

Abstract
High-valent non-heme FeIV–oxido species are key intermediates in biological oxidation, and their properties are proposed to be influenced by the unique microenvironments present in protein active sites. Microenvironments are regulated by non-covalent interactions, such as hydrogen bonds (H-bonds) and electrostatic interactions; however, there is little quantitative information about how these interactions affect crucial properties of high valent metal–oxido complexes. To address this knowledge gap, we introduced a series of FeIV–oxido complexes that have the same S = 2 spin ground state as those found in nature and then systematically probed the effects of non-covalent interactions on their electronic, structural, and vibrational properties. The key design feature that provides access to these complexes is the new tripodal ligand [poat]3−, which contains phosphinic amide groups. An important structural aspect of [FeIVpoat(O)]− is the inclusion of an auxiliary site capable of binding a Lewis acid (LAII); we used this unique feature to further modulate the electrostatic environment around the Fe–oxido unit. Experimentally studies confirmed that H-bonds and LAII s can interact directly with the oxido ligand in FeIV–oxido complexes, which weakens the Fe=O bond and has an impact on the electronic structure. We found that relatively large vibrational changes in the Fe–oxido unit correlate with small structural changes that could be difficult to measure, especially within a protein active site. Our work demonstrates the important role that non-covalent interactions have on the properties of metal complexes; these interactions need to be considered when developing effective oxidants.
Graphical Abstract
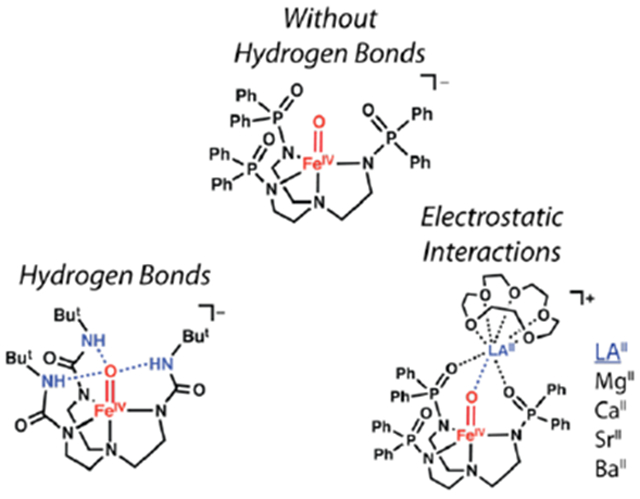
Introduction
Metal–oxido units within the active sites of proteins are important intermediates in catalytic cycles.1–9 Their significance is exemplified in non-heme oxygenases with mononuclear Fe sites, whose competent oxidant responsible for cleaving the unactivated C–H bonds of substrates is known to be an FeIV–oxido species.10–14 Magnetic studies have shown that these intermediates have an S = 2 spin ground state, which is suggested to be an important property for function.15–17 Efforts to understand the roles of these intermediates in catalysis have led to the preparation of synthetic FeIV–oxido complexes, of which there are now several examples.18–24 However, nearly all of these synthetic complexes have S = 1 spin ground states, and it is difficult to directly correlate their properties to those of the intermediates detected in proteins.11,25,26 Far fewer examples of FeIV–oxido complexes with S = 2 spin ground states exist,27–34 and only two of those complexes have been structurally characterized.28,30 There is thus still a need for well-defined systems whose structural and physical properties can be readily tuned and investigated.
One approach to modulating the properties of metal–oxido species is to regulate their local environments.11,35,36 The unique microenvironments that exist within the active sites of proteins illustrate how essential properties can be controlled through this approach. In protein active sites, non-covalent interactions are the major force that influences the microenvironment, with hydrogen bonds (H-bonds) being the most common type of non-covalent interaction. The exact influences that H-bonds have on structure and function are still emerging, but in many instances, these bonds are proposed to directly involve M–oxido complexes via M=O⋯H–X interactions.4,37–39 Furthermore, H-bonds serve to regulate the electrostatic properties of an active site and thus aid in governing key processes such as the transfer of protons and electrons.
Another popular approach to modulating microenvironment is to change the electrostatic properties around M–oxido species by treating them with redox-inactive Lewis acids (LAs) such as Group II metal ions and Sc3+ ions.40–49 The premise that these auxiliary metal ions affect function has been previously substantiated in synthetic systems. For instance, work by Agapie examined the effects of LAs on the properties of manganese-oxido clusters.50–52 Complimentary work by Lau showed that in the presence of a LA, the rate of alkane oxidation by metal–oxido systems increased.53 In addition, Fukuzumi demonstrated that the rate of electron transfer in a Co–porphyrin system was correlated to the Lewis acidity of various redox-inactive metal ions.54,55 Subsequent work from Fukuzumi and Nam reported that the rate of electron transfer involving an FeIV–oxido complex was dependent on redox-inactive ions,56 and this correlation led them to report crystallographic evidence of a complex containing an FeIV–(μ-O)–ScIII core.57 However, computational studies by Swart58 and an experimental reinvestigation of this complex by Que found that it should be formulated as an FeIII–(μ-O)–ScIII species.40 These findings highlight some of the current uncertainties that surround the addition of LAs to M–oxido complexes. Although there is strong evidence that these ions enhance the reactivity of M–oxido complexes, there is less information about how they interact with M–oxido units and about their effects on molecular and electronic structures. There is thus a need to develop molecular systems that are capable of forming FeIV=O---LA cores whose properties can be evaluated directly.
In this report, we describe the preparation of a new high-spin FeIV–oxido complex and its binding of LAII (LAII = MgII, CaII, SrII, and BaII) in the presence of 15-crown-5 to produce a series of heterometallic complexes (denoted [FeIVpoat(O)---LAII]+). To prepare these complexes, we designed a new tripodal ligand, [poat]3−, which contains three phosphinic amido groups and readily forms [FeIVpoat(O)]− (Figure 1A). This complex has local C3 symmetry, which also allows its properties to be compared to those of similar FeIV complexes, particularly [FeIVH3buea(O)]−,30,31 which we reported previously (Figure 1B). We are unaware of any other synthetic systems that allow the influences of non-covalent interactions around M–oxido complexes to be so definitively examined and compared. Our findings show that H-bonds cause large changes in the bonding within the FeIV–oxido unit. Moreover, the experimental and computational evidence presented supports the binding of LAIIs to the FeIV=O unit (Figure 1C), which was correlated to changes in the structural and electronic properties. Finally, our results enable the first direct comparison of how LAIIs and H-bonds affect the electronic and vibrational properties of FeIV=O complexes.
Figure 1.
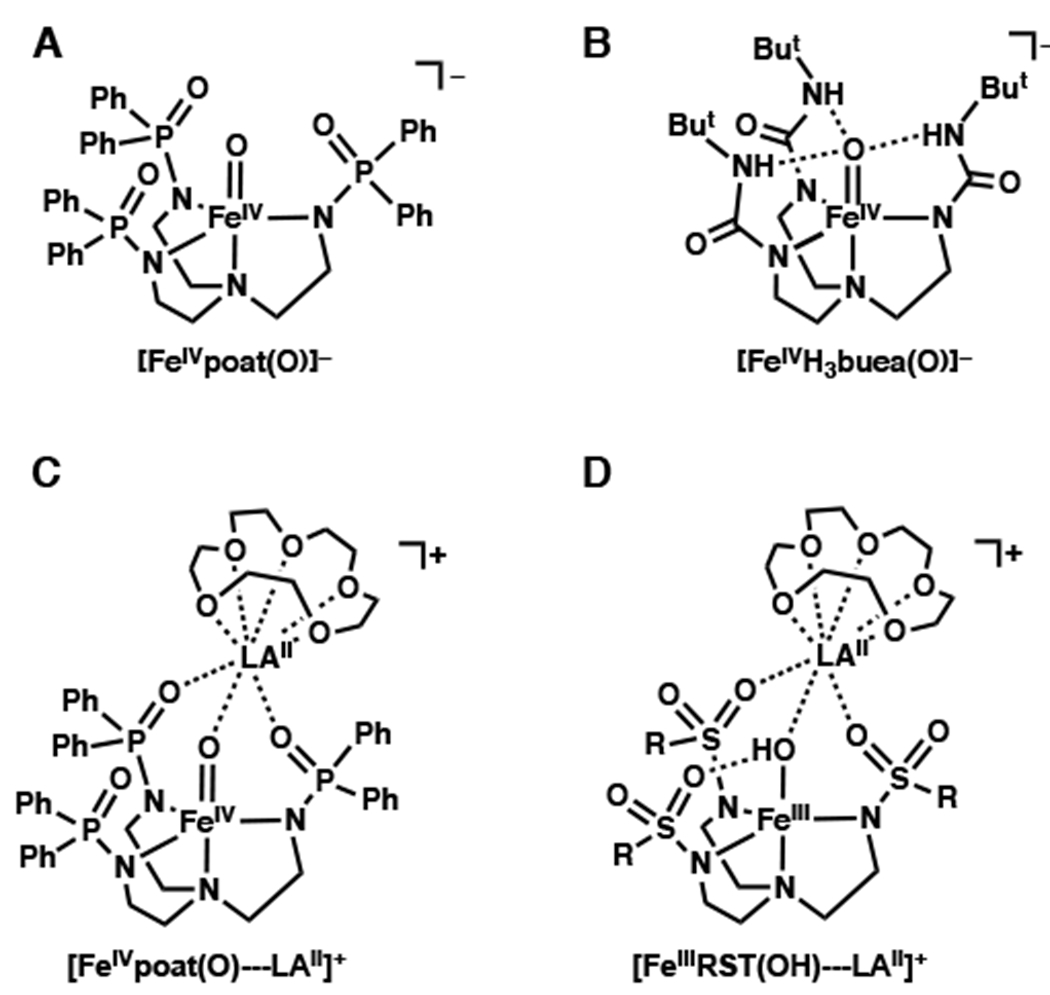
Relevant structures of complexes for this study.
Results and Discussion
Design Considerations.
In our laboratory, previous work has investigated the use of the urea-containing tripodal ligand [H3buea]3− (Figure 1B) to prepare high-spin Fen–oxido complexes (n = 3+ and 4+).30,31,59 A key design component of this ligand is the presence of intramolecular H-bond donors that we have argued are crucial for the isolation of these Fen–oxido complexes. To further evaluate the effects of these intramolecular H-bonding interactions on FeIV–oxido species, we prepared the ligand [poat]3−, which contains deprotonated phosphinic amide groups instead of ureas. The design of this ligand was inspired by the work of Stephen60 and the ability of the ligand to stabilize high-valent complexes relies on the fact that deprotonated phosphinic amides and ureas produce comparable ligand fields.61 However, the local environments that surround the Fe–oxido centers in complexes of [poat]3− are different from those in complexes of [H3buea]3− because the [poat]3− ligand lacks the intramolecular H-bond donors contained in [H3buea]3−. Thus, our preparation of [FeIVpoat(O)]− provides the ability to directly determine the influence of H-bonds on the properties of FeIV–oxido centers by establishing an analog without these non-covalent interactions.
The structure of [FeIVpoat(O)]− also includes an auxiliary site composed of two P=O units and the oxido ligand (Figure 1C) to which a second metal ion can be coordinated. This design principle was first incorporated in complexes of our tripodal ligands with sulfonamido groups ([RST]3−, Figure 1D) that produced a variety of discrete heterobimetallic complexes with MIII-(μ-OH)-LAII cores (MIII = Mn, Fe, Co, Ga).62–64 In these systems, two S=O groups and the hydroxido ligand form an auxiliary site that is capable of facially binding a second metal ion. However, our studies found that sulfonamido groups do not provide a strong enough ligand field to stabilize more highly oxidized complexes.61,64,65 We reasoned that redesign of the ligand to [poat]3− would provide access to an FeIV–oxido species capable of supporting a LAII at its auxiliary binding site and allow us to evaluate changes in the structural and spectroscopic properties of the FeIV–oxido species upon LAII binding.
Preparation and Properties of [FeIVpoat(O)]−.
The [FeIVpoat(O)]− complex was produced from [K(18c6)2][FeIIpoat], whose properties are consistent with a four-coordinate FeII complex having an S = 2 spin ground state (Scheme 1, see experimental section).64,66 We found that 18-crown-6 (18c6) increases the solubility of this FeII complex and sequesters the K+ counter ion to prevent its interaction with the P=O moieties of the [poat]3− ligand. [K(18c6)2][FeIIpoat] was treated with isopropyl 2-iodoxybenzoate (IBX-iPr) in a DMF:THF mixture at −60°C (Scheme 1), and spectrophotometric monitoring of this reaction showed the growth of several new absorption bands (Figure 2). Importantly, the feature that appeared at λmax (εM) = 925 nm (170), which is associated with a d-d transition, is indicative of a high-valent FeIV–oxido species (details below).67 This assignment is supported by the Mössbauer spectra of [FeIVpoat(O)]−, which showed signals with an 57Fe isomer shift (δ) typical of species with an FeIV–oxido center (Figure 3). The displayed simulations are global least-square fits for the parameters given in the figure legend. The simulations include a minority S = 5/2 FeIII species that accounts for 20% of the total iron in the sample. The A tensor is comparable to that of [FeIVH3buea(O)]− with the anisotropic component being dominated by the spin-dipolar contributions which had one tensor component twice the magnitude of the others (ASD = (+4, +4, −8) T).31 These properties are indicative of an FeIV complex that has an S = 2 spin ground state and an unoccupied dz2 orbital.
Scheme 1.
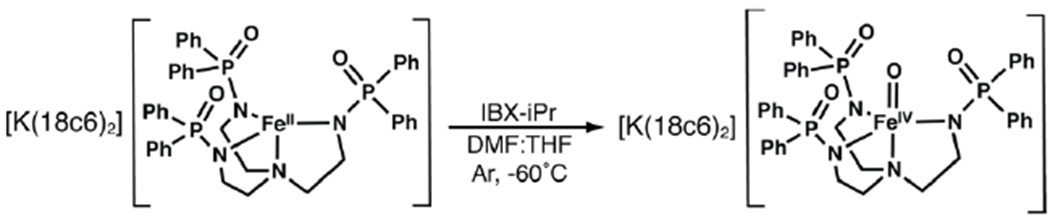
Preparative route to [FeIVpoat(O)]−.
Figure 2.
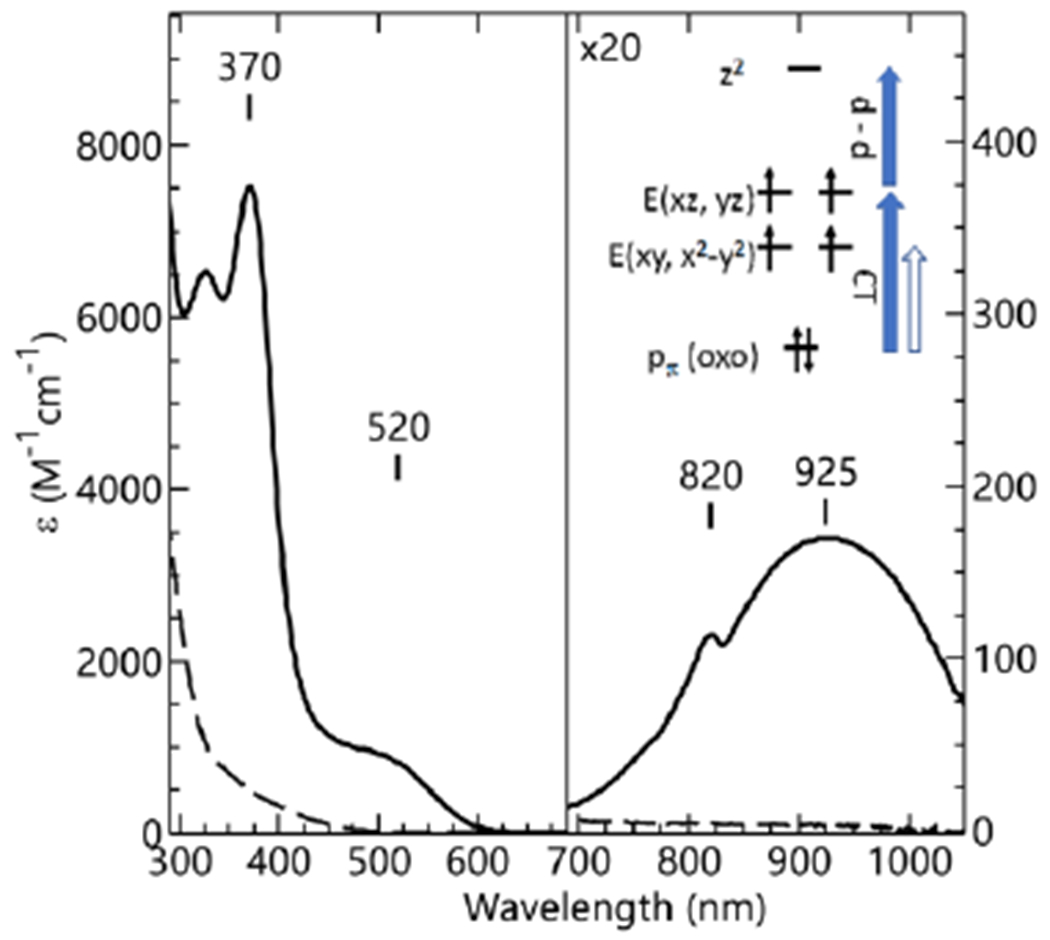
Electronic absorption spectra for the generation of [FeIVpoat(O)]− (solid) from the treatment of [FeIIpoat]− (dashed) with IBX-iPr at −60 °C in a DMF:THF mixture. Inset: magnification of the low-energy region. The inset shows die orbitals involved in the electronic transitions.
Figure 3.
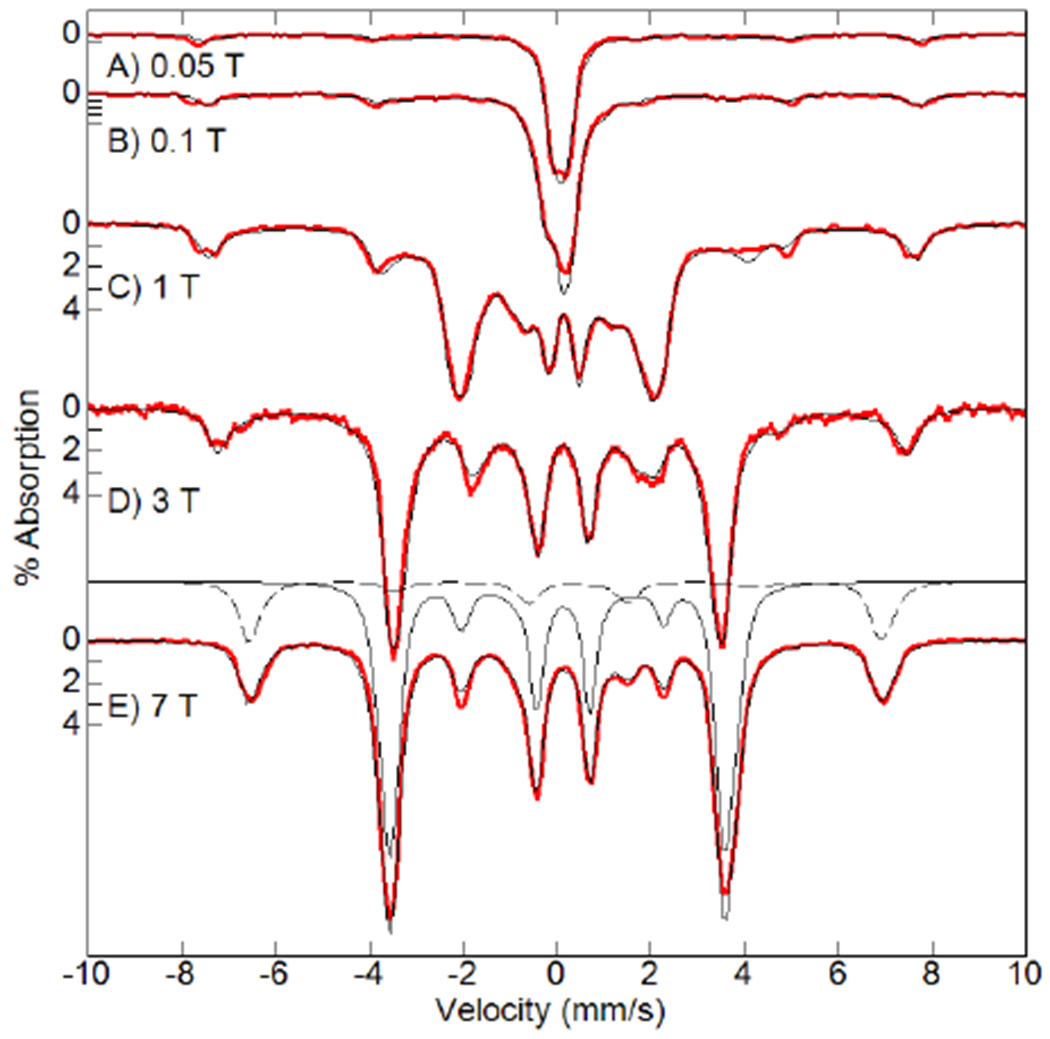
Mössbauer spectra of [FeIVpoat(O)]− (red traces, 4.2 K) and simulations (black traces) at the magnetic fields listed. Each simulation is the sum of S = 2 (80%) and S = 5/2 (20%) species. One set of simulations of the individual species is shown for the spectrum recorded at 7 T (solid trace is S = 2 and dashed trace is S = 5/2). S = 2 parameters: D = +5.1(3) cm−1, E/D = 0.03, A = (−15 −16 −28) T, δ = 0.11 mm/s, ΔEQ = +0.27 mm/s, η = 0.04. S = 5/2 parameters: D = +1.0 cm−1, E/D = 0.33, Aiso = −20 T, δ = 0.32 mm/s, ΔEQ = 0.80 mm/s, η = 0.03.
The vibrations of the FeIV–oxido unit of [FeIVpoat(O)]− were probed using 57Fe nuclear resonance vibrational spectroscopy (NRVS). This method detects only the vibrational modes associated with the motions of the 57Fe center in a complex and thus is highly sensitive and selective.68–70 The NRVS-derived 57Fe partial vibrational density of states (PVDOS) spectrum of [FeIVpoat(16O)]− shows a prominent peak at 843 cm−1 that shifts to 812 cm−1 in [FeIVpoat(18O)]− (Figures 4, S1). The observed shift for the two isotopomers is close to the predicted shift of 39 cm−1 obtained from a harmonic Fe–O oscillator model. Negative-mode electrospray ionization mass spectrometry (ESI-MS) provides further support for the formulation [FeIVpoat(16O)]−, as it shows an ion peak at m/z = 815.2 that shifts by two mass units in [FeIVpoat(18O)]− (Figure S2).
Figure 4.
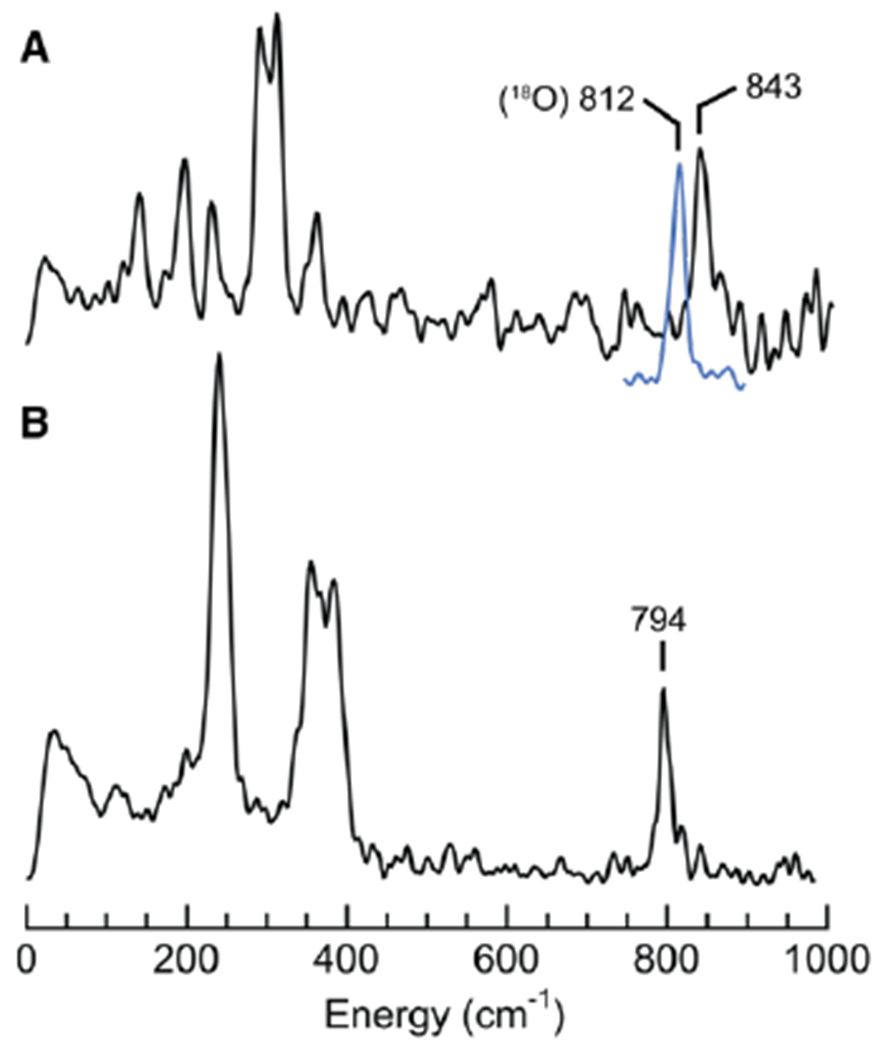
57Fe PVDOS spectra of [FeIVpoat(O)]− (A) and, for comparison, [FeIVH3buea(O)]− (B).71 Samples were ~25 mM 57Fe in a DMF:THF mixture. The blue spectrum is of [FeIVpoat(18O)]−.
Effects of H-Bonds.
As described above, the spectroscopic investigation of [FeIVpoat(O)]− and of the previously reported [FeIVH3buea(O)]− complex allows the influence of intramolecular H-bonds on the electronic properties of the FeIV–oxido unit to be evaluated through comparison of these two complexes. Our Mössbauer studies demonstrate that the H-bonds of [FeIVH3buea(O)]− affect the electronic properties, as the isomer shift of [FeIVpoat(O)]− (δ = 0.11(1) mm/s) is significantly higher than that of [FeIVH3buea(O)]− (δ = 0.02 mm/s, Table 1).31 Isomer shifts are related to the electron density at the Fe nucleus (ρ(0)) by the equation δ = α ρ(0) + constant, in which α has a negative value.71,72 The presence of H-bond donors proximal to the oxido ligand in [FeIVH3buea(O)]− lowers the amount of electron donation from the oxido ligand into the 3d orbitals. This lower electron density in the 3d orbitals reduces the shielding of the s electrons associated with the Fe center, which in turn, increases ρ(0). In contrast, the oxido ligand is not involved in forming H-bonds in [FeIVpoat(O)]− and more electron density is donated from the oxido ligand into the 3d orbitals. This difference causes a lower ρ(0) value and higher isomer shift relative to [FeIVH3buea(O)]−.
Table 1.
Experimental and DFT Results for the FeIV–oxido Complexes.a
| Complex | δb | ΔEQb | Dc | E/D | νFe–Oc |
|---|---|---|---|---|---|
| [FeIVH3buea(O)]−d | 0.02 | +0.43 | +4.7 | 0.03 | 794 |
| [FeIVpoat(O)]− | 0.11(1) [0.12] |
+0.27(1)) [0.21] |
+4.5(5) | 0.014(2) | 843 [832] |
| [FeIVpoat(O)---MgII]+ | 0.02(1) [0.03] |
+0.43(1) [0.41] |
+4.5(5) | 0.054(2) | 822 [823] |
| [FeIVpoat(O)---CaII]+ | 0.03(1) [0.05] |
+0.42(1) [0.39] |
+4.5(5) | 0.067(2) | 818 [812] |
| [FeIVpoat(O)---SrII]+ | 0.04(1) [0.05] |
+0.42(1) [0.39] |
+4.5(5) | 0.069(2) | 818 [810] |
| [FeIVpoat(O)---BaII]+ | 0.06(1) [0.05] |
+0.39(1) [0.37] |
+4.5(5) | 0.070(2) | 818 [821] |
Differences are also observed in the quadrupole splitting terms (ΔEQ) of the two FeIV–oxido complexes, in which [FeIVH3buea(O)]− has a larger value (ΔEQ = 0.43 mm/s) than [FeIVpoat(O)]− (ΔEQ = 0.27(1) mm/s). This difference can once more be understood in terms of how H-bonds affect the amount of electron density donated by the oxido ligand to the FeIV center. A free, high-spin FeIV ion is known to have a large and positive value for ΔEQ that is approximately 4 mm/s.71 However, in FeIV–oxido complexes, ΔEQ is reduced because the oxido ligand donates electron density into the unoccupied dz2 and singly occupied dxz, dyz orbitals of the FeIV center. Thus, any changes in the contributions of the oxido ligand to the bonding within the FeIV–oxido unit should then be reflected in a commensurate change in ΔEQ. From this analysis, the larger value of ΔEQ measured for [FeIVH3buea(O)]− is consistent with the involvement of the oxido ligand in H-bonds, which reduces the amount of electron density donated by the oxido ligand into orbitals on the Fe center that give a net negative contribution to ΔEQ.
The changes in the experimentally observed Mössbauer parameters are consistent with vibrational studies that show the bonding within the FeIV–oxido unit is stronger in [FeIVpoat(O)]− than in [FeIVH3buea(O)]− because [FeIVpoat(O)]− lacks intramolecular H-bonds. A comparison of the vibrational properties of the two complexes supports this finding. We recently reported the 57Fe PVDOS spectrum of [FeIVH3buea(O)]−,73 which contained an oxygen-isotopically sensitive band at 794 cm−1 that was assigned to νFe–O. The energy of this vibration is 49 cm−1 lower than that found for [FeIVpoat(O)]− (Figure 4). We further used Badger’s rule, an empirical formula that relates bond length to vibrational frequency (eq 1), to evaluate how the FeIV–Ooxido bond lengths vary within [FeIVpoat(O)]− and [FeIVH3buea(O)]−.74,75 We have already determined the values of CFe–O (56.69) and dFe–O (1.038) using a training set composed of related Fe–oxido and Fe–hydroxido complexes with tripodal ligands that include [H3buea]3−.75 From this analysis, we found FeIV–Ooxido bond lengths of 1.70 Å for [FeIVH3buea(O)]− and 1.67 Å for [FeIVpoat(O)]−. These values trend in the predicted way, with a longer bond length calculated for the weaker Fe–O bond of [FeIVH3buea(O)]−, but the difference between the values is small. Thus, we sought additional experimental evidence from extended X-ray absorption fine structure (EXAFS) studies on [FeIVpoat(O)]− (Figures S3 & S4A, Table S1). An FeIV–Ooxido bond length of 1.65 Å was found by EXAFS for [FeIVpoat(O)]−, which is again only slightly shorter than that measured for [FeIVH3buea(O)]− by X-ray diffraction methods (FeIV–Ooxido = 1.680(1) Å).
| (1) |
EPR Studies.
The [FeIVpoat(O)]− complex displays EPR signals in parallel mode (Brf ||B) with features from transitions observed within the excited |±1> (at g = 4) and|±2> (at g=8) doublets (Figure 5). Similar spectroscopic properties were previously reported for [FeIVH3buea(O)]− (Table 1),30,31 but whereas [FeIVH3buea(O)]− exhibited a sharp feature at g = 8, this feature is nearly absent in the spectrum of [FeIVpoat(O)]−. The intensities of the transitions within the |±1> and |±2> doublets are proportional to (E/D)2 and (E/D)4, respectively; thus, small changes in the E/D value have a substantial impact on the intensities of the signals.76 Both complexes have small E/D values that indicate nearly axial coordination environments around the Fe centers (Table 1). The quantitative simulations of the spectra (Figure 5) demonstrated that the lower E/D value of 0.014 for [FeIVpoat(O)]− resulted in a relatively weak signal at g = 8, whereas the g = 4 signal is relatively more intense owing to the smaller zero-field splitting of the |±1> doublet.
Figure 5.
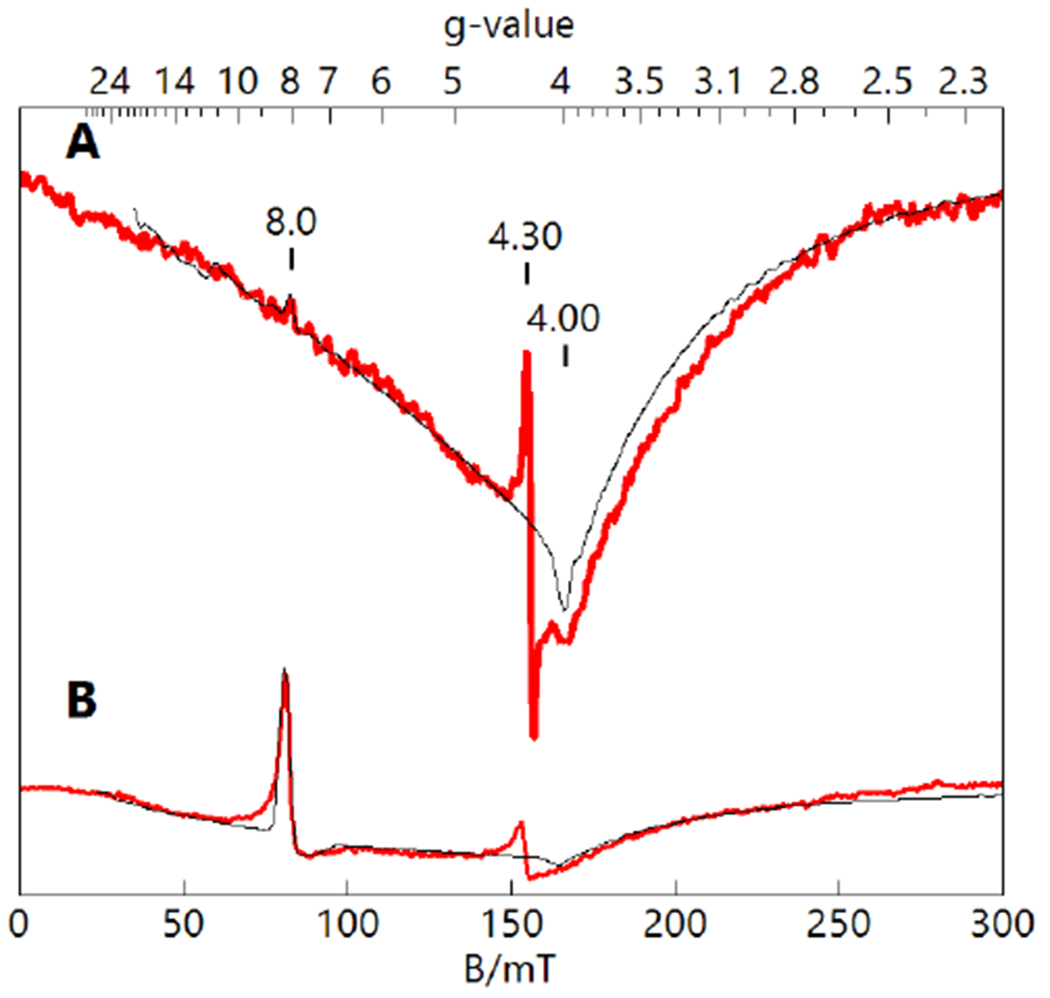
Parallel-mode EPR spectra (red) and simulations (black) of [FeIVpoat(O)]− (A, 10 mM in DMF:THF) and [FeIVH3buea(O)]− (B, 10 mM in DMF).30 The simulation parameters for S = 2 species are listed in Table 1. The signals at g = 4.3 are FeIII minority species. Instrumental parameters: 12 K, 9.33 GHz, 2mW.
Effects of LAIIs.
In addition to serving as an analog of [FeIVH3buea(O)]− without H-bond donors in the secondary coordination sphere, [FeIVpoat(O)]− possesses the unique ability to bind a second metal ion through an auxiliary binding site that involves the FeIV–oxido unit. Based on the strong influence of H-bonds discussed above, the binding of LAIIs to [FeIVpoat(O)]− should alter the electrostatic character of the secondary coordination sphere and therefore cause detectable changes in the spectroscopic properties. To test this premise, [FeIVpoat(O)]− was treated with several Group II metal ions in the presence of 15-crown-5 at −60°C to produce the series of [FeIVpoat(O)---LAII]+ complexes.
Mössbauer and EPR Studies.
The direct interaction of the LAIIs with the FeIV–oxido unit was indicated from Mössbauer studies, which changed the isomer shift and quadrupole splitting parameters for the [FeIVpoat(O)---LAII]+ adducts generated at −80°C (Table 1, Figures S5). These changes can be explained using the same arguments described for the effects of H-bonds on the bonding within the FeIV–O unit. The interaction of a LAII with the oxido ligand would partially reduce the amount of electron density donated into the empty orbitals of the FeIV center and should be reflected in commensurate changes in the values of δ and ΔEQ (see above). For instance, the parameters δ = 0.02(1) mm/s and ΔEQ = 0.43(1) mm/s measured for [FeIVpoat(O)---MgII]+ are different from those measured for [FeIVpoat(O)]− (Table 1) and identical to those recorded for [FeIVH3buea(O)]−, which we have shown to contain intramolecular H-bonds involving the oxido ligand. Moreover, the parameters for the series of [FeIVpoat(O)---LAII]+ adducts follow a trend that can be explained by a decrease in the acidity of the LAII, where the adduct having the lowest acidity, [FeIVpoat(O)---BaII]+, displays values most similar to those for [FeIVpoat(O)]−.
The binding of LAIIs to [FeIVpoat(O)]− was also monitored using parallel-mode EPR spectroscopy (Figure 6). Recall that [FeIVpoat(O)]− has nearly axial symmetry, which results in a relatively weak signal for the transition from the |±2> doublet at g = 8 (Figure 5). Based on DFT structures that show an asymmetric binding of the crown ether to the [poat]3− ligand (see below), it is anticipated that the [FeIVpoat(O)---LAII]+ species have more rhombic character than [FeIVpoat(O)]−, which increases the E/D values and the intensities of the signal at g = 8. These expected changes are indeed observed for each [FeIVpoat(O)---LAII]+ adduct, where the spectra display a prominent feature at g = 8 (Figure 6). The values of E/D for these heterometallic complexes were determined from simulations and found to be larger than that measured for [FeIVpoat(O)]− (Table 1).
Figure 6.
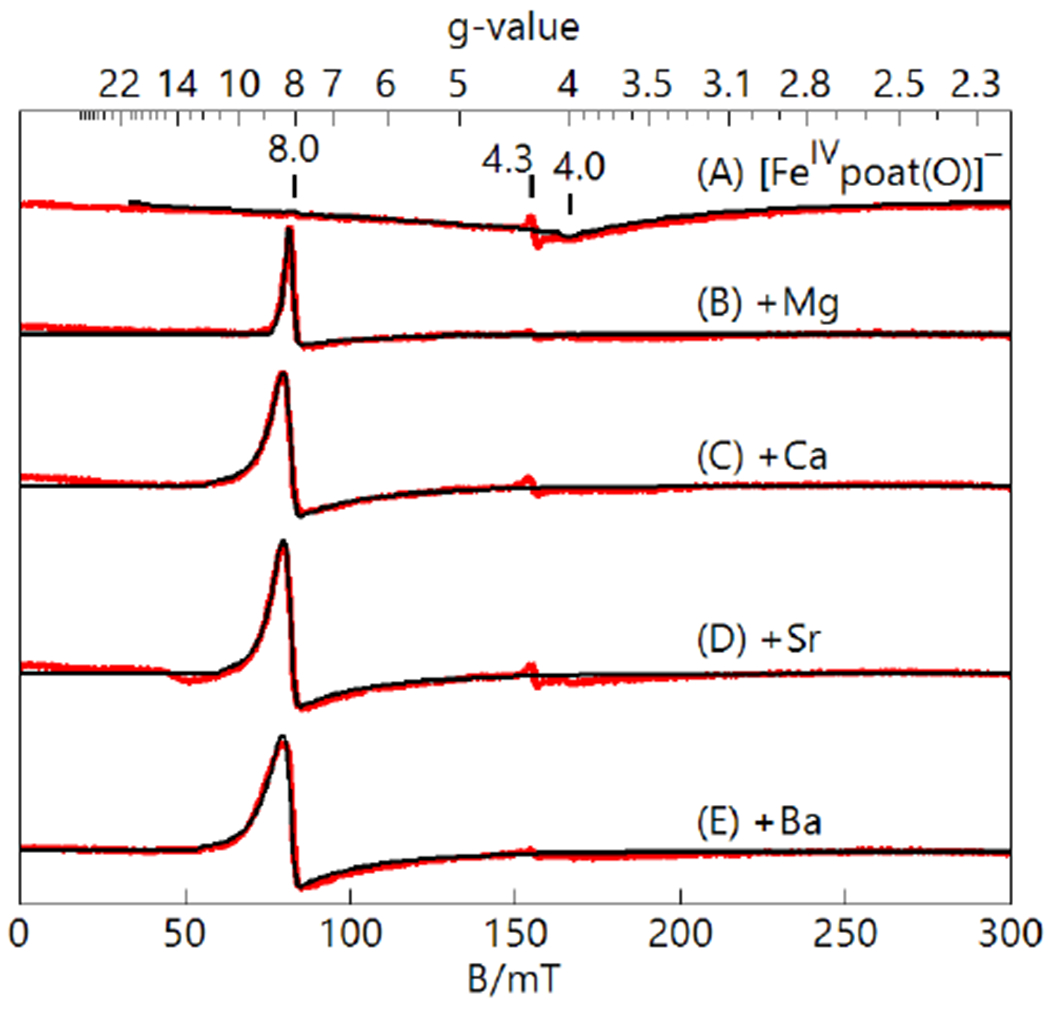
Parallel-mode EPR spectra (red) and simulations (black) of [FeIVpoat(O)]− (A) and the [FeIVpoat(O)---LAII]+ adducts (B-E), all 10 mM in DMF:THF. The simulation parameters for S = 2 species are listed in Table 1. The signal at g=4.3 is an FeIII minority species. Instrumental parameters: 12 K, 9.32 GHz, 2mW.
Electronic Absorption Studies.
The electronic absorption spectra of FeIV–oxido complexes have been extensively studied by Solomon and coworkers and include an analysis of [FeIVTMG-tren(O)]2+ (TMG-tren, 1,1,1-tris{2-[N2-(1,1,3,3-tetramethylguanidino)]ethyl}amine).67 This complex is similar to [FeIVpoat(O)]− in that it also contains a tripodal ligand, is C3 symmetric, and has an S = 2 spin ground state. We have used the findings for [FeIVTMG-tren(O)]2+ to guide our analysis of the absorption spectra for [FeIVpoat(O)]− and the [FeIVpoat(O)---LAII]+ complexes. The electronic absorption spectrum of [FeIVpoat(O)]− features a high energy band at λmax = 370 nm (εM = 7500) that is assigned to ligand-to-metal charged-transfer transition (LMCT) from the Oπ → E{xz,yz} transition (Figures 2 inset). A second LMCT transition is observed as a shoulder at λmax = 520 nm that is from the Oπ → E{xy,x2-y2} transition. The lower intensity of the feature at λmax = 520 nm is attributed to the electric dipole transition moment for this transition being smaller than for the Oπ → E{xz,yz} transition. As mention above, the absorption spectrum of [FeIVpoat(O)]− also contains a band at λmax = 925 nm that is from a d-d transition (5A → 5E). In addition, the small feature at λmax = 825 nm is assigned to a Fano interference of the spin forbidden 5A → 3E transition, which is a perturbation of the 5A → 5E band arising from the spin–orbit coupling of the 5E and 3E states (Figure S6).67,77
The binding of LAIIs to [FeIVpoat(O)]− has an effect on the absorption properties (Figure 7A) which further supports that the FeIV–oxido unit is directly interacting with the LAIIs. Both the LMCT and d-d bands are affected and their changes in energy correlate with the acidity of the Lewis acid. For the d-d transition, the band shift to higher energies upon the binding of a LAII and shows a linear correlation between the energy of the transition and the acidity of the LAII (Figure 7).78,79 The d-d band shift could be a function of both the Fe–oxido bond and the distortion of the equatorial ligands caused by the interaction with the crown ether. However, the shift of the dd band for [FeIVpoat(O)---MgII]+ is close to that of the [FeIVH3buea(O)]3−,30 which suggests that the Fe–oxido bond is the primary determinant of the position for this band. For the LMCT transition, the band shift to lower energies upon binding of stronger LAIIs and a linear correlation is also observed (Figure S7). These opposite trends can be explained by a downward shift in energy of the E{xz,yz} level when the LAIIs bind to [FeIVpoat(O)]−. The sum of the LMCT and d-d energies (Figure S7, Table 2) is approximately constant within the series and thus the main difference between the electronic levels of [FeIVpoat(O)]− and [FeIVpoat(O)---LAII]+ is the shift of the E{xz,yz} level (Figure S8). This interpretation is consistent with Fano interference associated with the 5A → 3E transition not being affected because it is independent of the E{xz,yz} level shift. Also evident in the spectra for the [FeIVpoat(O)---LAII]+ complexes is a weaker Fano interference at higher energies (λmax = 725-735 nm) about the assignment of which we will not speculate here. Finally, we note that there are increases in the intensity and width of d-d band that could be caused by the splitting of the E{xz,yz} doublet. These changes indicate an increasing distortion away from C3 symmetry with an increase in the acidity of the LAII.
Figure 7.
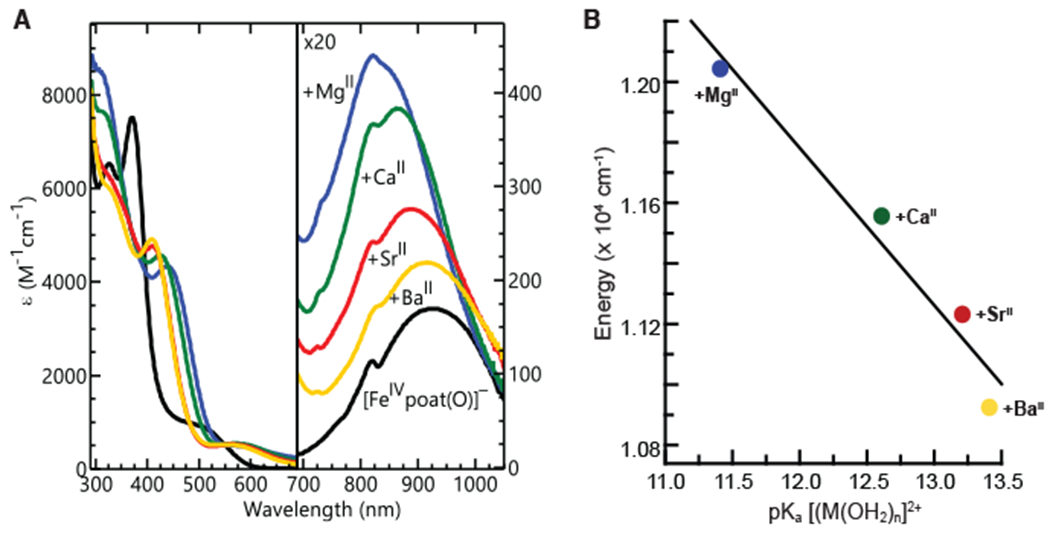
Electronic absorption spectra of [FeIVpoat(O)]− and the [FeIVpoat(O)---LAII]+ adducts at −60°C in a DMF:THF mixture (A), and a plot of the energy of the d-d transitions versus the pKa of [LA(OH2)n]2+ (B). The pKa of the appropriate complex ion, [LA(OH2)n]2+, in water is used to assess the acidity of the LAs.50,52,78,79
Table 2.
Energies of the Absorption Bands in [FeIVpoat(O)]−, [FeIVpoat(O)---LAII]+, and [FeIVTMG-tren(O)]2+
NRVS and XAS Studies.
The [FeIVpoat(O)---LAII]+ adducts were further evaluated by NRVS and XAS. The νFe–O band in these species is at least 18 cm−1 lower in energy than that in the parent [FeIVpoat(O)]− complex (Figure 8&S1 and Table 1) – such a lowering of νFe–O is expected for the direct interaction of the LAII with the Fe–O unit. However, the shifts in energy upon adduct formation are relatively small and do not compare with the difference in νFe–O between [FeIVpoat(O)]− and [FeIVH3buea(O)]−. In addition, the vibrational energies of the LAII adducts are the same within experimental error.
Figure 8.
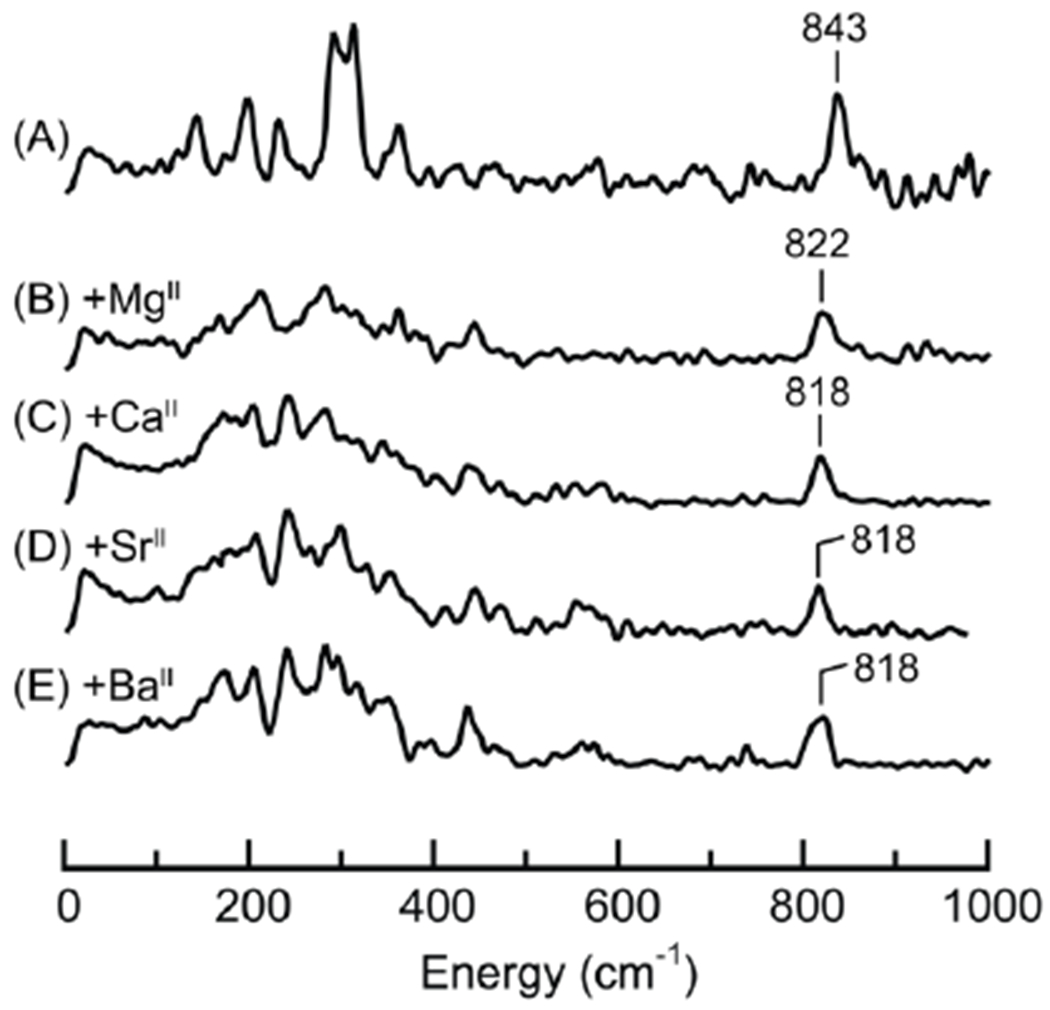
57Fe PVDOS spectra of [FeIVpoat(O)]− (A) and the [FeIVpoat(O)---LAII]+ adducts (B-E). Samples were ~20 mM 57Fe in a DMF:THF mixture. The peaks corresponding to the Fe–O vibrations are labelled with their corresponding energies.
XANES spectra were recorded for [FeIVpoat(O)]−, [FeIVpoat(O)---MgII]+, and [FeIVpoat(O)---CaII]+, and they show similar Fe–K edge energies at 7124.2, 7125.6, and 7125.3 eV, respectively; these values are within the range previously reported for FeIV complexes (Figure 9A&B).29,80,81 Within this subset of complexes, a distinct decrease in pre-edge peak area is observed upon the addition of a LAII, with area values of 32 units for [FeIVpoat(O)]− and of 25 and 28 units for the MgII and CaII adducts, respectively (Figure 9C). This trend in pre-edge peak area is consistent with the direct interaction of the LAIIs with the FeIV–O unit, which would cause a decrease in the mixing of the 3p and 3d orbitals at the FeIV center and less intense transitions from the 1s to 3d orbitals. EXAFS analysis allows the bond distances in the three complexes to be compared (Figures S4B&C, Tables 2&S1). In particular, as expected, the Fe–Ooxido bond lengthens to 1.67 Å in [FeIVpoat(O)---MgII]+ and [FeIVpoat(O)---CaII]+ from 1.65 Å in [FeIVpoat(O)]−, but this change is too small to enable a definitive assessment. For comparison, a Badger’s rule analysis estimated Fe–O bond lengths of approximately 1.68 Å for these two heterometallic complexes (Table 2).
Figure 9.
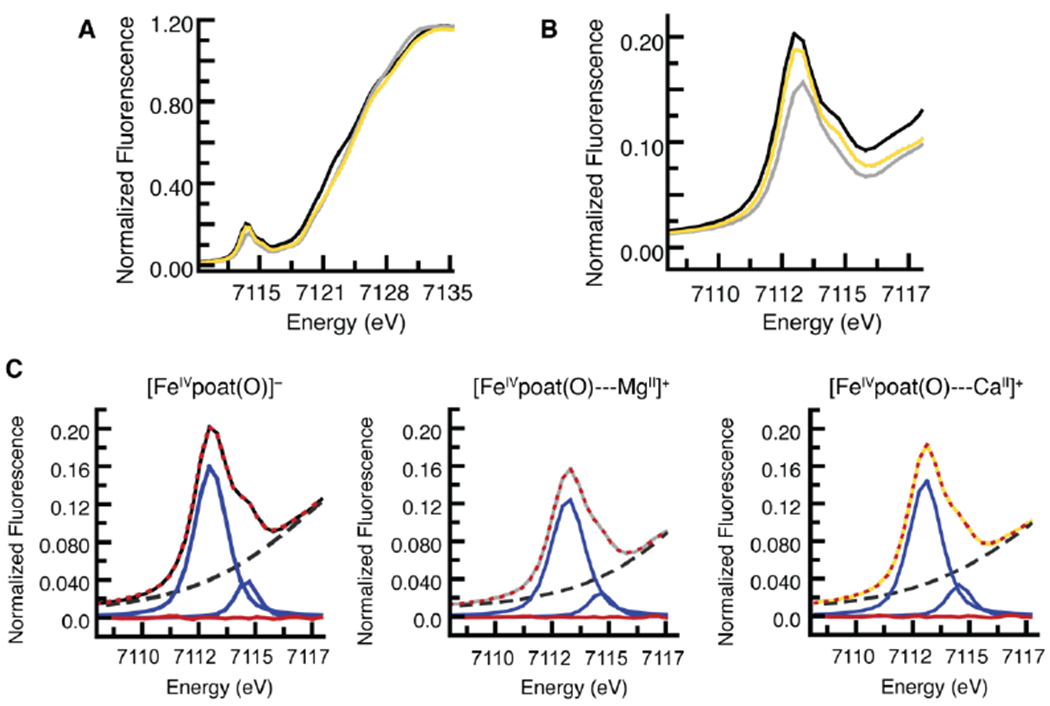
Experimentally observed pre-edge and XANES of the XAS spectra for [FeIVpoat(O)]− (black), [FeIVpoat(O)---MgII]+ (gray), and [FeIVpoat(O)---CaII]+ (gold) (A); the pre-edge region (B); and analysis of the pre-edge peaks (C). Experimental data in C are represented by the same colored spectra as in B, the best fits as dotted red lines, the modeled baselines as black dashed lines, the fitted component peaks as blue lines, and the residuals as red lines.
Computational Studies.
Density functional theory calculations were performed to obtain optimized structures of the [FeIVpoat(O)]− complex and the [FeIVpoat(O)---LAII]+ adducts (Figures 10&S9, Table S2). The structures reveal that the LAII forms bonds to the oxido ligand, two P=O groups of the [poat]3− ligand, and O atoms of the crown ether. In some cases, not all the O atoms of the crown ether are coordinated to the LAII which agrees with the known coordination properties of these group 2 metal ions. For example, DFT suggests that the MgII center in [FeIVpoat(O)---MgII]+ is six-coordinate with the crown ether serving as a tridentate ligand (Figure 10B). We used DFT to compute both the Mössbauer parameters and νFe–O, and the results of the calculations are in good agreement with the experimental results, in terms of both numerical accuracy and predicting the trends (Table 1). For instance, the calculated δ value drops significantly from 0.12 mm/s to 0.03 mm/s upon the binding of the MgII ion to [FeIVpoat(O)]−, a near match to what was observed experimentally. In addition, the calculated ΔEQ values for these two complexes differ by 0.20 mm/s, which is comparable to the experimental change of 0.16 mm/s. To demonstrate that the direct Fe=O⋯MgII interaction is essential for lowering the value of δ, we performed a single point DFT calculation on a modified version for [FeIVpoat(O)---MgII]+ in which the Mg2+ ion and crown ether were removed without changing the positions of the remaining atoms. A δ = 0.15 mm/s was found for this structure, a value that is close to δ = 0.12 mm/s for the optimized structure of the [FeIVpoat(O)]−. The DFT calculations also reveal that the coordination of a LAII lowers the energy of νFe–O by 9–22 cm−1 (Table 1); for comparison, shifts that range between 17 and 21 cm−1 were deduced from NRVS. Both the experimental and DFT-calculated Fe–oxido frequencies are smaller than the frequencies expected based on Badger’s rule when using the Fe–O bond distances taken from the optimized structures (~55 cm−1).
Figure 10.
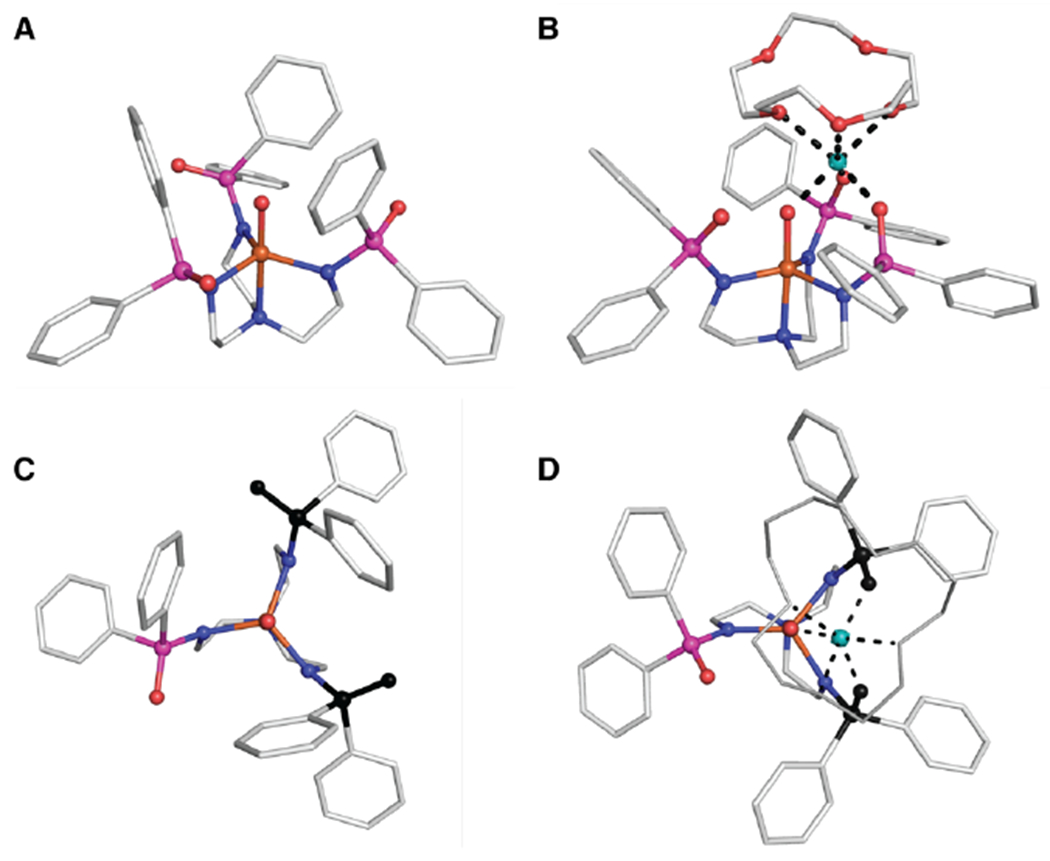
DFT-optimized structures of [FeIVpoat(O)]− (A), [FeIVpoat(O)---MgII]+ (B), view along the C3 axis for [FeIVpoat(O)]− (C), and view along the C3 axis for [FeIVpoat(O)---MgII]+ (D). FeIV ions are colored in orange, O atoms in red, N atoms in blue, P atoms in magenta, C atoms in white, and MgII ions in teal. Hydrogen atoms are omitted for clarity. In C and D, the two P=O units that change positions are highlighted in black. Hydrogen atoms are omitted for clarity and the 15-crown-5 unit in D is represented by grey sticks.
The DFT structures also revealed that [poat]3− ligand distorts from C3 to C1 symmetry to bind a LAII. Specifically, two of the P=O units rotate such that they can form the tridentate binding site with the oxido ligand to coordinate the LAII (Figure 10C&D). These large structural changes led us to consider whether the shifts in the absorption bands in [FeIVpoat(O)]− are not only the result of the Fe=O⋯LAII interaction but may also in part be mediated by the distortions of the equatorial ligands. We have ruled out this possibility for the following reasons. The DFT structures show that the distortion of the Fe=O unit in the [FeIVpoat(O)---LAII]+ adducts are nearly independent of LAII; for instance, the Fe–O⋯LAII angle is approximately 135° for all the complexes. The large changes in the LMCT and d-d bands that were observed are more likely the result of the direct interaction of the LAII with the Fe=O moiety, the strongest effect being for [FeIVpoat(O)---MgII]+, which has the shortest FeO⋯LAII distance. This premise is consistent with the conclusion inferred from DFT calculations that the trend in the isomer shift is the result of direct FeO⋯LAII interaction and not of distortions of the equatorial ligand (see above). In addition, the electrostatic properties of the LAIIs should affect the energies of the orbitals within the Fe–oxido unit that include linear combinations of the dxz, dyz, and dz2 orbitals and the oxido p orbitals: dxz + cx px, dyz + cy py, and dz2 + cz pz. This effect depends on the Fe–O⋯LAII angle with those close to 180° favoring a lowering of the admixed dz2 (decreasing the d-d energy) and those close to 90° causing a lowering of the admixed dxz or dyz (increasing the d-d energy). The experimental data suggest that the lowering of the E{xz,yz} level prevails over the lowering of the dz2 level, resulting in a larger d-d energy and consequently an Fe–O⋯LAII angle biased towards 90°.
Summary and Conclusions
The preparation of [FeIVpoat(O)]− was achieved using a newly designed tripodal ligand that creates a primary coordination sphere capable of stabilizing high-valent metal centers. Its spectroscopic properties and correlated optimized structure from DFT supported the symmetrical arrangement of the [poat]3− ligand around the Fe–oxido unit. Importantly, our findings were consistent with [FeIVpoat(O)]− having an S = 2 spin ground state that matches the spin states of intermediates found in non-heme oxygenases.16 These types of synthetic complexes are rare with [FeIVpoat(O)]− being only the sixth known example.27–34 What distinguishes [FeIVpoat(O)]− is that it provides the unique opportunity to study the influences of H-bonds and electrostatic interactions on high-valent metal–oxido complexes. First, this complex served as an analog of [FeIVH3buea(O)]− that lacks intramolecular H-bond donors, which allowed us to examine the effects of H-bonds on the electronic structure of an FeIV–oxido complex through a comparative investigation of [FeIVpoat(O)]− and [FeIVH3buea(O)]−. Our previous reports on [FeIVH3buea(O)]− detailed its molecular structure determined from XRD measurements, its electronic structure determined from EPR, Mössbauer, and absorption spectroscopies, and its vibrational properties determined from FTIR and NRVS spectroscopies. These data led us to conclude that intramolecular H-bonds formed with the FeIV–oxido unit, but we were unable to gauge the magnitude of their effect on the properties of this complex. We have now learned that the intramolecular H-bonds modulate the electron density of the oxido ligand and weaken the bond within the FeIV–oxido unit. These effects were identified using Mossbauer spectroscopy and correlated with NRVS vibrational studies, in which a decrease of 49 cm−1 was observed in νFe–O from [FeIVpoat(O)]− to [FeIVH3buea(O)]−. A Badger’s rule analysis showed that these changes in electronic structure resulted in only a modest change of approximately 0.02 Å in the Fe–O bond length, a change that was also supported by EXAFS measurements.
We further probed the electrostatic effects of FeIV–oxido units by examining the influences of LAIIs on the properties of [FeIVpoat(O)]−. In our design of [FeIVpoat(O)]−, we employed an auxiliary metal ion binding site composed of two P=O units of [poat]3– and the oxido ligand. The presence of this tridentate auxiliary site provides an advantage over other FeIV–oxido complexes for which the binding of LAIIs is assumed to occur only at the oxido ligand. This proposed monodentate coordination of a LAII is inherently weak because an oxido ligand bound to a high-valent transition metal ion is electrophilic and thus would be a poor ligand to metal ions. Thus, it has proven difficult to experimentally assess the exact binding mode of LAII s in other FeIV–oxido complexes.
The binding of LAIIs to [FeIVpoat(O)]− was screened using absorption and EPR spectroscopies. Both methods showed effects that were consistent with the formation of [FeIVpoat(O)---LAII]+ adducts. We observed a blue shift in the d-d band and a red shift in the LMCT band that correlated with the acidity of the LAIIs. Further confirmation of a direct interaction between LAII and the oxido ligand comes from Mössbauer spectroscopy. Significant differences were observed in the Mössbauer parameters of the [FeIVpoat(O)---LAII]+ adducts compared to those of [FeIVpoat(O)]−. These differences were again linked to changes in the electron donation of the oxido ligand into the vacant dz2 orbital and the singly occupied dxz and dyz orbitals of the FeIV center that were caused by the binding of the LAs. Within the series of [FeIVpoat(O)---LAII]+ adducts, the shifts in Mössbauer parameters trended toward those found for [FeIVH3buea(O)]−. In fact, the Mössbauer properties of [FeIVpoat(O)---MgII]+ were identical to those of [FeIVH3buea(O)]−, indicating that the binding of a MgII ion is comparable to the formation of three intramolecular H-bonds in [FeIVH3buea(O)]−. Because we know that the H-bonds in [FeIVH3buea(O)]− involve the oxido ligand, we concluded that the MgII ion, and the other LAIIs, also directly coordinate to the oxido ligand in [FeIVpoat(O)]− through its auxiliary binding site. Optimized structures of the [FeIVpoat(O)---LAII]+ adducts supported this premise and provided structural and spectroscopic values that were closely aligned with those found experimentally. Moreover, the results of vibrational and XAS pre-edge studies were consistent with the binding of LAIIs to [FeIVpoat(O)]− via interactions with the oxido ligand.
There has been discussion in the recent literature about how best to manipulate the properties of Fe–oxido complexes in order to influence the formal oxidation and spin states of the iron center.13,18,26,56,81–83 Most previous investigations have made changes within the primary coordination sphere and examined how covalent interactions to the Fe–oxido unit can be correlated to functional changes. In contrast, the primary coordination sphere in our system remained nearly constant, and we instead modulated the non-covalent interactions within the secondary coordination sphere of the FeIV–oxido center to effect change. Our findings show that these non-covalent interactions significantly influence the properties of metal–oxido species. Our experimental confirmation that H-bonds and LAIIs can interact with the oxido ligand in FeIV–oxido complexes showed the direct effects that these interactions have on the bonding within the Fe–oxido unit and the electronic structure. Furthermore, our results provide an explanation for why Fe–oxido species formed in the gas phase have different properties than those prepared in the condensed phase83–86—namely, that the different microenvironments present around the Fe–oxido units in these two phases can cause changes in the electronic and structural properties. Thus, we argue that caution should be taken when comparing Fe–oxido complexes prepared in different phases. Within a broader context, our work supports the premise that non-covalent interactions are instrumental in tuning the properties within protein active sites and therefore could be used in a similar manner within synthetic constructs to produce more functional oxidants.
Experimental
Physical Methods.
Physical Methods.
Absorption, EPR, and Mossbauer Spectroscopic Methods.
Electronic absorption spectra were recorded in a 1-cm cuvette on an 8453 Agilent UV-Vis spectrometer equipped with an Unisoku Unispeks cryostat. X-band EPR spectra were recorded on a Bruker ELEXSYS spectrometer equipped with an Oxford ESR-910 liquid helium cryostat and a Bruker bimodal cavity for the generation of microwave fields parallel and transverse to the applied magnetic field. The quantification of all signals was performed relative to a CuEDTA spin standard prepared from a copper atomic absorption standard (Sigma-Aldrich). The microwave frequency was calibrated with a frequency counter, and the magnetic field was measured with an NMR gaussmeter. The sample temperature was calibrated against a calibrated cernox sensor (Lakeshore CX-1050) mounted inside an EPR tube. A modulation frequency of 100 kHz was used for all EPR spectra. Mossbauer spectra were recorded on either a variable field or a weak-field spectrometer operating in a constant acceleration mode in a transmission geometry using Janis Research Inc. cryostats that allow for a variation in temperature from 4 to 300 K. One of the dewars housed a superconducting magnet that allowed for the application of magnetic fields up to 8 T parallel to the γ-radiation. Isomer shifts are reported relative to Fe metal at 298 K. The simulation software SpinCount was written by one of the authors.87
The software diagonalizes the electronic terms of the spin Hamiltonian:
where the parameters have the usual definitions,88 and performs least-squares fitting of simulations to the spectra. The quantitative simulations are generated with consideration of all intensity factors, which allows the computation of simulated spectra for a specified sample concentration.
Nuclear Resonance Vibrational Spectroscopic Methods.
The 57Fe NRVS data were recorded using published procedures on multiple occasions at beamline 3-ID at the Advanced Photon Source (APS).70,89 The incident flux provided by the beamline is ~2 × 109 photons/s in a 1 meV bandwidth centered at 14.4125 keV in a 1 mm (vertical) × 3 mm (horizontal) spot. The monochromators used in the experiment consisted of a water-cooled diamond (1,1,1) double crystal with 1.1 eV bandpass followed by two separate Si(4,0,0) and Si(10,6,4) channel-cut crystals in a symmetric geometry. During the measurements, samples were maintained at low temperatures using a closed-cycle helium cryostat. The temperature for individual spectra was calculated using the ratio of anti-Stokes to Stokes intensity according to S(−E) = S(E) exp(−E/kT) and was generally in the range of 40 K to 80 K. Spectra were recorded between −40 meV and 120 meV in 0.25-meV steps. Delayed nuclear fluorescence and iron K fluorescence (from internal conversion) were recorded with a single avalanche photodiode detector (APD) with a 1 cm2 detection area. Each scan required approximately 50 minutes, and all scans were added and normalized to the intensity of the incident beam. The 57Fe partial vibrational density of state (PVDOS) was extracted from the raw NRVS data using the PHOENIX software package.90
X-ray Absorption Spectroscopic Methods.
XAS experiments were performed at the Stanford Synchrotron Radiation Laboratory (SSRL) on beam line 7-3 at 13 K. Fe K-edge data were collected using a Si(220) φ=0° double-crystal monochromator with a 9.0 keV cutoff for harmonic rejection. Data were collected in fluorescence mode with a Canberra 30-element Ge solid-state detector. To limit photoreduction of the samples, only first scans were averaged into the final data sets (exposure time ~35 minutes). Fe K-edge data for the both all species were comprised of 7 first scans. Energies were calibrated using an iron foil (7111.2 eV), and edge energies were obtained from the first derivative of the data with 1.0 eV smoothing and a third-order polynomial in the program EXAFSPAK.91 The Fe K-edge data sets were fit over the region k = 3–14 A°−1 using EXAFSPAK91 with ab initio phases and amplitudes generated with the program FEFF v8.40.92 The Fe K-edge fits for the three iron species were comprised of the first, second, third, fourth, and fifth shell atoms. All Debye-Waller factors were treated as free parameters. The scale factor, S0, was set to 0.9. Monochromator glitches in the Fe K-edge data sets at approximately k = 12.5 Aΰ-1 were removed using a cubic polynomial fit to the data. No other modifications to the raw data were performed.
DFT Calculations.
The DFT calculations performed with the hybrid functional B3LYP,93,94 using the program Gaussian ’09.95 The calculations included geometry optimizations and property calculations for the frequencies and 57Fe hyperfine parameters. The triple-zeta basis set 6-311G was used for all atoms except for the phosphorus atoms and the alkaline earth atoms X.96 For the P atoms the polarization-function-augmented basis set 6-311G* was adopted to allow for a proper description of the pentavalency of these atoms. As 6-311G* is not available for the heavier alkaline earth metals, the pseudo-potential Stuttgard-Dresden basis set SDD was used, also for the lighter members of the series to avoid possible basis set effects on the property trends along the LA series.97–99 To correct for the systematic overestimation of the frequencies obtained with the employed functional/basis set combination, the calculated frequencies were multiplied with a factor of 0.9.74 The quadrupole splittings, ΔEQ, were evaluated from the calculated eigenvalues of the electric-field gradient tensor, −Vii, i = x, y, z, using the expression , where ℓ is the unit charge, is the nuclear quadrupole moment for 57Fe, and η is the asymmetry parameter, which is defined as (Vxx − Vyy)/Vzz. By defining x, y and z such that | Vxx | ≤ | Vyy | ≤ | Vzz |, η is confined to the range [0,1]. By adopting the consensus value , the calculated Vii values, given in atomic units, convert to the velocity scale by multiplication with −1.6 mm s−1/AU. In all systems considered here, the eigenvector corresponding to Vzz is closely aligned with the Fe-O axis. The 57Fe isomer shift was evaluated from the electron density at the nucleus using the calibration of Vrajmasu.100
Geometry optimizations on [FeIVpoat(O)]− were initiated with the XRD structure of [FeIIIpoat]. The geometry optimization of [FeIVpoat(O)---MgII]+ began from a structure that was assembled with the aid of the visualization program GaussView by manually combining the optimized structures of the metallated crown ether moiety, [MgII15c5]2+, with the optimized structure of [FeIVpoat(O)]−. The structure avoided steric interactions but allowed the MgII ion to interact with the oxido ligand and the O-atoms of two of the P=O groups. The optimizations of the geometries of the other members of the [FeIVpoat(O)---LAII]+ were obtained in a stepwise manner via replacement of the LAII.
Preparative Methods.
General Synthetic Procedures.
All manipulations, unless otherwise stated, were completed under an argon atmosphere in a VAC drybox. Solvents were sparged with argon and dried over columns containing Q-5 and molecular sieves. All reagents were purchased from commercial suppliers and used as received unless otherwise noted. Potassium hydride as a 30% suspension in mineral oil was filtered and washed five times each with Et2O and pentane and dried under vacuum. IBX-iPr,101,102 15c5Ca(OTf)2,62 15c5Mg(OTf)2,103 15c5Sr(OTf)2,64 and 15c5Ba(OTf)2104 were prepared according to literature procedures.
H3poat.
To a solution of tris(2-aminoethyl) amine (tren) (1.12 g, 7.66 mmol) and triethylamine (11.65 g, 115.1 mmol) in 80 mL of tetrahydrofuran (THF), diphenyl phosphinic chloride (5.51 g, 23.3 mmol) in 20 mL of THF was added dropwise while stirring. Once the addition was complete, 20 mL of THF was added to rinse the addition funnel. After the addition funnel was removed, the round bottom flask was covered with a glass stopper and left to stir overnight. After filtering the white precipitate (Et3NHCl), the solvent was removed, and the residue was dried under vacuum. Diethyl ether (Et2O) (125 mL) was added to the resulting oil to give a white powder (82%), which was collected on a medium porosity glass-fritted funnel, washed twice with ether (40 mL), and dried for several hours under vacuum. 1H NMR (400 MHz, CDCl3, ppm): 2.44 (6H, br t), 2.81 (6H, q, J = 6.6 Hz), 5.64 (3H, q, J = 10.1 Hz, NH), 7.43 (12H, d, J = 8.9 Hz, o-Ar), 7.51 (6H, t, J = 8.9 Hz, p-Ar), 7.68 (12H, t, J = 9.8, m-Ar). 31P NMR (162 MHz, CDCl3, ppm): 21.6; FTIR (KBr, selected bands cm−1): 3260, 3130, 3261; 2970, 2941, 2870, 2790, 2604, 2497, 1590, 1438, 1188, 1122, 724, 697, 517. HRMS (ES+, m/z): Exact mass calculated for C42H45N4O3P3: 746.27, Found: 746.85.
K[FeIIpoat].
To a solution of H3poat (200 mg, 0.269 mmol) in anhydrous THF (6 mL) was added potassium hydride (KH) (33 mg, 0.81 mmol), and the reaction was allowed to proceed until gas evolution ceased and all solids were dissolved. To the light-yellow solution was added FeII(OAc)2 (47 mg, 0.26 mmol). The solution was stirred for 25 minutes and then filtered through a medium fritted glass funnel to remove insoluble material. Light yellow crystals (208 mg, 93%) were afforded by vapor diffusion or the layering of diethyl ether (Et2O) into or on top of the solution of THF. Elemental analysis calcd. (found) for K[FeIIpoat] (C42H42FeKN4O3P3): C, 60.15 (60.17); H, 5.05 (4.91); N, 6.68 (6.66) %. X-band, parallel-mode EPR (DMF:THF, 10 K) g = 9.44. Mössbauer (DMF:THF, 4K) δ = 1.02 mm/s; ΔEQ = 2.66 mm/s.
Low-temperature Solution Studies using [FeIIpoat]−
In a typical experiment, a 20-mM solution of K[FeIIpoat] (17 mg, 0.020 mmol) with 18-crown-6 ether (11 mg, 0.042 mmol), added to increase solubility, was prepared in the desired solvent (1 mL) at room temperature and kept in a −35 °C freezer for the duration of the experiment. A 30-μL aliquot of stock the FeII complex (0.60 μmol) was added via air-tight syringe to the solvent mixture (3 mL) in a 1-cm quartz cuvette to give the desired concentration for oxidation experiments (0.2 mM). The cuvette was then sealed with a rubber septum and precooled to the desired temperature in an 8453 Agilent UV-vis spectrophotometer equipped with an Unisoku Unispeks cryostat. The solution of metal complex was allowed to equilibrate to the desired temperature for at least 15 min. Stock solutions of other reagents were prepared at concentrations between 20 and 50 mM in the same solvent and added via gas-tight syringe to an aliquot of (18c6)2K[FeIIpoat]. In typical reaction, the 30-μL aliquot of (18c6)2K[FeIIpoat] (see above) was treated with a 33-μL aliquot of IBX-iPr stock solution (20 mM, 0.66 μmol, 1.1 equiv.) to afford (18c6)2K[FeIIpoat(O)], which could be convert to the bimetallic complexes via addition of a 30-μL aliquot of [15c5LAII](OTf)2 (20 mM, 0.60 μmol, 1.0 equiv.). Reactions were initially monitored spectrophotometrically by UV-vis spectroscopy. Preparations of the [FeIVpoat(O)---LAII]+ complexes were monitored spectrophotometrically and we found no changes in the absorption spectra after the addition of more than 1 equiv of the LA salt to suggest discrete bimetallic species were formed (Figure S10 for [FeIVpoat(O)---SrII]+). Samples for EPR studies were prepared in an similar manner in a −80 °C bath with the following modifications: an 80-μL aliquot of the (18c6)2K[FeIIpoat] stock solution (45 mM, 3.6 μmol ) was added to a 150-μL THF:DMF solution of IBX-iPr (30 mM, 4.5 μmol, 1.3 equiv.) in a pre-chilled EPR tube (~−80°C). Formation of the bimetallic involved the addition of the appropriate [15c5LAII](OTf)2 salt as 40-μL aliquots (3.8 μmol, 1.1 equiv) to (18c6)2K[FeIVpoat(O)]. Mössbauer samples were prepared in an analogous manner to those for EPR studies but in a cold-well of a drybox using 96% 57Fe-enriched K[57FeIIpoat]. Multiple attempts to grow single crystals in various solvents and temperatures for X-ray diffraction measurements were unsuccessful.
Preparation of Low-temperature Solution NRVS Samples.
Solution NRVS sample holders were prepared from Mössbauer sample holders by cutting a 2 × 6 mm slot out of the bottom and covering the hole with kapton tape. The NRVS sample holder was cooled in the cold well of a drybox to −196 °C. A 40-mM solution of 96% 57Fe-enriched K[FeIIpoat]− (34 mg, 0.041 mmol) was prepared in a DMF:THF mixture (1 mL) containing 18-crown-6 ether (22 mg, 0.083 mmol) to increase solubility. The NRVS sample holder was cooled in the cold well of a drybox to −196 °C. Additionally, a stock solution of IBX-iPr (40 mM) was prepared in a 2:1 DMF:THF mixture and kept in a −80 °C cold well for the duration of the experiment. An aliquot of (18c6)2K[FeIIpoat] (300 μL) was added to one equivalent of IBX-iPr (300 μL). A sample of [FeIVpoat(O)]− (500 μL of the 20 mM solution) was added to the pre-cooled NRVS sample holder via syringe and allowed to freeze completely. The sample was then transferred from the drybox and quickly placed in a storage container pre-cooled to 77 K. Samples were analyzed for purity using Mössbauer spectroscopy prior to NRVS data collection. Analogous procedures were followed for [FeIVpoat(O)---LAII]+ adducts with the subsequent addition of 100 μL (183 mM, 18.3 μmol) of 15c5Ca(OTf)2, 15c5Mg(OTf)2, 15c5Sr(OTf)2 and 15c5Ba(OTf)2 respectively.
Preparation of Low-temperature Solution XAS Samples.
Solution XAS samples were prepared in a similar manner as that employed for the solution NRVS samples described above with the following modifications: a 30-mM solution of K[FeIIpoat] (50 mg, 0.060 mmol), of which 3 mM was prepared with 96% 57Fe-enriched [FeIIpoat]−, was prepared in a DMF:THF mixture (2 mL) with 18-crown-6 ether (32 mg, 0.12 mmol) added to increase solubility. Samples were analyzed for purity using Mössbauer spectroscopy prior to XAS data collection. Analogous procedures were followed for the [FeIVpoat(O)---MgII]+ and [FeIVpoat(O)---CaII]+ adducts with the subsequent addition of 500 μL (130 mM, 0.065 mmol) of 15c5Ca(OTf)2 or 15c5Mg(OTf)2, respectively. Solution XAS sample holders were prepared from Mössbauer sample holders by cutting off the bottom and covering the resulting hole with kapton tape.
Labelling Studies.
Preparation of 18O Labeled Samples.
H218O (50 μL, 2.7 mmol) was added to a solution of 16O IBX-iPr (0.013 g, 0.040 mmol) in D3COD (1 mL) and stirred for 30 min. All volatiles were removed in vacuo. The residue was redissolved in D3COD (1 mL), treated with a second iteration of H218O (50 μL, 2.7 mmol), stirred for an additional 30 min, and then reduced under pressure to a white solid that was used without further purification. [FeIVpoat(18O)]− was prepared according to the low-temperature procedure described above, in which the 18O source was 18O IBX-iPr.
Supplementary Material
Table 3.
Structural Parameters of the FeIV–oxido Complexes
| Complex | Fe–Oa,b | Fe–Oa,c | Fe–Oa,d |
|---|---|---|---|
| [FeIVpoat(O)]− | 1.65 | 1.67 | 1.64 |
| [FeIVpoat(O)---MgII]+ | 1.67 | 1.68 | 1.67 |
| [FeIVpoat(O)---CaII]+ | 1.67 | 1.69 | 1.66 |
| [FeIVpoat(O)---SrII]+ | — | 1.69 | 1.66 |
| [FeIVpoat(O)---BaII]+ | — | 1.69 | 1.66 |
Å;
from EXAFS;
from Badger’s rule;
from DFT
ACKNOWLEDGMENT
The authors acknowledge the NIH (GM050781 to A.S.B., GM077387 to GM101390 to M.T.G., and GM125924 to Y.G.) for funding. The NRVS data were recorded at the Advanced Photon Source (APS), Argonne National Laboratory (proposal GUP-50088 and GUP-60939). The use of APS is supported by the Department of Energy.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publication website at DOI: https://doi.org/10.1021/jacs.0c03085
The following file is available free of charge.
Figures S1–S10and Tables S1–S3 (PDF)
References
- (1).Solomon EI; Brunold TC; Davis MI; Kemsley JN; Lee S-K; Lehnert N; Neese F; Skulan AJ; Yang Y-S; Zhou J Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev 2000, 100, 235–350. [DOI] [PubMed] [Google Scholar]
- (2).Que L; Tolman WB Biologically Inspired Oxidation Catalysis. Nature 2008, 455, 333–340. [DOI] [PubMed] [Google Scholar]
- (3).Borovik AS Role of Metal-Oxo Complexes in the Cleavage of C-H Bonds. Chem. Soc. Rev 2011, 40, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Cook SA; Hill EA; Borovik AS Lessons from Nature: A Bio-Inspired Approach to Molecular Design. Biochemistry 2015, 54, 4167–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Shook RL; Borovik AS Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation. Inorg. Chem 2010, 49, 3646–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Krest CM; Onderko EL; Yosca TH; Calixto JC; Karp RF; Livada J; Rittle J; Green MT Reactive Intermediates in Cytochrome P450 Catalysis. J. Biol. Chem 2013, 288, 17074–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Huang X; Groves JT Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev 2018, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Poulos TL Heme Enzyme Structure and Function. Chem. Rev 2014, 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Moody PCE; Raven EL The Nature and Reactivity of Ferryl Heme in Compounds I and II. Acc. Chem. Res 2018, 51, 427–435. [DOI] [PubMed] [Google Scholar]
- (10).Costas M; Mehn MP; Jensen MP; Que L Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev 2004, 104, 939–986. [DOI] [PubMed] [Google Scholar]
- (11).Cook SA; Borovik AS Molecular Designs for Controlling the Local Environments around Metal Ions. Acc. Chem. Res 2015, 48, 2407–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Nam W Synthetic Mononuclear Nonheme Iron-Oxygen Intermediates. Acc. Chem. Res 2015, 48, 2415–2423. [DOI] [PubMed] [Google Scholar]
- (13).Guo M; Corona T; Ray K; Nam W Heme and Nonheme High-Valent Iron and Manganese Oxo Cores in Biological and Abiological Oxidation Reactions. ACS Cent. Sci 2019, 5, 13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kovaleva EG; Lipscomb JD Versatility of Biological Non-Heme Fe(II) Centers in Oxygen Activation Reactions. Nat. Chem. Biol 2008, 4, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Price JC; Barr EW; Tirupati B; Bollinger JM; Krebs C The First Direct Characterization of a High-Valent Iron Intermediate in the Reaction of an α-Ketoglutarate-Dependent Dioxygenase: A High-Spin Fe(IV) Complex in Taurine/α-Ketoglutarate Dioxygenase (TauD) from Escherichia Coli. Biochemistry 2003, 42, 7497–7508. [DOI] [PubMed] [Google Scholar]
- (16).Krebs C; Galonić Fujimori D; Walsh CT; Bollinger JM Non-Heme Fe(IV)–Oxo Intermediates. Acc. Chem. Res 2007, 40, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Galonić Fujimori D; Barr EW; Matthews ML; Koch GM; Yonce JR; Walsh CT; Bollinger JM; Krebs C; Riggs-Gelasco PJ Spectroscopic Evidence for a High-Spin Br-Fe(IV)-Oxo Intermediate in the α-Ketoglutarate-Dependent Halogenase CytC3 from Streptomyces. J. Am. Chem. Soc 2007, 129, 13408–13409. [DOI] [PubMed] [Google Scholar]
- (18).Que L The Road to Non-Heme Oxoferryls and Beyond. Acc. Chem. Res 2007, 40, 493–500. [DOI] [PubMed] [Google Scholar]
- (19).Nam W Dioxygen Activation by Metalloenzymes and Models. Acc. Chem. Res 2007, 40, 465. [Google Scholar]
- (20).Meyer S; Klawitter I; Demeshko S; Bill E; Meyer F A Tetracarbene-Oxoiron(FV) Complex. Angew. Chem. Int. Ed 2013, 52, 901–905. [DOI] [PubMed] [Google Scholar]
- (21).Sahu S; Zhang B; Pollock CJ; Dürr M; Davies CG; Confer AM; Ivanović-Burmazović I; Siegler MA; Jameson GNL; Krebs C; Goldberg DP Aromatic C-F Hydroxylation by Nonheme Iron(FV)-Oxo Complexes: Structural, Spectroscopic, and Mechanistic Investigations. J. Am. Chem. Soc 2016, 138, 12791–12802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chanda A; Shan X; Chakrabarti M; Ellis WC; Popescu DL; De Oliveira FT; Wang D; Que L; Collins TJ; Munck E; Bominaar EL (TAML)FeIV=O Complex in Aqueous Solution: Synthesis and Spectroscopic and Computational Characterization. Inorg. Chem 2008, 47, 3669–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chantarojsiri T; Sun Y; Long JR; Chang CJ Water-Soluble Iron(IV)-Oxo Complexes Supported by Pentapyridine Ligands: Axial Ligand Effects on Hydrogen Atom and Oxygen Atom Transfer Reactivity. Inorg. Chem 2015, 54, 5879–5887. [DOI] [PubMed] [Google Scholar]
- (24).Rana S; Biswas JP; Sen A; Clemancey M; Blondin G; Latour JM; Rajaraman G; Maiti D Selective C-H Halogenation over Hydroxylation by Non-Heme Iron(IV)-Oxo. Chem. Sci 2018, 9, 7843–7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Puri M; Que L Toward the Synthesis of More Reactive S = 2 Non-Heme Oxoiron(IV) Complexes. Acc. Chem. Res 2015, 48, 2443–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang D; Ray K; Collins MJ; Farquhar ER; Frisch Jonathan R; Gomez L; Jackson TA; Kerscher M; Waleska A; Comba P; Costas M; Que L Nonheme Oxoiron(IV) Complexes of Pentadentate N5 Ligands: Spectroscopy, Electrochemistry, and Oxidative Reactivity. Chem. Sci 2012, 4, 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pestovsky O; Stoian S; Bominaar EL; Shan X; Münck E; Que L; Bakac A Aqueous FeIV=O: Spectroscopic Identification and Oxo-Group Exchange. Angew. Chem. Int. Ed 2005, 44, 6871–6874. [DOI] [PubMed] [Google Scholar]
- (28).England J; Guo Y; Farquhar ER; Young VGJ; Munck E; Que L The Crystal Structure of a High-Spin Oxoiron(IV) Complex and Characterization of Its Self-Decay Pathway. J. Am. Chem. Soc 2010, 132, 8635–8644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).England J; Martinho MM; Farquhar ER; Frisch JR; Bominaar EL; Münck E; Que L A Synthetic High-Spin Oxoiron(IV) Complex: Generation, Spectroscopic Characterization, and Reactivity. Angew. Chem. Int. Ed 2009, 48, 3622–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lacy DC; Gupta R; Stone KL; Greaves J; Ziller JW; Hendrich MP; Borovik AS Formation, Structure, and EPR Detection of a High Spin FeIV-Oxo Species Derived from Either an FeIII-Oxo or FeIII-OH Complex. J. Am. Chem. Soc 2010, 132, 12188–12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gupta R; Lacy DC; Bominaar EL; Borovik AS; Hendrich MP Electron Paramagnetic Resonance and Mössbauer Spectroscopy and Density Functional Theory Analysis of a High-Spin FeIV-Oxo Complex. J. Am. Chem. Soc 2012, 134, 9775–9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bigi JP; Harman WH; Lassalle-Kaiser B; Robles DM; Stich TA; Yano J; Britt RD; Chang CJ A High-Spin Iron(IV)–Oxo Complex Supported by a Trigonal Nonheme Pyrrolide Platform. J. Am. Chem. Soc 2012, 134, 1536–1542. [DOI] [PubMed] [Google Scholar]
- (33).England J; Guo Y; Van Heuvelen KM; Cranswick MA; Rohde GT; Bominaar EL; Munck E; Que L A More Reactive Trigonal-Bipyramidal High-Spin Oxoiron(IV) Complex with a Cis-Labile Site. J. Am. Chem. Soc 2011, 133, 11880–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Biswas AN; Puri M; Meier KK; Oloo WN; Rohde GT; Bominaar EL; Münck E; Que L Modeling TauD-J: A High-Spin Nonheme Oxoiron(IV) Complex with High Reactivity toward C-H Bonds. J. Am. Chem. Soc 2015, 137, 2428–2431. [DOI] [PubMed] [Google Scholar]
- (35).Barman SK; Jones JR; Sun C; Hill EA; Ziller JW; Borovik AS Regulating the Basicity of Metal-Oxido Complexes with a Single Hydrogen Bond and Its Effect on C-H Bond Cleavage. J. Am. Chem. Soc 2019, 141, 11142–11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Shook RL; Borovik AS The Effects of Hydrogen Bonds on Metal-Mediated O2 Activation and Related Processes. Chem. Commun 2008, 6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chreifi G; Baxter EL; Doukov T; Cohen AE; McPhillips SE; Song J; Meharenna YT; Soltis SM; Poulos TL Crystal Structure of the Pristine Peroxidase Ferryl Center and Its Relevance to Proton-Coupled Electron Transfer. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Casadei CM; Gumiero A; Metcalfe CL; Murphy EJ; Basran J; Concilio MG; Teixeira SCM; Schrader TE; Fielding AJ; Ostermann A; Blakeley MP; Raven EL; Moody PCE Neutron Cryo-Crystallography Captures the Protonation State of Ferryl Heme in a Peroxidase. Science (80-.). 2014, 345, 193–197. [DOI] [PubMed] [Google Scholar]
- (39).Kwon H; Basran J; Casadei CM; Fielding AJ; Schrader TE; Ostermann A; Devos JM; Aller P; Blakeley MP; Moody PCE; Raven EL Direct Visualization of a Fe(IV)-OH Intermediate in a Heme Enzyme. Nat. Commun 2016, 7, 13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Prakash J; Rohde GT; Meier KK; Jasniewski AJ; Van Heuvelen KM; Münck E; Que L Spectroscopic Identification of an FeIII Center, Not FeIV, in the Crystalline Sc-O-Fe Adduct Derived from [FeIV(O)(TMC)]2+. J. Am. Chem. Soc 2015, 137, 3478–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Leeladee P; Baglia RA; Prokop KA; Latifi R; De Visser SP; Goldberg DP Valence Tautomerism in a High-Valent Manganese-Oxo Porphyrinoid Complex Induced by a Lewis Acid. J. Am. Chem. Soc 2012, 134, 10397–10400. [DOI] [PubMed] [Google Scholar]
- (42).Baglia RA; Dürr M; Ivanović-Burmazović I; Goldberg DP Activation of a High-Valent Manganese-Oxo Complex by a Nonmetallic Lewis Acid. Inorg. Chem 2014, 53, 5893–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zaragoza JPT; Baglia RA; Siegler MA; Goldberg DP Strong Inhibition of O-Atom Transfer Reactivity for MnIV(O)(π-Radical-Cation)(Lewis Acid) versus MnV(O) Porphyrinoid Complexes. J. Am. Chem. Soc 2015, 137, 6531–6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Baglia RA; Krest CM; Yang T; Leeladee P; Goldberg DP High-Valent Manganese-Oxo Valence Tautomers and the Influence of Lewis/Bronsted Acids on C-H Bond Cleavage. Inorg. Chem 2016, 55, 10800–10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Park J; Morimoto Y; Lee YM; Nam W; Fukuzumi S Metal Ion Effect on the Switch of Mechanism from Direct Oxygen Transfer to Metal Ion-Coupled Electron Transfer in the Sulfoxidation of Thioanisoles by a Non-Heme Iron(IV)–Oxo Complex. J. Am. Chem. Soc 2011, 133, 5236–5239. [DOI] [PubMed] [Google Scholar]
- (46).Hong S; Lee YM; Sankaralingam M; Vardhaman AK; Park YJ; Cho K. Bin; Ogura T; Sarangi R; Fukuzumi S; Nam WA Manganese(V)-Oxo Complex: Synthesis by Dioxygen Activation and Enhancement of Its Oxidizing Power by Binding Scandium Ion. J. Am. Chem. Soc 2016, 138, 8523–8532. [DOI] [PubMed] [Google Scholar]
- (47).Sharma N; Jung J; Ohkubo K; Lee YM; El-Khouly ME; Nam W; Fukuzumi S Long-Lived Photoexcited State of a Mn(IV)-Oxo Complex Binding Scandium Ions That Is Capable of Hydroxylating Benzene. J. Am. Chem. Soc 2018, 140, 8405–8409. [DOI] [PubMed] [Google Scholar]
- (48).Sankaralingam M; Lee YM; Pineda-Galvan Y; Karmalkar DG; Seo MS; Jeon SH; Pushkar Y; Fukuzumi S; Nam W Redox Reactivity of a Mononuclear Manganese-Oxo Complex Binding Calcium Ion and Other Redox-Inactive Metal Ions. J. Am. Chem. Soc 2019, 141, 1324–1336. [DOI] [PubMed] [Google Scholar]
- (49).Rice DB; Grotemeyer EN; Donovan AM; Jackson TA Effect of Lewis Acids on the Structure and Reactivity of a Mononuclear Hydroxomanganese(III) Complex. Inorg. Chem 2020, 59, 2689–2700. [DOI] [PubMed] [Google Scholar]
- (50).Tsui EY; Kanady JS; Agapie T Synthetic Cluster Models of Biological and Heterogeneous Manganese Catalysts for O2 Evolution. Inorg. Chem 2013, 52, 13833–13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tsui EY; Tran R; Yano J; Agapie T Redox-Inactive Metals Modulate the Reduction Potential in Heterometallic Manganese–Oxido Clusters. Nat. Chem 2013, 5, 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Tsui EY; Agapie T Reduction Potentials of Heterometallic Manganese-Oxido Cubane Complexes Modulated by Redox-Inactive Metals. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 10084–10088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Yiu S-M; Man W-L; Lau T-C Efficient Catalytic Oxidation of Alkanes by Lewis Acid/[OsVI(N)Cl4] –Using Peroxides as Terminal Oxidants. Evidence for a Metal-Based Active Intermediate. J. Am. Chem. Soc 2008, 130, 10821–10827. [DOI] [PubMed] [Google Scholar]
- (54).Fukuzumi S; Ohkubo K Quantitative Evaluation of Lewis Acidity of Metal Ions Derived from the g Values of ESR Spectra of Superoxide: Metal Ion Complexes in Relation to the Promoting Effects in Electron Transfer Reactions. Chem. Eur. J 2000, 6, 4532–4535. [DOI] [PubMed] [Google Scholar]
- (55).Ohkubo K; Menon SC; Orita A; Otera J; Fukuzumi S Quantitative Evaluation of Lewis Acidity of Metal Ions with Different Ligands and Counterions in Relation to the Promoting Effects of Lewis Acids on Electron Transfer Reduction of Oxygen. J. Org. Chem 2003, 68, 4720–4726. [DOI] [PubMed] [Google Scholar]
- (56).Morimoto Y; Kotani H; Park J; Lee YM; Nam W; Fukuzumi S Metal Ion-Coupled Electron Transfer of a Nonheme Oxoiron(IV) Complex: Remarkable Enhancement of Electron-Transfer Rates by Sc3+. J. Am. Chem. Soc 2011, 133, 403–405. [DOI] [PubMed] [Google Scholar]
- (57).Fukuzumi S; Morimoto Y; Kotani H; Naumov P; Lee YM; Nam W Crystal Structure of a Metal Ion-Bound Oxoiron(IV) Complex and Implications for Biological Electron Transfer. Nat. Chem 2010, 2, 756–759. [DOI] [PubMed] [Google Scholar]
- (58).Swart M A Change in the Oxidation State of Iron: Scandium Is Not Innocent. Chem. Commun 2013, 49, 6650–6652. [DOI] [PubMed] [Google Scholar]
- (59).MacBeth CE; Golombek AP; Young VG; Yang C; Kuczera K; Hendrich MP; Borovik AS O2 Activation by Nonheme Iron Complexes: A Monomeric Fe(III)-Oxo Complex Derived from O2. Science (80-. ). 2000, 289, 938–941. [DOI] [PubMed] [Google Scholar]
- (60).Sgro MJ; Stephan DW Frustrated Lewis Pair Inspired Carbon Dioxide Reduction by a Ruthenium Tris(Aminophosphine) Complex. Angew. Chem. Int. Ed 2012, 51, 11343–11345. [DOI] [PubMed] [Google Scholar]
- (61).Oswald VF; Weitz AC; Biswas S; Ziller JW; Hendrich MP; Borovik AS Manganese–Hydroxido Complexes Supported by a Urea/Phosphinic Amide Tripodal Ligand. Inorg. Chem 2018, 57, 13341–13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Park YJ; Ziller JW; Borovik AS The Effects of Redox-Inactive Metal Ions on the Activation of Dioxygen: Isolation and Characterization of a Heterobimetallic Complex Containing a MnIII–(μ-OH)–CaII Core. J. Am. Chem. Soc 2011, 133, 9258–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Lacy DC; Park YJ; Ziller JW; Yano J; Borovik AS Assembly and Properties of Heterobimetallic CoII/III/CaII Complexes with Aquo and Hydroxo Ligands. J. Am. Chem. Soc 2012, 134, 17526–17535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Park YJ; Cook SA; Sickerman NS; Sano Y; Ziller JW; Borovik AS Heterobimetallic Complexes with M(III)-(μ-OH)-M(II) Cores (M(III) = Fe, Mn, Ga; M(II) = Ca, Sr, and Ba): Structural, Kinetic, and Redox Properties. Chem. Sci 2013, 4, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Cook SA; Ziller JW; Borovik AS Iron(II) Complexes Supported by Sulfonamido Tripodal Ligands: Endogenous versus Exogenous Substrate Oxidation. Inorg. Chem 2014, 53, 11029–11035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Sickerman NS; Peterson SM; Ziller JW; Borovik AS Synthesis, Structure and Reactivity of Fe II/III –NH3 Complexes Bearing a Tripodal Sulfonamido Ligand. Chem. Commun 2014, 50, 2515–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Srnec M; Wong SD; England J; Que L; Solomon EI π-Frontier Molecular Orbitals in S = 2 Ferryl Species and Elucidation of Their Contributions to Reactivity. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 14326–14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Sage JT; Paxson C; Wyllie GRA; Sturhahn W; Durbin SM; Champion PM; Alp EE; Scheidt WR Nuclear Resonance Vibrational Spectroscopy of a Protein Active-Site Mimic. J. Phys. Condens. Matter 2001, 13, 7707–7722. [Google Scholar]
- (69).Kohn VG; Chumakov AI DOS: Evaluation of Phonon Density of States from Nuclear Resonant Inelastic Absorption. Hyperfme Interact. 2000, 125, 205–221. [Google Scholar]
- (70).Sturhahn W Nuclear Resonant Spectroscopy. J. Phys. Condens. Matter 2004, 16, S497–S530. [Google Scholar]
- (71).Gütlich P; Eckhard B; Trautwein AX Mössbauer Spectroscopy and Transition Metal Chemistry, 1st ed.; Springer-Verlag Berlin Heidelberg, 2011. [Google Scholar]
- (72).Debrunner PG Mössbauer Spectroscopy of Iron Porphyrins In Physical Bioinorganic Chemistry Series 4. Iron Porphyrins Part III; Lever ABP, Gray HB, Eds.; VCH Publishers, Inc.: New York, 1989; pp 137–234. [Google Scholar]
- (73).Hill EA; Weitz AC; Onderko E; Romero-Rivera A; Guo Y; Swart M; Bominaar EL; Green MT; Hendrich MP; Lacy DC; Borovik AS Reactivity of an Fe(IV)–Oxo Complex with Protons and Oxidants. J. Am. Chem. Soc 2016, 138, 13143–13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Green MT Application of Badger’s Rule to Heme and Non-Heme Iron-Oxygen Bonds: An Examination of Ferryl Protonation States. J. Am. Chem. Soc 2006, 128, 1902–1906. [DOI] [PubMed] [Google Scholar]
- (75).Weitz AC; Hill EA; Oswald VF; Bominaar EL; Borovik AS; Hendrich MP; Guo Y Probing Hydrogen Bonding Interactions to Iron-Oxido/Hydroxido Units by 57Fe Nuclear Resonance Vibrational Spectroscopy. Angew. Chemie - Int. Ed 2018, 57, 16010–16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Hendrich MP; Debrunner PG Integer-Spin Electron Paramagnetic Resonance of Iron Proteins. Biophys. J 1989, 56, 489–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Fano U Effects of Configuration Interaction on Intensities and Phase Shifts. Phys. Rev 1961, 124, 1866–1878. [Google Scholar]
- (78).Perrin DD. Dissociation Constant of Inorganic Acids and Bases in Aqueous Solution; Pergamon: New York, NY, 1982. [Google Scholar]
- (79).Herbert DE; Lionetti D; Rittle J; Agapie T Heterometallic Triiron-Oxo/Hydroxo Clusters: Effect of Redox-Inactive Metals. J. Am. Chem. Soc 2013, 135, 19075–19078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Rohde J-U; Torelli S; Shan X; Lim MH; Klinker EJ; Kaizer J; Chen K; Nam W; Que L Structural Insights into Nonheme Alkylperoxoiron(III) and Oxoiron(IV) Intermediates by X-Ray Absorption Spectroscopy. J. Am. Chem. Soc 2004, 126, 16750–16761. [DOI] [PubMed] [Google Scholar]
- (81).Jackson TA; Rohde J-U; Seo MS; Sastri CV; DeHont R; Stubna A; Ohta T; Kitagawa T; Munck E; Nam W; Que L Axial Ligand Effects on the Geometric and Electronic Structures of Nonheme Oxoiron(IV) Complexes. J. Am. Chem. Soc 2008, 130, 12394–12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Jasniewski AJ; Que L Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev 2018, 118, 2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Andris E; Navrátil R; Jašík J; Puri M; Costas M; Que L; Roithovaá J Trapping Iron(III)-Oxo Species at the Boundary of the “Oxo Wall”: Insights into the Nature of the Fe(III)-O Bond. J. Am. Chem. Soc 2018, 140, 14391–14400. [DOI] [PubMed] [Google Scholar]
- (84).Borrell M; Andris E; Navrátil R; Roithová J; Costas M Characterized Cis-FeV(O)(OH) Intermediate Mimics Enzymatic Oxidations in the Gas Phase. Nat. Commun 2019, 10, 901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Andris E; Jašík J; Gómez L; Costas M; Roithová J Spectroscopic Characterization and Reactivity of Triplet and Quintet Iron(IV) Oxo Complexes in the Gas Phase. Angew. Chemie 2016, 128, 3701–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Andris E; Navrátil R; Jašík J; Terencio T; Srnec M; Costas M; Roithová J Chasing the Evasive Fe=O Stretch and the Spin State of the Iron(IV)-Oxo Complexes by Photodissociation Spectroscopy. J. Am. Chem. Soc 2017, 139, 2757–2765. [DOI] [PubMed] [Google Scholar]
- (87).Petasis DT; Hendrich MP Quantitative Interpretation of Multifrequency Multimode EPR Spectra of Metal Containing Proteins, Enzymes, and Biomimetic Complexes. Methods Enyymol. 2015, 563, 171–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Abragam A; Bleaney B. Electron Paramagnetic Resonance of Transition Ions, Reprint.; Oxford University Press: Oxford, 2012. [Google Scholar]
- (89).Toellner TS Monochromatization of Synchrotron Radiation for Nuclear Resonant Scattering Experiments. Hyperfine Interact. 2000, 125, 3–28. [Google Scholar]
- (90).Sturhahn W CONUSS and PHOENIX: Evaluation of Nuclear Resonant Scattering Data. Hyperfine Interact. 2000, 125, 149–172. [Google Scholar]
- (91).George GN EXAFSPAK. 2000. [Google Scholar]
- (92).Ankudinov AL; Ravel B; Rehr JJ; Conradson SD Real-Space Multiple-Scattering Calculation and Interpretation of X-Ray-Absorption near-Edge Structure. Phys. Rev. B 1998, 58, 7565–7576. [Google Scholar]
- (93).Becke AD A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys 1993, 98, 1372–1377. [Google Scholar]
- (94).Lee C; Yang W; Parr RG Development of the Colic-Salvetti Correlation-Energy into a Functional of the Electron Density Formula. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- (95).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M ;Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Keith T; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N;Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas O; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ; Gaussian 09. Gaussian, Inc.: Wallingford, CT: 2013. [Google Scholar]
- (96).Binning RC; Curtiss LA Compact Contracted Basis Sets for Third-Row Atoms: Ga–Kr. J. Comput. Chem 1990, 11, 1206–1216. [Google Scholar]
- (97).Fuentealba P; Preuss H; Stoll H; Von Szentpály L A Proper Account of Core-Polarization with Pseudopotentials: Single Valence-Electron Alkali Compounds. Chem. Phys. Lett 1982, 89, 418–422. [Google Scholar]
- (98).Fuentealba P; Von Szentpály L; Stoll H; Fraschio FX; Preuss H Pseudopotential Calculations Including Core-Valence Correlations: Alkali Compounds. J. Mol. Struct 1983, 93, 213–219. [Google Scholar]
- (99).Fuentealba P; Stoll H; Von Szentpaly L; Schwerdtfeger P; Preuss H On the Reliability of Semi-Empirical Pseudopotentials: Simulation of Hartree-Fock and Dirac-Fock Results. J. Phys. B At. Mol. Phys 1983, 16, L323–L328. [Google Scholar]
- (100).Vrajmasu V; Munck E; Bominaar EL Density Functional Study of the Electric Hyperfine Interactions and the Redox-Structural Correlations in the Cofactor of Nitrogenase. Analysis of General Trends in 57Fe Isomer Shifts. Inorg. Chem 2003, 42, 5974–5988. [DOI] [PubMed] [Google Scholar]
- (101).Zhdankin VV; Litvinov DN; Koposov AY; Luu T; Ferguson MJ; McDonald R; Tykwinski RR Preparation and Structure of 2-Iodoxybenzoate Esters: Soluble and Stable Periodinane Oxidizing Reagents. Chem. Commun 2004, 263, 106–107. [DOI] [PubMed] [Google Scholar]
- (102).Zhdankin VV; Koposov AY; Litvinov DN; Ferguson MJ; McDonald R; Luu T; Tykwinski RR Esters of 2-Iodoxybenzoic Acid: Hypervalent Iodine Oxidizing Reagents with a Pseudobenziodoxole Structure. J. Org. Chem 2005, 70, 6484–6491. [DOI] [PubMed] [Google Scholar]
- (103).Sanchez ER; Gessel MC; Groy TL; Caudle MT Interaction of Biotin with Mg-O Bonds: Bifunctional Binding and Recognition of Biotin and Related Ligands by the Mg(15-Crown-5)2+ Unit. J. Am. Chem. Soc 2002, 124, 1933–1940. [DOI] [PubMed] [Google Scholar]
- (104).Wallen CM; Palatinus L; Bacsa J; Scarborough CC Hydrogen Peroxide Coordination to Cobalt(II) Facilitated by Second-Sphere Hydrogen Bonding. Angew. Chem 2016, 128, 12081–12085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.