies associated with metabolic perturbations and impaired mitochondrial bioenergetics, as the ability of iPSCs for self-renewal and differentiation makes them an ideal platform for investigating disease pathogenesis in a controlled in vitro environment. Continuing progress will help elucidate novel mechanistic pathways, and discover novel therapies for preventing the onset and progression of heart failure, thereby advancing a new era of personalized therapeutics for improving health outcomes in patients with cardiomyopathy.
Keywords: Cardiomyopathy, Metabolism, Bioenergetics, Human-induced pluripotent stem cells
Graphical Abstract
1. Introduction
Heart failure is one of the leading causes of death and disability globally, affecting >37.7 million individuals worldwide.1,2 A leading cause of heart failure is cardiomyopathy, which is defined as a disease of myocardium associated with cardiac dysfunction.3 They can be classified into two groups based on the aetiology. Primary cardiomyopathies have genetic, acquired, or mixed aetiology, whereas secondary cardiomyopathies develop as a result of an extra-cardiovascular cause (e.g. systemic or multi-organ disease) that results in different phenotypes, including dilated, hypertrophic, or restrictive patterns. Currently, there are no specific treatments for these disorders, with existing disease management strategies mainly focused on delaying the progression of cardiomyopathies into HF.4–6 Effective treatment options for cardiomyopathies have remained elusive partly due to the lack of experimental models that are able to recapitulate the diseases accurately.
Several animal studies have provided evidence that metabolic perturbations and/or mitochondrial dysfunction are key contributors to various cardiomyopathies. However, differences in clinical manifestation, penetrance, and severity in human cardiomyopathies make animal models less suitable for studying these conditions. The logical approach would therefore be to perform investigational and interventional studies on human heart tissue, such as primary cardiomyocytes obtained directly from the heart. This however requires invasive procedures and access to such samples is limited. Coupled with their limited proliferative ability, it is nearly impossible to conduct meaningful long-term studies on primary human heart tissue. With the advent of human-induced pluripotent stem cells (hiPSCs),7 now it may be possible to overcome these shortcomings.
In this review article, we discuss current research on cardiomyopathies with a special emphasis on metabolic and bioenergetic perturbations that underpin diabetic cardiomyopathy (DMCM), dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), anthracycline cardiomyopathy, peripartum cardiomyopathy (PPCM), and mitochondrial cardiomyopathies. We also discuss the use of hiPSCs to model these cardiomyopathies, allowing new insights into perturbed metabolism and impaired bioenergetics. The ultimate goal of the field is to elucidate novel mechanistic pathways to enable the discovery of novel therapies for preventing the onset and progression of HF, thereby opening up a new era of personalized therapeutics for improving health outcomes in patients with cardiomyopathy.
2. Cardiac bioenergetics and metabolism
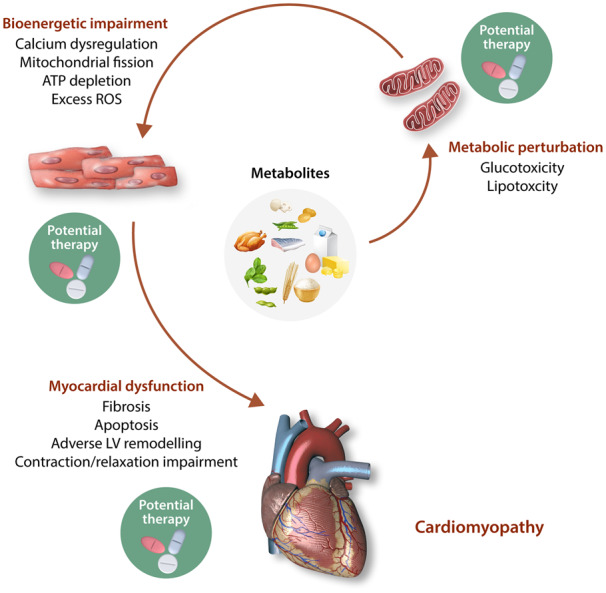
The heart is a constant mechanical pump with high-energy demands. Therefore, the disruption of energy production due to impaired bioenergetics and/or perturbed substrate utilization have detrimental effects on cardiac contractile function. Cardiac bioenergetics has three components: (i) cardiomyocyte uptake and utilization of substrate and entry of metabolites into the Krebs cycle; (ii) oxidative phosphorylation (OXPHOS) by the mitochondrial electron transport carrier; and (iii) transfer and utilization of ATP that involves creatine kinase energy shuttle.8 Conditions or diseases that perturb one or more of these components may impair cardiac contractile function, leading to clinical manifestations of certain cardiomyopathies.
Early studies have shown significant changes in energy substrate utilization during heart development. The foetal heart has a greater preference for carbohydrates, including glucose, lactate, and pyruvate as compared to the adult heart.9 The foetal heart also has a lower capacity for OXPHOS, relying mostly on glycolysis for ATP production; this is advantageous due to the lower arterial oxygen content in the foetus relative to adults. Moreover, the foetal heart can take advantage of the lactate-rich environment created by the placenta.10,11 In the neonatal period, there is a transition in metabolism from glycolysis to OXPHOS to accommodate the increasing energy demands of the maturing heart. The high-energy demands of the adult heart are met mainly by mitochondrial OXPHOS, which contributes to >95% of the ATP produced in the heart, with glycolysis contributing the remaining 5%.12 The adult heart relies on fatty acids as the primary fuel for OXPHOS.13,14 Fatty acids are transported to the heart as free fatty acids (FFA) bound to albumin or as fatty acids released from triglycerides in chylomicrons or very-low-density lipoprotein, with the majority being derived from chylomicron triglycerides.15 FFAs are taken up by cardiomyocytes through passive diffusion or protein-mediated transport.16 A more detailed account of the metabolic pathways present in cardiomyocytes can be found in Lopaschuk et al.’s17 review.
When challenged with pathological or physiological stresses, the heart demonstrates metabolic flexibility by switching its substrate preference to carbohydrates, lipids, amino acids, or ketone bodies for continual ATP production. This metabolic plasticity is characteristic of a healthy heart. Cardiac substrate metabolism extends beyond energy production as the resultant metabolites can serve as secondary messengers and disrupt proper contractile function.14,18 Prolonged metabolic shifts are associated with cardiomyopathies such as ischaemic heart disease,19 cardiac hypertrophy,20 atrial fibrillation,21 and HF.12,13,16,20 Maladaptive metabolic switching can therefore lead to impaired bioenergetics that contributes to the pathophysiology of HF20 (Figure 1).
Figure 1.

Schematic illustration of metabolic substrate preferences in cardiomyocytes under healthy vs. diseased conditions. Under healthy conditions, both fatty acids and glucose are able to enter the cell via the CD36 fatty acid translocase and GLUT4 transporter, respectively. Both substrates can enter the mitochondria and undergo OXPHOS; however, there is a preference for fatty acid β-oxidation as this process generates more ATP than glucose oxidation, which is critical for efficient energy production. In diabetic cardiomyopathy, GLUT4 membrane translocation is impaired, and as a result, glucose uptake is reduced. Increased fatty acid entry and β-oxidation then results in proton leak, ROS formation, and lipotoxicity, with eventual energy depletion. In dilated cardiomyopathy (DCM), an increase in glucose uptake and oxidation is accompanied by reduced fatty acid uptake and β-oxidation. Despite glucose being the more energy-efficient substrate, glucose oxidation is unable to produce sufficient energy to keep up with the demanding workload of a failing heart. In hypertrophic cardiomyopathy (HCM) and mitochondrial cardiomyopathy, much like in DCM, there is a metabolic switch towards preferential utilization of glucose. In HCM, however, an increase in glycolysis with concurrent reduction in glucose oxidation, results in uncoupling between glucose entry and oxidation. This leads to suboptimal OXPHOS and insufficient energy production. In anthracycline cardiomyopathy and peripartum cardiomyopathy, fatty acid and glucose cellular uptake and oxidation have yet to be elucidated. While ROS formation has been identified as a common occurrence, disrupted mitochondrial structure has been observed in anthracycline cardiomyopathy. Both cardiomyopathies are associated with impaired OXPHOS, leading to insufficient energy production.
3. Diabetic cardiomyopathy
Diabetic cardiomyopathy was first described in 1972, and has been defined as left ventricular (LV) dysfunction that occurs in the diabetic patient independent of recognized cardiovascular risk factors, such as coronary atherosclerosis, valvular heart disease, hypertension, and other congenital heart diseases.22,23 It is characterized by both morphological and functional changes. On the functional level, DMCM results in reduced LV systolic function, LV stiffness, and diastolic dysfunction, as well as clinical HF. On the structural level, DMCM is characterized by cardiomyocyte hypertrophy and interstitial fibrosis, and lipid accumulation in cardiomyocytes.24 Although the pathogenesis of DMCM is incompletely understood, it has been mainly attributed to impaired cardiac metabolism secondary to reduced glucose uptake seen in both type 1 and type 2 diabetes mellitus.25 Reduced glucose uptake, glycolysis, and glucose oxidation in diabetic hearts are likely caused by decreased expression and translocation of glucose transporter 4 (GLUT4), as well as impaired pyruvate decarboxylation.26 Both expression and membrane translocations of GLUT4 (the major isoform expressed in adult cardiomyocytes) were found to be reduced in cardiomyocytes of diabetic animal models.27,28 GLUT4 deficiency is attributable to impaired insulin-stimulated PI3K/Akt signalling that contributes to the pathogenesis of DMCM.27,29 Conversely, the activation of PI3K/Akt pathway was shown to be beneficial in DMCM.30
Given that cardiovascular events contribute to the leading cause of mortality in diabetic patients,18 further understanding on how insulin-resistance (a hallmark feature of diabetes) leads to DMCM is important. An early study revealed that insulin and islet transplantation in mildly diabetic rats was able to improve glucose metabolism and LV function, suggesting a possible link between metabolic alterations and DMCM.31 The incidence of perturbed metabolism in DMCM was further validated in a clinical study that found a correlation between increased insulin resistance and severe HF and decreased survival.32 When investigating FoxO family of transcription factors, researchers found FoxO proteins to be constantly activated in the hearts of diabetic mice, which resulted in insulin resistance and cardiomyopathy. Consistently, isolated murine cardiomyocytes with depleted FoxO1 showed metabolic substrate shift from FFAs to glucose, with subsequent improvements in cardiac function and responsiveness to insulin.33
Overexpression of cardiac-specific MG53 (a striated muscle-specific E3 ubiquitin ligase) was found to induce DMCM in mice.34 In addition to inducing insulin resistance, the MG53 transgenic mice showed impaired glucose uptake, increased lipid accumulation, cardiomyocyte hypertrophy, interstitial fibrosis, and cardiac dysfunction. The study showed that MG53 contributes to DMCM by (i) destabilizing the insulin receptor and insulin receptor substrate 1, leading to insulin resistance, and (ii) up-regulating PPAR-α and target genes, resulting in lipid accumulation and lipotoxicity.35 Interestingly, as this study showed suppression of glycolysis and enhancement of lipid metabolism with activation of PPAR-α, it was contrary to what was observed in dilated cardiomyopathy.36 The increased lipid metabolism phenomenon is indicative of the Randle cycle whereby, increased fatty acid oxidation is associated with decreased glucose oxidation. The former metabolic pathway results in the accumulation of acetyl-CoA which can inhibit pyruvate dehydrogenase, while increasing citrate levels via the Krebs cycle. The resulting increase in citrate and ATP/ADP ratio, decreases the enzymatic activity of phosphofructokinase and glucose metabolism.37 The Randle cycle has long been postulated as a potential mechanism for type 2 diabetes mellitus38 and it has also been speculated that increased fatty acid oxidation would lead to oxygen wastage, as relatively more oxygen would be required as compared to glucose oxidation.39 It is also possible that increased lipid metabolism could lead to oxidative stress, resulting in mitochondrial dysfunction and eventual cardiac cell death via apoptosis. This hypothesis is supported by a proof-of-concept study showing that metallothionein (a group of intracellular metal-chelating proteins) was able to attenuate cardiac apoptosis, thereby providing significant protection against the development of DMCM.40
In type 2 diabetes mellitus, increased circulating palmitate, a common saturated fatty acid, not only contributes to insulin resistance41 but also cardiac dysfunction in DMCM through the Akt/GSK3β/β-catenin pathway.42 Elevated FFA can be a result of augmented dietary fat intake and adipose tissue lipolysis. When fatty acid uptake surpasses fatty acid oxidation, fatty acids can be sequestered in cardiomyocytes as lipid droplets in the form of triglycerides and fatty acid metabolites such as diacylglycerols, long-chain acyl-CoAs, and acylcarnitines that contribute to cardiomyopathy. More than a decade ago, elevated FFAs were found to be associated with insulin resistance and myocardial dysfunction. In addition to damaging the plasma membrane and ion channels, elevated FFAs can lead to an increase in mitochondrial uncoupling proteins, resulting in a reduction in mitochondrial membrane potential and consequently impaired OXPHOS in cardiomyocytes.39
A causative relationship between myocardial lipid accumulation and impaired myocardial contractility has been suggested by human studies using non-invasive magnetic resonance spectroscopy.43,44 Excess fatty acids can exert deleterious effects through (i) production of reactive oxygen species (ROS), leading to oxidative stress; (ii) uncoupling of OXPHOS; and (iii) opening of the mitochondrial permeability transition pore and initiation of apoptosis.45 Since cellular uptake of fatty acids occurs via either passive diffusion, or protein carrier-mediated transport via fatty acid translocase (FAT)/CD36, several rodent studies have shown that sarcolemmal overexpression of CD36 contributes to diabetes-mediated cardiotoxicity, and is associated with increased fatty acid uptake, triglyceride accumulation, and contractile dysfunction. Conversely, several studies have shown that the restriction of fatty acid delivery and uptake provides protection against cardiotoxicity associated with insulin resistance/diabetes.43 For instance, inhibition of CD36 in rat cardiomyocytes was shown to confer protection on the cellular level,46 while CD36 deficiency conferred protection against cardiac steatosis and cardiomyopathy in PPAR-α overexpressing mice.47 Taken together, rodent studies have suggested that the inhibition of CD36 could be a therapeutic intervention for DMCM.
Evidence for the involvement of mitochondrial dysfunction was described recently, wherein mitophagy was shown to be important for cardiac function during high-fat diet-induced DMCM.48,49 When several transgenic mice were randomly assigned to normal or high-fat diet for 2 months, researchers found that high-fat diet induced Atg7-dependent mitophagy in the early stage of DMCM, which was also partly mediated by Parkin. Mitophagy in the heart was found to be activated in a time-dependent manner in response to high-fat diet consumption. Given that mitophagy serves as a quality control mechanism for functional mitochondria, defective mitophagy would result in impaired bioenergetics and lipotoxicity, thereby exacerbating the DMCM phenotype as seen in atg7−/− mice.50
Many diabetic studies employ the use of streptozotocin-induced diabetic murine models. Streptozotocin destroys insulin-producing β cells of the pancreas, recapitulating type 1, as opposed to type 2 diabetes mellitus. This approach avoids confounders such as obesity and impaired leptin signalling.51 Recently, researchers have developed an alternative murine model of DMCM using a combination of low-dose of streptozotocin and high-fat diet over longer periods of time. The murine model at 26 weeks produces LV dysfunction, characteristic of patients with DMCM, with no change in total body weight.52
4. Dilated cardiomyopathy
Dilated cardiomyopathy is one of the most prevalent forms of cardiomyopathy, affecting 1 in 2500 individuals globally53 and is one of the leading causes of HF.54 It can be primary or secondary (e.g. infiltrative or autoimmune), and is characterized by dilatation and impaired contraction of either the left or both ventricles; however, the extent of myocardial dysfunction is not exclusively attributable to abnormal loading or ischaemia.5,55 The major genomic determinants of DCM were found to be titin truncating and lamin A/C mutations.56 Recent studies have also implicated epigenetics in the development of DCM.57,58
Metabolic perturbations in DCM have been reported in early studies that examined hearts of DCM patients along with normal control hearts and found reduction in cytochrome content as well as cytochrome-mediated enzymatic activity in DCM.59 Furthermore, autoimmunity against myocardial ADP/ATP carrier of the inner mitochondrial membrane was found to contribute to the pathophysiology of DCM, by diminishing cardiac function via impairment of energy production.60 This was further validated in guinea pig and murine models which showed that immune response directed to the ADP/ATP carrier would perturb energy metabolism and consequently lead to mitochondrial dysfunction.61 Further evidence of mitochondrial dysfunction in DCM was elucidated in MnSOD-deficient mice, which have an absence of MnSOD (an intramitochondrial enzyme responsible for scavenging free radicals) that leads to neonatal lethal DCM, likely through the unchecked superoxide produced as a by-product of OXPHOS.62 Similarly, the deficiency in another enzyme important for mitochondrial ROS scavenging, TXNRD2, was found to contribute to DCM, likely through increased oxidative stress.63 Clinical studies have demonstrated metabolic alterations in DCM that are characterized by decreased fatty acid metabolism with increased myocardial glucose metabolism similar to animal models of HF.36
ANT, a dimeric protein complex that mediates the transport of ADP and ATP across the inner mitochondrial membrane, serves important roles in ATP production (via OXPHOS) and consumption.64 ANTs are encoded by three different genes with ANT1 being mainly expressed in tissues with limited regenerative capacity such as the heart, skeletal muscle, and brain. ANT2 is expressed in most tissues, and is the prevalent isoform in the kidney, liver, and spleen.65 Lastly, ANT3 has properties of a housekeeping gene and is the least transcribed. Consistent with its tissue-specific expression in heart, researchers have shown that by knocking out ANT1 in mice, initial mechanical dysfunction was followed by a decline in LV systolic function, which suggested that mitochondrial energy deficiency was sufficient to cause DCM, further validating the critical role of perturbed energy metabolism in DCM pathophysiology.66 Contrary to this, increased ANT1 and decreased ANT2 expression in human heart biopsies has been associated with the development of DCM.67 Similarly, an early clinical study highlighted that altered isoform expression of ANT in DCM hearts (increased ANT1 and decreased ANT2) affects ADP/ATP translocation across the inner mitochondrial membrane, leading to perturbed energy metabolism and contributing to cardiac dysfunction.64
A recent study has shown that proteins such as NAD+ and sirtuins play a role in the development of DCM.68 Besides serving as a key coenzyme in fuel oxidation and mitochondrial ATP production, as well as being the precursor of NADPH, which is needed for anabolic pathways and scavenging ROS, NAD+ is also an important substrate for sirtuins.69 Through NAD+-dependent deacetylation of lysine residues modified by Ac-CoA-derived acyl groups, sirtuins regulate nuclear gene expression and mitochondrial enzyme activities, and have been reported to have cardioprotective effects.69 Levels of myocardial NAD+ were found to be depressed in murine models of non-ischaemic DCM (generated by inactivation of Serum Response Factor). Treatment with nicotinamide riboside significantly preserved cardiac function through enhanced citrate and Ac-CoA metabolism in addition to anti-oxidative effects, presenting a potential therapy in DCM, particularly as nicotinamide riboside is orally available.68
5. Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy, a common inherited disease affecting 1 in 500 individuals, exhibits an autosomal dominant pattern of inheritance, and is characterized by LV hypertrophy with diastolic dysfunction.70,71 Following the discovery of mutations in MYH7 (MYH7R403Q) in HCM patients,72 several other gene mutations have been identified, including mutations in MYBPC3 that encodes for cMyBP-C.73 HCM has since been associated with >1400 mutations occurring in 11 or more genes encoding for contractile or sarcomere complex proteins.74–76 More recently, a study showed that mutations in the Ig domains of titin are associated with HCM in both fish and humans.77 Most of these mutations result in asymmetric hypertrophy of the interventricular septum, which could cause LV outflow tract obstruction that is secondary to subaortic stenosis.78
Perturbed cardiac bioenergetics was described in HCM as early as three decades ago.79 When investigating cardiac energetics in subjects with mutations in three different familial HCM genes, the cardiac phosphocreatine to ATP ratio was found to be reduced by 30% in HCM subjects, irrespective of the harboured mutation.80 Interestingly, this bioenergetic deficit was also observed in genotype-positive subjects who had yet to develop hypertrophy, supporting a role for altered bioenergetics in disease progression. Furthermore, heterozygous mice with αMyHCR403Q mutation (orthologous to human βMHCR403Q that causes lethal forms of HCM) showed increased energetic demands and reduction in free energy released from ATP hydrolysis, which likely contributed to diastolic dysfunction.81 Mutations implicated in HCM have been shown to increase sarcomere Ca2+ sensitivity, ATPase activity, and overall energetic cost of cardiomyocyte contraction, thus leading to the idea that energy deficiency contributes to the pathophysiology of HCM. In support of this, a phase II clinical trial documented improvements in cardiac bioenergetics (as seen in increased myocardial ratios of phosphocreatine to ATP), alleviation of diastolic dysfunction, and increased exercise capacity in HCM patients following treatment with perhexiline, a metabolic substrate modulator which promotes utilization of carbohydrates for myocardial energy through inhibition of carnitine-palmitoyltransferase.82 This study provides further evidence that metabolic perturbations play crucial roles in HCM development. Furthermore, a recent study incorporated three transgenic murine models that exhibited a graded decrease in cMyBP-C.83 While the severity of the cardiomyocyte phenotype was found to be correlated with cMyBP-C levels, treatment with a pharmacological inhibitor of myosin ATPase alleviated the relaxation defects and led to a dose-dependent attenuation of hypercontractility.83
Apart from sarcomere mutations, researchers found that genes involved in mitochondrial function could also be associated with HCM (e.g. ELAC2 mutants).84 Mutations in ELAC2, which encodes for mitochondrial RNase Z, were found to impair mitochondrial tRNA 3′ end processing and consequently resulted in diminished mitochondrial function.85 Mitochondrial involvement in HCM was also highlighted in another study.86 GTPBP3 is a highly conserved tRNA modifying enzyme involved in the biosynthesis of 5-taurinomethyl group of tRNAGlu, tRNAGln, tRNALys, tRNATrp, tRNALeu(UUR) in human mitochondria.87–89 GTPBP3 deficiency in zebrafish resulted in decreased mitochondrial translation and impaired OXPHOS, in addition to being associated with HCM. The defective taurine modification and impaired mitochondrial translation were also seen in HCM patient-derived mutant fibroblasts containing GTPBP3 mutations.90,91 Recently, researchers generated a gtpbp3ko zebrafish model that successfully recapitulated the clinical phenotypes of HCM patients having GTPBP3 mutations, highlighting the crucial role of aberrant nucleotide alterations of mitochondrial tRNAs in the pathogenesis of HCM.86
Interestingly, the metabolic perturbations involved in HCM extend beyond the heart. Metabolic crosstalk between the heart and liver was elucidated using a previously established transgenic HCM murine model expressing rat αMyHCR403Q with deletion, Δaa468–527. The mice showed reduced FAT CD36 and decreased AMPK activity, which lead to decreased myocardial ATP and triglycerides. However, increased levels of oleic acids and triglycerides in circulating very-low-density lipoproteins were observed in the liver.92 While this metabolic shift led to increased gluconeogenesis in the liver, inhibition of gluconeogenesis improved LV architecture and function, thereby demonstrating how metabolic perturbations in other organs could contribute to HCM pathogenesis.92
6. Anthracycline cardiomyopathy
Anthracyclines are a class of chemotherapeutic agents which exert anti-tumour effects through: (i) interaction with DNA gyrase and topoisomerase IIβ, and intercalating DNA to cause double-strand breaks in chromosomal DNA, thereby inhibiting transcription and replication leading to cell-cycle arrest and apoptosis; (ii) modulation of signal transduction pathways such as Fas ligand pathway to inhibit cellular growth; (iii) generation of ROS that causes DNA damage and lipid peroxidation.93 Anthracyclines are effective and widely used for leukaemia94,95 and a variety of solid tumours, including breast cancer, the leading cause of cancer mortality in women.96 Though it serves as one of the most effective chemotherapeutics, it is also the most toxic. Anthracycline-induced cardiotoxicity is associated with progressive DCM in a dose-dependent manner and often presents with LV dysfunction.96,97 As anthracycline-induced cardiotoxicity is often progressive and irreversible,96 it can increase mortality due to HF, despite the patient surviving cancer.97 Though the exact mechanism of anthracycline-induced cardiotoxicity has yet to be determined, studies have shown that anthracyclines can impair intracellular Ca2+ signalling and pro-survival signalling pathways as well as disrupt cardiac progenitor cells.98 Furthermore, anthracyclines can accumulate in the mitochondria of cardiomyocytes, disrupting OXPHOS and leading to ROS formation, which can result in endomyocardial interstitial fibrosis and vacuolation.93 Thus, it is widely believed that the generation of ROS, which results in increased apoptosis, disruption of mitochondrial structure and function in addition to energy depletion, plays a crucial role in the pathogenesis of anthracycline-induced cardiotoxicity.98
7. Peripartum cardiomyopathy
Peripartum cardiomyopathy, which was first described in the 1930s,99 is idiopathic in nature, and presents with LV systolic dysfunction and symptoms of HF between the final month of pregnancy and the first 5 months post-partum.100 It occurs at 1 in 2187 live births in USA,101 and although it has relatively low incidence, the prognosis can be poor with cardiogenic shock.102 Although the exact mechanism of PPCM has yet to be elucidated, it is a non-ischaemic cardiomyopathy that often occurs in the presence of predictable stressors.103 The familial occurrence, its varying prevalence among different regions and ethnicities, as well as genome-wide association studies, suggest that genetics may play an important role in the development of PPCM.104 Metabolic perturbations and increased oxidative stress in the heart during pregnancy also appear to contribute to the pathogenesis.103,104
Murine models of PPCM have been generated by genetic ablation of the STAT3 transcription factor, which drives transcription of genes encoding antioxidant (e.g. MnSOD) and anti-apoptotic (e.g. BCL-XL) proteins.105–107 These mutant mice, which are referred to as STAT3-CKO mice, recapitulate syndromes similar to PPCM, with a fraction of mice dying of HF within the first 3 weeks after delivery. Post-partum STAT3-CKO mice exhibit significant reductions in myocardial capillary density, compared to wild-type and non-pregnant STAT3-CKO mice. Subsequently, researchers have associated this phenotype with the secretion of prolactin which, ironically promotes angiogenesis.108 Serum of patients with PPCM, however, show increased levels of cleaved prolactin which has strong anti-angiogenic effects, prompting researchers to describe a pathway whereby, the lack of STAT3 in cardiomyocytes, leads to reduced levels of MnSOD, which in turn causes the accumulation of ROS and increases the expression of cathepsin D, which cleaves prolactin.109,110 Given that cardiomyopathy in STAT3-CKO mice could be prevented by treatment with bromocriptine (a drug that inhibits prolactin secretion), researchers managed to demonstrate that decreased myocardial capillary density contributes largely to the development of PPCM.105,110 Further studies have shown that cleaved prolactin could induce the expression of miR-146a, which subsequently inhibits proliferation of endothelial cells and promotes cell death, in addition to being transported into cardiomyocytes through exosomes, where it decreased ERBB4 levels and perturbed cardiomyocyte metabolism.110 Consequently, treating STAT3-CKO mice with antisense oligonucleotides or cholesterol-modified antago-miRs directed against miR-146a, alleviated PPCM, suggesting that miR-146a could be a potential therapeutic target against PPCM.111
A subsequent study has also used similar murine models for PPCM (mice with cardiomyocyte-specific deletion of Stat3) to investigate the benefit of β1-adrenergic receptor (β1-AR) agonists for treatment of acute HF in PPCM.112In vivo analysis using β1-AR stimulation by isoprenaline in Stat3-KO mouse cardiomyocytes showed severe cardiac dysfunction with high mortality, likely through the disruption of glucose uptake and subsequent energy depletion, oxidative stress, and cellular death, suggesting that β1-AR stimulation in PPCM may lead to irreversible HF. The adverse effects by β1-AR agonist could be attenuated by β1-AR blocker metoprolol, as well as glucose-uptake-promoting drugs, perhexiline and etomoxir.112 Importantly, while energy depletion in cardiomyocytes was shown to exacerbate the disease phenotype, rescue with perhexiline resulted in an alleviated disease phenotype, highlighting the significance of perturbed cardiac metabolism in the development of PPCM. Given that cardiac STAT3 expression is found to be reduced in patients with PPCM,105 these studies confirm a causal role of STAT3-deficiency in the development of PPCM. Furthermore, the adverse effect by β1-AR agonists in murine models of PPCM has shed light on the controversial use of dobutamine (β1-AR agonist) in treating acute HF in PPCM. Instead, the use of β1-AR agonists should be contraindicated in PPCM.
8. Mitochondrial cardiomyopathies
Mitochondrial cardiomyopathies are characterized by diffuse ventricular hypertrophy, with progressive diastolic dysfunction and HF with preserved ejection fraction.113 They arise as one of the most common clinical manifestations in mitochondrial diseases.114 Mitochondrial diseases are characterized by mitochondrial dysfunction due to mutations in mtDNA or nuclear genes that are essential for mtDNA maintenance; thus, mitochondrial diseases can exhibit autosomal-dominance or be recessive, X-linked, or maternally inherited.115 Mitochondrial dysfunction can lead to energetic depletion through impaired OXPHOS and mitochondrial Ca2+ regulation, which are central for excitation–contraction coupling.116 Consequently, mitochondrial dysfunction often affects organs with high-energy demands such as cardiac muscles,117 resulting in cardiomyopathies. Mitochondrial dysfunction extends beyond metabolic perturbations as the mitochondrion is involved in adaptive thermogenesis, ion homoeostasis, innate immunity, generation of ROS, and regulation of apoptosis.118
The involvement of mitochondrial dysfunction in the development of cardiomyopathy was first observed more than two decades ago. Utilizing endocardial biopsies from infants, researchers showed significant decreases in the activity of complex I and/or complex IV relative to complex II and III in idiopathic HCM.119 Further investigations using transmitochondrial hybrids demonstrated the pathogenicity of mtDNA mutations.119 Later on, a murine model for mitochondrial myopathy and cardiomyopathy was described by knocking out the heart/muscle isoform of Ant1. Without ADP/ATP transport across the inner mitochondrial membrane, the absence of ANT1 resulted in reduced ADP-stimulated mitochondria respiration and defective OXPHOS in the heart.120 The Ant1-deficient mice exhibited cardiac hypertrophy associated with mitochondrial proliferation similar to human patients with mitochondrial cardiomyopathy. This study showed for the first time that mitochondrial bioenergetic insufficiency could lead to HCM.120
Transgenic murine models have also been used to investigate gene therapy approaches in the prevention and treatment of mitochondrial cardiomyopathy. Friedreich’s ataxia, a mitochondrial disease caused by diminished frataxin (an essential mitochondrial protein required for the biosynthesis of iron–sulphur clusters), often presents with HCM. Patients often exhibit defective mitochondrial OXPHOS, bioenergetic imbalance, deficient iron–sulphur cluster enzymes, and mitochondrial iron overload.121–123 Excessive iron in cardiomyocytes can lead to the generation of free radicals via the Fenton reaction, resulting in oxidative stress. Consistently, iron accumulation in the myocardium has been associated with diastolic dysfunction in the early stages of DCM.124 Recently, mitochondrial iron overload has been postulated to contribute to oxidative stress, mitochondrial damage, and in the development of cardiomyopathy.125 By knocking out frataxin in mice, researchers generated a model that recapitulates most features of Friedreich’s ataxia-associated cardiomyopathy.126 Interestingly, intravenous administration of adeno-associated virus rh10 vector expressing human frataxin prevented the development of cardiomyopathy, but more importantly, administration after onset of HF was able to reverse the cardiomyopathy at functional, cellular, and molecular levels within a few days. Murine cardiomyocytes with severe energy depletion and ultrastructural disorganization were shown to be rescued by this gene therapy approach.126
In other studies, researchers showed that ablation of Tfam (a nucleus-encoded protein required for mtDNA transcription), led to reduced mitochondrial mass in murine hearts but, interestingly, also resulted in increased ROS, ultimately resulting in lethal DCM in neonatal mice. The elimination of Tfam was found to prevent the expression of 13 polypeptides encoded by mtDNA, including components of enzyme complexes required for mitochondrial OXPHOS. Mutant foetal cardiomyocytes did not exhibit energy deficiency, most likely due to the foetal heart relying mainly on glycolysis for energy production. In contrast, the mutant neonatal mice exhibited perturbed mitochondrial biogenesis, morphology, and function. The study demonstrated that Tfam inactivation results in cardiomyopathy through ROS-activated DNA damage, whereas ROS scavenging or WEE1 kinase inhibition could rescue the cardiomyopathy phenotype in vivo, thereby providing a proof-of-concept therapy for mitochondrial cardiomyopathy. Interestingly, although Tfam inactivation did not affect murine cardiomyocyte maturation as indicated by the unchanged Myh6/Myh7 gene expression ratio, it significantly affected intracellular Ca2+ handling and contraction, establishing a link between mitochondrial perturbation and cardiac dysfunction.127
Defective mitochondrial protein synthesis has also been associated with mitochondrial cardiomyopathy. Mitochondrial protein translation, essential for assembly of the OXPHOS components, requires mitochondrial tRNA to be charged with the cognate amino acids by mitochondrial aminoacyl-tRNA synthetase except for glutaminyl mitochondrial tRNA (mt-tRNAGln), which is indirectly charged via a transamidation reaction requiring GatCAB aminoacyl-tRNA amidotransferase complex.128,129 Post-mortem examination of infants (from five different families) with suspected severe mitochondrial cardiomyopathy revealed cardiomyocytes with extensive mitochondrial proliferation and abnormal respiratory chain enzymes. Sequencing results indicated at least one missense mutation that leads to residual GatCAB activity. The pathogenic variant in GatCAB aminoacyl-tRNA amidotransferase complex was speculated to prevent normal OXPHOS enzyme activity, but was not sufficient to cause embryonic lethality. Immunofluorescence staining failed to reveal co-localization of QARS (another protein involved in the aminoacylation of mt-tRNAGln) with mitochondrial protein TOM20, thereby suggesting absence of aminoacylation of mt-tRNAGln and that charging of mt-tRNAGln is dependent on GatCAB complex.130 A functional assay was performed in Saccharomyces cerevisiae, whereby mutagenesis of orthologous genes into pathogenic variants revealed defective OXPHOS along with decreased oxygen consumption rate. This confirmed the importance of GatCAB complex and highlighted how pathogenic variants impair its function, thereby diminishing the amount of charged mt-tRNAGln and resulting in perturbed mitochondrial translation of nearly all mtDNA-encoded OXPHOS subunits.130
Transgenic mice have also been used to evaluate potential therapies for mitochondrial cardiomyopathy. AOX, a non-mammalian enzyme, is capable of bypassing the blockade of the complex III-to-IV segments of the mitochondrial OXPHOS machinery.131,132 By preventing lethal mitochondrial cardiomyopathy, transgenic expression of Ciona intestinalis AOX extended the median survival time from 210 to 590 days in complex III-deficient mice (knock-in of Bcs1lS78). BCS1L is a mitochondrial inner membrane translocase essential for Rieske iron–sulphur protein topogenesis and incorporation into complex III (cIII). Mutations in BCS1L presents the most common cause of cIII deficiency, leading to GRACILE syndrome that consists of foetal growth restriction, aminoaciduria, cholestasis, liver iron overload, lactic acidosis, and early death during infancy.133–135Bcs1lS78G homozygous knock-in mice recapitulate many features of GRACILE syndrome, with a short survival time of 35 days, due to lethal cardiomyopathy with prominent fibrosis in the heart, as well as in the liver and kidneys.136 Interestingly, apart from the increased lifespan, cIII-deficient mice with transgenic expression of AOX showed normal-sized hearts with the absence of fibrosis throughout their lives, thereby demonstrating AOX is able to rescue lethal cardiomyopathy. The rescue was correlated with preservation of mitochondrial ultrastructure, metabolic stress relief through up-regulation of genes involved in mitochondrial function (including TFAM), and restoration of cardiac mitochondrial respiration. However, the effects of AOX did not extend to ROS damage. Taken together, AOX was shown to rescue mitochondrial cardiomyopathy through respiratory chain bypass, transferring electrons from quinols to oxygen without proton translocation, thus maintaining electron flow when complex III-to-IV segments of mitochondrial OXPHOS machinery are defective. This study provides evidence that AOX is able to attenuate cIII- or cIV-related pathological development, and is potentially translatable to patients with cIII- or cIV-blockade and associated mitochondrial cardiomyopathy using gene therapy approaches.136
Mitochondrial structural abnormalities are common in mitochondrial diseases. Electron microscopy of a patient with mitochondrial cardiomyopathy, secondary to mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome, has revealed morphosis of multi-layered mitochondria that is believed to contribute to the pathophysiology of MELAS.137 Furthermore, endomyocardial biopsy from a 46-year-old man with congestive heart failure revealed a novel point mutation in mtDNA with cardiomyocytes exhibiting deformed mitochondria of various shapes (spherical, oval, and enlarged forms). Transmission electron microscopy revealed that the enlarged mitochondria had fused with neighbouring normal-sized mitochondria, which may serve as a compensatory mechanism for functional impairment by mutations in mtDNA.138
9. Insights provided by hiPSCs
Animals have contributed to some of the greatest achievements in medicine in the 19th and 20th centuries,139 and continue to play an important role in biomedical research. However, animal models have their limitations, which mainly include interspecies differences in pathophysiology as well as the length of time involved in generation and validation. For instance, the generation of type 2 diabetes mellitus mice using low-dose streptozotocin and high-fat diet requires 18 weeks to induce type 2 diabetes mellitus (3 days of intraperitoneal injection of streptozotocin with 18 weeks of high-fat diet),52 whereas type 2 diabetes mellitus rat model requires 8 weeks to induce type 2 diabetes mellitus (8 weeks of high-fat diet followed by one dose of streptozotocin).140 Moreover, the murine model does not recapitulate certain hallmarks of DMCM, including LV systolic dysfunction and LV hypertrophy.52 Furthermore, in patients with DCM, a switch in titin isoform expression has been described in which failing hearts were found to have a larger fraction of the more compliant N2BA titin isoform. This change in isoform expression reduces myocardial stiffness and affects diastolic filling.141,142 This isoform switch was not reflected in animal models, such as the experimental tachycardia-related cardiomyopathy (a model for DCM and HF) using dogs; instead, the study showed a markedly reduced expression of the N2BA titin isoform.143 By comparison, a murine model of DCM (Bmal1−/− mice) found increased levels of the stiffer N2B isoform and reduction of the more compliant N2BA isoform,144 demonstrating interspecies differences in the pathogenesis of DCM.
Additionally, the major myosin isoforms are differentially expressed in humans vs. mice, as β-myosin is the predominant form in humans and α-myosin (encoded by MYH6) in mice. Consequently, murine studies evaluating the causal mutations in β-myosin in HCM patients induced these mutations on a MYH6 backbone. Although mutant MYH6 was sufficient to induce pathogenic phenotypes, the contractile kinetics is significantly different between the two isoforms,145–147 and thus caution is advised when interpreting these results. The discovery of hiPSCs may circumvent these limitations and provide new insights into the pathophysiology underlying cardiomyopathies. The capacity of hiPSCs to retain patient-specific information, including genetic and epigenetic characteristics,148 makes them suitable candidates for modelling cardiac diseases.149–153
Assessing the contractile properties of hiPSC-derived cardiomyocytes (hiPSC-CMs) is an important parameter when evaluating a disease phenotype. Assessing contractile function at the cellular level may not accurately reflect the contractile properties at the tissue level. However, this can be overcome by using 3D engineered heart tissue (EHT).154–158 Researchers have described methods to generate EHT using collagen- and fibrin-based matrix between flexible silicone posts. The EHT is capable of spontaneous contraction and force development. Furthermore, this approach can show morphological maturation as well as metabolic maturation with increased fatty acid oxidation and reduced anaerobic glycolysis. Importantly, the 3D EHT has a higher mitochondrial mass compared to 2D cultures of hiPSC-CMs and similar mitochondrial proteomic profiles compared to adult human cardiomyocytes.155
Similarly, when hiPSC-CMs and human dermal fibroblasts were mixed together in a fibrin hydrogel and allowed to polymerize around pillars, the resulting stretching motion created passive tension within the EHT, leading to cardiomyocyte elongation and alignment. Physical conditioning via electrical stimulation with increasing intensity over time led to an adult-like gene expression profile, with remarkably organized ultrastructure, physiological sarcomere length, dense mitochondria, transverse tubules, OXPHOS, functional calcium handling, and a positive force-frequency relationship in the EHT.159 The authors did however observe slower development of the electromechanical properties which were at a lower maturation state than the adult human myocardium. Nevertheless, the notion that 3D cultures result in enhanced structural and functional maturity, as compared to 2D cultures is consistent across several studies.160,161 With regard to metabolic maturity, when whole-transcriptome and C-metabolic flux analysis were performed on 2D and 3D cultures, the latter was found to have decreased levels of glycolytic and lipid biosynthesis genes with concurrent increase in OXPHOS genes. These 3D aggregates also demonstrated lower glycolytic and fatty acid synthesis flux, with increased TCA-cycle activity, indicative of enhanced metabolic maturity on the molecular and fluxome level in comparison to 2D cultures.160
9.1 Recapitulating cardiac metabolism
As highlighted in this review, metabolic perturbations play key roles in the pathophysiology of various cardiomyopathies. As such, when modelling cardiomyopathies, it is essential for hiPSCs to recapitulate maladaptive metabolic alterations. One of the early limitations of hiPSC-CMs to model complex diseases was their immature phenotype.162 More recently, researchers have used various methods to facilitate the maturation process with the aim of attaining a more adult-like phenotype. These include prolonged culture,163,164 electrical/mechanical stimulation,155,165–167 modulation of the extracellular matrix,168,169 and the use of microRNAs.170 Marked changes in metabolic preference have been reported during development, as foetal cardiomyocytes transition from a primarily carbohydrate-based metabolism to a fatty acid-based metabolism in adulthood, which is accompanied by the maturation of mitochondrial processes.11 Recognizing that cellular metabolism plays an important role in the pathogenesis of cardiac diseases as well as during cardiomyocyte development, recent studies have placed emphasis on attaining hiPSC-CM maturation through modulation of the metabolic environment.
Healthy human adult cardiomyocytes derive the majority of their energy requirements through fatty acid oxidation, but can also exhibit metabolic plasticity by metabolizing glucose or other substrates when fatty acid oxidation is impaired or unavailable.171 This has been demonstrated by studies showing that activation of PPAR-α (a major transcriptional regulator in fatty acid metabolism) induces an adult-like metabolism as well as mitochondrial maturation with active OXPHOS in hiPSC-CMs, without initiating pathological phenotypes.171 To circumvent the absence of physiological factors that would normally be present in the heart, researchers have also identified several postnatal factors that could improve the functional properties of hiPSC-CMs in serum-free culture. These factors included triiodothyronine (T3), insulin-like growth factor 1 (IGF-1), and glucocorticoid dexamethasone. T3 was found to increase the resting membrane potential, which is essential for cardiomyocyte excitability and contractility, whereas IGF-1 and dexamethasone provided synergistic effects to stimulate cellular bioenergetics and generate traction force.172 Other studies also found that inhibition of HIF-1α and the downstream target, LDHA, led to a metabolic shift from anaerobic glycolysis to OXPHOS in hiPSC-CMs, with enhanced mitochondrial content and ATP levels. The metabolic maturation of hiPSC-CMs was accompanied by functional maturation, as well as improved sarcomere length and contractility.173 A recent study found that combining the treatment of the three separate studies (PPAR-α activation, HIF-1α inhibition, and three postnatal factors T3, IGF-1, and dexamethasone) resulted in a significant improvement in the metabolic and functional maturation of hiPSC-CMs.174 Furthermore, our group demonstrated that direct supplementation of fatty acids (palmitate/oleate) can promote metabolic maturation in hiPSC-CMs, as evident from the enhanced mitochondrial remodelling, oxygen consumption rates, and ATP production. Further bioenergetic analysis showed increased mitochondrial OXPHOS along with ultra-structural enhancements, suggesting that fatty acids induce both metabolic and structural maturation.175 These findings were validated in two independent studies showing similar results.176,177 Taken together, new strategies that facilitate the metabolic and functional maturation of hiPSC-CMs have shown promising results, highlighting the feasibility of hiPSC-CMs to recapitulate adult-like metabolic phenotypes, making them applicable to model cardiomyopathies with underlying metabolic perturbations.
9.2 Modelling perturbed metabolism in cardiomyopathies
In addition to retaining patient genetic and epigenetic information, hiPSCs allow for the generation of isogenic pairs, offering appropriate controls to be compared against. This is an advantage over animal models or primary cell lines in which appropriate controls are often unavailable, leaving the results confounded by heterogeneity. Isogenic pairs are particularly useful when the pathogenesis of the disease involves a genetic background and epigenetics, such as in cases of HCM.178 Researchers generated 11 isogenic variants in three hiPSC lines with c. C9123T-MYH7 (p.R453C-βMHC) substitution mutation and found that the R453C-βMHC mutation alone was sufficient to cause severe pathology, regardless of genetic background. Results from ATP, calcium handling, and transcriptomic assays on the isogenic hiPSC-CMs supported the energy depletion hypothesis, which postulates that sarcomere disorganization leads to inefficient ATP utilization, resulting in increased energy demands. Interestingly, the study showed that mitochondrial content was unchanged, which the authors attributed to the increased energy demand from similar mitochondrial contents that could result in increased ROS and cellular stress, as seen in mitochondrial cardiomyopathy.179 Similarly, the phospholamban mutation, PLNR9C, has been implicated in DCM. Using clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 technology to insert a hereditary PLNR9C mutation, researchers were able to generate isogenic hiPSC-CMs. Transcriptomic analysis showed up-regulation of genes involved in glycolysis and down-regulation of genes involved in fatty acid β-oxidation, including PPAR-α, demonstrating a metabolic shift from aerobic to anaerobic metabolism. Further transcriptomic analysis also showed that miRNA perturbations may contribute to metabolic disruption.180
Studies using patient-derived hiPSCs have also provided insights into Barth syndrome, an X-linked genetic disorder caused by Tafazzin gene (TAZ) mutation and characterized by mitochondrial cardiomyopathy. hiPSC-CMs generated from a patient with Barth syndrome showed increased mitochondrial fragmentation, as well as mitochondrial functional impairment, due to depletion of mature cardiolipin. Interestingly, the study showed that the contractile dysfunction in Barth syndrome was not a result of a global cellular energy deficit, but was due to impairment of the sarcomere assembly. The sarcomere disorganization was attributed to increased generation of ROS, which upon suppression resulted in improved metabolism, sarcomere organization, and contractile properties in diseased hiPSC-CMs. Therefore, strategies that enhance ROS scavenging may be beneficial for patients with Barth syndrome.181
Human iPSC-CMs have been reported to interrogate rare and less-studied diseases, such as DCM with ataxia (DCMA), a Barth syndrome-like condition. DCMA, a form of mitochondrial cardiomyopathy, is caused by a splice mutation in the DNAJC19 gene, which has been proposed to facilitate the import of proteins into the mitochondria of cardiomyocytes.182 When hiPSC-CMs were generated from DCMA patients, researchers observed mitochondrial defects, including fragmentation and abnormal mitochondrial morphology associated with an imbalanced isoform ratio of OPA1, a mitochondrial fusion protein. Interestingly, treatment with SS-31, a member of Szeto-Schiller peptides that target the inner mitochondrial membrane and protects mitochondrial cristae, was able to restore the OPA1 isoform ratio balance and rescue mitochondrial abnormalities, thereby providing further insight into the underlying mechanism of DCMA and more importantly, highlighting SS-31 as a potential therapeutic compound for DCMA.183
Other mechanistic insights brought forth by hiPSC-CMs include the assessment of mitochondrial mutations in HCM. Previously, mtDNA mutations were proposed to be involved in maternally inherited HCM, however, due to the lack of appropriate models, the underlying pathogenesis remained inconclusive. When assessing hiPSC-CMs generated from patients with maternally inherited HCM, researchers were able to demonstrate that the m.2336T>C mutation in the mitochondrial 16S rRNA gene (MT-RNR2) resulted in mitochondrial dysfunction, ultrastructural defects, reduction in ATP/ADP ratio and mitochondrial membrane potential which culminated in an HCM-like phenotype.114
The PRKAG2 gene encodes for regulator γ2 subunit of AMPK (a critical modulator of cellular energy homoeostasis). Hence, mutations within this gene give rise to cardiomyopathy characterized by progressive cardiac conduction disorder and ventricular hypertrophy. Evidence of excessive glycogen deposition and increased AMPK activities in PRKAG2 mutation-associated cardiomyopathy was found using hiPSC-CMs derived from patients with PRKAG ZW/ZJ. The study also showed that AMPK inhibition was able to reduce the glycogen accumulation and rescue cardiac remodelling in PRKAG2 cardiac syndrome.184
Lastly, hiPSC-CMs have been reported to retain the epigenetic background implicated in DMCM. Researchers generated hiPSC-CMs from patients with type 2 diabetes mellitus at different disease progression states to serve as an in vitro platform for compound screening. The patient-derived hiPSC-CMs successfully recapitulated the pathological hallmarks of DMCM, including glucose restriction, induced distortion of cell structure, lipid accumulation, oxidative stress, and cellular hypertrophy. The hiPSC-CMs were able to develop a DMCM phenotype in the absence of a diabetic stimulus, showing the genetic/epigenetic predisposition towards DMCM. Remarkably, the patient-derived hiPSC-CMs exhibited varying severity of pathological phenotypes that correlated with the original clinical status of the patient185 which led to the postulation that fast-progressing diabetes may have underlying epigenetic involvement, however, given that the study provided limited genetic/epigenetic data, such conclusions would warrant independent confirmation.
9.3 Advantages of hiPSCs
Research using hiPSCs has several advantages (Figure 2), one of which involves the availability of required starting materials. hiPSCs could be generated from many different somatic cell types, including fibroblasts and peripheral blood mononuclear cells, which are easily accessible and require minimal invasive procedures. This circumvents the need for invasive procedures, such as endomyocardial biopsies. Furthermore, as the parental hiPSCs can be maintained in culture indefinitely, cardiomyocytes could be generated when required, using a number of established protocols.186–188 This is highly advantageous compared to the process of rodent cardiomyocyte isolation, which is laborious and highly dependent on the skills of individual researchers (e.g. in obtaining cell number and viability) and the variable methods used for isolation. Most importantly, because hiPSCs contain the genetic makeup of the individual from which they were generated, the underlying predisposition towards any disease can be recapitulated in a petri-dish. This feature makes hiPSCs an ideal platform to study the pathophysiology of cardiac diseases, including metabolic perturbations. As an in vitro assaying platform, hiPSC-CMs allow for the manipulation of environmental substrates easily through the use of different culture media. For instance, a diabetic environment could be created in as little as 2 days with all accompanying pathological features,185 compared to up to 18 weeks in an animal model.52 Similarly, the time needed to generate hiPSC-CM models for HCM189 or DCM151 is much shorter compared to conventional animal models.190–193 Finally, another appealing aspect of using hiPSC-CMs especially when modelling perturbed metabolism and mitochondrial dysfunction is their compatibility with various cellular assays which assess fuel preference and bioenergetics with relative ease. For example, measurement of oxygen consumption rate and extracellular acidification rate can provide a good indication of energy metabolism under both basal and stressed environments. Similarly, analysis of acylcarnitines and TCA-cycle intermediates can provide insight into fuel preference and energy flux under healthy and diseased conditions. Imaging studies using mitochondrial dyes (e.g. TMRM, JC-1) can also provide information on mitochondria membrane potential and when coupled with ROS, ATP, and ADP determination assays, can help interrogate impaired substrate selectivity and/or impaired bioenergetics which could be a major contributor to disease pathology.
Figure 2.

Schematic illustration summarizing the advantages and disadvantages of hiPSCs and animal models in terms of identifying novel targets and personalized therapies.
In a landmark clinical trial (EMPA-REG OUTCOME), empagliflozin, a sodium-glucose co-transporter-2 (SGLT-2) inhibitor was reported to lower CV-related death and reduce hospitalization for HF.194 In light of these positive outcomes, investigators have attempted to identify the mode of action for empagliflozin. In a recent study, when exposed to a diabetic-like environment comprising high-glucose and insulin-free culture conditions, hiPSC-CMs demonstrated cellular hypertrophy and reduced contractility with accompanying increase in SGLT1 and SGLT2 expressions.195 Interestingly, empagliflozin treatment pre- or post-high-glucose exposure was found to attenuate the high-glucose-induced cardiac abnormalities, with no effect on cell viability nor on the glycolytic capacity of hiPSC-CMs. Since SGLT1 and SGLT2 expression was found to be down-regulated post-treatment, this led to the postulation that empagliflozin may exert protective effects directly through the reduction of SGLT1 and SGLT2.195
The findings in hiPSC-CMs are consistent with reports that associate an increase in cardiac SGLT1 with DMCM,196 however, the postulation that empagliflozin works via down-regulation of SGLT-1 and SGLT-2 should be viewed with caution. While SGLT1 has been found to be expressed in human cardiomyocytes,196,197 the expression of SGLT2 in the heart remains debatable.198–201 Though the mechanisms by which SGLT-2 inhibitors exert cardioprotective effects are yet to be identified, several hypotheses have been proposed which includes, natriuretic and diuretic effects202–205 as well as inhibition of the myocardial Na+/H+ exchanger which results in improved myocardial calcium handling.206,207 It would therefore seem that the beneficial effects exerted by SGLT-2 inhibitors is multifaceted, affecting various tissues and organs, including the kidney208–211 and vasculature.212,213
Human iPSC-CMs also offer a far more accurate platform for toxicity studies in drug development compared to conventional approaches. A library of hiPSCs was generated from patients with various hereditary cardiac disorders, including DCM (TNNT2R173W) and HCM (MYH7R663H). The hiPSC-CMs retained pathogenic phenotypes; for instance, hiPSC-CMs derived from HCM patients showed cellular enlargement and sarcomere disorganization and those derived from DCM patients also exhibited sarcomere disorganization. Transcriptomic assays showed that these disease-specific hiPSC-CMs expressed all cardiac ion channels of adult LV cardiac tissue, suggesting retention of electrophysiology of the adult human heart. Given that DCM and HCM patients are more susceptible to drug-induced cardiotoxicity, it is particularly important to recapitulate this drug-susceptibility in the toxicity assay. By recapitulating the clinical susceptibility of high-risk patient populations to drug-induced cardiotoxicity, a patient-derived hiPSC-CM library offers a vastly superior platform for toxicity studies.214
The potential for scalability of hiPSC-CMs in 3D culture215 together with the recent characterization of comparative electrophysiology216 offer a unique platform for high-throughput phenotypic screening of drugs and assaying for cardiotoxicity. In a recent study, 21 FDA-approved tyrosine kinase inhibitors (TKIs) were screened for cardiotoxicity using hiPSC-CMs derived from 11 healthy individuals and 2 patients undergoing cancer treatment.217 A majority of TKIs was found to promote cardiotoxicity, with some having more profound effects than others, affecting several cardiovascular cell types via inhibition of VEGFR2/PDGFR signalling. Importantly, the cardiotoxic effect of TKIs could be reversed via insulin/IGF1 signalling. Through the use of hiPSC-CMs, the researchers were able to establish a ‘cardiac safety index’ for TKI cardiotoxicity,217 further substantiating the use of hiPSC-CMs in high-throughput cardiotoxicity screens.
In an attempt to establish a hazard score system for cardiac electrical liabilities, researchers demonstrated that calcium transient screening assays are able to identify drugs (from 587 new chemical entities) that target various cardiac ion channels and receptors, thereby providing a comprehensive drug profile on cardiovascular safety.218 A more recent study has also demonstrated the potential for hiPSC-CMs to provide functional screening of 105 small molecules for pro-regenerative benefits.219 When treated with selected compounds, high-throughput proteomic analysis of 3D cardiac organoids showed synergistic activation of the mevalonate pathway and cell-cycle network, providing mechanistic insight into the small molecules being tested. These studies exemplify the versatility and robustness of the high-throughput approach offered by hiPSC-CMs, making them highly appropriate for preclinical drug development.
Apart from phenotypic screens where the target is unknown, hiPSC-CMs can also be used as a platform for known target screens. MAP4K4 has been reported to be activated in HF in both human and rodent models, and likely contributes to the pathological presentation through interactions with TAK1, JNK, and p38 MAPK.220,221 When hiPSC-CMs were treated with hydrogen peroxide, researchers were able to model ischaemic injury and subsequently establish the cardioprotective benefits of an initial screen hit and an optimized analogue, DMX-5804 (a small molecule inhibitor) which targets MAP4K4.222 Consistent with the in vitro observations, DMX-5804 was found to reduce ischaemia-reperfusion injury by >50% in a mouse ischaemia-reperfusion injury model, thereby highlighting the use of hiPSC-CMs for known target screens.
In addition to phenotypic/target screens and cardiotoxicity studies, experimental studies with hiPSC-CMs can be undertaken in parallel with clinical trials to gather additional information, which may have a direct influence on the related clinical study. A recent study has validated the application of hiPSC-CMs in identifying patient-specific drugs for cardiomyopathy. The proof-of-concept study used hiPSC-CMs derived from a patient with HCM who had a rare mutation in the gene encoding α-actinin 2 (ACTN2T247M). The resulting EHTs recapitulated hallmarks of HCM, including increased L-type Ca2+ current. Diltiazem, a L-type Ca2+ channel blocker, was found to decrease force amplitude, relaxation, and action potential duration in HCM hiPSC-CMs, when compared to the isogenic control. These findings were later translated to patient care. Consistent with the results from the hiPSC-CM assay, treatment with diltiazem reduced the prolonged QT intervals in the HCM-affected son and sister of the index patient, but did not alter the QT intervals in healthy controls. Taken together, the study provides evidence that ACTN2 missense mutations cause HCM on the cellular level and that patient-derived hiPSC-CMs offer a promising platform for personalized therapy in cardiomyopathies.96
9.4 Limitations of hiPSCs
Although hiPSC-CMs are highly suitable for modelling human cardiomyopathies, they do possess some limitations. Culture conditions may have profound effects on disease phenotype as observed previously. Researchers found that serum could increase cellular surface area of healthy hiPSC-CMs, but not hiPSC-CMs derived from HCM patients.223 The reduced phenotypic differences between control and disease-associated hiPSC-CMs may in turn underestimate the effect of compounds in drug-related studies. Furthermore, the undefined nature of serum and associated batch batch-to-batch variations can potentially hinder the establishment of a disease model, leading to inconsistencies in mechanistic studies and drug screening, thereby necessitating the requirement for defined/serum-free cultures. An alternative could be the use of serum-free media and though this may not represent physiological conditions, it does provide a controlled environment not marred by undefined factors, which is a critical prerequisite when attempting to determine compound efficacy and safety. To circumvent the absence of physiological factors and improve the functional properties of hiPSC-CMs in serum-free culture, supplementation with postnatal factors could be considered.172
Human iPSCs contain the genetic/epigenetic makeup of the individual from which they were generated, potentially enabling them to recapitulate underlying predispositions towards any disease. However, it is important to note that such genetic/epigenetic states can be influenced during the reprogramming process and culture conditions. Genetic variations in hiPSCs can lead to unexpected phenotypes in hiPSC-derived progenies as highlighted in a previous study which attempted to model X-linked Lesch–Nyhan syndrome.224 In this study, while early passage hiPSCs were found to retain the inactive X-chromosome, long-term culture resulted in the erosion of the inactive X-chromosome, an effect that could not be reversed through differentiation or further reprogramming.224 These findings are consistent with other reports which suggest long-term culture of hiPSCs to be associated with karyotype abnormalities.7,225–227 Epigenetic variations in hiPSCs, on the other hand, are thought to be attributable to ‘epigenetic memory’ of somatic origin228 and/or culture conditions.229,230 While epigenetic memory is proposed to influence the differentiation potential of hiPSCs, it has also been suggested that the methylation patterns of somatic origin can be erased with increasing cell passage number. It is therefore crucial to optimize the passage number and regularly evaluate the genetic/epigenetic status of the patient-derived hiPSCs, prior to using them in downstream studies.
The purity of hiPSC-CMs is another factor that needs to be considered. High purity of hiPSC-CMs can be obtained via several methods, such as negative selection using lactate. This approach exploits the metabolic plasticity of cardiomyocytes to produce energy from several substrates, including lactate. By simply substituting the culture medium to a glucose-depleted but lactate-enriched medium, an almost pure cardiomyocyte population could be obtained.231 Though this strategy is a simple, cost-effective solution capable of removing residual undifferentiated hiPSCs and generating a purified hiPSC-CM population,232 downstream repercussions could occur when hiPSC-CMs are subjected to an anaerobic substrate. We recently described an alternative method for purifying hiPSC-CMs through the use of a sheath-less acoustic fluorescence-activated cell sorting device that can circumvent the use of an anaerobic substrate.233
10. Conclusions
Maladaptive metabolic alterations are not limited to mitochondrial cardiomyopathies, and as highlighted in this review, most cardiomyopathies do include elements of perturbed metabolism and impaired bioenergetics. Given that mitochondrial OXPHOS contributes to over 90% of the adult heart’s energy requirements, impaired OXPHOS can result in drastic energy deficiency in the heart. Consistently, defective mitochondrial OXPHOS from both fatty acid β-oxidation and glucose oxidation is seen in HF.234 To compensate for this, increased glycolysis may provide a source of energy in the failing heart, but is unable to completely replace the deficit in mitochondrial ATP production.235 Therefore, hiPSC-CM models capable of recapitulating the maladaptive metabolic changes and impaired bioenergetics found in cardiomyopathies can provide much needed new insights into disease progression. In addition, patient-derived hiPSC-CM models possess several key advantages in terms of their ability to retain important aspects of cardiomyopathies. These include, but are not limited to: (i) underlying perturbed metabolism; (ii) genetic predisposition; and (iii) epigenetic factors. Moreover, hiPSC-CMs provide a unique platform for drug screening and toxicology studies. Finally, given their ability to model diseases in a human context, the use of hiPSC-CMs can provide unprecedented knowledge into the pathophysiology of cardiomyopathies by providing appropriate and robust disease models that are capable of interrogating cardiomyopathies at the cellular level, with the ultimate aim of identifying novel targets for improved clinical outcomes in patients with cardiomyopathies.
Conflict of interest: none declared.
Funding
C.J.A.R. was supported by the Singapore Ministry of Health’s National Medical Research Council under its Open Fund-Young Individual Research Grant (OF-YIRG)—(NMRC/OFYIRG/0073/2018) and through the National Health Innovation Centre Singapore under its Innovation to Develop Grant (NHIC-I2S-1811007). S.C. was supported by the Fudan University Shanghai Medical College under its Exchange Program Scholarship for Postgraduate Student (2019201). D.J.H. was supported by the British Heart Foundation (CS/14/3/31002), the National Institute for Health Research University College London Hospitals Biomedical Research Centre, Duke-National University Singapore Medical School, Singapore Ministry of Health’s National Medical Research Council under its Clinician Scientist-Senior Investigator Scheme (NMRC/CSA-SI/0011/2017) and Collaborative Centre Grant Scheme (NMRC/CGAug16C006), and the Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2016-T2-2-021). This article is based upon work from COST Action EU-CARDIOPROTECTION CA16225 supported by COST (European Cooperation in Science and Technology).
References
- 1. Farré N, Vela E, Clèries M, Bustins M, Cainzos-Achirica M, Enjuanes C, Moliner P, Ruiz S, Verdú-Rotellar JM, Comín-Colet J.. Real world heart failure epidemiology and outcome: a population-based analysis of 88,195 patients. PLoS One 2017;12:e0172745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ziaeian B, Fonarow GC.. Epidemiology and aetiology of heart failure. Nat Rev Cardiol 2016;13:368–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McKenna WJ, Maron BJ, Thiene G.. Classification, epidemiology, and global burden of cardiomyopathies. Circ Res 2017;121:722–730. [DOI] [PubMed] [Google Scholar]
- 4. Brieler J, Breeden MA, Tucker J.. Cardiomyopathy: an overview. Am Fam Physician 2017;96:640–646. [PubMed] [Google Scholar]
- 5. Jefferies J, Towbin J.. Dilated cardiomyopathy. Lancet 2010;375:752–762. [DOI] [PubMed] [Google Scholar]
- 6. Seferović PM, Polovina M, Bauersachs J, Arad M, Gal TB, Lund LH, Felix SB, Arbustini E, Caforio ALP, Farmakis D, Filippatos GS, Gialafos E, Kanjuh V, Krljanac G, Limongelli G, Linhart A, Lyon AR, Maksimović R, Miličić D, Milinković I, Noutsias M, Oto A, Oto Ö, Pavlović SU, Piepoli MF, Ristić AD, Rosano GMC, Seggewiss H, Ašanin M, Seferović JP, Ruschitzka F, Čelutkiene J, Jaarsma T, Mueller C, Moura B, Hill L, Volterrani M, Lopatin Y, Metra M, Backs J, Mullens W, Chioncel O, de Boer RA, Anker S, Rapezzi C, Coats AJS, Tschöpe C.. Heart failure in cardiomyopathies: a position paper from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2019;21:553–576. [DOI] [PubMed] [Google Scholar]
- 7. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S.. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 8. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med 2007;356:1140–1151. [DOI] [PubMed] [Google Scholar]
- 9. Fisher DJ, Heymann MA, Rudolph AM.. Myocardial consumption of oxygen and carbohydrates in newborn sheep. Pediatr Res 1981;15:843–846. [DOI] [PubMed] [Google Scholar]
- 10. Burd LI, Jones MD Jr, Simmons MA, Makowski EL, Meschia G, Battaglia FC.. Placental production and foetal utilisation of lactate and pyruvate. Nature 1975;254:710–711. [DOI] [PubMed] [Google Scholar]
- 11. Lopaschuk GD, Collins-Nakai RL, Itoi T.. Developmental changes in energy substrate use by the heart. Cardiovasc Res 1992;26:1172–1180. [DOI] [PubMed] [Google Scholar]
- 12. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Kruger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, Kelly DP.. The failing heart relies on ketone bodies as a fuel. Circulation 2016;133:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carley AN, Lewandowski ED.. Triacylglycerol turnover in the failing heart. Biochim Biophys Acta 2016;1861:1492–1499. [DOI] [PubMed] [Google Scholar]
- 14. Ritterhoff J, Tian R.. Metabolism in cardiomyopathy: every substrate matters. Cardiovasc Res 2017;113:411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Niu YG, Hauton D, Evans RD.. Utilization of triacylglycerol-rich lipoproteins by the working rat heart: routes of uptake and metabolic fates. J Physiol 2004;558:225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fukushima A, Milner K, Gupta A, Lopaschuk GD.. Myocardial energy substrate metabolism in heart failure: from pathways to therapeutic targets. Curr Pharm Des 2015;21:3654–3664. [DOI] [PubMed] [Google Scholar]
- 17. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC.. Myocardial fatty acid metabolism in health and disease. Physiol Rev 2010;90:207–258. [DOI] [PubMed] [Google Scholar]
- 18. Zamora M, Villena JA.. Contribution of impaired insulin signaling to the pathogenesis of diabetic cardiomyopathy. Int J Mol Sci 2019;20. pii: E2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD.. Targeting fatty acid and carbohydrate oxidation—a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta 2011;1813:1333–1350. [DOI] [PubMed] [Google Scholar]
- 20. Horton JL, Davidson MT, Kurishima C, Vega RB, Powers JC, Matsuura TR, Petucci C, Lewandowski ED, Crawford PA, Muoio DM, Recchia FA, Kelly DP.. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight 2019;4. pii: 124079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jie QQ, Li G, Duan JB, Li XB, Yang W, Chu YP, Yu SD, Liu XY, Wang CY, Liu FF, Ze F, Huang YW, Chen Y, Ding YS, Guo JH, Wu L.. Remodeling of myocardial energy and metabolic homeostasis in a sheep model of persistent atrial fibrillation. Biochem Biophys Res Commun 2019;517:8–14. [DOI] [PubMed] [Google Scholar]
- 22. Wu B, Huang XY, Li L, Fan XH, Li PC, Huang CQ, Xiao J, Gui R, Wang S.. Attenuation of diabetic cardiomyopathy by relying on kirenol to suppress inflammation in a diabetic rat model. J Cell Mol Med 2019;23:7651–7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jia G, DeMarco VG, Sowers JR.. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol 2016;12:144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Battiprolu PK, Gillette TG, Wang ZV, Lavandero S, Hill JA.. Diabetic cardiomyopathy: mechanisms and therapeutic targets. Drug Discov Today Dis Mech 2010;7:e135–e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jia G, Whaley-Connell A, Sowers JR.. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 2018;61:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bugger H, Abel ED.. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014;57:660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang JP, Huang SS, Deng JY, Hung LM.. Impairment of insulin-stimulated Akt/GLUT4 signaling is associated with cardiac contractile dysfunction and aggravates I/R injury in STZ-diabetic rats. J Biomed Sci 2009;16:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Desrois M, Sidell RJ, Gauguier D, King LM, Radda GK, Clarke K.. Initial steps of insulin signaling and glucose transport are defective in the type 2 diabetic rat heart. Cardiovasc Res 2004;61:288–296. [DOI] [PubMed] [Google Scholar]
- 29. Qi K, Zhong J.. LncRNA HOTAIR improves diabetic cardiomyopathy by increasing viability of cardiomyocytes through activation of the PI3K/Akt pathway. Exp Ther Med 2018;16:4817–4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hou N, Mai Y, Qiu X, Yuan W, Li Y, Luo C, Liu Y, Zhang G, Zhao G, Luo JD.. Carvacrol attenuates diabetic cardiomyopathy by modulating the PI3K/AKT/GLUT4 pathway in diabetic mice. Front Pharmacol 2019;10:998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stroedter D, Schmidt T, Bretzel RG, Federlin K.. Glucose metabolism and left ventricular dysfunction are normalized by insulin and islet transplantation in mild diabetes in the rat. Acta Diabetol 1995;32:235–243. [DOI] [PubMed] [Google Scholar]
- 32. Witteles RM, Tang WH, Jamali AH, Chu JW, Reaven GM, Fowler MB.. Insulin resistance in idiopathic dilated cardiomyopathy: a possible etiologic link. J Am Coll Cardiol 2004;44:78–81. [DOI] [PubMed] [Google Scholar]
- 33. Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, Shelton JM, Gerard RD, Rothermel BA, Gillette TG, Lavandero S, Hill JA.. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest 2012;122:1109–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Song R, Peng W, Zhang Y, Lv F, Wu HK, Guo J, Cao Y, Pi Y, Zhang X, Jin L, Zhang M, Jiang P, Liu F, Meng S, Zhang X, Jiang P, Cao CM, Xiao RP.. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature 2013;494:375–379. [DOI] [PubMed] [Google Scholar]
- 35. Liu F, Song R, Feng Y, Guo J, Chen Y, Zhang Y, Chen T, Wang Y, Huang Y, Li CY, Cao C, Zhang Y, Hu X, Xiao RP.. Upregulation of MG53 induces diabetic cardiomyopathy through transcriptional activation of peroxisome proliferation-activated receptor alpha. Circulation 2015;131:795–804. [DOI] [PubMed] [Google Scholar]
- 36. Davila-Roman VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, Gropler RJ.. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol 2002;40:271–277. [DOI] [PubMed] [Google Scholar]
- 37. Martins AR, Nachbar RT, Gorjao R, Vinolo MA, Festuccia WT, Lambertucci RH, Cury-Boaventura MF, Silveira LR, Curi R, Hirabara SM.. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: importance of the mitochondrial function. Lipids Health Dis 2012;11:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shuldiner AR, McLenithan JC.. Genes and pathophysiology of type 2 diabetes: more than just the Randle cycle all over again. J Clin Invest 2004;114:1414–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pilz S, Marz W.. Free fatty acids as a cardiovascular risk factor. Clin Chem Lab Med 2008;46:429–434. [DOI] [PubMed] [Google Scholar]
- 40. Cai L, Wang Y, Zhou G, Chen T, Song Y, Li X, Kang YJ.. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol 2006;48:1688–1697. [DOI] [PubMed] [Google Scholar]
- 41. Hu W, Ross J, Geng T, Brice SE, Cowart LA.. Differential regulation of dihydroceramide desaturase by palmitate versus monounsaturated fatty acids implications for insulin resistance. J Biol Chem 2011;286:16596–16605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ying Y, Zhu H, Liang Z, Ma X, Li S.. GLP1 protects cardiomyocytes from palmitate-induced apoptosis via Akt/GSK3b/b-catenin pathway. J Mol Endocrinol 2015;55:245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. D’Souza K, Nzirorera C, Kienesberger PC.. Lipid metabolism and signaling in cardiac lipotoxicity. Biochim Biophys Acta 2016;1861:1513–1524. [DOI] [PubMed] [Google Scholar]
- 44. Szczepaniak LS, Victor RG, Orci L, Unger RH.. Forgotten but not gone: the rediscovery of fatty heart, the most common unrecognized disease in America. Circ Res 2007;101:759–767. [DOI] [PubMed] [Google Scholar]
- 45. Rial E, Rodríguez-Sánchez L, Gallardo-Vara E, Zaragoza P, Moyano E, González-Barroso MM.. Lipotoxicity, fatty acid uncoupling and mitochondrial carrier function. Biochim Biophys Acta 2010;1797:800–806. [DOI] [PubMed] [Google Scholar]
- 46. Angin Y, Steinbusch LK, Simons PJ, Greulich S, Hoebers NT, Douma K, van Zandvoort MA, Coumans WA, Wijnen W, Diamant M, Ouwens DM, Glatz JF, Luiken JJ.. CD36 inhibition prevents lipid accumulation and contractile dysfunction in rat cardiomyocytes. Biochem J 2012;448:43–53. [DOI] [PubMed] [Google Scholar]
- 47. Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, Kelly DP.. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res 2007;100:1208–1217. [DOI] [PubMed] [Google Scholar]
- 48. Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN, Liang Q.. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem 2013;288:18077–18092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, Watanabe T, Morishita K, Okada H, Kawasaki M, Seishima M, Minatoguchi S.. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy 2015;11:1146–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J.. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res 2019;124:1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wanrooy BJ, Kumar KP, Wen SW, Qin CX, Ritchie RH, Wong C.. Distinct contributions of hyperglycemia and high-fat feeding in metabolic syndrome-induced neuroinflammation. J Neuroinflammation 2018;15:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tate M, Prakoso D, Willis AM, Peng C, Deo M, Qin CX, Walsh JL, Nash DM, Cohen CD, Rofe AK, Sharma A, Kiriazis H, Donner DG, De Haan JB, Watson AMD, De Blasio MJ, Ritchie RH.. Characterising an alternative murine model of diabetic cardiomyopathy. Front Physiol 2019;10:1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shi X, Chen R, Zhang Y, Yun J, Brand-Arzamendi K, Liu X, Wen XY.. Zebrafish heart failure models: opportunities and challenges. Amino Acids 2018;50:787–798. [DOI] [PubMed] [Google Scholar]
- 54. Zhang L, Wu YJ, Zhang SL.. Circulating lncRNA MHRT predicts survival of patients with chronic heart failure. J Geriatr Cardiol 2019;16:818–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nomura S. Genetic and non-genetic determinants of clinical phenotypes in cardiomyopathy. J Cardiol 2019;73:187–190. [DOI] [PubMed] [Google Scholar]
- 56. Cheedipudi SM, Matkovich SJ, Coarfa C, Hu X, Robertson MJ, Sweet M, Taylor M, Mestroni L, Cleveland J, Willerson JT, Gurha P, Marian AJ.. Genomic reorganization of lamin-associated domains in cardiac myocytes is associated with differential gene expression and DNA methylation in human dilated cardiomyopathy. Circ Res 2019;124:1198–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gilsbach R, Schwaderer M, Preissl S, Grüning BA, Kranzhöfer D, Schneider P, Nührenberg TG, Mulero-Navarro S, Weichenhan D, Braun C, Dreßen M, Jacobs AR, Lahm H, Doenst T, Backofen R, Krane M, Gelb BD, Hein L.. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat Commun 2018;9:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Meder B, Haas J, Sedaghat-Hamedani F, Kayvanpour E, Frese K, Lai A, Nietsch R, Scheiner C, Mester S, Bordalo DM, Amr A, Dietrich C, Pils D, Siede D, Hund H, Bauer A, Holzer DB, Ruhparwar A, Mueller-Hennessen M, Weichenhan D, Plass C, Weis T, Backs J, Wuerstle M, Keller A, Katus HA, Posch AE.. Epigenome-wide association study identifies cardiac gene patterning and a novel class of biomarkers for heart failure. Circulation 2017;136:1528–1544. [DOI] [PubMed] [Google Scholar]
- 59. Buchwald A, Till H, Unterberg C, Oberschmidt R, Figulla HR, Wiegand V.. Alterations of the mitochondrial respiratory chain in human dilated cardiomyopathy. Eur Heart J 1990;11:509–516. [DOI] [PubMed] [Google Scholar]
- 60. Schulze K, Becker BF, Schauer R, Schultheiss HP.. Antibodies to ADP-ATP carrier–an autoantigen in myocarditis and dilated cardiomyopathy–impair cardiac function. Circulation 1990;81:959–969. [DOI] [PubMed] [Google Scholar]
- 61. Schultheiss HP. Disturbance of the myocardial energy metabolism in dilated cardiomyopathy due to autoimmunological mechanisms. Circulation 1993;87:IV43–IV48. [PubMed] [Google Scholar]
- 62. Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ.. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 1995;11:376–381. [DOI] [PubMed] [Google Scholar]
- 63. Sibbing D, Pfeufer A, Perisic T, Mannes AM, Fritz-Wolf K, Unwin S, Sinner MF, Gieger C, Gloeckner CJ, Wichmann HE, Kremmer E, Schafer Z, Walch A, Hinterseer M, Nabauer M, Kaab S, Kastrati A, Schomig A, Meitinger T, Bornkamm GW, Conrad M, von Beckerath N.. Mutations in the mitochondrial thioredoxin reductase gene TXNRD2 cause dilated cardiomyopathy. Eur Heart J 2011;32:1121–1133. [DOI] [PubMed] [Google Scholar]
- 64. Dörner A, Giessen S, Gaub R, Siestrup HG, Schwimmbeck PL, Hetzer R, Poller W, Schultheiss H-P.. An isoform shift in the cardiac adenine nucleotide translocase expression alters the kinetic properties of the carrier in dilated cardiomyopathy. Eur J Heart Fail 2006;8:81–89. [DOI] [PubMed] [Google Scholar]
- 65. Dorner A, Olesch M, Giessen S, Pauschinger M, Schultheiss HP.. Transcription of the adenine nucleotide translocase isoforms in various types of tissues in the rat. Biochim Biophys Acta 1999;1417:16–24. [DOI] [PubMed] [Google Scholar]
- 66. Narula N, Zaragoza MV, Sengupta PP, Li P, Haider N, Verjans J, Waymire K, Vannan M, Wallace DC.. Adenine nucleotide translocase 1 deficiency results in dilated cardiomyopathy with defects in myocardial mechanics, histopathological alterations, and activation of apoptosis. JACC Cardiovasc Imaging 2011;4:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dorner A, Pauschinger M, Schwimmbeck PL, Kuhl U, Schultheiss HP.. The shift in the myocardial adenine nucleotide translocator isoform expression pattern is associated with an enteroviral infection in the absence of an active T-cell dependent immune response in human inflammatory heart disease. J Am Coll Cardiol 2000;35:1778–1784. [DOI] [PubMed] [Google Scholar]
- 68. Diguet N, Trammell SAJ, Tannous C, Deloux R, Piquereau J, Mougenot N, Gouge A, Gressette M, Manoury B, Blanc J, Breton M, Decaux JF, Lavery GG, Baczko I, Zoll J, Garnier A, Li Z, Brenner C, Mericskay M.. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation 2018;137:2256–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bindu S, Pillai VB, Gupta MP.. Role of sirtuins in regulating pathophysiology of the heart. Trends Endocrinol Metab 2016;27:563–573. [DOI] [PubMed] [Google Scholar]
- 70. Viswanathan SK, Sanders HK, McNamara JW, Jagadeesan A, Jahangir A, Tajik AJ, Sadayappan S.. Hypertrophic cardiomyopathy clinical phenotype is independent of gene mutation and mutation dosage. PLoS One 2017;12:e0187948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Semsarian C, Ingles J, Maron MS, Maron BJ.. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015;65:1249–1254. [DOI] [PubMed] [Google Scholar]
- 72. Geisterfer-Lowrance AAT, Kass S, Tanigawa G, Vosberg H-P, McKenna W, Seidman CE, Seidman JG.. A molecular basis for familial hypertrophic cardiomyopathy: a β cardiac myosin heavy chain gene missense mutation. Cell 1990;62:999–1006. [DOI] [PubMed] [Google Scholar]
- 73. Hossain MB, Elbeck Z, Siga H, Knöll R.. Myosin binding protein-C and hypertrophic cardiomyopathy: role of altered C10 domain. Cardiovasc Res 2019;115:1943–1945. [DOI] [PubMed] [Google Scholar]
- 74. Makavos G, Κairis C, Tselegkidi M-E, Karamitsos T, Rigopoulos AG, Noutsias M, Ikonomidis I.. Hypertrophic cardiomyopathy: an updated review on diagnosis, prognosis, and treatment. Heart Fail Rev 2019;24:439–459. [DOI] [PubMed] [Google Scholar]
- 75. Maron BJ, Maron MS.. Hypertrophic cardiomyopathy. The Lancet 2013;381:242–255. [DOI] [PubMed] [Google Scholar]
- 76. Lafreniere-Roula M, Bolkier Y, Zahavich L, Mathew J, George K, Wilson J, Stephenson EA, Benson LN, Manlhiot C, Mital S.. Family screening for hypertrophic cardiomyopathy: is it time to change practice guidelines? Eur Heart J 2019;40:3672–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Higashikuse Y, Mittal N, Arimura T, Yoon SH, Oda M, Enomoto H, Kaneda R, Hattori F, Suzuki T, Kawakami A, Gasch A, Furukawa T, Labeit S, Fukuda K, Kimura A, Makino S.. Perturbation of the titin/MURF1 signaling complex is associated with hypertrophic cardiomyopathy in a fish model and in human patients. Dis Model Mech 2019;12. pii: dmm041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McKenna WJ, Moon JC, Sulaiman A.. Understanding the myocardial architecture of hypertrophic cardiomyopathy for clinical care. J Am Coll Cardiol 2019;73:2503–2505. [DOI] [PubMed] [Google Scholar]
- 79. Grover-McKay M, Schwaiger M, Krivokapich J, Perloff JK, Phelps ME, Schelbert HR.. Regional myocardial blood flow and metabolism at rest in mildly symptomatic patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 1989;13:317–324. [DOI] [PubMed] [Google Scholar]
- 80. Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, McKenna WJ, Ostman-Smith I, Clarke K, Watkins H.. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol 2003;41:1776–1782. [DOI] [PubMed] [Google Scholar]
- 81. Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG, Ingwall JS.. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. J Clin Invest 1998;101:1775–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Abozguia K, Elliott P, McKenna W, Phan TT, Nallur-Shivu G, Ahmed I, Maher Abdul R, Kaur K, Taylor J, Henning A, Ashrafian H, Watkins H, Frenneaux M.. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation 2010;122:1562–1569. [DOI] [PubMed] [Google Scholar]
- 83. Toepfer CN, Wakimoto H, Garfinkel AC, McDonough B, Liao D, Jiang J, Tai AC, Gorham JM, Lunde IG, Lun M, Lynch TLt, McNamara JW, Sadayappan S, Redwood CS, Watkins HC, Seidman JG, Seidman CE.. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci Transl Med 2019;11. pii: eaat1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Haack TB, Kopajtich R, Freisinger P, Wieland T, Rorbach J, Nicholls TJ, Baruffini E, Walther A, Danhauser K, Zimmermann FA, Husain RA, Schum J, Mundy H, Ferrero I, Strom TM, Meitinger T, Taylor RW, Minczuk M, Mayr JA, Prokisch H.. ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am J Hum Genet 2013;93:211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Saoura M, Powell CA, Kopajtich R, Alahmad A, Al-Balool HH, Albash B, Alfadhel M, Alston CL, Bertini E, Bonnen PE, Bratkovic D, Carrozzo R, Donati MA, Di Nottia M, Ghezzi D, Goldstein A, Haan E, Horvath R, Hughes J, Invernizzi F, Lamantea E, Lucas B, Pinnock KG, Pujantell M, Rahman S, Rebelo-Guiomar P, Santra S, Verrigni D, McFarland R, Prokisch H, Taylor RW, Levinger L, Minczuk M.. Mutations in ELAC2 associated with hypertrophic cardiomyopathy impair mitochondrial tRNA 3’-end processing. Hum Mutat 2019;40:1731–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chen D, Zhang Z, Chen C, Yao S, Yang Q, Li F, He X, Ai C, Wang M, Guan MX.. Deletion of Gtpbp3 in zebrafish revealed the hypertrophic cardiomyopathy manifested by aberrant mitochondrial tRNA metabolism. Nucleic Acids Res 2019;47:5341–5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chen D, Li F, Yang Q, Tian M, Zhang Z, Zhang Q, Chen Y, Guan MX.. The defective expression of gtpbp3 related to tRNA modification alters the mitochondrial function and development of zebrafish. Int J Biochem Cell Biol 2016;77:1–9. [DOI] [PubMed] [Google Scholar]
- 88. Villarroya M, Prado S, Esteve JM, Soriano MA, Aguado C, Pérez-Martínez D, Martínez-Ferrandis JI, Yim L, Victor VM, Cebolla E, Montaner A, Knecht E, Armengod M-E.. Characterization of human GTPBP3, a GTP-binding protein involved in mitochondrial tRNA modification. Mol Cell Biol 2008;28:7514–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Li X, Guan MX.. Identification and characterization of mouse GTPBP3 gene encoding a mitochondrial GTP-binding protein involved in tRNA modification. Biochem Biophys Res Commun 2003;312:747–754. [DOI] [PubMed] [Google Scholar]
- 90. Kopajtich R, Nicholls TJ, Rorbach J, Metodiev MD, Freisinger P, Mandel H, Vanlander A, Ghezzi D, Carrozzo R, Taylor RW, Marquard K, Murayama K, Wieland T, Schwarzmayr T, Mayr JA, Pearce SF, Powell CA, Saada A, Ohtake A, Invernizzi F, Lamantea E, Sommerville EW, Pyle A, Chinnery PF, Crushell E, Okazaki Y, Kohda M, Kishita Y, Tokuzawa Y, Assouline Z, Rio M, Feillet F, Mousson de Camaret B, Chretien D, Munnich A, Menten B, Sante T, Smet J, Regal L, Lorber A, Khoury A, Zeviani M, Strom TM, Meitinger T, Bertini ES, Van Coster R, Klopstock T, Rotig A, Haack TB, Minczuk M, Prokisch H.. Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am J Hum Genet 2014;95:708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Asano K, Suzuki T, Saito A, Wei FY, Ikeuchi Y, Numata T, Tanaka R, Yamane Y, Yamamoto T, Goto T, Kishita Y, Murayama K, Ohtake A, Okazaki Y, Tomizawa K, Sakaguchi Y, Suzuki T.. Metabolic and chemical regulation of tRNA modification associated with taurine deficiency and human disease. Nucleic Acids Res 2018;46:1565–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Magida JA, Leinwand LA.. Metabolic crosstalk between the heart and liver impacts familial hypertrophic cardiomyopathy. EMBO Mol Med 2014;6:482–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jasra S, Anampa J.. Anthracycline use for early stage breast cancer in the modern era: a review. Curr Treat Options Oncol 2018;19:30. [DOI] [PubMed] [Google Scholar]
- 94. Lipshultz SE, Colan SD, Gelber RD, Perez-Atayde AR, Sallan SE, Sanders SP.. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N Engl J Med 1991;324:808–815. [DOI] [PubMed] [Google Scholar]
- 95. Mort MK, Sen JM, Morris AL, DeGregory KA, McLoughlin EM, Mort JF, Dunn SP, Abuannadi M, Keng MK.. Evaluation of cardiomyopathy in acute myeloid leukemia patients treated with anthracyclines. J Oncol Pharm Pract 2020;26:680–687. [DOI] [PubMed] [Google Scholar]
- 96. Prondzynski M, Lemoine MD, Zech AT, Horvath A, Di Mauro V, Koivumaki JT, Kresin N, Busch J, Krause T, Kramer E, Schlossarek S, Spohn M, Friedrich FW, Munch J, Laufer SD, Redwood C, Volk AE, Hansen A, Mearini G, Catalucci D, Meyer C, Christ T, Patten M, Eschenhagen T, Carrier L.. Disease modeling of a mutation in alpha-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol Med 2019;11:e11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chung W-B, Youn H-J.. Pathophysiology and preventive strategies of anthracycline-induced cardiotoxicity. Korean J Intern Med 2016;31:625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kim AS, Bergmann SR.. Anthracycline-induced cardiomyopathy: the search continues. J Nucl Cardiol 2017;24:265–267. [DOI] [PubMed] [Google Scholar]
- 99. Meadows W. Idiopathic myocardial failure in the last trimester of pregnancy and the puerperium. Circulation 1957;15:903–914. [DOI] [PubMed] [Google Scholar]
- 100. Sliwa K, Fett J, Elkayam U.. Peripartum cardiomyopathy. The Lancet 2006;368:687–693. [DOI] [PubMed] [Google Scholar]
- 101. Masoomi R, Shah Z, Arany Z, Gupta K.. Peripartum cardiomyopathy: an epidemiologic study of early and late presentations. Pregnancy Hypertens 2018;13:273–278. [DOI] [PubMed] [Google Scholar]
- 102. Lund LH, Grinnemo KH, Svenarud P, van der Linden J, Eriksson MJ.. Myocardial recovery in peri-partum cardiomyopathy after continuous flow left ventricular assist device. J Cardiothorac Surg 2011;6:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Cowgill JA, Francis SA, Sawyer DB.. Anthracycline and peripartum cardiomyopathies. Circ Res 2019;124:1633–1646. [DOI] [PubMed] [Google Scholar]
- 104. Lee YZJ, Judge DP.. The role of genetics in peripartum cardiomyopathy. J Cardiovasc Trans Res 2017;10:437–445. [DOI] [PubMed] [Google Scholar]
- 105. Hilfiker-Kleiner D, Kaminski K, Podewski E, Bonda T, Schaefer A, Sliwa K, Forster O, Quint A, Landmesser U, Doerries C, Luchtefeld M, Poli V, Schneider MD, Balligand JL, Desjardins F, Ansari A, Struman I, Nguyen NQ, Zschemisch NH, Klein G, Heusch G, Schulz R, Hilfiker A, Drexler H.. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell 2007;128:589–600. [DOI] [PubMed] [Google Scholar]
- 106. Negoro S, Kunisada K, Fujio Y, Funamoto M, Darville MI, Eizirik DL, Osugi T, Izumi M, Oshima Y, Nakaoka Y, Hirota H, Kishimoto T, Yamauchi-Takihara K.. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation 2001;104:979–981. [DOI] [PubMed] [Google Scholar]
- 107. Bolli R, Stein AB, Guo Y, Wang OL, Rokosh G, Dawn B, Molkentin JD, Sanganalmath SK, Zhu Y, Xuan YT.. A murine model of inducible, cardiac-specific deletion of STAT3: its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J Mol Cell Cardiol 2011;50:589–597. [DOI] [PubMed] [Google Scholar]
- 108. Reuwer AQ, Nowak-Sliwinska P, Mans LA, van der Loos CM, von der Thüsen JH, Twickler MTB, Spek CA, Goffin V, Griffioen AW, Borensztajn KS.. Functional consequences of prolactin signalling in endothelial cells: a potential link with angiogenesis in pathophysiology? J Cell Mol Med 2012;16:2035–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Patten IS, Rana S, Shahul S, Rowe GC, Jang C, Liu L, Hacker MR, Rhee JS, Mitchell J, Mahmood F, Hess P, Farrell C, Koulisis N, Khankin EV, Burke SD, Tudorache I, Bauersachs J, del Monte F, Hilfiker-Kleiner D, Karumanchi SA, Arany Z.. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature 2012;485:333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Halkein J, Tabruyn SP, Ricke-Hoch M, Haghikia A, Nguyen NQ, Scherr M, Castermans K, Malvaux L, Lambert V, Thiry M, Sliwa K, Noel A, Martial JA, Hilfiker-Kleiner D, Struman I.. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J Clin Invest 2013;123:2143–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Yang Y, Rodriguez JE, Kitsis RN.. A microRNA links prolactin to peripartum cardiomyopathy. J Clin Invest 2013;123:1925–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Stapel B, Kohlhaas M, Ricke-Hoch M, Haghikia A, Erschow S, Knuuti J, Silvola JM, Roivainen A, Saraste A, Nickel AG, Saar JA, Sieve I, Pietzsch S, Muller M, Bogeski I, Kappl R, Jauhiainen M, Thackeray JT, Scherr M, Bengel FM, Hagl C, Tudorache I, Bauersachs J, Maack C, Hilfiker-Kleiner D.. Low STAT3 expression sensitizes to toxic effects of beta-adrenergic receptor stimulation in peripartum cardiomyopathy. Eur Heart J 2017;38:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. St-Pierre G, Steinberg C, Dubois M, Sénéchal M.. What the cardiologist should know about mitochondrial cardiomyopathy? Can J Cardiol 2019;35:221–224. [DOI] [PubMed] [Google Scholar]
- 114. Li S, Pan H, Tan C, Sun Y, Song Y, Zhang X, Yang W, Wang X, Li D, Dai Y, Ma Q, Xu C, Zhu X, Kang L, Fu Y, Xu X, Shu J, Zhou N, Han F, Qin D, Huang W, Liu Z, Yan Q.. Mitochondrial dysfunctions contribute to hypertrophic cardiomyopathy in patient iPSC-derived cardiomyocytes with MT-RNR2 mutation. Stem Cell Rep 2018;10:808–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ylikallio E, Suomalainen A.. Mechanisms of mitochondrial diseases. Ann Med 2012;44:41–59. [DOI] [PubMed] [Google Scholar]
- 116. Sommakia S, Houlihan PR, Deane SS, Simcox JA, Torres NS, Jeong M-Y, Winge DR, Villanueva CJ, Chaudhuri D.. Mitochondrial cardiomyopathies feature increased uptake and diminished efflux of mitochondrial calcium. J Mol Cell Cardiol 2017;113:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wallace DC. A mitochondrial bioenergetic etiology of disease. J Clin Invest 2013;123:1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Koopman WJ, Willems PH, Smeitink JA.. Monogenic mitochondrial disorders. N Engl J Med 2012;366:1132–1141. [DOI] [PubMed] [Google Scholar]
- 119. Zeviani M, Mariotti C, Antozzi C, Fratta GM, Rustin P, Prelle A.. OXPHOS defects and mitochondrial DNA mutations in cardiomyopathy. Muscle Nerve 1995;18:S170–S174. [DOI] [PubMed] [Google Scholar]
- 120. Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC.. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet 1997;16:226–234. [DOI] [PubMed] [Google Scholar]
- 121. Lodi R, Rajagopalan B, Blamire AM, Cooper JM, Davies CH, Bradley JL, Styles P, Schapira AH.. Cardiac energetics are abnormal in Friedreich ataxia patients in the absence of cardiac dysfunction and hypertrophy: an in vivo 31P magnetic resonance spectroscopy study. Cardiovasc Res 2001;52:111–119. [DOI] [PubMed] [Google Scholar]
- 122. Rotig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P.. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet 1997;17:215–217. [DOI] [PubMed] [Google Scholar]
- 123. Michael S, Petrocine SV, Qian J, Lamarche JB, Knutson MD, Garrick MD, Koeppen AH.. Iron and iron-responsive proteins in the cardiomyopathy of Friedreich’s ataxia. Cerebellum 2006;5:257–267. [DOI] [PubMed] [Google Scholar]
- 124. Das SK, Zhabyeyev P, Basu R, Patel VB, Dyck JRB, Kassiri Z, Oudit GY.. Advanced iron-overload cardiomyopathy in a genetic murine model is rescued by resveratrol therapy. Biosci Rep 2018;38. pii: BSR20171302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Gordan R, Wongjaikam S, Gwathmey JK, Chattipakorn N, Chattipakorn SC, Xie LH.. Involvement of cytosolic and mitochondrial iron in iron overload cardiomyopathy: an update. Heart Fail Rev 2018;23:801–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Perdomini M, Belbellaa B, Monassier L, Reutenauer L, Messaddeq N, Cartier N, Crystal RG, Aubourg P, Puccio H.. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat Med 2014;20:542–547. [DOI] [PubMed] [Google Scholar]
- 127. Zhang D, Li Y, Heims-Waldron D, Bezzerides V, Guatimosim S, Guo Y, Gu F, Zhou P, Lin Z, Ma Q, Liu J, Wang DZ, Pu WT.. Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circ Res 2018;122:74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Nagao A, Suzuki T, Katoh T, Sakaguchi Y, Suzuki T.. Biogenesis of glutaminyl-mt tRNAGln in human mitochondria. Proc Natl Acad Sci U S A 2009;106:16209–16214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Echevarría L, Clemente P, Hernández-Sierra R, Gallardo ME, Fernández-Moreno MA, Garesse R.. Glutamyl-tRNAGln amidotransferase is essential for mammalian mitochondrial translation in vivo. Biochem J 2014;460:91–101. [DOI] [PubMed] [Google Scholar]
- 130. Friederich MW, Timal S, Powell CA, Dallabona C, Kurolap A, Palacios-Zambrano S, Bratkovic D, Derks TGJ, Bick D, Bouman K, Chatfield KC, Damouny-Naoum N, Dishop MK, Falik-Zaccai TC, Fares F, Fedida A, Ferrero I, Gallagher RC, Garesse R, Gilberti M, González C, Gowan K, Habib C, Halligan RK, Kalfon L, Knight K, Lefeber D, Mamblona L, Mandel H, Mory A, Ottoson J, Paperna T, Pruijn GJM, Rebelo-Guiomar PF, Saada A, Sainz B, Salvemini H, Schoots MH, Smeitink JA, Szukszto MJ, ter Horst HJ, van den Brandt F, van Spronsen FJ, Veltman JA, Wartchow E, Wintjes LT, Zohar Y, Fernández-Moreno MA, Baris HN, Donnini C, Minczuk M, Rodenburg RJ, Van Hove JLK.. Pathogenic variants in glutamyl-tRNA(Gln) amidotransferase subunits cause a lethal mitochondrial cardiomyopathy disorder. Nat Commun 2018;9:4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. McDonald A, Vanlerberghe G.. Branched mitochondrial electron transport in the Animalia: presence of alternative oxidase in several animal phyla. IUBMB Life 2004;56:333–341. [DOI] [PubMed] [Google Scholar]
- 132. El-Khoury R, Kemppainen KK, Dufour E, Szibor M, Jacobs HT, Rustin P.. Engineering the alternative oxidase gene to better understand and counteract mitochondrial defects: state of the art and perspectives. Br J Pharmacol 2014;171:2243–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Fellman V, Rapola J, Pihko H, Varilo T, Raivio KO.. Iron-overload disease in infants involving fetal growth retardation, lactic acidosis, liver haemosiderosis, and aminoaciduria. Lancet 1998;351:490–493. [DOI] [PubMed] [Google Scholar]
- 134. Fernandez-Vizarra E, Zeviani M.. Nuclear gene mutations as the cause of mitochondrial complex III deficiency. Front Genet 2015;6:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Visapaa I, Fellman V, Vesa J, Dasvarma A, Hutton JL, Kumar V, Payne GS, Makarow M, Van Coster R, Taylor RW, Turnbull DM, Suomalainen A, Peltonen L.. GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am J Hum Genet 2002;71:863–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Rajendran J, Purhonen J, Tegelberg S, Smolander O‐P, Mörgelin M, Rozman J, Gailus‐Durner V, Fuchs H, Hrabe de Angelis M, Auvinen P, Mervaala E, Jacobs HT, Szibor M, Fellman V, Kallijärvi J.. Alternative oxidase-mediated respiration prevents lethal mitochondrial cardiomyopathy. EMBO Mol Med 2019;11. pii: e9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ikeda M, Sezutsu Y, Sakamoto T, Ide T.. Electron microscopy reveals morphosis of multi-layered mitochondria in the myocardium of a patient with mitochondrial cardiomyopathy. Eur Heart J 2016;37:1372–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kanzaki Y, Terasaki F, Okabe M, Otsuka K, Katashima T, Fujita S, Ito T, Kitaura Y.. Giant mitochondria in the myocardium of a patient with mitochondrial cardiomyopathy: transmission and 3-dimensional scanning electron microscopy. Circulation 2010;121:831–832. [DOI] [PubMed] [Google Scholar]
- 139. Pawlik WW. The significance of animals in biomedical research. Folia Med Cracov 1998;39:175–182. [PubMed] [Google Scholar]
- 140. Guo XX, Wang Y, Wang K, Ji BP, Zhou F.. Stability of a type 2 diabetes rat model induced by high-fat diet feeding with low-dose streptozotocin injection. J Zhejiang Univ Sci B 2018;19:559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL.. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004;110:155–162. [DOI] [PubMed] [Google Scholar]
- 142. Makarenko I, Opitz CA, Leake MC, Neagoe C, Kulke M, Gwathmey JK, del Monte F, Hajjar RJ, Linke WA.. Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ Res 2004;95:708–716. [DOI] [PubMed] [Google Scholar]
- 143. Jaber WA, Maniu C, Krysiak J, Shapiro BP, Meyer DM, Linke WA, Redfield MM.. Titin isoforms, extracellular matrix, and global chamber remodeling in experimental dilated cardiomyopathy: functional implications and mechanistic insight. Circ Heart Fail 2008;1:192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Lefta M, Campbell KS, Feng HZ, Jin JP, Esser KA.. Development of dilated cardiomyopathy in Bmal1-deficient mice. Am J Physiol Heart Circ Physiol 2012;303:H475–H485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Deacon JC, Bloemink MJ, Rezavandi H, Geeves MA, Leinwand LA.. Erratum to: identification of functional differences between recombinant human alpha and beta cardiac myosin motors. Cell Mol Life Sci 2012;69:4239–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Lowey S, Bretton V, Gulick J, Robbins J, Trybus KM.. Transgenic mouse alpha- and beta-cardiac myosins containing the R403Q mutation show isoform-dependent transient kinetic differences. J Biol Chem 2013;288:14780–14787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Malmqvist UP, Aronshtam A, Lowey S.. Cardiac myosin isoforms from different species have unique enzymatic and mechanical properties. Biochemistry 2004;43:15058–15065. [DOI] [PubMed] [Google Scholar]
- 148. Shao K, Koch C, Gupta MK, Lin Q, Lenz M, Laufs S, Denecke B, Schmidt M, Linke M, Hennies HC, Hescheler J, Zenke M, Zechner U, Saric T, Wagner W.. Induced pluripotent mesenchymal stromal cell clones retain donor-derived differences in DNA methylation profiles. Mol Ther 2013;21:240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flügel L, Dorn T, Goedel A, Höhnke C, Hofmann F, Seyfarth M, Sinnecker D, Schömig A, Laugwitz K-L.. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med 2010;363:1397–1409. [DOI] [PubMed] [Google Scholar]
- 150. Mehta A, Sequiera GL, Ramachandra CJ, Sudibyo Y, Chung Y, Sheng J, Wong KY, Tan TH, Wong P, Liew R, Shim W.. Re-trafficking of hERG reverses long QT syndrome 2 phenotype in human iPS-derived cardiomyocytes. Cardiovasc Res 2014;102:497–506. [DOI] [PubMed] [Google Scholar]
- 151. Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, Pavlovic A, Lin S, Chen R, Hajjar RJ, Snyder MP, Dolmetsch RE, Butte MJ, Ashley EA, Longaker MT, Robbins RC, Wu JC.. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med 2012;4:130ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, Abilez OJ, Hu S, Ebert AD, Navarrete EG, Simmons CS, Wheeler M, Pruitt B, Lewis R, Yamaguchi Y, Ashley EA, Bers DM, Robbins RC, Longaker MT, Wu JC.. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013;12:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Viswanathan SK, Puckelwartz MJ, Mehta A, Ramachandra CJA, Jagadeesan A, Fritsche-Danielson R, Bhat RV, Wong P, Kandoi S, Schwanekamp JA, Kuffel G, Pesce LL, Zilliox MJ, Durai UNB, Verma RS, Molokie RE, Suresh DP, Khoury PR, Thomas A, Sanagala T, Tang HC, Becker RC, Knoll R, Shim W, McNally EM, Sadayappan S.. Association of cardiomyopathy with MYBPC3 D389V and MYBPC3Delta25bpIntronic deletion in South Asian descendants. JAMA Cardiol 2018;3:481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Tiburcy M, Hudson JE, Balfanz P, Schlick S, Meyer T, Chang Liao ML, Levent E, Raad F, Zeidler S, Wingender E, Riegler J, Wang M, Gold JD, Kehat I, Wettwer E, Ravens U, Dierickx P, van Laake LW, Goumans MJ, Khadjeh S, Toischer K, Hasenfuss G, Couture LA, Unger A, Linke WA, Araki T, Neel B, Keller G, Gepstein L, Wu JC, Zimmermann WH.. Defined engineered human myocardium with advanced maturation for applications in heart failure modeling and repair. Circulation 2017;135:1832–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Ulmer BM, Stoehr A, Schulze ML, Patel S, Gucek M, Mannhardt I, Funcke S, Murphy E, Eschenhagen T, Hansen A.. Contractile work contributes to maturation of energy metabolism in hiPSC-derived cardiomyocytes. Stem Cell Rep 2018;10:834–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Breckwoldt K, Letuffe-Breniere D, Mannhardt I, Schulze T, Ulmer B, Werner T, Benzin A, Klampe B, Reinsch MC, Laufer S, Shibamiya A, Prondzynski M, Mearini G, Schade D, Fuchs S, Neuber C, Kramer E, Saleem U, Schulze ML, Rodriguez ML, Eschenhagen T, Hansen A.. Differentiation of cardiomyocytes and generation of human engineered heart tissue. Nat Protoc 2017;12:1177–1197. [DOI] [PubMed] [Google Scholar]
- 157. Mills RJ, Titmarsh DM, Koenig X, Parker BL, Ryall JG, Quaife-Ryan GA, Voges HK, Hodson MP, Ferguson C, Drowley L, Plowright AT, Needham EJ, Wang QD, Gregorevic P, Xin M, Thomas WG, Parton RG, Nielsen LK, Launikonis BS, James DE, Elliott DA, Porrello ER, Hudson JE.. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc Natl Acad Sci U S A 2017;114:E8372–E8381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Shadrin IY, Allen BW, Qian Y, Jackman CP, Carlson AL, Juhas ME, Bursac N.. Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat Commun 2017;8:1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, Morikawa K, Teles D, Yazawa M, Vunjak-Novakovic G.. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 2018;556:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Correia C, Koshkin A, Duarte P, Hu D, Carido M, Sebastiao MJ, Gomes-Alves P, Elliott DA, Domian IJ, Teixeira AP, Alves PM, Serra M.. 3D aggregate culture improves metabolic maturation of human pluripotent stem cell derived cardiomyocytes. Biotechnol Bioeng 2018;115:630–644. [DOI] [PubMed] [Google Scholar]
- 161. Branco MA, Cotovio JP, Rodrigues CAV, Vaz SH, Fernandes TG, Moreira LM, Cabral JMS, Diogo MM.. Transcriptomic analysis of 3D cardiac differentiation of human induced pluripotent stem cells reveals faster cardiomyocyte maturation compared to 2D. Sci Rep 2019;9:9229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Keung W, Boheler KR, Li RA.. Developmental cues for the maturation of metabolic, electrophysiological and calcium handling properties of human pluripotent stem cell-derived cardiomyocytes. Stem Cell Res Ther 2014;5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Kamakura T, Makiyama T, Sasaki K, Yoshida Y, Wuriyanghai Y, Chen J, Hattori T, Ohno S, Kita T, Horie M, Yamanaka S, Kimura T.. Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ J 2013;77:1307–1314. [DOI] [PubMed] [Google Scholar]
- 164. Lundy SD, Zhu WZ, Regnier M, Laflamme MA.. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev 2013;22:1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Lieu DK, Fu JD, Chiamvimonvat N, Tung KC, McNerney GP, Huser T, Keller G, Kong CW, Li RA.. Mechanism-based facilitated maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Arrhythm Electrophysiol 2013;6:191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, Jiang J, Masse S, Gagliardi M, Hsieh A, Thavandiran N, Laflamme MA, Nanthakumar K, Gross GJ, Backx PH, Keller G, Radisic M.. Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods 2013;10:781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ruan JL, Tulloch NL, Razumova MV, Saiget M, Muskheli V, Pabon L, Reinecke H, Regnier M, Murry CE.. Mechanical stress conditioning and electrical stimulation promote contractility and force maturation of induced pluripotent stem cell-derived human cardiac tissue. Circulation 2016;134:1557–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Herron TJ, Rocha AM, Campbell KF, Ponce-Balbuena D, Willis BC, Guerrero-Serna G, Liu Q, Klos M, Musa H, Zarzoso M, Bizy A, Furness J, Anumonwo J, Mironov S, Jalife J.. Extracellular matrix-mediated maturation of human pluripotent stem cell-derived cardiac monolayer structure and electrophysiological function. Circ Arrhythm Electrophysiol 2016;9:e003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Fong AH, Romero-Lopez M, Heylman CM, Keating M, Tran D, Sobrino A, Tran AQ, Pham HH, Fimbres C, Gershon PD, Botvinick EL, George SC, Hughes CC.. Three-dimensional adult cardiac extracellular matrix promotes maturation of human induced pluripotent stem cell-derived cardiomyocytes. Tissue Eng Part A 2016;22:1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. White MC, Pang L, Yang X.. MicroRNA-mediated maturation of human pluripotent stem cell-derived cardiomyocytes: towards a better model for cardiotoxicity? Food Chem Toxicol 2016;98:17–24. [DOI] [PubMed] [Google Scholar]
- 171. Wen JY, Wei CY, Shah K, Wong J, Wang C, Chen HS.. Maturation-based model of arrhythmogenic right ventricular dysplasia using patient-specific induced pluripotent stem cells. Circ J 2015;79:1402–1408. [DOI] [PubMed] [Google Scholar]
- 172. Birket MJ, Ribeiro MC, Kosmidis G, Ward D, Leitoguinho AR, van de Pol V, Dambrot C, Devalla HD, Davis RP, Mastroberardino PG, Atsma DE, Passier R, Mummery CL.. Contractile defect caused by mutation in MYBPC3 revealed under conditions optimized for human PSC-cardiomyocyte function. Cell Rep 2015;13:733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Hu D, Linders A, Yamak A, Correia C, Kijlstra JD, Garakani A, Xiao L, Milan DJ, van der Meer P, Serra M, Alves PM, Domian IJ.. Metabolic maturation of human pluripotent stem cell-derived cardiomyocytes by inhibition of HIF1alpha and LDHA. Circ Res 2018;123:1066–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Gentillon C, Li D, Duan M, Yu WM, Preininger MK, Jha R, Rampoldi A, Saraf A, Gibson GC, Qu CK, Brown LA, Xu C.. Targeting HIF-1alpha in combination with PPARalpha activation and postnatal factors promotes the metabolic maturation of human induced pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol 2019;132:120–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ramachandra CJA, Mehta A, Wong P, Ja K, Fritsche-Danielson R, Bhat RV, Hausenloy DJ, Kovalik JP, Shim W.. Fatty acid metabolism driven mitochondrial bioenergetics promotes advanced developmental phenotypes in human induced pluripotent stem cell derived cardiomyocytes. Int J Cardiol 2018;272:288–297. [DOI] [PubMed] [Google Scholar]
- 176. Yang X, Rodriguez ML, Leonard A, Sun L, Fischer KA, Wang Y, Ritterhoff J, Zhao L, Kolwicz SC Jr, Pabon L, Reinecke H, Sniadecki NJ, Tian R, Ruohola-Baker H, Xu H, Murry CE.. Fatty acids enhance the maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cell Rep 2019;13:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Horikoshi Y, Yan Y, Terashvili M, Wells C, Horikoshi H, Fujita S, Bosnjak ZJ, Bai X.. Fatty acid-treated induced pluripotent stem cell-derived human cardiomyocytes exhibit adult cardiomyocyte-like energy metabolism phenotypes. Cells 2019;8:1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Ingles J, Burns C, Bagnall RD, Lam L, Yeates L, Sarina T, Puranik R, Briffa T, Atherton JJ, Driscoll T, Semsarian C.. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet 2017;10. pii: e001620. [DOI] [PubMed] [Google Scholar]
- 179. Mosqueira D, Mannhardt I, Bhagwan JR, Lis-Slimak K, Katili P, Scott E, Hassan M, Prondzynski M, Harmer SC, Tinker A, Smith JGW, Carrier L, Williams PM, Gaffney D, Eschenhagen T, Hansen A, Denning C.. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur Heart J 2018;39:3879–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Ceholski DK, Turnbull IC, Kong CW, Koplev S, Mayourian J, Gorski PA, Stillitano F, Skodras AA, Nonnenmacher M, Cohen N, Bjorkegren JLM, Stroik DR, Cornea RL, Thomas DD, Li RA, Costa KD, Hajjar RJ.. Functional and transcriptomic insights into pathogenesis of R9C phospholamban mutation using human induced pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol 2018;119:147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, Yuan H, Jiang D, Zhang D, Zangi L, Geva J, Roberts AE, Ma Q, Ding J, Chen J, Wang DZ, Li K, Wang J, Wanders RJ, Kulik W, Vaz FM, Laflamme MA, Murry CE, Chien KR, Kelley RI, Church GM, Parker KK, Pu WT.. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med 2014;20:616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Davey KM, Parboosingh JS, McLeod DR, Chan A, Casey R, Ferreira P, Snyder FF, Bridge PJ, Bernier FP.. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J Med Genet 2005;43:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Rohani L, Machiraju P, Sabouny R, Meng G, Liu S, Zhao T, Iqbal F, Wang X, Ravandi A, Wu JC, Khan A, Shutt T, Rancourt D, Greenway SC.. Reversible mitochondrial fragmentation in iPSC-derived cardiomyocytes from children with DCMA, a mitochondrial cardiomyopathy. Can J Cardiol 2020;36:554–563. [DOI] [PubMed] [Google Scholar]
- 184. Zhan Y, Sun X, Li B, Cai H, Xu C, Liang Q, Lu C, Qian R, Chen S, Yin L, Sheng W, Huang G, Sun A, Ge J, Sun N.. Establishment of a PRKAG2 cardiac syndrome disease model and mechanism study using human induced pluripotent stem cells. J Mol Cell Cardiol 2018;117:49–61. [DOI] [PubMed] [Google Scholar]
- 185. Drawnel FM, Boccardo S, Prummer M, Delobel F, Graff A, Weber M, Gerard R, Badi L, Kam-Thong T, Bu L, Jiang X, Hoflack JC, Kiialainen A, Jeworutzki E, Aoyama N, Carlson C, Burcin M, Gromo G, Boehringer M, Stahlberg H, Hall BJ, Magnone MC, Kolaja K, Chien KR, Bailly J, Iacone R.. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep 2014;9:810–821. [DOI] [PubMed] [Google Scholar]
- 186. Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP.. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A 2012;109:E1848–E1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD, Wu JC.. Chemically defined generation of human cardiomyocytes. Nat Methods 2014;11:855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Mehta A, Ramachandra CJ, Sequiera GL, Sudibyo Y, Nandihalli M, Yong PJ, Koh CH, Shim W.. Phasic modulation of Wnt signaling enhances cardiac differentiation in human pluripotent stem cells by recapitulating developmental ontogeny. Biochim Biophys Acta 2014;1843:2394–2402. [DOI] [PubMed] [Google Scholar]
- 189. Wu H, Yang H, Rhee JW, Zhang JZ, Lam CK, Sallam K, Chang ACY, Ma N, Lee J, Zhang H, Blau HM, Bers DM, Wu JC.. Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients. Eur Heart J 2019;40:3685–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Freeman LM, Rush JE, Stern JA, Huggins GS, Maron MS.. Feline hypertrophic cardiomyopathy: a spontaneous large animal model of human HCM. Cardiol Res 2017;8:139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Zheng M, Pan F, Liu Y, Li Z, Zhou X, Meng X, Liu L, Ge S.. Echocardiographic strain analysis for the early detection of myocardial structural abnormality and initiation of drug therapy in a mouse model of dilated cardiomyopathy. Ultrasound Med Biol 2017;43:2914–2924. [DOI] [PubMed] [Google Scholar]
- 192. Gannon MP, Link MS.. Phenotypic variation and targeted therapy of hypertrophic cardiomyopathy using genetic animal models. Trends Cardiovasc Med 2019. pii: S1050-1738(19)30159-8. [DOI] [PubMed] [Google Scholar]
- 193. Shen LJ, Lu S, Zhou YH, Li L, Xing QM, Xu YL.. Developing a rat model of dilated cardiomyopathy with improved survival. J Zhejiang Univ Sci B 2016;17:975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE, EMPA-REG OUTCOME Investigators.. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015;373:2117–2128. [DOI] [PubMed] [Google Scholar]
- 195. Ng KM, Lau YM, Dhandhania V, Cai ZJ, Lee YK, Lai WH, Tse HF, Siu CW.. Empagliflozin ammeliorates high glucose induced-cardiac dysfuntion in human iPSC-derived cardiomyocytes. Sci Rep 2018;8:14872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Banerjee SK, McGaffin KR, Pastor-Soler NM, Ahmad F.. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc Res 2009;84:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Zhou L, Cryan EV, D’Andrea MR, Belkowski S, Conway BR, Demarest KT.. Human cardiomyocytes express high level of Na+/glucose cotransporter 1 (SGLT1). J Cell Biochem 2003;90:339–346. [DOI] [PubMed] [Google Scholar]
- 198. von Lewinski D, Rainer PP, Gasser R, Huber MS, Khafaga M, Wilhelm B, Haas T, Machler H, Rossl U, Pieske B.. Glucose-transporter-mediated positive inotropic effects in human myocardium of diabetic and nondiabetic patients. Metabolism 2010;59:1020–1028. [DOI] [PubMed] [Google Scholar]
- 199. Di Franco A, Cantini G, Tani A, Coppini R, Zecchi-Orlandini S, Raimondi L, Luconi M, Mannucci E.. Sodium-dependent glucose transporters (SGLT) in human ischemic heart: a new potential pharmacological target. Int J Cardiol 2017;243:86–90. [DOI] [PubMed] [Google Scholar]
- 200. Vrhovac I, Balen Eror D, Klessen D, Burger C, Breljak D, Kraus O, Radovic N, Jadrijevic S, Aleksic I, Walles T, Sauvant C, Sabolic I, Koepsell H.. Localizations of Na(+)-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch 2015;467:1881–1898. [DOI] [PubMed] [Google Scholar]
- 201. Lambert R, Srodulski S, Peng X, Margulies KB, Despa F, Despa S.. Intracellular Na+ concentration ([Na+]i) is elevated in diabetic hearts due to enhanced Na+-glucose cotransport. J Am Heart Assoc 2015;4:e002183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Kim Y, Babu AR.. Clinical potential of sodium-glucose cotransporter 2 inhibitors in the management of type 2 diabetes. Diabetes Metab Syndr Obes 2012;5:313–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, Fagan NM, Woerle HJ, Johansen OE, Broedl UC, von Eynatten M.. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 2014;129:587–597. [DOI] [PubMed] [Google Scholar]
- 204. Petrykiv S, Sjostrom CD, Greasley PJ, Xu J, Persson F, Heerspink H.. Differential effects of dapagliflozin on cardiovascular risk factors at varying degrees of renal function. Clin J Am Soc Nephrol 2017;12:751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Yasui A, Lee G, Hirase T, Kaneko T, Kaspers S, von Eynatten M, Okamura T.. Empagliflozin induces transient diuresis without changing long-term overall fluid balance in japanese patients with type 2 diabetes. Diabetes Ther 2018;9:863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Hammoudi N, Jeong D, Singh R, Farhat A, Komajda M, Mayoux E, Hajjar R, Lebeche D.. Empagliflozin improves left ventricular diastolic dysfunction in a genetic model of type 2 diabetes. Cardiovasc Drugs Ther 2017;31:233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Baartscheer A, Schumacher CA, Wust RC, Fiolet JW, Stienen GJ, Coronel R, Zuurbier CJ.. Empagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits. Diabetologia 2017;60:568–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Heerspink HJL, Karasik A, Thuresson M, Melzer-Cohen C, Chodick G, Khunti K, Wilding JPH, Garcia Rodriguez LA, Cea-Soriano L, Kohsaka S, Nicolucci A, Lucisano G, Lin FJ, Wang CY, Wittbrodt E, Fenici P, Kosiborod M.. Kidney outcomes associated with use of SGLT2 inhibitors in real-world clinical practice (CVD-REAL 3): a multinational observational cohort study. Lancet Diabetes Endocrinol 2020;8:27–35. [DOI] [PubMed] [Google Scholar]
- 209. Heerspink HJ, Perkins BA, Fitchett DH, Husain M, Cherney DZ.. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 2016;134:752–772. [DOI] [PubMed] [Google Scholar]
- 210. Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, Johansen OE, Woerle HJ, Broedl UC, Zinman B, Investigators E-R.. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016;375:323–334. [DOI] [PubMed] [Google Scholar]
- 211. Skrtic M, Cherney DZ.. Sodium-glucose cotransporter-2 inhibition and the potential for renal protection in diabetic nephropathy. Curr Opin Nephrol Hypertens 2015;24:96–103. [DOI] [PubMed] [Google Scholar]
- 212. Zimlichman R. Treatment of hypertension and metabolic syndrome: lowering blood pressure is not enough for organ protection, new approach-arterial destiffening. Curr Hypertens Rep 2014;16:479. [DOI] [PubMed] [Google Scholar]
- 213. Chilton R, Tikkanen I, Cannon CP, Crowe S, Woerle HJ, Broedl UC, Johansen OE.. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes Metab 2015;17:1180–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, Diecke S, Sallam K, Knowles JW, Wang PJ, Nguyen PK, Bers DM, Robbins RC, Wu JC.. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 2013;127:1677–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Chen VC, Ye J, Shukla P, Hua G, Chen D, Lin Z, Liu JC, Chai J, Gold J, Wu J, Hsu D, Couture LA.. Development of a scalable suspension culture for cardiac differentiation from human pluripotent stem cells. Stem Cell Res 2015;15:365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Wells SP, Waddell HM, Sim CB, Lim SY, Bernasochi GB, Pavlovic D, Kirchhof P, Porrello ER, Delbridge LMD, Bell JR.. Cardiomyocyte functional screening: interrogating comparative electrophysiology of high-throughput model cell systems. Am J Physiol Cell Physiol 2019;317:C1256–C1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, Churko JM, Kitani T, Wu H, Holmstrom A, Matsa E, Zhang Y, Kumar A, Fan AC, Del Alamo JC, Wu SM, Moslehi JJ, Mercola M, Wu JC.. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med 2017;9. pii: eaaf2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Kopljar I, Lu HR, Van Ammel K, Otava M, Tekle F, Teisman A, Gallacher DJ.. Development of a human iPSC cardiomyocyte-based scoring system for cardiac hazard identification in early drug safety de-risking. Stem Cell Rep 2018;11:1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Mills RJ, Parker BL, Quaife-Ryan GA, Voges HK, Needham EJ, Bornot A, Ding M, Andersson H, Polla M, Elliott DA, Drowley L, Clausen M, Plowright AT, Barrett IP, Wang QD, James DE, Porrello ER, Hudson JE.. Drug screening in human PSC-cardiac organoids identifies pro-proliferative compounds acting via the mevalonate pathway. Cell Stem Cell 2019;24:895–907.e896. [DOI] [PubMed] [Google Scholar]
- 220. Jacquet S, Nishino Y, Kumphune S, Sicard P, Clark JE, Kobayashi KS, Flavell RA, Eickhoff J, Cotten M, Marber MS.. The role of RIP2 in p38 MAPK activation in the stressed heart. J Biol Chem 2008;283:11964–11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Zhang D, Gaussin V, Taffet GE, Belaguli NS, Yamada M, Schwartz RJ, Michael LH, Overbeek PA, Schneider MD.. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med 2000;6:556–563. [DOI] [PubMed] [Google Scholar]
- 222. Fiedler LR, Chapman K, Xie M, Maifoshie E, Jenkins M, Golforoush PA, Bellahcene M, Noseda M, Faust D, Jarvis A, Newton G, Paiva MA, Harada M, Stuckey DJ, Song W, Habib J, Narasimhan P, Aqil R, Sanmugalingam D, Yan R, Pavanello L, Sano M, Wang SC, Sampson RD, Kanayaganam S, Taffet GE, Michael LH, Entman ML, Tan TH, Harding SE, Low CMR, Tralau-Stewart C, Perrior T, Schneider MD.. MAP4K4 inhibition promotes survival of human stem cell-derived cardiomyocytes and reduces infarct size in vivo. Cell Stem Cell 2019;24:579–591.e512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Dambrot C, Braam SR, Tertoolen LG, Birket M, Atsma DE, Mummery CL.. Serum supplemented culture medium masks hypertrophic phenotypes in human pluripotent stem cell derived cardiomyocytes. J Cell Mol Med 2014;18:1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Mekhoubad S, Bock C, de Boer AS, Kiskinis E, Meissner A, Eggan K.. Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 2012;10:595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Mayshar Y, Ben-David U, Lavon N, Biancotti JC, Yakir B, Clark AT, Plath K, Lowry WE, Benvenisty N.. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 2010;7:521–531. [DOI] [PubMed] [Google Scholar]
- 226. Gore A, Li Z, Fung HL, Young JE, Agarwal S, Antosiewicz-Bourget J, Canto I, Giorgetti A, Israel MA, Kiskinis E, Lee JH, Loh YH, Manos PD, Montserrat N, Panopoulos AD, Ruiz S, Wilbert ML, Yu J, Kirkness EF, Izpisua Belmonte JC, Rossi DJ, Thomson JA, Eggan K, Daley GQ, Goldstein LS, Zhang K.. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011;471:63–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Martins-Taylor K, Nisler BS, Taapken SM, Compton T, Crandall L, Montgomery KD, Lalande M, Xu RH.. Recurrent copy number variations in human induced pluripotent stem cells. Nat Biotechnol 2011;29:488–491. [DOI] [PubMed] [Google Scholar]
- 228. Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LIR, Yabuuchi A, Takeuchi A, Cunniff KC, Hongguang H, Mckinney-Freeman S, Naveiras O, Yoon TJ, Irizarry RA, Jung N, Seita J, Hanna J, Murakami P, Jaenisch R, Weissleder R, Orkin SH, Weissman IL, Feinberg AP, Daley GQ.. Epigenetic memory in induced pluripotent stem cells. Nature 2010;467:285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Newman AM, Cooper JB.. Lab-specific gene expression signatures in pluripotent stem cells. Cell Stem Cell 2010;7:258–262. [DOI] [PubMed] [Google Scholar]
- 230. Newman AM, Cooper JB.. AutoSOME: a clustering method for identifying gene expression modules without prior knowledge of cluster number. BMC Bioinformatics 2010;11:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, Egashira T, Seki T, Muraoka N, Yamakawa H, Ohgino Y, Tanaka T, Yoichi M, Yuasa S, Murata M, Suematsu M, Fukuda K.. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2013;12:127–137. [DOI] [PubMed] [Google Scholar]
- 232. Hemmi N, Tohyama S, Nakajima K, Kanazawa H, Suzuki T, Hattori F, Seki T, Kishino Y, Hirano A, Okada M, Tabei R, Ohno R, Fujita C, Haruna T, Yuasa S, Sano M, Fujita J, Fukuda K.. A massive suspension culture system with metabolic purification for human pluripotent stem cell-derived cardiomyocytes. Stem Cells Transl Med 2014;3:1473–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Li P, Liang M, Lu X, Chow JJM, Ramachandra CJA, Ai Y.. Sheathless acoustic fluorescence activated cell sorting (aFACS) with high cell viability. Anal Chem 2019;91:15425–15435. [DOI] [PubMed] [Google Scholar]
- 234. Mori J, Zhang L, Oudit GY, Lopaschuk GD.. Impact of the renin-angiotensin system on cardiac energy metabolism in heart failure. J Mol Cell Cardiol 2013;63:98–106. [DOI] [PubMed] [Google Scholar]
- 235. Hsu YR, Yogasundaram H, Parajuli N, Valtuille L, Sergi C, Oudit GY.. MELAS syndrome and cardiomyopathy: linking mitochondrial function to heart failure pathogenesis. Heart Fail Rev 2016;21:103–116. [DOI] [PubMed] [Google Scholar]