

Abstract
The complex yet interrelated connections between cancer metabolism, gene expression, and oncogenic driver genes have the potential to identify novel biomarkers and drug targets with prognostic and therapeutic value. Here we effectively integrated metabolomics and gene expression data from breast cancer mouse models through a novel unbiased correlation-based network analysis. This approach identified 35 metabolite and 34 gene hubs with the most network correlations. These hubs have prognostic value and are likely integral to tumor metabolism and breast cancer. The gene hub Aquaporin-7 (Aqp7), a water and glycerol channel, was identified as a novel regulator of breast cancer. AQP7 was prognostic of overall survival in breast cancer patients. In mouse breast cancer models, reduced expression of Aqp7 caused reduced primary tumor burden and lung metastasis. Metabolomics and complex lipid profiling of cells and tumors with reduced Aqp7 revealed significantly altered lipid metabolism, glutathione metabolism, and urea/arginine metabolism compared to controls. These data identify AQP7 as a critical regulator of metabolic and signaling responses to environmental cellular stresses in breast cancer, highlighting AQP7 as a potential cancer-specific therapeutic vulnerability.
Keywords: Aquaporin, breast cancer, tumor metabolism, correlation-based network analysis
Graphical Abstract
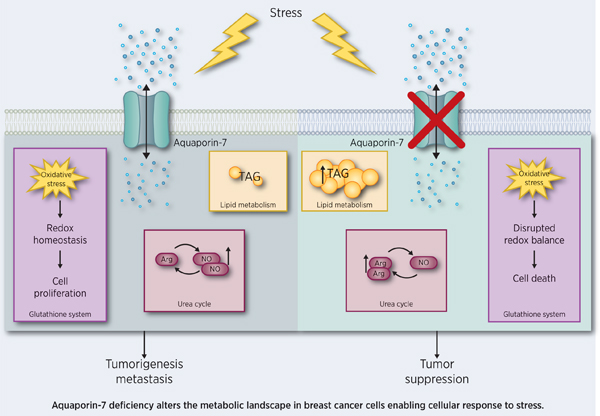
Introduction
Recent research established a new understanding of the Warburg effect and other metabolic pathways of cancer (1–3). Even with these advances in understanding cancer metabolism, therapeutic targeting of cancer metabolism has been difficult, mainly due to the complex, interrelated nature of metabolic pathways. Thus, understanding not only the factors that distinguish cancer metabolism from normal metabolism but also the crosstalk between metabolic pathways, gene expression, and the microenvironment is crucial for designing metabolism-based cancer therapies.
Aquaporins facilitate the passive transport of water across plasma membranes and are candidate regulators of tumor metabolism. A subset of these membrane proteins, known as aquaglyceroporins, transport glycerol, water, and other small molecules (4). Aquaporin-7 (human AQP7/mouse Aqp7) is a transmembrane aquaglyceroporin and member of the aquaporin family (5–7). AQP7 also transports hydrogen peroxide, ammonia, urea, and arsenite (8–13).
AQP7 has been particularly well studied in adipose tissue (6,7,14,15). During high energy demands and metabolic stress, lipolysis increases and converts triglycerides (TAG) into free fatty acids (FFA) and glycerol. AQP7 controls the efflux of glycerol under these conditions. The exported glycerol then is taken up by other cells and used as a backbone for energy needs during high energy demands.
Aqp7 deficiency in animal models is associated with adipocyte hypertrophy, increased glycerol and TAG accumulation, insulin resistance, and increased obesity in both mice and humans (6,7,14,15). Since obesity and the mammary adipose microenvironment promote aggressive breast cancers in both rodents and humans, Aqp7 may contribute to breast cancer pathogenesis (16,17). While other aquaporins regulate breast cancer invasion, Aqp7 has not been investigated in tumor pathogenesis (18–20).
Materials and Methods
Animals
Mice were maintained under pathogen-free conditions in the University of Notre Dame Freimann Life Sciences animal facility. Animal experiments were conducted in accordance with the protocol guidelines approved by the Institution Animal Care and Use Committee (15-10-2724). Details on orthotopic, contralateral, and tail vein models are discussed in Supplementary Methods.
Cell Culture
All cells were maintained at 37°C in humidified incubators with 5% CO2 at atmospheric oxygen levels. 4T1 cells were a gift from Dr. Siyuan Zhang, University of Notre Dame. EpH4 and NMuMG cells were a gift from Dr. Zena Werb, University of California, San Francisco. 4T1 cells were grown in RPMI1640 media (Sigma R6504) with 10% FBS. EpH4 cells were cultured in DMEM High Glucose media (Sigma D5648) with 5% FBS. NMuMG cells were cultured in DMEM High Glucose media with 10% FBS and 1 μg/mL insulin. All cells were routinely tested for mycoplasma by PCR (Genlantis MY01050) and colorimetric detection kit (InvivoGen rep-pt1) according to manufacturer’s directions. Quality control of the cell lines was maintained by continual authentication of morphology and growth rate and were used less than 16 passages. Mouse cell lines were not authenticated genetically.
Detailed information on various in vitro experiments - proliferation, scratch, 3-D organoid and contact inhibition assays, cell size, gene knockdown, dependence, ROS, NO detection, urea and NADP+ levels, glycerol quantification and stress tolerance assays can be found in the Supplementary Methods.
RNA Extraction and Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
For RNA extraction, cultured cells were scraped from culture dishes and washed with PBS before flash freezing with liquid nitrogen. Flash frozen tissue samples were ground with a pestle and mortar on liquid nitrogen. RNA was isolated with RNA Bee (Amsbio, CS-104B) following the manufacturer’s instructions. RNA analysis, cDNA synthesis and RT-qPCR were carried out as described in Supplementary Methods.
Western Blot, Immunofluorescence and IHC
Western blot, immunofluorescence and IHC conducted as described in the Supplementary Methods.
Metabolism Experiments by Seahorse Assays
Glycolysis (Seahorse 103020-100) and mitochondrial stress test (Seahorse 103015-100) were conducted following the manufacturer’s instructions using the XFe-96 analyzer from Seahorse Bioscience. Results from analysis were normalized to cell count. See Supplementary Methods for details.
Metabolomics and Complex Lipid Profiling
Cell and tumor sample preparation for metabolomics and complex lipid profiling according to previously described protocol (21). See Supplementary Methods for additional details.
Oil Red O Staining
Lipids and triglycerides were stained using Oil Red O (Sigma-Aldrich, O0625-25G). See Supplementary Methods for details.
MALDI Imaging
Tumor tissues were embedded in gelatin and then sectioned into 10 µm −20 µm slices. Alternating slices were collected during cryosection for Matrix-assisted laser desorption/ionization-mass spectrometry imaging (MALDI-IMS) and H&E staining. MALDI-MSI spectra were carried out on an UltrafleXtreme (Bruker Daltonics, Billerica, MA). Depending on mounting reagent different MALDI methods were used. Data was processed using FlexImaging 4.1(Bruker Daltonics, Billerica, MA). All spectra were normalized to total ion count. See Supplementary Methods for details.
Statistical Analysis
Statistical analyses were calculated with the program R (http://cran.r-project.org/). The metabolomics raw data were analyzed by significance tests and classification analysis, as indicated. For pair-wise comparisons, we used Welch’s t-tests and/or Wilcoxon’s rank sum tests, as indicated. For statistical designs comparing more than two samples, we used ANOVA (e.g., repeated measures ANOVA). Gene-Metabolite Correlation Analysis: Mouse gene expressions were acquired from (22) and processed to contain only mouse model data, then filtered to exclude genes with a small dynamic range (supplemental methods). We used the linear regression model to measure the relationships between two groups. We tested whether vs . P values were obtained and used for network construction via Cytoscape.
Results
Network analysis of metabolite and gene expression profiles
We previously identified a panel of oncogene-specific metabolites through metabolomics analysis of breast tumors collected from several breast cancer transgenic mouse models (21). One difficulty in the analysis of large datasets is that, due to the complexity of the interactions between components of the metabolic and transcriptomic network (such as signal amplification, feedback loops, etc.), the metabolites that changed most in concentration levels may not be the most important drivers in the whole network.
To address this, we generated unbiased discovery-based statistical tools to capture novel interactions between metabolic profiling and gene expression data using correlation-based network analysis. To do this, we integrated our previous metabolomics dataset with published gene expression data (22) to develop a higher order understanding of the connections between the individual components of our data. Correlations between these datasets might highlight potentially important metabolites and genes of tumor metabolism in breast cancer. If a gene or metabolite has many other factors that change with it, then the gene/metabolite might be a driver of these changes or could be a common intermediate factor at the crossroads between these pathways. Unlike previous studies integrating metabolomics with gene expression, our method did not rely on fitting metabolites and genes to existing metabolic and gene regulation pathways, which offered us the unique opportunity to integrate the data in an unbiased, discovery-based manner (23–29).
We used the biological replicates from both the metabolomics and gene expression datasets to analyze similarities between samples across cohorts. Specifically, metabolites were assigned correlation values based on their degree of similarity within individual groups (i.e., transgenic mouse groups) (Figure 1A). Using our correlation analysis, we produced three integrated networks: (1) Metabolite-Metabolite network (Met-Met), (2) Metabolite-Gene network (Met-Gene), and (3) Gene-Gene network (Gene-Gene) (Figure 1B, Table S1).
Figure 1. Network correlation study reveals metabolite and gene hubs of breast cancer.
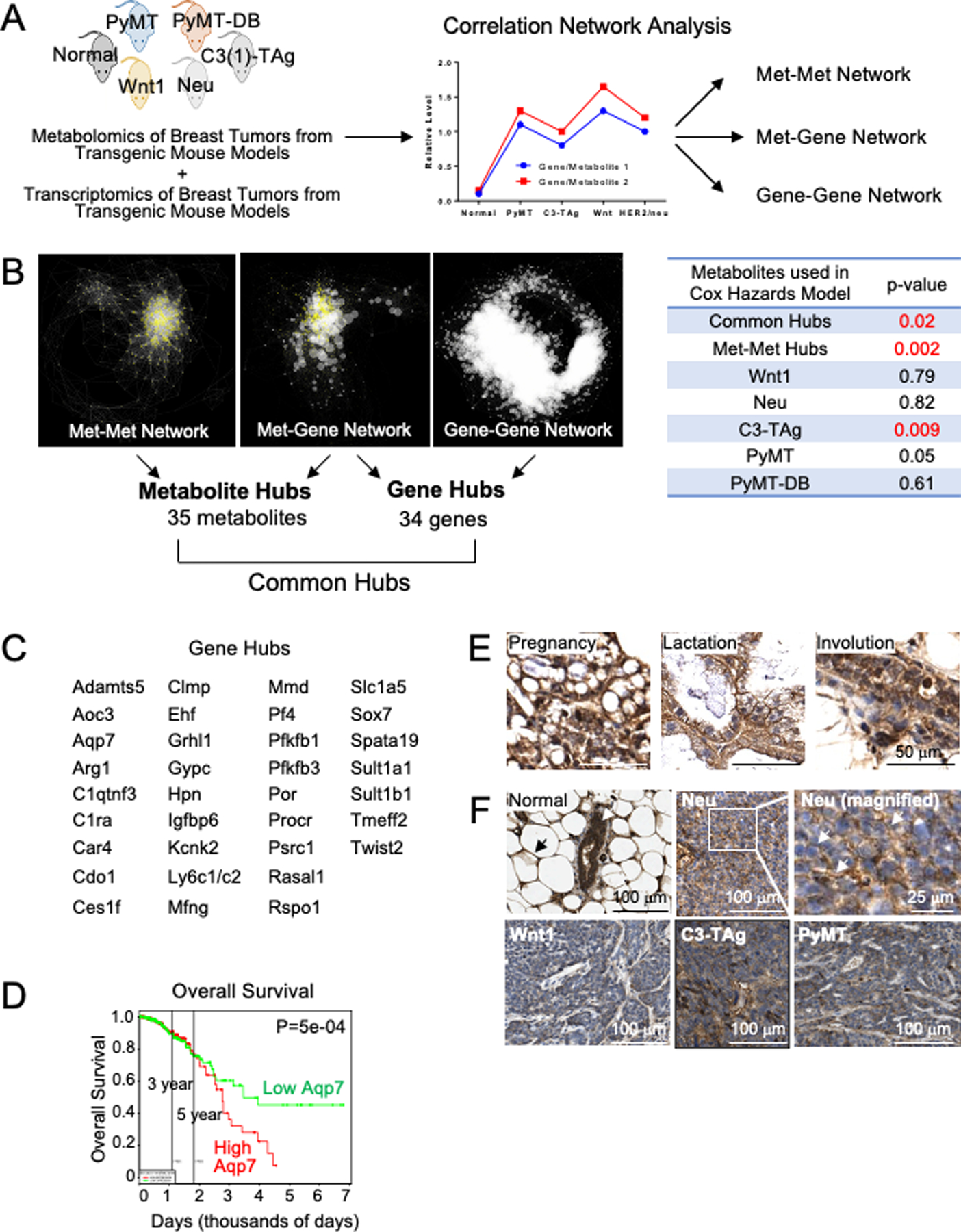
(A) Study Design. Metabolomics and transcriptomics data from tumors of transgenic mice were incorporated in a correlation network analysis. The graph is an illustration representing a positive correlation between cohorts. Analysis generated three networks. (B) (Left) Visualization of networks. Network nodes included metabolites (yellow triangles) and genes (white circles). The node size indicates the node number connected to it. Bigger nodes indicate more correlated metabolites/genes. The length of the edges connecting to nodes was determined by p-values, with a shorter distance indicating a lower p-value. (Left) Metabolite-metabolite (Met-Met) network; (Middle) Metabolite-gene (Met-Gene) network; (Right) Gene-gene network. (Right) Patient survival prediction with the metabolite hubs and oncogene-specific metabolites. (C) Common gene hubs. (D) Overall survival in breast cancer patients. The patient cohort was divided into two groups based on gene expression, with the median AQP7 gene expression separating the two groups. PROGgeneV2 generated these plots (31). 297 samples were in each cohort. (E–F) IHC of AQP7 in (E) healthy mammary glands at the indicated developmental stages and in mammary gland tumors from transgenic mouse models (F) compared to a healthy mouse mammary gland (top left). Arrows in normal point to adipocytes (black) and epithelium (white) and in Neu point to membrane localized AQP7.
Metabolite and gene hubs identified by correlated network analysis
Our network analysis identified key nodes, or “hubs,” of the metabolites/genes with the most degrees (i.e., the most correlations between metabolites/genes). A particular gene/metabolite was defined as a hub if it had more than twice the average number of correlated metabolite/gene. Most of the hubs clustered together because they are heavily interconnected (Figure 1B). Significantly, this approach identified interactions of metabolites that are not captured by current databases, such as KEGG and HMDB (Table S1). The Met-Met network was constructed exclusively with metabolomics data, with each node representing one metabolite. These hubs primarily included amino acids, carbohydrates, and nucleotide metabolites (Figure S1A). The Met-Gene network was constructed by integrating our metabolomics data with published mouse gene expression data (22), and the Gene-Gene network was constructed exclusively with published gene expression data for the tumor models used for our metabolomics (22).
We compared the differing metabolite hubs in the Met-Met and Met-Gene networks, normalized against total number of metabolites detected in each pathway. Met-Gene networks were enriched with more lipids than Met-Met networks (Figure S1A). We hypothesized that Met-Met network metabolites represented metabolites with a more complex metabolic flux, while the metabolites of the Met-Gene network represented metabolites under tight gene control, suggesting lipids are under tighter gene control than amino acids and carbohydrates.
The hubs in the Gene-Gene network predominantly segregated into two groups (Figure 1B). Analysis with the bioinformatics tool DAVID (30) on the genes from each group revealed one group enriched for glycoprotein and plasma membrane genes and another group enriched for cell cycle and DNA replication genes. The gene hubs in the Met-Gene network were enriched for glycoprotein, signaling, and plasma membrane genes.
From the networks, we identified the subset of metabolites in common between the networks (“metabolite and gene hubs”). The metabolite hubs in common between the Met-Met and Met-Gene networks included 35 metabolites that we sorted into their respective metabolic super pathways (e.g., amino acid) and sub-pathways (e.g., glutamate) (Table S1). Interestingly, this list of common metabolite hubs differed from metabolites highly enriched in tumors (21). The metabolite hubs included many metabolites of amino acid and lipid super pathways, while the enriched tumor metabolites included primarily amino acid and carbohydrate super pathways.
We also identified 41 unique gene accession numbers in common between the Met-Gene and Gene-Gene networks (“gene hubs”), which correspond to 34 unique genes (Figure 1C, complete list including gene accession numbers in Table S1).
Metabolic and gene hubs are prognostic of survival
To determine if our metabolic hubs have clinical significance, we conducted the same prognostic analysis as with the model-specific metabolites identified in our previous study (Figure 1B, Table S2A) (21). Common hub and Met-Met hub metabolic signatures both were prognostic of survival with high statistical significance (P=0.02 and P=0.002, respectively, based on a likelihood ratio test of the top ten signatures combined) and had limited overlap in metabolites. The prognostic metabolite signatures consisted of mostly gamma-glutamyl amino acids and phospholipids, while the Met-Met hub metabolite signatures also included nucleotide metabolites. We also examined signatures composed of fewer than ten metabolites and found seven metabolites per metabolic signature were sufficient for prognostic value for both common hubs (P=0.01) and Met-Met hubs (P=0.01). The prognostic potential of these metabolites demonstrates the power of this discovery-based network analysis.
To determine the clinical prognostic potential of the gene hubs, we used gene expression data of breast cancer patients (PROGgeneV2) to see if the hubs are prognostic indicators (31). Out of the 34 gene hubs, 30 were in the dataset, and all 30 were prognostic indicators of patient overall survival (Table S2B). The gene hubs included targets already implicated in breast cancer progression (e.g., Procr, Sox7) and additional targets that have not yet been identified as functionally contributing to breast cancer or tumor metabolism (e.g., Aqp7, Psrc1, Car4). As a proof of concept, we evaluated Aquaporin-7 (AQP7/Aqp7), whose roles in cancer metabolism and breast cancer were not yet known.
Aqp7 was chosen for several reasons. In our network analysis, it was one of the few genes whose accession number appeared as a common hub more than once, which we focused on initially. From these common hubs, we wanted to find genes with an impact on metabolism and whose impact can drive or inhibit cancer. The other genes that appeared twice were Pfkfb3, Psrc1, Mmd, Clmp, Car4, and Ehf. Pfkfb3 was already relatively well-studied in metabolism; the functions in metabolism of Psrc1, Mmd, Clmp, and Ehf were practically unknown at the beginning of the study. Left with Car4 and Aqp7, we chose Aqp7 due to its roles in glycerol transport and lipid homeostasis and that a few other members of the aquaporin family impact cancer progression. Additionally, mammary glands are an adipocyte-rich tissue, so metabolic genes with an impact on lipid metabolism were attractive to our lab to study.
Aquaporin-7 (AQP7) is a prognostic marker of overall survival and metastasis in breast cancer patients
We investigated AQP7 gene expression for prognostic value in breast cancer patients. Patients with breast tumors from the TCGA Breast Cancer database with high AQP7 had reduced overall survival compared to tumors with low AQP7 (P=5×10−4, Log-rank test) (PROGgeneV2) (31) (Figure 1D). Therefore, high AQP7 expression in breast tumors is a biomarker of poor prognosis in these patients, with high AQP7 being prognostic of reduced survival.
AQP7 gene expression highly correlates with lipid synthesis genes in breast tumor tissues
To identify genes that best correlate with AQP7 gene expression in human breast cancer patient tumor samples, we used the online breast cancer database cBioportal and two published gene expression datasets of breast tumors (TCGA, METABRIC) (Table S3) (32,33). Many of the top gene hits included lipid metabolism genes and had extremely high Pearson’s and Spearman’s correlation coefficients (0.7–0.96) across both TCGA and METABRIC datasets. The list includes genes with a role during milk production and lipid synthesis, including genes regulating lipid droplet transport (PLIN1, PLIN4, CIDEC/FSP27, FABP4), fatty acid import (CD36/FAT), inhibitors of lipolysis (CIDEA, G0S2), lipid biogenesis (GPD1), TAG/glycerolipid synthesis (GPAM), and hormonal regulation of appetite (leptin). These data suggest AQP7 affects multiple facets of lipid metabolism.
AQP7 protein is dynamically expressed and localized in the epithelium and adipocytes of developing mammary gland and breast tumor tissue
We investigated the transcription, protein expression, and localization of Aqp7 in normal mouse mammary tissue during mammary gland development and in mouse breast tumors (Figure 1E–F). By RT-qPCR, Aqp7 was expressed at lower levels in mouse tumors than in normal mammary tissue (Figure S1B), consistent with Aqp7 expression in published microarray datasets (22). By RT-PCR, Aqp7 expression levels were highest in C3-TAg compared to the other tumor models.
We investigated AQP7 protein localization by immunohistochemistry (IHC) in normal mammary and breast tumor tissues. AQP7 protein was prominently expressed in both normal adult mouse mammary gland epithelium and in adipocytes (Figure 1F). During lactation, AQP7 localization to the plasma membrane was particularly prominent compared to at other stages of development (Figure 1E).
In contrast, AQP7 protein levels were lower in all tumors than in normal tissue, similar to that seen by RT-qPCR, and AQP7 protein localization was heterogeneous and varied across tumor types (Figure 1F). Wnt1 tumors expressed low levels of AQP7 but higher levels in more differentiated tumor tissue, where it localized to the epithelium most proximal to the stroma. Neu and PyMT tumors expressed low levels of AQP7. However, AQP7 often localized to the plasma membrane of tumor cells.
These data indicate that AQP7 is expressed at the highest levels in normal mammary epithelium and stromal adipocytes, while overall levels are reduced but heterogeneous in expression and localization in mouse epithelium in breast tumors. Also, the prominent membrane localization of AQP7 during both lactation and breast tumors leaves open the possibility of overlapping AQP7 functions during lactation and cancer or that AQP7 protein may be regulated by localization.
Aqp7 deficiency decreases proliferation, blocks contact inhibition, and increases migration in normal mouse mammary epithelial cells (MECs)
To investigate the requirements for AQP7, we knocked down Aqp7 expression using multiple Aqp7 shRNAs in normal mouse mammary epithelial cell lines (NMuMG, EpH4) and in a metastatic mouse breast cancer cell line (4T1) and tested Aqp7 expression by RT-qPCR and western (Figure 2A–B, S1C–D), which supported Aqp7 knockdown (Aqp7 KD).
Figure 2. AQP7 affects proliferation, migration, and invasive branching in culture.
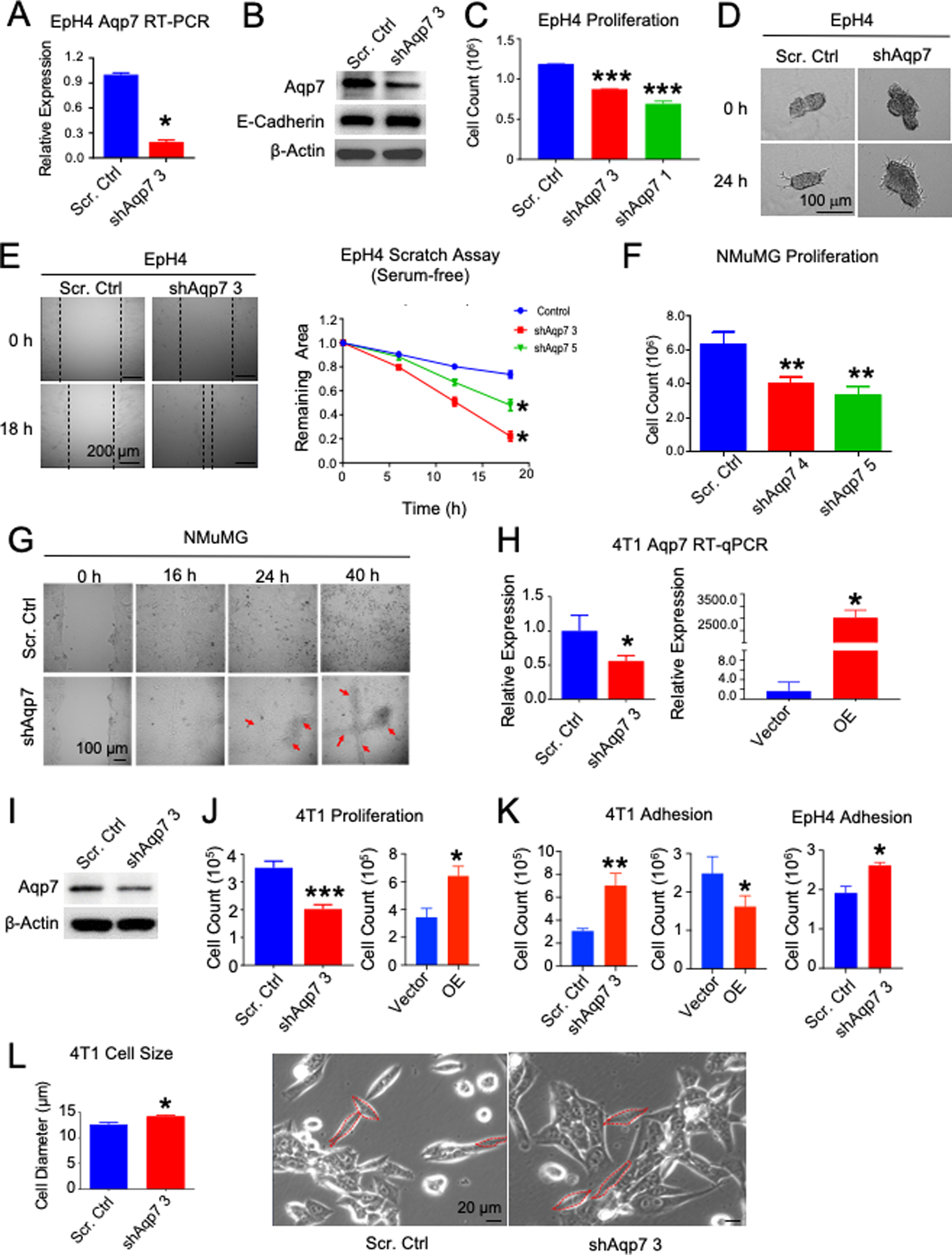
Aqp7 KD by shRNA (shAqp7) or scrambled control (Scr. Ctrl) in EpH4 and NMuMG MECs ((A)–(F)) and metastatic breast cancer cell line 4T1 ((G)–(L)). (A) RT-qPCR and (B) western analysis of EpH4 cells. (C) Proliferation, (D) Branching assay, and (E) Scratch assay (Left) images and (Right) quantification of EpH4 cells. (F) Proliferation and (G) scratch assay by time-lapse microscopy of NMuMG cells. Arrows indicate reduced contact inhibition. (H) RT-qPCR and (I) western blot of AQP7 expression in 4T1 cells. (J) Proliferation and (K) adhesion assay in 4T1 and EpH4 cells and 4T1 AQP7 overexpression compared to vector control. (L) Cell size of 4T1 cells measured in cells shown in dashed red lines (right). *, P<0.05, **, P<0.01, ***, P<0.001, unpaired t test.
To determine if Aqp7 contributes to invasive growth phenotypes, we examined the consequences of reduced expression of Aqp7 by shRNA in normal MECs, NMuMG and EpH4 cells. Aqp7 KD reduced proliferation in both (Figure 2C, 2F, S1D). In EpH4 cells, Aqp7 KD increased migration potential by scratch assay, marked by closing the scratch gap significantly faster than controls, but did not reduce E-cadherin, suggesting that AQP7 is not acting through epithelial-to-mesenchymal transition (EMT) (Figure 2B, E). Aqp7 KD also significantly increased invasive branching of cell spheroids compared to controls by 3D branching assay (Figure 2D).
In NMuMG cells, Aqp7 KD also decreased contact inhibition compared to control, which developed contact inhibition once confluent in 2D culture (Figure 2G). Aqp7 KD cells continued growing on top of other cells and formed cell foci, whereas control cells did not (Figure 2G, S1F).
Together, reduced expression of Aqp7 promotes migration and decreases proliferation and contact inhibition in MECs. Aqp7 KD alone was insufficient to make normal MECs tumorigenic, as injections of NMuMG cells with Aqp7 KD failed to initiate tumors in mouse mammary glands.
Aqp7 deficiency decreases proliferation and enhances contact inhibition and adhesion in metastatic mouse cancer cells
Similar to EpH4 and NMuMG cells, reduced Aqp7 expression in 4T1 cells also decreased proliferation but had the opposite effects on contact inhibition as in NMuMGs (Figure 2H–J, S1G–H). Similar to EpH4 cells, Aqp7 KD increased adhesion and cell size (Figure 2K–L, S1E). These Aqp7 KD cells formed fewer cell foci, suggesting increased contact inhibition compared to control cells (Figure S1H).
To make sure that the effects were related to Aqp7 expression, we also overexpressed Aqp7 (Aqp7 OE) in 4T1 cells (Figure 2H). Aqp7 overexpression in 4T1 cells increased proliferation and cell foci formation but reduced adhesion and contact inhibition (Figure 2J–K, S1H).
Aqp7 deficiency inhibits primary tumor growth and lung metastasis in vivo
We next tested the requirements for AQP7 on tumor formation by transplantation of Aqp7 KD and control 4T1 cells by contralateral injection, orthotopic injection, and tail vein injection. By contralateral and single orthotopic mammary gland injection of 4T1 cells, tumors formed with Aqp7 KD cells were smaller than with control cells (Figure 3A–G, S2A). By tail vein injection, both single and clustered Aqp7 KD cells formed significantly decreased numbers of lung metastases than controls (Figure 3H–K). Therefore, AQP7 in breast cancer MECs significantly promotes and is required for breast cancer growth and metastasis.
Figure 3. Aqp7 knockdown in metastatic breast cancer cells inhibits primary tumor growth and metastasis in vivo.
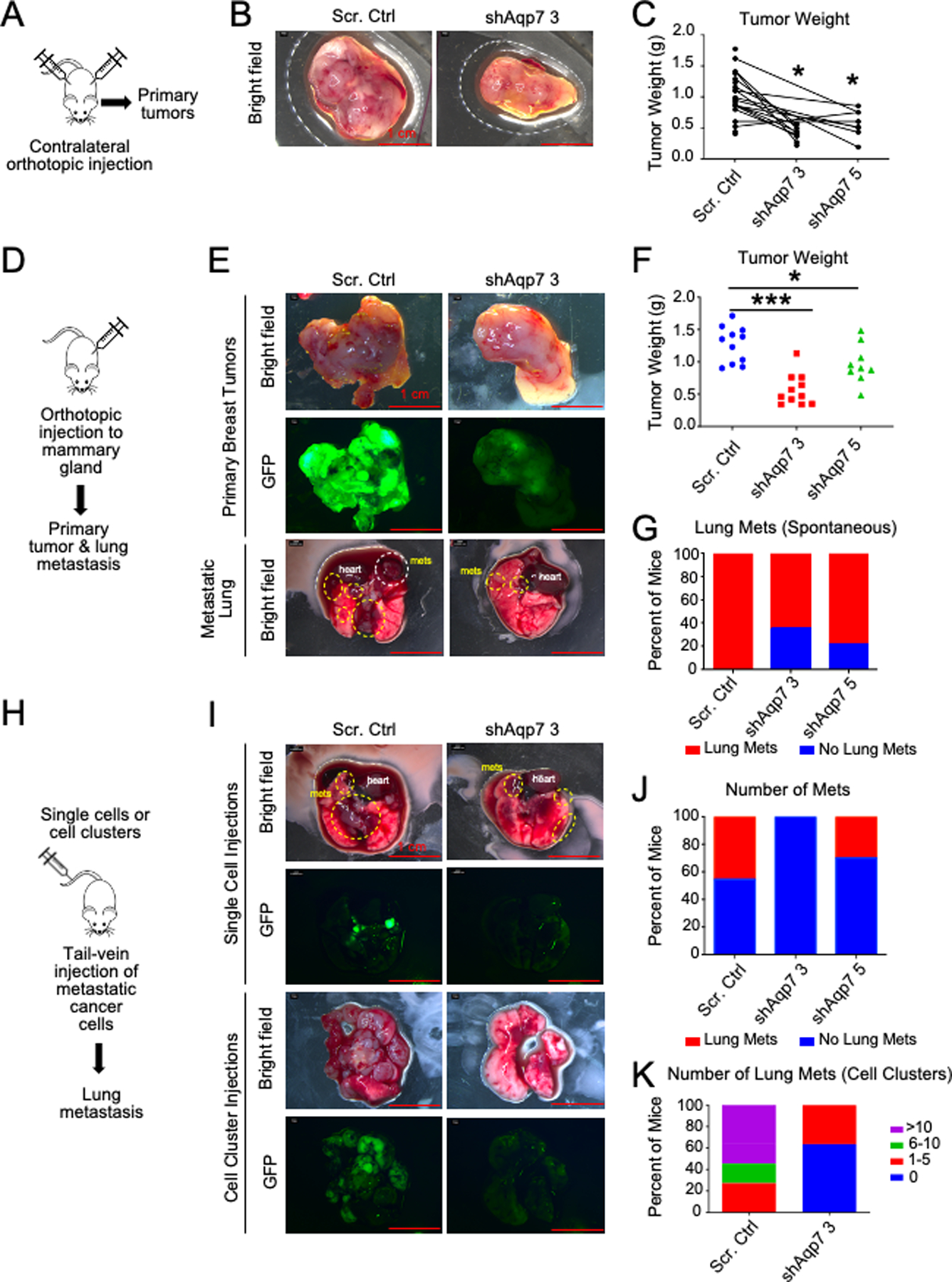
(A–C) Contralateral orthotopic injection of 4T1 cells with Aqp7 KD or scrambled control. (A) Overview of contralateral orthotopic injection. (B) Representative brightfield images of primary tumors. (C) Tumor weight at collection. Lines connect tumors growing on the same mouse. (D–G) Orthotopic injection of 4T1 cells with Aqp7 KD or scrambled control. (D) Overview of orthotopic injection. (E) Brightfield and GFP images of primary tumors and lungs. Metastases are circled. (F–G) Quantification of tumor weight (F) and lung metastasis. (G). (H–K) Tail vein injection of 4T1 cells with Aqp7 KD or scrambled control (H) Overview of tail vein injection. (I) Representative images of lungs three weeks after tail vein injection with single cells (top) or cell clusters (bottom). Metastases are circled. (J-K) Quantification of lung metastasis after single cell (J) and cell cluster (K) tail vein injections at the time of collection.
AQP7 regulates multiple metabolic pathways
To identify the metabolic reprogramming regulated by AQP7, we quantified the metabolites and complex lipids that changed in response to silencing Aqp7 expression in EpH4 and 4T1 cells and in 4T1 breast tumors (Figure 4A, S2B, Table S4). Principal component analysis separated all samples into two distinct clusters based on Aqp7 expression, indicating that AQP7 significantly impacts the metabolome (Figure S2C). Furthermore, random forest analysis identified the top differentiating metabolites to belong to lipid, amino acid, nucleotide metabolism, and peptides (Figure S2D–E).
Figure 4. Aqp7 knockdown leads to changes in lipid metabolism and carbohydrate metabolism.
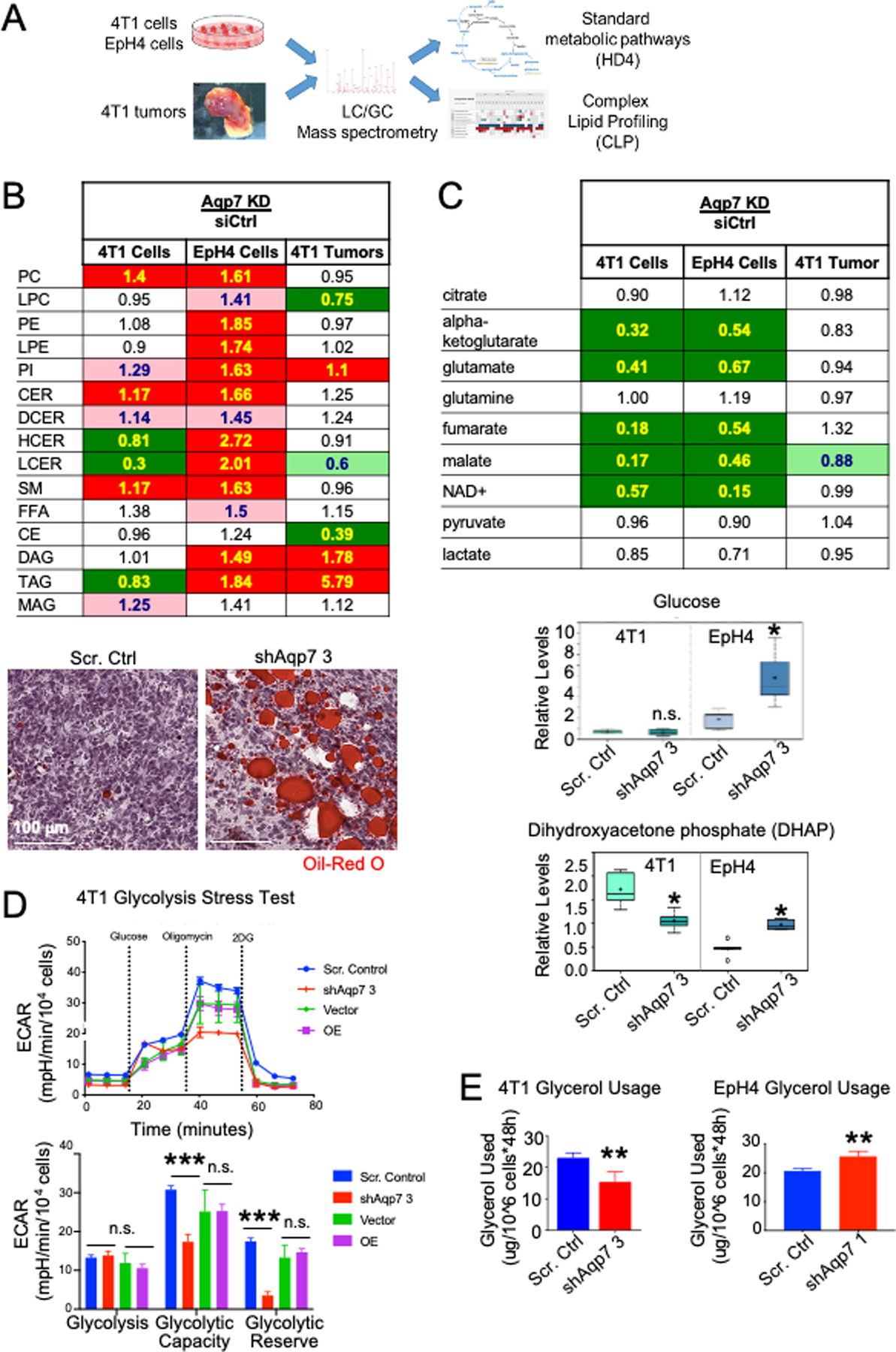
(A) Experimental overview of standard metabolomics and complex lipid profiling conducted on Aqp7 KD and scrambled control samples from EpH4 and 4T1 MECs and 4T1 tumors. (B) (Top) Total complex lipid levels in cell lines and tumors. Numbers indicate the ratio of lipid levels in Aqp7 KD compared to scrambled control. In B-C, cell colors indicate significant increase (red; p<0.05, Welch’s t-test), decrease (green) in the knockdown group, or approaching significance (faded; 0.05<p<0.10). (Bottom) Oil Red O staining of tumors of scrambled control and Aqp7 KD 4T1 cells. (C) (Top) TCA cycle metabolomics of cell lines and tumors. (Bottom) Glucose and DHAP levels in 4T1 and EpH4 cells. (D) Glycolysis stress test. Representative assay quantification of ECAR in 4T1 Aqp7 KD or OE, compared to controls (scrambled and vector, respectively). Error bars depict s.d. of four independent wells from a representative tracing from three independent experiments. (E) Glycerol usage in EpH4 and 4T1 cells after Aqp7 KD or scrambled control.
AQP7 dramatically alters lipid metabolism in cells and tumors
Since Aqp7 knockout mice accumulated FFA and TAG in adipose tissue, we expected AQP7-dependent changes in lipid metabolism (6). The phospholipids phosphatidylcholine (PC) and phosphatidylethanolamine (PE) were among the lipids present in the highest concentrations in all cell line groups followed by FFA, sphingomyelins (SM), TAG, and cholesterol esters (CE). However, Aqp7 KD had different effects on lipid composition in 4T1 and EpH4 cells (Figure 4B, S2B). In 4T1 cells, Aqp7 KD decreased TAG, increased PC and ceramides (CER), and FFA remained unaffected, while in EpH4 cells, Aqp7 KD significantly increased TAG and DAG and trended for higher FFA.
In 4T1 tumors, TAG, PC, and PE lipids were the most represented lipids in both control and Aqp7 KD tumors, followed by FFA, SM, and CE. TAG and diacylglycerols (DAG) were significantly higher in Aqp7 KD than control tumors, consistent with Aqp7 regulating fatty acid metabolism via glycerol and glycerol 3-phosphate metabolism. Lipid related gene expression was unaffected by RT-qPCR after Aqp7 deficiency, suggesting the altered lipids were unlikely transcriptionally regulated based (Figure S3D). Consistently, lipid accumulation in Aqp7 KD tumors was detectable by Oil Red O staining (Figure 4B).
CE accumulation correlates with increased aggressiveness and proliferation in various cancers, including breast (34–36). CE significantly decreased following Aqp7 KD in tumors, though total cholesterol was unchanged according to standard metabolomics data. These data suggest that tumors with decreased Aqp7 expression are less aggressive, have less metastatic potential, and accumulate TAG and FFA and decrease CE.
AQP7 regulates carbohydrate and glycerol metabolism
Glucose uptake and aerobic glycolysis are frequently upregulated in tumors to support energy needs and to provide biosynthetic precursors (e.g., pentose phosphate pathway for nucleotide synthesis). Aqp7 deficiency dramatically altered carbohydrate metabolism, including glycolysis and the TCA cycle, in both 4T1 and EpH4 cells (Figure 4C, Table S4). In 4T1 cells and tumors, Aqp7 KD did not alter levels of glucose or glycolytic end products (e.g., pyruvate, lactate) but reduced dihydroxyacetone phosphate (DHAP), an important metabolite in TAG synthesis (Figure 4C). In contrast, in EpH4 cells, Aqp7 KD increased glucose and DHAP levels. The differing DHAP levels in 4T1 and EpH4 cells are notable because DHAP links carbohydrate and lipid metabolism via glycerol and glycerol 3-phosphate (G3P). Interestingly, 4T1 cells had elevated levels of glycolysis intermediates, such as 3-phosphoglycerate and 2-phosphoglycerate, as well as phosphoenolpyruvate, which may be products from glycerol and DHAP (Table S4).
Given the changes observed in a number of metabolites in the glycolytic pathway in 4T1 and EpH4 cells, we determined if these changes were due to glycolytic rate. We assessed changes in extracellular acidification rate (ECAR), a measure of glycolytic function. In 4T1 and EpH4 cells, basal glycolysis was not different between Aqp7 KD and control cells (Figure 4D, S3A). Interestingly, Aqp7 KD in 4T1 cells exhibited a significant reduction in glycolytic capacity and reserve, whereas the EpH4 cells had a significant increase in ECAR for glycolytic capacity and reserve. Aqp7 OE rescued glycolytic functions in both cell lines, evidenced by a reversal in ECAR.
Glycerol-3-phosphate dehydrogenase (GPDH) catalyzes the reversible conversion of DHAP to glycerol 3-phosphate and uses NAD+/NADH as a coenzyme. Nicotinamide adenine dinucleotide (NAD+) plays an essential role in energy metabolism and redox status. After Aqp7 KD, both cell lines had significantly lower NAD+, which appears to be due to decreased nicotinamide salvage pathway in Aqp7 KD cells (Figure 4C).
Glycerol levels were unaltered after Aqp7 KD in cell lines and tumors. This is surprising, since AQP7 is an aquaglyceroporin hypothesized to increase intracellular glycerol levels after Aqp7 KD, similar to that seen in adipose tissue of knockout mice. Glycerol 3-phosphate was unchanged in tumors but decreased in both KD cell lines (Table S4).
To test our hypothesis that glycerol usage (through conversion to DHAP) differed between 4T1 and EpH4 cells and to account for metabolic differences between these cells, we measured the glycerol consumption levels in cell culture media, both before and after cell culture. Glycerol usage after Aqp7 KD increased in EpH4 cells but decreased in 4T1 cells (Figure 4E). These data suggest Aqp7 mediates glycerol usage in 4T1 and EpH4 cells and is reflected by DHAP levels. Additionally, Aqp7 KD decreased NAD+ levels and may affect additional energy and redox metabolism.
AQP7 sensitizes cells to oxidizing environments
AQP7 transports and removes hydrogen peroxide (H2O2) from cells (11). Glutathione is a small molecular weight thiol with antioxidant properties. The glutathione system plays an important role in antioxidant defense, redox-homeostasis, protein folding, and detoxification of drugs. Glutathione exists in reduced and oxidized states. Under normal conditions, glutathione is found at greater levels in its reduced (GSH) than its oxidized (GSSG) form, creating a reducing environment. Interestingly, GSH, GSSG, and many precursors and glutathione conjugates decreased after Aqp7 KD in cell lines and tumors (Figure 5A). In contrast, oxidized metabolites, such as methionine sulfoxide, cysteine-glutathione disulfide, and cystine (in tumors) increased in Aqp7 KD cell lines and tumors, suggesting that AQP7 may inhibit oxidative stress by preventing accumulation of oxidizing metabolites and may alter the redox status of breast cancer cells and tumors.
Figure 5. Aqp7 knockdown leads to decreased oxidative stress tolerance.
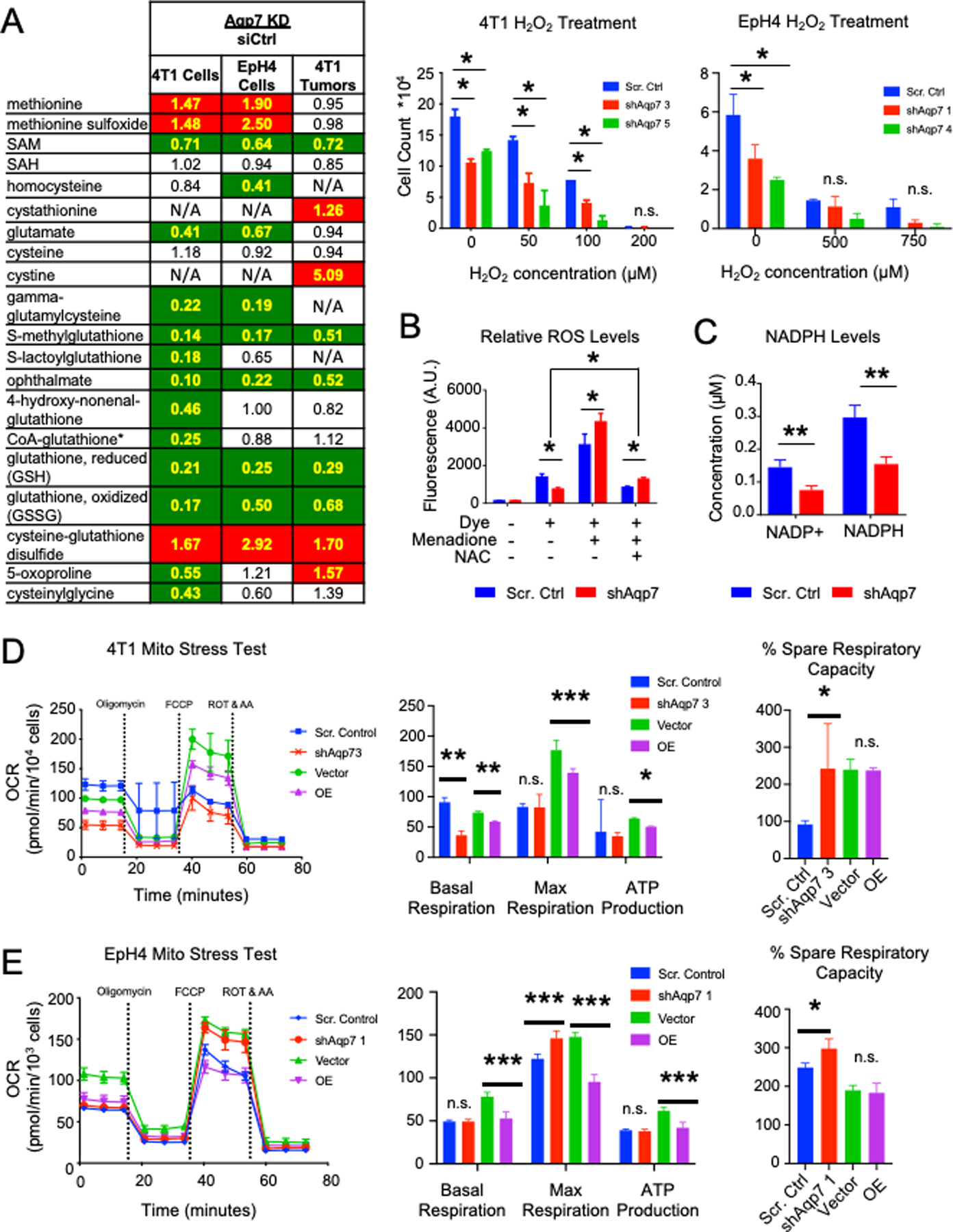
(A) (Left) Metabolomics of glutathione and related metabolites following Aqp7 KD compared to scrambled control in cells and tumors. Numbers indicate the ratio of metabolite levels in Aqp7 KD compared to scrambled control. Cell colors are described in Figure 4. (Right) Oxidative stress tolerance assay reveals lowered tolerance to H2O2 following Aqp7 KD in 4T1 and EpH4 cells. (B) ROS levels in 4T1 scrambled control and Aqp7 KD cells after treatment with ROS inducer menadione or N-acetyl cysteine supplementation. (C) NADP+ and NADPH levels in 4T1 Aqp7 KD cells compared to scrambled control. (D–E) Mitochondrial stress test. Representative assays showing changes in OCR in 4T1 (D) and EpH4 (E) cells and measurement quantification. Error bars depict s.d. of four independent wells from a representative tracing from three independent experiments.
Since glutathione levels decreased after Aqp7 KD, we assessed the cellular redox requirements for AQP7. First, we assessed the ability of Aqp7 KD cells to withstand oxidative stress induced by H2O2 in 4T1 and EpH4 (Figure 5A). 4T1 Aqp7 KD cells were less viable and more sensitive to oxidative stress induced by H2O2 than controls, whereas no difference in cell viability was observed in EpH4 cells. Furthermore, Aqp7 KD increased expression of both glutathione synthesis related gene glutathione synthetase (Gss) and glutathione-utilizing genes, such as glutathione peroxidase 4 (Gpx4) and glutathione s-transferase P1 (Gstp1) (Figure S3E). However, Gstp1 and Gss expression levels were inversely related in EpH4 cells and 4T1 cells.
We determined if the decreased glutathione in Aqp7 KD cells was from impaired glutathione synthesis and decreased basal ROS levels. We measured the ROS levels in 4T1 and EpH4 cells with live cell ROS detection dye CellROX by flow cytometry (Figure 5B, S3B). Basal ROS levels decreased in 4T1 Aqp7 KD cells, which could be counterintuitive to our hypothesis of an impaired glutathione synthesis pathway. However, upon ROS induction with menadione, the ROS levels reversed with elevated ROS levels in Aqp7 KD cells. Additionally, even after the addition of glutathione precursor N-acetylcysteine (NAC) after menadione, Aqp7 KD cells maintained elevated ROS levels compared to basal levels (no menadione), while basal ROS decreased even lower in control cells. However, in EpH4 cells we observed no difference in basal ROS levels or in ROS after menadione or NAC treatment in Aqp7 KD cells (Figure S3B).
These data suggest that Aqp7 maintains both glutathione synthesis and usage but eventually decreases oxidative stress tolerance in metastatic cancer cells. Whereas decreased Aqp7 expression does not damage basal ROS scavenging ability, it decreases cell survival after oxidative stress.
Reduced GSH is regenerated in cells from oxidized GSSG by glutathione reductase, consuming NAPDH in the process. We measured intracellular NADP+ and NADPH in 4T1 cells to determine if Aqp7 impacts the NADP+/NADPH ratio, which is used as a readout of cellular redox state. While the concentration of NADP+ and NADPH decreased in Aqp7 KD cells compared to control cells (Figure 5C), the ratio of NADP+ to NADPH did not change (Figure S3C). These data suggest Aqp7 is required for the production of glutathione but not for the recycling of GSSG to GSH.
Mitochondria are a major source of ROS. We assessed key parameters of mitochondrial function by directly measuring the oxygen consumption rate (OCR) in 4T1 and EpH4 cells (Figure 5D–E). After Aqp7 KD, ATP production did not differ, but percent spare respiratory capacity increased in both 4T1 and EpH4 cells. Although the maximal respiration did not differ, we observed a significant decrease in basal respiration. Aqp7 KD in 4T1 cells resulted reduced the basal but not the maximal respiration. However, we observed the opposite effects in EpH4 cells.
AQP7 regulates arginine/urea metabolism and nitric oxide production
Arginine precursors, such as aspartate and arginino-succinate, decreased in both cell lines and tumors after Aqp7 KD compared to controls (Figure 6A). However, arginine itself increased in both cell cultures but decreased in vivo, likely due to differential amino acid availability and/or uptake from the microenvironment (37,38).
Figure 6. Aqp7 knockdown leads to changes in urea and arginine metabolism.
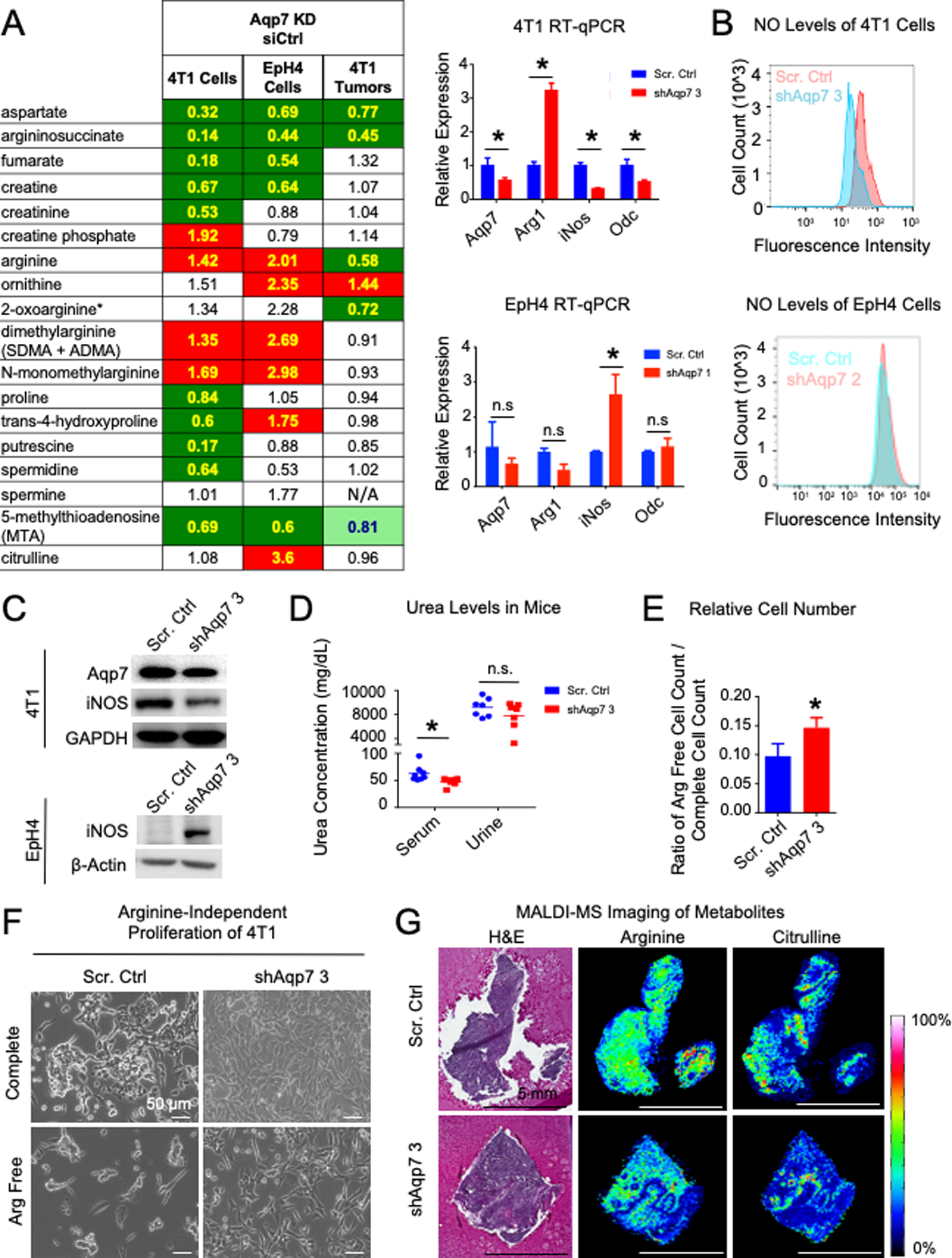
(A) (Left) Metabolomics of urea/arginine metabolism with decreased Aqp7 expression. Cell colors are described in Figure 4. (Right) RT-qPCR of arginine metabolism genes. (B) Nitric oxide levels following Aqp7 KD in 4T1 and EpH4 cells. (C) Western analysis of arginine metabolism genes after AQP7 KD in 4T1 and EpH4 cells. (D) Urea levels in serum and urine in mice injected with 4T1 Scr. Control or Aqp7 KD cells. (E–F) Proliferation of 4T1 Aqp7 KD cells and scrambled control cells in arginine-free media. (G) (Left) H&E and (Right) MALDI-MS images of arginine and citrulline in 4T1 tumors.
Downstream of arginine, ornithine production increased and polyamine synthesis decreased after Aqp7 KD. These data suggest altered arginine synthesis and utilization by enzymes of the urea-producing pathway via the enzyme Arginase-1 (Arg1) and the NO-producing pathway via the enzyme nitric oxide synthase-2 (Nos2/iNos). We measured the expression levels of urea metabolism enzymes Arg1 and iNos (inducible nitrogen oxide synthase) and related enzymes by RT-qPCR (Figure 6A, S3F). Arg1 increased and iNos decreased after Aqp7 KD in 4T1 cells, while the opposite was observed in EpH4 cells (Figure 6A). iNOS protein expression also decreased in 4T1 cells (Figure 6C). Ornithine decarboxylase (Odc) gene expression also decreased, consistent with the reduced polyamine synthesis observed by metabolomics.
Nitric oxide (NO) is an important regulator of cancer metabolism, with both tumor suppressive and promoting abilities through impacting many signaling pathways and epigenetics (39). To monitor urea metabolism, we measured NO production in 4T1 and EpH4 cells with a live cell dye (Figure 6B). NO levels decreased in 4T1 Aqp7 KD cells compared to controls, consistent with decreased iNOS expression, and did not change in EpH4 cells.
Serum urea levels in mice bearing Aqp7 KD tumors decreased significantly compared to mice bearing control tumors, while urine levels were not significantly different (Figure 6D).
We assessed the ability of 4T1 cells to grow in arginine-free conditions. Since arginine is required to produce NO to support proliferation, control cells should be more dependent on arginine than Aqp7 KD cells, which produce less NO. Indeed, in arginine free media, the Aqp7 KD cells proliferated as fast as control cells (Figure 6E–F).
Citrulline is produced from arginine by the enzyme iNOS and can be produced from ornithine by the enzyme ornithine transcarbamoylase. With MALDI-MS imaging, we visualized arginine and citrulline localization within mouse tumors derived from 4T1 orthotopic implantation (Figure 6G). Arginine accumulation was fairly homogenous across both epithelium and stroma in control tumors but localized more prominently to the epithelium in Aqp7 KD tumors. Similarly, citrulline accumulation overlapped with arginine accumulation in control tumors, spreading across cancer epithelium and stroma, but overlapped with low arginine accumulation in Aqp7 KD tumors, more concentrated within the stromal regions.
AQP7 regulates p38, EGFR, and mTOR cell signaling
To assess the requirements for AQP7 in key signaling pathways, we evaluated protein expression of signaling molecules by western analysis from both 4T1 and EpH4 lysates (Figure 7A). In 4T1 cells, phospho-p38 but not p38 protein levels decreased in Aqp7 KD compared to control. Aqp7 KD also significantly lowered the levels of phosphorylated ERK1/2. Both phosphorylated and total EGFR decreased in 4T1 Aqp7 KD cells compared to control cells. Phosphorylated AKT increased while total AKT levels decreased slightly in 4T1 Aqp7 KD cells. Both phosphorylated and non-phosphorylated mTOR expression increased after Aqp7 KD compared to control. Downstream of mTOR, phospho-p70S6K but not p70S6K increased in Aqp7 KD compared to control.
Figure 7. Aqp7 knockdown leads to changes in PI3K/Akt/mTOR and p38 MAPK signaling.
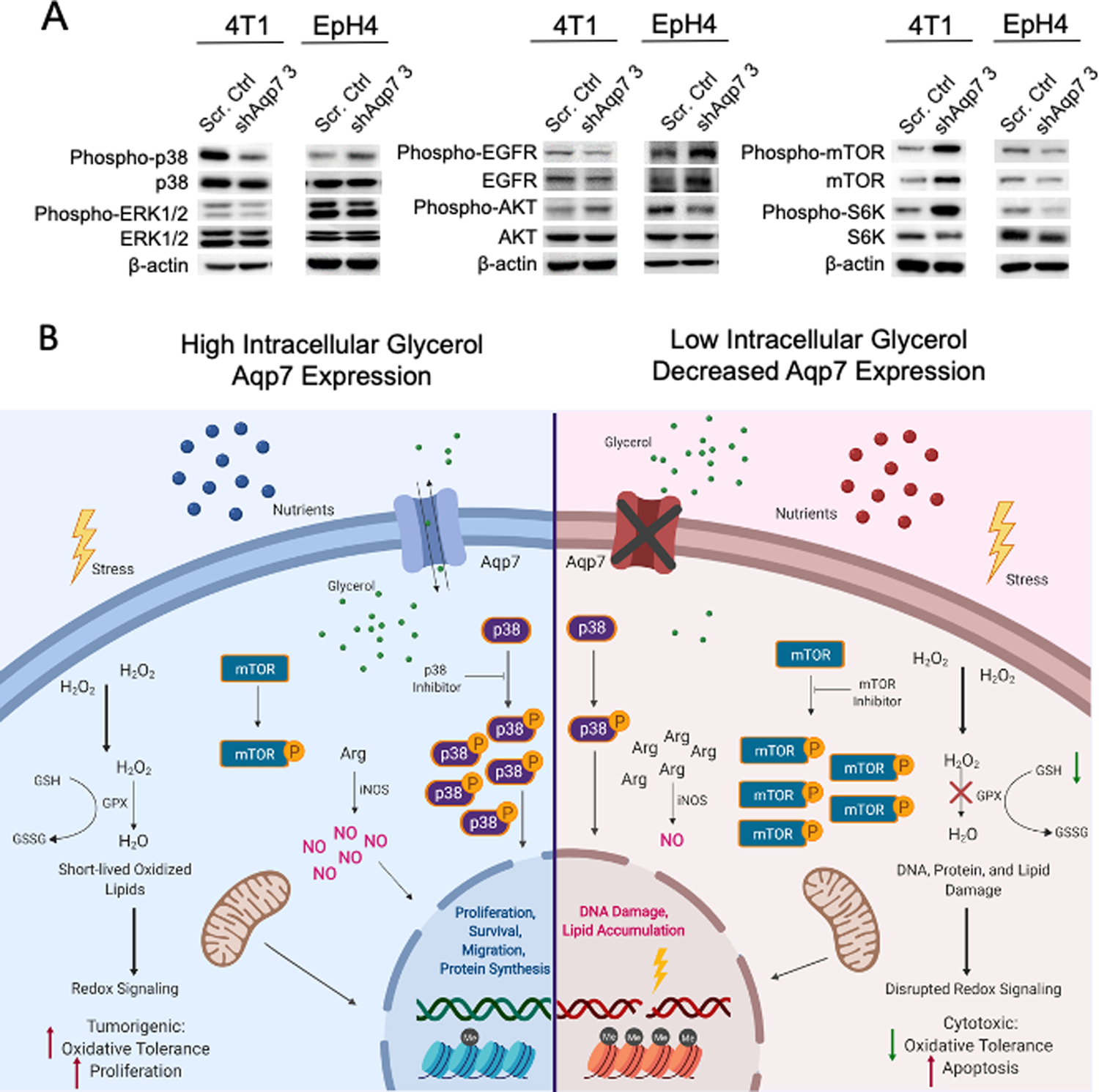
(A) Western analysis of indicated proteins in 4T1 and EpH4 cells. (B) Model of AQP7 function during breast cancer and metabolome reprogramming. Aberrant AQP7 expression alters lipid metabolism, carbohydrate metabolism, nucleotide synthesis, and urea/arginine metabolism. With high intracellular glycerol or active AQP7, cells increase p38 signaling, NO, and oxidative tolerance. With low intracellular glycerol conditions or decreased AQP7 expression, cells increase phospho-mTOR signaling, making them potentially more susceptible to mTOR inhibitors.
In EpH4 cells, some signaling pathways were inversely related to 4T1 cells. Phosphorylated p38 MAPK increased in EpH4 Aqp7 KD cells compared to control cells. Also, relative Aqp7-dependent differences in phosphorylated EGFR, AKT, mTOR, and S6K in EpH4 cells were inversely related to 4T1 cells.
Discussion
We developed a correlation-based network analysis to identify metabolites and genes important in breast cancer metabolism. By integrating metabolomics and gene expression data, we identified both metabolic and gene hubs that had prognostic value in breast cancer patients and are predicted to be key in breast cancer and/or breast cancer metabolism, making them worth future investigation. The identification of AQP7 as a critical and novel regulator of breast cancer reinforces the value of using correlated network analysis to analyze large datasets. Reduced Aqp7 expression inhibits breast cancer proliferation and metastasis and profoundly alters the metabolomic profile of breast cell lines and tumors but affects breast cancer cells and normal MECs differently. Metabolically, AQP7 is required for carbohydrate metabolism, complex lipid biosynthesis, urea/arginine metabolism, redox balance, amino acid metabolism, and nucleotide metabolism. AQP7 differentially regulates cancer metabolic pathways via PI3K/AKT/mTOR and p38 MAPK to respond to nutrient and stress conditions (Figure 7B).
The identification of unique gene and metabolite hubs by correlated network analysis is an approach that to our knowledge has not been used to integrate breast metabolite and gene expression data. Recently, multiple mathematical methods have been developed for integration of metabolomics and transcriptomics data based on pathway analysis (23–25,27–29). While each differ in their exact methodology, these guided integration techniques usually map gene expression according to gene ontology (GO) terms, or, more recently, directly to the enzymes of each step of known metabolic pathways. The advantage of these integration techniques is that they add information about the flux of metabolic activities into metabolomics data, which are simply “snapshots” of metabolic activities. However, these methods are biased by existing databases used in the analysis and do not leave room for alternative moonlighting or neomorphic functions.
Our informatics analysis has two main advantages. First, compared to previous guided approaches of integrating metabolomics and transcriptomics (23–29), our method analyzes metabolites and genes, including those whose metabolic functions are not yet understood and not yet incorporated into current pre-defined pathways. This allows the analysis to discover novel metabolites and genes important in tumor metabolism. The disadvantage is the requirement for multiple groups of independent data. If groups of data are only separated by a related factor (e.g., expression levels of a gene), then the final results would be biased towards genes related to the factor and skew the analysis. Second, our analysis provides greater predictive power. Compared to other analyses that compare levels of metabolites/genes between two groups of samples (e.g., treated/untreated, normal/cancer) and then match this change to pre-defined pathways, we increased the comparison to five groups. This increased the statistical power and allowed us to assess with greater certainty which genes/metabolites were linked with each other and were important in cancer.
Although not all identified gene hubs may represent master regulators, they represent key points in cancer metabolism and sit at critical pathway junctions. We suggest that each hub plays a role as: 1.) master regulators, 2.) key sensors that sense and regulate metabolic changes of genes/metabolites, 3.) key products from multiple converged pathways, either metabolic or transcriptional, or 4.) crucial material for producing other products. Identifying pathways that the hubs affect gives us the opportunity to perturb those pathways to identify key regulators of these pathways.
Interestingly, in normal cells, reduced Aqp7 expression promotes migration and branching in normal cells, but in metastatic cells, reduced Aqp7 expression inhibits cancer progression, both in culture and in vivo. Since AQP7 functions as a channel for glycerol, a possible explanation is that AQP7 is a gatekeeper of glycerol availability. In 4T1s, AQP7 KD cells used less glycerol than control, while the opposite was observed in EpH4 cells. This is consistent with AQP7-dependent glycerol availability and usage as potential causes of phenotypic differences between cell lines. In addition, metabolic differences between mammary cell lines and tumor tissue derived from the same cells may reflect contributions by the tumor microenvironment or interchange/flux of metabolites between the cancer and stromal cells.
This study is the first to quantify global metabolites affected by AQP7 in breast cancer, both in culture and in vivo. Our results indicate knocking down AQP7 dramatically altered metabolism in culture, significantly downregulating many major metabolites in amino acid metabolism (especially glutathione metabolism), TCA cycle, and nucleotide synthesis, consistent with decreased proliferation. Fewer metabolites were significantly altered in vivo, possibly reflecting the tissue heterogeneity.
Though citrate was unaltered by decreased Aqp7 expression, other TCA cycle intermediates (α-ketoglutarate, fumarate, and malate) decreased significantly in cells and may reflect decreased glutaminolysis, as suggested by the decreased glutamate in both cell lines, and changes in NAD+. Glutaminolysis, where glutamine is uptaken by cells, converted to glutamate, and shuttled into the TCA cycle as α-ketoglutarate, is especially important in cells such as cancer cells that use a large portion of their citrate for fatty acid synthesis. Decreased intermediate concentrations could also reflect low levels of NAD+, which would inhibit production of TCA intermediates, since NADH production from the TCA cycle requires NAD+ as a substrate. Furthermore, fumarate, an oncometabolite in many cancers, regulates epigenetics by inhibiting histone demethylases and TET enzymes (40,41). Lowered fumarate and oncometabolite 2HG could be another mechanism by which AQP7 regulates tumor growth.
AQP7 regulates the lipidome, likely via AQP7’s role in glycerol transport. Aqp7 knockout in adipocytes generated TAG from accumulated glycerol, which is converted to glycerol-3-phosphate and TAG (6). While glycerol levels were unchanged in 4T1 or EpH4 cells after Aqp7 KD, glycerol-3-phosphate levels decreased by either reduced synthesis or increased usage. Lipid metabolism, specifically the increased TAG, DAG, and FFA in EpH4 cells, mimicked the changes observed in adipose tissue of Aqp7 KO mice. This effect was cell type specific, since in 4T1 cells, TAGs decreased in Aqp7 KD cells. Directional DHAP changes in these cell lines may explain these differences. The availability of external sources of fatty acids and intracellular de novo fatty acid synthesis likely contributed to lipidome reprogramming. While 4T1 cells decreased TAG after Aqp7 KD, TAG accumulated significantly in vivo. These differences may be due to the availability of FFA for TAG synthesis, which would impact the downstream signaling pathways and anabolic enzymes. Compared to 4T1 cells, EpH4 cells increased most complex lipid species after Aqp7 KD, consistent with increased glycerol usage, proliferation, and migration. Finally, while the exact functions of CE in cancers remain poorly understood, CEs are associated with tumor growth in various cancers, including breast (34–36). Consistently, CE levels dramatically decreased in Aqp7 KD tumors, which also had decreased tumor burden. Therefore, AQP7 plays an important role regulating lipid metabolism.
NADPH and NADP+ measurements of 4T1 cells show that, while reduced Aqp7 expression significantly lowered both NADPH and NADP+ levels, the NADPH/NADP+ ratio did not change. AQP7’s impact on cellular redox was evident under increased oxidative stress. ROS increased significantly under an ROS inducer in 4T1 AQP7 KD than control cells. This has implications for the AQP7-dependent differences observed in cancer versus normal MECs. Increased ROS in cancer cells could tip the balance towards cell death, whereas the same ROS level increase in normal cells may actually boost proliferation (1). The increased ROS in AQP7 KD cells may reflect trapped H2O2, an important signaling molecule (11), which lowers glutathione levels and increases the sensitivity to ROS inducers.
Dysregulation in mitochondrial function may explain why AQP7 KD decreases oxidative tolerance in metastatic but not in normal MECs. These results of differences between cancer and normal cells suggest a potential vulnerability induced by AQP7, and targeting AQP7 may be a therapeutic strategy.
AQP7’s modulation of urea/arginine metabolism is surprising but not wholly unexpected. AQP7 transports nitrogen-containing molecules ammonia and urea (8–10,12,13). AQP7 KD could inhibit this transport. In 4T1 cells, AQP7 KD greatly decreased the levels of NAG (N-acetyl glutamate), a crucial metabolite in the process of carbamoyl phosphate synthesis from ammonia in ammonia detoxification. Besides entering the urea cycle to form citrulline, carbamoyl phosphate is used in pyrimidine synthesis, and pyrimidine-related metabolites also decrease after AQP7 KD in 4T1 cells compared to control cells. Other precursors to the urea cycle, including aspartate and argininosuccinate, also decrease. Additionally, intracellular arginine levels increased after AQP7 KD, but downstream metabolites, such as polyamines, creatine, and proline, decreased. These data suggest decreased arginine usage after Aqp7 deficiency, potentially due to fewer available precursors. Correspondingly, iNOS activity decreased in Aqp7 KD cells. Therefore, AQP7 reprograms the urea/arginine metabolism to utilize less arginine and produce less NO, an important signaling molecule in tumor growth (42,43). The AQP7-dependent metabolic changes may be induced by AQP7 transport function. AQP7 could directly control urea/arginine metabolism through its transport of ammonia and urea.
Aqp7 deficiency reprogrammed multiple metabolic signaling pathways. In particular, Aqp7 deficiency in metastatic cancer cells increased mTOR signaling and decreased p38 signaling, while in normal cells the deficiency decreased mTOR signaling and increased p38 signaling.
AQP7 acts as a gatekeeper of external nutrients and cellular stress to regulate signaling pathways required for adapting to nutrient availability, such as mTOR signaling. Since AQP7 transports a variety of molecules, the changes in urea/arginine and nucleotide metabolism could be due to the transport of these often-overlooked molecules (8–13). Additionally, cancer cells are versatile in nutrient utilization, even using ammonia as a nutrient (44).
Aqp7 deficiency/inhibition may create a potential metabolic vulnerability that can be exploited therapeutically to overcome resistance, particularly in endocrine therapy resistance with elevated mTOR signaling. For example, oxidative stress is required for efficacy with mTOR inhibitors (45). Our study shows that Aqp7 deficiency sensitizes cancer cells to oxidative stress and increases mTOR signaling. These findings have implications for combination therapy of AQP7 inhibition and mTOR signaling in tumors resistant to mTOR inhibitors. Treatment with an AQP7 inhibitor in combination with mTOR inhibitors is suggested to induce sufficient cellular stresses to kill the cells and to regress tumors and will be the focus of future studies (46).
Supplementary Material
Statement of Significance.
Aquaporin-7 is identified as a critical regulator of nutrient availability and signaling that responds to cellular stresses, making it an attractive therapeutic target in breast cancer.
Acknowledgments
Special thanks to the Littlepage Lab and Zena Werb for ongoing support, encouragement, and critical reading of the manuscript, Dr. William Boggess, Notre Dame Mass Spectrometry and Proteomics Facility, for his help with this project, and George Lemieux and Jason Herschkowitz for helpful conversations at the start of this project. This work was supported by generous funding from the Walther Foundation for Cancer Research, the SAS Cancer Research Foundation, the National Institutes of Health (R01GM096767-N.J.D., R33CA206922-L.E.L., T32GM075762-V.C., M.C.M.V., R35GM136334-N.J.D.), the International Fellowship from AAUW, and the Notre Dame Advanced Diagnostics and Therapeutics Discovery Fund. V.C. and M.C.M.V. are supported by the Chemistry-Biochemistry-Biology Interface (CBBI) Program at the University of Notre Dame. L.E.L. has also been supported by American Cancer Society Research Scholar Award, Indiana CTSI Young Investigator Award, and Mary Kay Foundation. The graphical abstract and the model were created with BioRender.com.
Footnotes
Conflict of interest disclosure statement. The authors disclose no potential conflicts of interest.
References
- 1.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168:657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verkman AS. Aquaporins at a glance. J Cell Sci. 2011;124:2107–12. [DOI] [PubMed] [Google Scholar]
- 5.Kishida K, Kuriyama H, Funahashi T, Shimomura I, Kihara S, Ouchi N, et al. Aquaporin adipose, a putative glycerol channel in adipocytes. J Biol Chem. 2000;275:20896–902. [DOI] [PubMed] [Google Scholar]
- 6.Hara-Chikuma M, Sohara E, Rai T, Ikawa M, Okabe M, Sasaki S, et al. Progressive adipocyte hypertrophy in aquaporin-7-deficient mice: Adipocyte glycerol permeability as a novel regulator of fat accumulation. J Biol Chem. 2005;280:15493–6. [DOI] [PubMed] [Google Scholar]
- 7.Hibuse T, Maeda N, Funahashi T, Yamamoto K, Nagasawa A, Mizunoya W, et al. Aquaporin 7 deficiency is associated with development of obesity through activation of adipose glycerol kinase. Proc Natl Acad Sci U S A. 2005;102:10993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishibashi K, Kuwahara M, Gu Y, Kageyama Y, Tohsaka A, Suzuki F, et al. Cloning and Functional Expression of a New Water Channel Abundantly Expressed in the Testis Permeable to Water, Glycerol, and Urea. J Biol Chem. 1997;272:20782–6. [DOI] [PubMed] [Google Scholar]
- 9.Liu Z, Shen J, Carbrey JM, Mukhopadhyay R, Agre P, Rosen BP. Arsenite transport by mammalian aquaglyceroporins AQP7 and AQP9. Proc Natl Acad Sci. 2002;99:6053–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu B, Beitz E. Aquaporins with selectivity for unconventional permeants. Cell Mol Life Sci. 2007;64:2413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bienert GP, Chaumont F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim Biophys Acta - Gen Subj. 2014;1840:1596–604. [DOI] [PubMed] [Google Scholar]
- 12.Geyer RR, Musa-Aziz R, Qin X, Boron WF. Relative CO2 /NH3 selectivities of mammalian aquaporins 0–9. Am J Physiol Physiol. 2013;304:C985–94. [DOI] [PubMed] [Google Scholar]
- 13.Yusupov M, Yan D, Cordeiro RM, Bogaerts A. Atomic scale simulation of H2O2 permeation through aquaporin: toward the understanding of plasma cancer treatment. J Phys D Appl Phys. 2018;51:125401. [Google Scholar]
- 14.Maeda N, Funahashi T, Hibuse T, Nagasawa A, Kishida K, Kuriyama H, et al. Adaptation to fasting by glycerol transport through aquaporin 7 in adipose tissue. Proc Natl Acad Sci U S A. 2004;101:17801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ceperuelo-Mallafré V, Miranda M, Chacón MR, Vilarrasa N, Megia A, Gutiérrez C, et al. Adipose tissue expression of the glycerol channel aquaporin-7 gene is altered in severe obesity but not in type 2 diabetes. J Clin Endocrinol Metab. 2007;92:3640–5. [DOI] [PubMed] [Google Scholar]
- 16.Kolb R, Phan L, Borcherding N, Liu Y, Yuan F, Janowski AM, et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun. 2016;7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017;5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jensen HH, Login FH, Koffman JS, Kwon TH, Nejsum LN. The role of aquaporin-5 in cancer cell migration: A potential active participant. Int J Biochem Cell Biol. 2016;79:271–6. [DOI] [PubMed] [Google Scholar]
- 19.Satooka H, Hara-Chikuma M. Aquaporin-3 Controls Breast Cancer Cell Migration by Regulating Hydrogen Peroxide Transport and Its Downstream Cell Signaling. Mol Cell Biol. 2016;36:1206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Ieso ML, Yool AJ. Mechanisms of Aquaporin-Facilitated Cancer Invasion and Metastasis. Front Chem. 2018;6:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai C, Arceo J, Arnold J, Sreekumar A, Dovichi NJ, Li J, et al. Metabolomics of oncogene-specific metabolic reprogramming during breast cancer. Cancer Metab. 2018;6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hollern DP, Andrechek ER. A genomic analysis of mouse models of breast cancer reveals molecular features ofmouse models and relationships to human breast cancer. Breast Cancer Res. 2014;16:R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910–4. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 24.Nam H, Chung BC, Kim Y, Lee KY, Lee D. Combining tissue transcriptomics and urine metabolomics for breast cancer biomarker identification. Bioinformatics. 2009;25:3151–7. [DOI] [PubMed] [Google Scholar]
- 25.Borgan E, Sitter B, Lingjærde OC, Johnsen H, Lundgren S, Bathen TF, et al. Merging transcriptomics and metabolomics - advances in breast cancer profiling. BMC Cancer. 2010;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brockmöller SF, Bucher E, Müller BM, Budczies J, Hilvo M, Griffin JL, et al. Integration of metabolomics and expression of glycerol-3-phosphate acyltransferase (GPAM) in breast cancer-link to patient survival, hormone receptor status, and metabolic profiling. J Proteome Res. 2012;11:850–60. [DOI] [PubMed] [Google Scholar]
- 27.Zhang G, He P, Tan H, Budhu A, Gaedcke J, Michael Ghadimi B, et al. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin Cancer Res. 2013;19:4983–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang X, Lin CC, Spasojevic I, Iversen ES, Chi JT, Marks JR. A joint analysis of metabolomics and genetics of breast cancer. Breast Cancer Res. 2014;16:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Auslander N, Yizhak K, Weinstock A, Budhu A, Tang W, Wang XW, et al. A joint analysis of transcriptomic and metabolomic data uncovers enhanced enzyme-metabolite coupling in breast cancer. Sci Rep. 2016;6:29662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang DW, Lempicki R a, Sherman BT. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 31.Goswami CP, Nakshatri H. PROGgene: gene expression based survival analysis web application for multiple cancers. J Clin Bioinforma. 2013;3:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signal. 2013;6:pl1–pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; [DOI] [PMC free article] [PubMed]
- 34.Paillasse MR, de Medina P, Amouroux G, Mhamdi L, Poirot M, Silvente-Poirot S. Signaling through cholesterol esterification: a new pathway for the cholecystokinin 2 receptor involved in cell growth and invasion. J Lipid Res. 2009;50:2203–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Gonzalo-Calvo D, López-Vilaró L, Nasarre L, Perez-Olabarria M, Vázquez T, Escuin D, et al. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: a molecular and clinicopathological study. BMC Cancer. 2015;15:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee SS-Y, Song B, Chen S, Li J, Konieczny SF, Liu X, et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene. 2016;35:6378–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geck RC, Toker A. Nonessential amino acid metabolism in breast cancer. Adv Biol Regul. 2016;62:11–7. [DOI] [PubMed] [Google Scholar]
- 38.Hosios AM, Hecht VC, Danai LV., Johnson MO, Rathmell JC, Steinhauser ML, et al. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev Cell. 2016;36:540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burke AJ, Sullivan FJ, Giles FJ, Glynn SA. The yin and yang of nitric oxide in cancer progression. Carcinogenesis. 2013;34:503–12. [DOI] [PubMed] [Google Scholar]
- 40.Yang M, Soga T, Pollard PJ, Adam J. The emerging role of fumarate as an oncometabolite. Front Oncol. 2012;2:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest. 2013;123:3652–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keshet R, Erez A. Arginine and the metabolic regulation of nitric oxide synthesis in cancer. Dis Model Mech. 2018;11:dmm033332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keshet R, Szlosarek P, Carracedo A, Erez A. Rewiring urea cycle metabolism in cancer to support anabolism. Nat Rev Cancer. 2018;18:634–45. [DOI] [PubMed] [Google Scholar]
- 44.Spinelli JB, Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, et al. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science (80- ). 2017;9305:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malone CF, Emerson C, Ingraham R, Barbosa W, Guerra S, Yoon H, et al. mTOR and HDAC Inhibitors Converge on the TXNIP/Thioredoxin Pathway to Cause Catastrophic Oxidative Stress and Regression of RAS-Driven Tumors. Cancer Discov. 2017;7:1450–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steelman LS, Martelli AM, Cocco L, Libra M, Nicoletti F, Abrams SL, et al. The therapeutic potential of mTOR inhibitors in breast cancer. Br. J. Clin. Pharmacol. 2016. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.