

Abstract
PLS is an extremely rare central nervous system degenerative disorder characterized by slowly progressive upper motor neuron loss leading to severe limb and bulbar dysfunction and disability. Although not necessarily life-shortening, PLS disease burden is substantial and improved symptomatic treatments are a major unmet need, especially for the often refractory spasticity that is a core feature of the syndrome. In Section 1, we describe clinical care needs and emphasize a highly personalized approach that can be best attained through multidisciplinary management. In Section 2, we describe progress in clinical trials in PLS that includes advances in symptomatic treatment, disease-modifying therapy, and emerging innovative trials.
Keywords: PLS
INTRODUCTION
Many amyotrophic lateral sclerosis (ALS) experts consider primary lateral sclerosis (PLS) to lie at one end of a spectrum of upper motor neuron degeneration. Unlike ALS, PLS often poses no primary threat to survival; however, the disease causes slowly progressive upper motor dysfunction that impacts all voluntary skeletal muscles. This results in often severe mobility impairment, upper limb and bulbar dysfunction. Those living with PLS require neurologists and wider multidisciplinary team members sufficiently knowledgeable to provide personalized care in this rare condition.
The first section of this review focuses on PLS care and management. In addition to all authors’ contribution, we are particularly fortunate to have input for this section from a retired academic neurologist and psychiatrist living with PLS. The second section focuses on therapeutic trials in which PLS has been hitherto neglected, partly due to its rarity but also uncertainty surrounding whether it shares the same pathogenic mechanisms linked to ALS. Diagnostic criteria for PLS have demanded a waiting period of at least 3 to 4 years before PLS can be confidently separated from upper motor neuron-predominant forms of ALS (1, 2). Recent consensus diagnostic criteria recognized the need to try to enable earlier therapeutic intervention and agreed upon a 2–4 year category of ‘Probable PLS’ (3).
Because of its slow, clinical progression, one presumes that a protracted observational period may be required for a clinical trial. At present, there are no diagnostic or prognostic biomarkers for PLS. Despite these obvious impediments, pioneering work to target spasticity, a main symptom of PLS, has been initiated. Symptomatic improvement is a crucial component of clinical trials. Another important component is testing disease-modifying medications. Although research is needed for biological approaches in PLS, new diagnostic criteria that can capture early PLS cases have already been established (3). To detect changes of progression in PLS more sensitively, a novel PLS-specific clinimetric scale, the PLSFRS, has been developed from the original ALSFRS (4).The necessary tools to undertake disease-modifying clinical trials in PLS appear to be at hand. In this review, we discuss how to conduct disease-modifying clinical trials in PLS, including conventional and innovative.
SECTION 1. CLINICAL CARE IN PLS
Background and Overall Goals of Clinical Care:
Treatment in PLS is aimed at minimizing symptoms and maximizing quality of life. Because few clinical trials of symptomatic treatments in PLS have been carried out, clinical experience treating neurological disorders with similar symptoms has been used to guide treatment. The ideal approach should be individualized for each patient with PLS to reach their goals for maintaining mobility, communication, and independence. The ideal treatment regimen includes medications, physical activity, psychosocial support, and when needed, durable equipment. Neurologists and rehabilitation specialists, including physical, occupational, and speech therapists, play key roles in determining treatment plans. Moreover, the optimal combination of treatments for each patient is likely to change over time as symptoms progress and should be periodically re-evaluated. Changes in treatment may be needed more frequently in the first years of symptoms as PLS patients often experience a plateau in symptom progression after several years (5).
Because a diagnosis of PLS often cannot be made without the passage of time, those patients must exist in a period of diagnostic uncertainty. Treating a patient’s symptoms (e.g., PT referral for gait disorder) in the absence of a firm diagnosis is appropriate and may strengthen the doctor-patient relationship. Providing accurate information about PLS as well as contact information for patient organizations, such as the Spastic Paraplegia Foundation, may also be beneficial (6). Use of complimentary therapies, including yoga and meditation, in the population have increased in recent years (7). An evidence-based review found varying degrees of efficacy of mindfulness meditation in reducing anxiety, depression, and stress (8) should symptoms accompany the wait for a diagnosis. If a diagnosis remains elusive, referral to the Undiagnosed Diseases Network is an option (9).
Person-Centered Approach Spasticity:
A person-centered approach to treating spasticity takes into account the individual’s goals of maintaining mobility and reducing discomfort from muscle stiffness (10). Medications are useful for reducing symptoms of spasticity (11). Effective doses for reducing spasticity can lead to sedation as an unwanted side effect. Patients need better options, pharmacologic and nonpharmacologic, to improve mobility by reducing spasticity. Exercise has been shown to be safe in ALS (12, 13) and has proven beneficial for improving activities of daily living, balance, mobility, and flexibility in other disorders such as stroke (14). Because different kinds of exercise lead to improvement in different domains (14), studies are needed to determine how best to compose a regimen of strengthening exercises, aerobic activity, active and passive stretching, and balance training for individuals with PLS..
Pseudobulbar Affect and Dysarthria:
Two other symptoms which may affect independence and social interactions are pseudobulbar affect (PBA) and dysarthria. Although tricyclic medications and dextromethorphan-quinidine (Nuedexta®) are effective treatments for PBA (15–17), each has potentially limiting side effects. In some patients, dextromethorphan-quinidine (Nuedexta®) may lead to improvements in speech (18). Dysarthria may increase under stress or emotionality, contributing to social and functional challenges. Voice banking or message banking are options that patients may consider. Although communication boards exist, typically tablets with pre-programmed phrases, they can be unwieldy. Another option is a text-to-speech app (e.g., Talk For Me (available in the App Store)) for individuals who are facile with a keyboard. Technology companies are working on apps that would permit people with dysarthria to speak into a smart phone, and the app would repeat the same phrase more clearly (19). This type of system would be a significant advancement, but apps are still in development with no release date in sight. Research is needed on speech recognition devices as well as on determining which speech therapy techniques work best.
Ambulation:
For many patients with PLS, walking is affected early, and combined with slowed reactions, often leads to falls. A home assessment for fall risks and safety measures is recommended. Equipment needs should also be assessed. As years go by, a patient’s need for gait aids often progresses from ambulation aids – hiking poles, cane(s), forearm crutches, and/or walker – to seated mobility aids – wheelchair, power scooter, and motorized wheelchair. Patients may use a combination of ambulation and seated mobility aides when appropriate. Other equipment, such as bedrails, lift-chairs, and large handled utensils may be beneficial in individual patients. Physicians should be alert to medical conditions that may be amenable to treatment such as osteopenia.
Multidisciplinary Care:
Comprehensive management can best be accomplished at ALS multidisciplinary Clinics, which offer expertise in PLS assessment, a team approach to treatment, and access to information on clinical trials in PLS. Such clinics are located in many places; there are about 100 clinics supported by the ALS Association or Muscular Dystrophy Association (MDA) in the US and equally as many clinics in Europe, UK, Australia with a few in Japan. Some patients with PLS, including those with spasticity as their main symptom, may prefer to be treated by their own neurologist (who may have neuromuscular fellowship training.) Some patients with PLS may feel uncomfortable attending clinics with patients with ALS since they progress more rapidly. We still recommend that patients with PLS be seen at an ALS Multidisciplinary Clinic when looking for comprehensive, multidisciplinary care in one location.
SECTION 2. THERAPEUTIC TRIALS
Symptomatic Treatment for Spasticity:
Ideally, clinical trials for PLS should be based on a scientific rationale. Because the cause(s) and even pathogeneses of PLS are currently unknown, current clinical trials focus on symptomatic, rather than disease modifying treatments. There are three general categories of hypotheses underlying symptomatic treatments. The first category involves enhancing existing corticospinal function, for example by improving conduction in axons with secondary demyelination (20). In other disorders with spasticity, it is known that maladaptive changes occur in subcortical and spinal neural circuits following the loss of corticospinal input. The second category involves interventions aimed at reducing spasticity and are hypothesized to enhance circuits for reciprocal inhibition thus reducing muscle co-contraction. The third category involves facilitating adaptive neuroplasticity in unaffected or less affected circuits outside the corticospinal system. This approach is particularly promising for PLS, given the relative sparing of motor pathways outside the corticospinal system and the long duration of disease. Facilitating spinal neuroplasticity, as has been piloted in spinal cord injury (21–24) and activating brainstem motor pathways, for example with rhythmic cueing (25) may be ways to improve ambulation in PLS. There is sufficient scientific rationale to support clinical trials using activity-based and non-invasive stimulation interventions in PLS to promote neuroplasticity.
Most therapies currently employed to ameliorate symptoms for patients with PLS are extrapolated from evidence and experience from treating patients with ALS and other diseases with UMN dysfunction, such as multiple sclerosis. Nevertheless, symptomatic treatments are needed to specifically address the needs of patients with PLS.
On-going Trial:
Currently, one symptomatic clinical trial for spasticity in patients with PLS is on-going. Patients with PLS are still being recruited to test the ability of Dalfampridine (Ampyra®) to improve walking speed. Dalfampridine is an extended release form of 4 aminopyridine, whose primary mechanism is thought to be a potassium channel blocker. Dalfampridine is hypothesized to enhance neuronal transmission in damaged axons. Patients with MS who took Dalfampridine showed improved walking speed. This study will determine if benefits of Dalfampridine can be realized by patients with PLS. The trial design is an open-label study and consistent improvement in function during the treatment phase is the primary endpoint.
The primary end point is to determine if walking speed consistently improves with treatment, the same measure used in the MS trials. The control group reflects the patient pre and post treatment. We also will assess safety in this patient population. We are including additional functional and quality of life measures to determine their applicability to PLS patients, while we work on a validated clinical tool. Further, we are studying cortical response to transcranial magnetic stimulation. Preliminary results confirm cortical inexcitability among patients with PLS. In this study, more than 50% of patients screened lacked a cortical response. Therefore, physiological markers of cortical excitability will not be applicable to all patients with PLS (see below). Improved neuroimaging and biochemical biomarkers will need to be developed in future studies.
Disease-Modifying Therapy:
The National Institute of Neurological Diseases and Stroke (NINDS) issued a request for applications (RFA) for “clinical trial readiness for rare neurodegenerative diseases” in 2017. There were a few basic requirements for this application. Typically, a multisite study would be needed since a single site can only recruit a limited number of patients. The study should also incorporate potential treatments and readily available biomarkers. Furthermore, diagnostic methodology and assessment techniques should be established. The NINDS’s key aim for this RFA was to enable the initiation of a clinical trial once the project was complete. PLS is obviously a rare neurodegenerative disease, and this RFA represented a great opportunity to investigate PLS for the development of future clinical trials. At this point, it is too early for our study group to obtain federal support for this particular clinical trial readiness grant for rare neurodegenerative diseases; however, in the near future, we should be able to consider pursuing this opportunity. We also should point out that although a very small number (approximately 15%) of definite PLS cases can be identified as other definable genetic diseases (26), PLS in general represents a well-defined disease entity. Fishman (2013) pointed out that performing small clinical trials in a small homogeneous group of patients with a rare disease is a promising strategy for future, successful clinical trials (27). In the next few sections, we describe necessary components of clinical trials in PLS.
Participant Selection:
In the study we should include participants who have spasticity symptoms at least 24 months or longer, insidious in onset but relentlessly progressive, usually beginning in the legs with stiffness and impaired walking or, less commonly, with difficulty speaking. The absence of LMN involvement is a key factor in distinction from ALS. Confusion most commonly arises from the finding of abnormal spontaneous activity (fibrillation potentials and positive sharp waves) on electromyography (EMG) and their significance. Consensus diagnostic criteria have been developed (3).
Clinical Assessments:
There had been no validated clinical assessments for PLS until the PLSFRS was established (4). The choice of clinical endpoints has been therefore based on face validity with scales developed in similar disease states. Assessments specifically designed to measure mobility and validated in other disease states showing the ability to be able to detect differences in walking between treated and untreated patients include the timed 25 foot walk test, the timed up and go (TUG) test, and the 2-minute endurance walk test. To detect changes in fine motor movements in the hands, the Purdue Pegboard Test is a validated test of fine motor skills. Upper motor neuron clinical motor function can be assessed by timed tapping of fingers and feet (See the review by Floeter in this Supplement). The ALSFRS-R is a validated tool for measuring decline in ALS patients over time and has been used for PLS. Clinical global impressions of subject and investigators are also a strong measure of degree of impact of treatment.
Biomarkers:
Quantitative laboratory measures of progression of PLS are also lacking, but many are being investigated. Among them are quantitative measures of brain MRI, MR spectroscopy, and transcranial magnetic stimulation (TMS). There are no known biologic markers for PLS that show change and sensitivity to change as the disease progresses.
Imaging:
A variety of methods to image the motor cortex are being investigated as possible biomarkers for PLS and to measure progression of upper motor neuron disease. T2 hyperintensity within the corticospinal tract or atrophy of the precentral gyrus are often seen but their presence is variable. The correlation between quantitative imaging deficits and clinical deficits is not clear. Alterations found in diffusion tensor imaging measures of the corticospinal tract were unchanged over time in small longitudinal study (28). T2 hypo-intensity in PLS patients and T2*/R2* susceptibility measures correlate with microglial iron deposition in the middle and deep cortical layers on autopsy. They also qualitatively correlate with disease severity and progression (28–30). Of interest in this technique is that changes can be appreciated in specific regions of the homunculus that can be correlated with the clinical syndrome (Fig. 1). Further, changes over time can be reflected in changes in quantitative susceptibility mapping (QSM) images to measure iron deposition. Although promising, validation with clinical progression are ongoing. Glial activation in patients with PLS has been identified but its role as a potential marker is uncertain (31).
Figure 1. Increased signal in the left arm and leg.
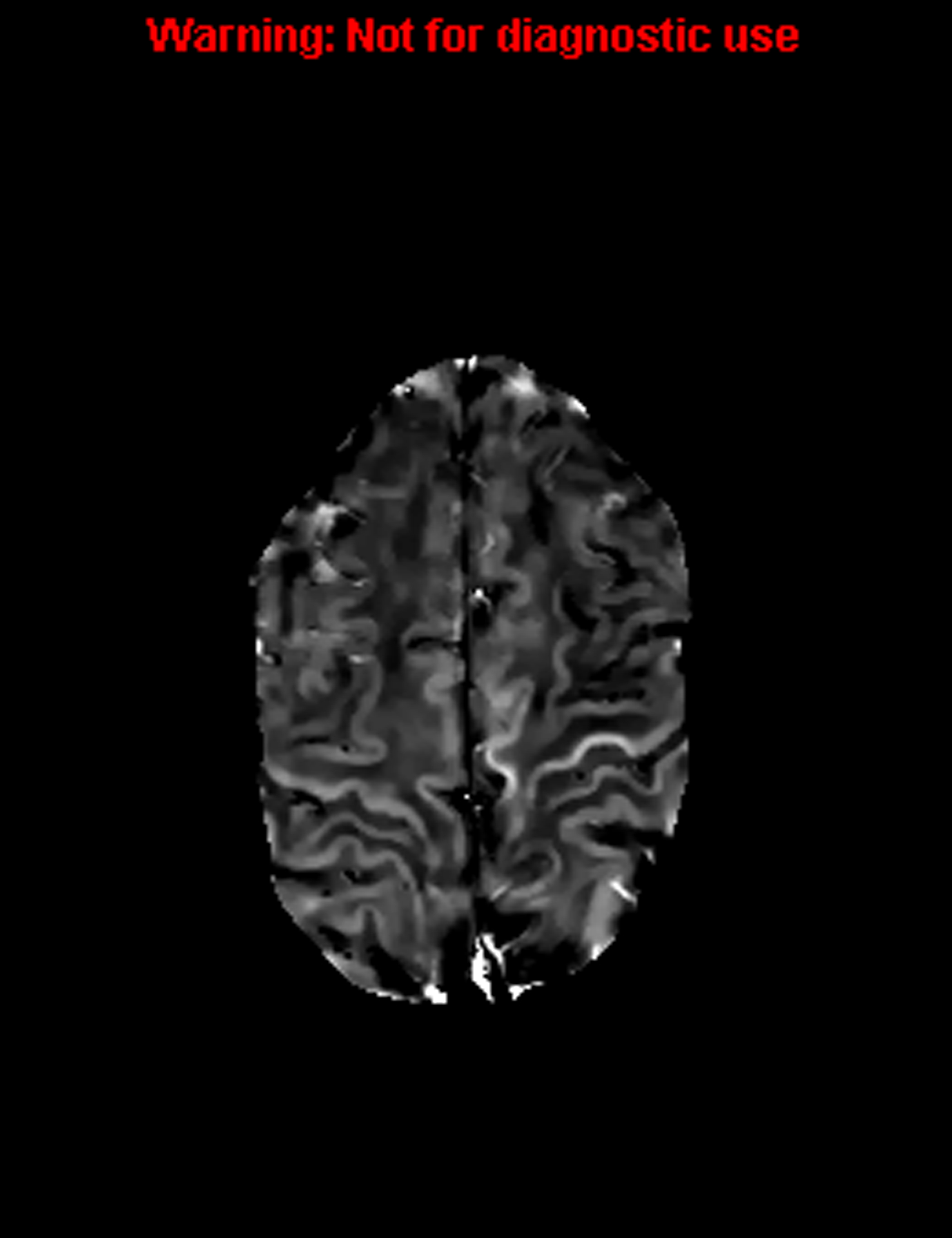
T2 hypo-intensity in PLS patients and T2*/R2* susceptibility measures correlate with microglial iron deposition in the middle and deep cortical layers on autopsy. The changes can be appreciated in specific regions of the homunculus that can be correlated with the clinical syndrome.
Magnetic Stimulation:
Changes in cortical excitability and loss of cortical motor neurons are the basis of the clinical deficits in PLS. Transcranial magnetic stimulation (TMS) has been used for more than 30 years as a means to test cortical excitability (32). There are several methods to assess motor cortex excitability. Resting motor threshold (RMT) is stimulus intensity needed to elicit motor responses in 50% or more of consecutive stimuli. An increased RMT or unresponsive motor cortex is present when there is no response recorded from a hand muscle when stimulating the motor cortex. For clinical trials, a reproducible change over time (in either direction, i.e., hyper or hypo excitable) would be a potential means for a validated measure of upper motor neuron deterioration or improvement. The cortical silent period (CSP) utilizes the fact that voluntary activity is inhibited after motor cortex stimulation and is measured from the onset of the motor evoked potential until the return of voluntary EMG activity. CSP is decreased in the hyper excitable motor cortex and increased in the inexcitable cortex. How these measures change with disease progression will dictate their utility in becoming a validated biomarker for UMN change in patients with PLS. Paired pulse studies are also used to measure cortical excitability and may be useful in measuring clinically relevant change (33, 34).
A combination of peripheral nerve and TMS using knowledge of impulse collision is the basis of the test called the triple stimulation technique (TST)(35, 36). The method requires a computer algorithm that is based on latencies between 3 stimulation sites: motor cortex, erbs point, and wrist. After appropriate impulse collisions, a quantitative estimate of the number of functioning cortical motor neurons is possible. When used in conjunction with neuroimaging, there is high correlation with the presence of upper motor neuron involvement. Change over time and correlation with clinical syndrome is yet to be demonstrated.
Clinical Trial Designs:
We need to rely on alternative trial methods. A cross-over design involves a small number of participants, although the design suffers from phase and carry-over effects. This design is used to see any identifiable effects in phase II studies. If it is used in PLS, it will take a minimum of 6 months and perhaps 12 months for one phase. A total of 24 months would be needed to complete a cross-over study in PLS. Another possibility is a clinical trial using historical controls. PLS cases constitute a small percentage of ALS. A personalized clinical trial (an N-of-1) design may also be feasible. PLS is a chronic and stable disease for which such a design may be ideal; however, it takes a long time to detect any meaningful changes with disease progression. If we use a 6-month, multiple cross-over trial, it will take at least 1.5 years to conduct three cross-over trials. This is a study design we need to consider (37). To design appropriate clinical trials, it is imperative to know the natural history of PLS.
INNOVATIVE TRIAL DESIGNS TO ACCELERATE THERAPY DEVELOPMENT FOR PLS
In recent years, investments in basic and translational research have led to several insights into the mechanisms that underlie ALS These discoveries, in turn, are feeding a robust pipeline of investigational products. Unfortunately, the traditional clinical trial paradigm (testing one product at a time in independent trials) is inefficient, expensive, and leads to prolonged timelines from discovery to testing. This context created the urgent need to innovate our clinical trial strategies to be able to test more agents, faster, and using less resources, which is especially critical in rare diseases such as ALS and PLS.
In parallel, recent advances in biostatistics and progress in regulatory policies enabled the development of innovative trial designs to accelerate therapy development (38). Among these novel designs, we will focus here on platform trials and basket trials. These designs have already proven successful in oncology (39), are viewed favorably by regulators and sponsors (38), and are rapidly being adopted in several fields of medicine, including the neurosciences (40, 41).
Platform trials are trials where multiple investigational products are tested in the context of a single disease (multiple drugs > one disease)(38, 42). These trials are “perpetual” in that they have no defined end date and leverage an infrastructure that remains open long-term to accommodate new experimental agents (42). The design of these trials is often adaptive: it is possible to continue to refine the trial design over time to include more efficient measures of disease progression as they become available (43). Furthermore, platform trials represent an opportunity to collect bio-samples and data using coordinated processes thus enabling disease-focused scientific discovery projects. This model has already been adopted by the ALS community with the launch of the HEALEY ALS Platform trial (44). The HEALEY ALS Platform Trial is testing multiple investigational products (both in parallel and sequentially). This collaborative trial is also facilitating the systematic collection of longitudinal bio-samples to support biomarker discovery efforts. Thus, in addition to evaluating promising investigational products, the trial is also a source of data and samples that will, in turn, contribute to our understanding of ALS mechanisms and help develop better outcome measures and biomarkers. The establishment of a PLS platform would help accelerate drug development and provide critical learnings for this understudied disease.
Basket trials are trials where a single investigational product is investigated in the context of multiple diseases (one drug > multiple diseases)(38, 39). One could imagine a scenario where an investigational product that has shown promise in preclinical models of neurodegeneration might have applicability to more than one neurodegenerative disease, e.g. to both ALS and PLS. Basket designs would be an efficient model to explore PLS as an indication alongside other disease cohorts.
SUMMARY AND DISCUSSION
In Section 1, we reviewed the current forefront of PLS care and management. Customized and personalized treatment based on knowledge and experience is always the best approach. Multidisciplinary ALS clinics, now established throughout many parts of the world, are clearly another important resource for those who have this rare disease. The services offered at these clinics are likely to benefit patients with PLS as well as ALS. Outcome studies in patient care and management are needed, so we can develop evidence-based management guidelines for patients with PLS.
In Section 2, we described an early, on-going symptomatic trial in patients with PLS. Better treatment for spasticity would greatly improve quality of life for those living with PLS. Therefore, a trial that focuses on spasticity in PLS is exciting and important. At the same time, there is an urgent need to develop clinical trials that test disease-modifying treatments. Clinical trials should be based on reasonable and compelling hypotheses about disease mechanisms. Obviously, we more research is needed, but current extensive investigations with neuroimaging, neurophysiology, early biomarkers studies, and genetic analyses (discussed thoroughly in this Supplement) are pointing to new plausible hypotheses in PLS. The recently established new diagnostic criteria that allow us to capture early PLS cases (3) and the development of the PLSFRS to assess patients’ disease progression more sensitively than the ALSFRS-R (4) represent important steps towards clinical trial readiness in PLS.
Supplementary Material
Acknowledgement:
Dr. Mary Kay Floeter received Intramural Program support from NINDS, NIH.
Contributor Information
Mary Kay Floeter, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, USA.
Deborah Warden, formerly of Departments of Neurology and Psychiatry, Uniformed Services University of the Health Sciences, Bethesda, MD, USA.
Dale Lange, Department of Neurology, Hospital for Special Surgery, Weill Cornell School of Medicine, New York, NY, USA.
James Wymer, Department of Neurology, University of Florida, Gainesville, Florida, USA..
Sabrina Paganoni, Healey Center for ALS at Mass General, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA.
Hiroshi Mitsumoto, Department of Neurology, Columbia University Irving Medical Center, New York, NY, USA.
REFERENCES
- 1.Gordon PH, Cheng B, Katz IB, Pinto M, Hays AP, Mitsumoto H, et al. The natural history of primary lateral sclerosis. Neurology. 2006;66(5):647–53. [DOI] [PubMed] [Google Scholar]
- 2.Pringle CE, Hudson AJ, Munoz DG, Kiernan JA, Brown WF, Ebers GC. Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain. 1992;115 (Pt 2):495–520. [DOI] [PubMed] [Google Scholar]
- 3.Turner MR, Barohn RJ, Corcia P, Fink JK, Harms MB, Kiernan MC, et al. Primary lateral sclerosis: consensus diagnostic criteria. J Neurol Neurosurg Psychiat. 2020; 91(4):373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitsumoto H, Chiuzan C, Gilmore M, Zhang Y, Simmons Z, Paganoni S, et al. Primary lateral sclerosis (PLS) functional rating scale: PLS-specific clinimetric scale. Muscle Nerve. 2019. [DOI] [PubMed] [Google Scholar]
- 5.Floeter MK, Wu T. Longitudinal evaluation of upper motor neuron burden scales in primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2020:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spastic Paraplegia Foundation, Inc. Spastic Paraplegia Foundation. https://sp-foundation.org. Accessed 12/13/2020.
- 7.National Center for Complimentary and Integrative Health. https://www.nccih.nih.gov/health/complementary-alternative-or-integrative-health-whats-in-a-name. Accessed 7/21/2020.
- 8.Goyal M, Singh S, Sibinga EM, Gould NF, Rowland-Seymour A, Sharma R, et al. Meditation programs for psychological stress and well-being: a systematic review and meta-analysis. JAMA Intern Med. 2014;174(3):357–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harvard University. Undiagnosed Diseases Network. https://undiagnosed.hms.harvard.edu/. Accessed 12/13/2020.
- 10.Turner-Stokes L, Ashford S, Esquenazi A, Wissel J, Ward AB, Francisco G, et al. A comprehensive person-centered approach to adult spastic paresis: a consensus-based framework. Eur J Phys Rehabil Med. 2018;54(4):605–17. [DOI] [PubMed] [Google Scholar]
- 11.Statland JM, Barohn RJ, Dimachkie MM, Floeter MK, Mitsumoto H. Primary Lateral Sclerosis. Neurol Clin. 2015;33(4):749–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bello-Haas VD, Florence JM, Kloos AD, Scheirbecker J, Lopate G, Hayes SM, et al. A randomized controlled trial of resistance exercise in individuals with ALS. Neurology. 2007;68(23):2003–7. [DOI] [PubMed] [Google Scholar]
- 13.Clawson LL, Cudkowicz M, Krivickas L, Brooks BR, Sanjak M, Allred P, et al. A randomized controlled trial of resistance and endurance exercise in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(3–4):250–8. [DOI] [PubMed] [Google Scholar]
- 14.Han P, Zhang W, Kang L, Ma Y, Fu L, Jia L, et al. Clinical Evidence of Exercise Benefits for Stroke. Adv Exp Med Biol. 2017;1000:131–51. [DOI] [PubMed] [Google Scholar]
- 15.Schiffer RB, Herndon RM, Rudick RA. Treatment of pathologic laughing and weeping with amitriptyline. N Engl J Med. 1985;312(23):1480–2. [DOI] [PubMed] [Google Scholar]
- 16.Brooks BR, Thisted RA, Appel SH, Bradley WG, Olney RK, Berg JE, et al. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial. Neurology. 2004;63(8):1364–70. [DOI] [PubMed] [Google Scholar]
- 17.Ahmed A, Simmons Z. Pseudobulbar affect: prevalence and management. Ther Clin Risk Manag. 2013;9:483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Green JR, Allison KM, Cordella C, Richburg BD, Pattee GL, Berry JD, et al. Additional evidence for a therapeutic effect of dextromethorphan/quinidine on bulbar motor function in patients with amyotrophic lateral sclerosis: A quantitative speech analysis. Br J Clin Pharmacol. 2018;84(12):2849–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munn D Schoolin’ Google in ALS 2019. [Available from: https://alsnewstoday.com/2019/01/29/google-dysarthria-als-speech-recognition/.
- 20.Medicine USNLo. Use of Dalfampridin in Primary Lateral Sclerosis 2019. [Available from: https://clinicaltrials.gov/ct2/show/NCT02868567?term=NCT02868567&draw=2&rank=1.
- 21.Behrman AL, Ardolino EM, Harkema SJ. Activity-Based Therapy: From Basic Science to Clinical Application for Recovery After Spinal Cord Injury. J Neurol Phys Ther. 2017;41 Suppl 3:S39–S45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angeli CA, Boakye M, Morton RA, Vogt J, Benton K, Chen Y, et al. Recovery of Over-Ground Walking after Chronic Motor Complete Spinal Cord Injury. N Engl J Med. 2018;379(13):1244–50. [DOI] [PubMed] [Google Scholar]
- 23.McDonald JW, Becker D, Sadowsky CL, Jane JA, Conturo TE, Schultz LM. Late recovery following spinal cord injury. Case report and review of the literature. J Neurosurg. 2002;97(2 Suppl):252–65. [DOI] [PubMed] [Google Scholar]
- 24.Dobkin BH. Do electrically stimulated sensory inputs and movements lead to long-term plasticity and rehabilitation gains? Curr Opin Neurol. 2003;16(6):685–91. [DOI] [PubMed] [Google Scholar]
- 25.Ghai S, Ghai I, Schmitz G, Effenberg AO. Effect of rhythmic auditory cueing on parkinsonian gait: A systematic review and meta-analysis. Sci Rep. 2018;8(1):506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitsumoto H, Nagy PL, Gennings C, Murphy J, Andrews H, Goetz R, et al. Phenotypic and molecular analyses of primary lateral sclerosis. Neurol Genet. 2015;1(1):e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fishman MC. Power of rare diseases: found in translation. Sci Transl Med. 2013;5(201):201ps11. [DOI] [PubMed] [Google Scholar]
- 28.Kwan JY, Meoded A, Danielian LE, Wu T, Floeter MK. Structural imaging differences and longitudinal changes in primary lateral sclerosis and amyotrophic lateral sclerosis. Neuroimage Clin. 2012;2:151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwan JY, Jeong SY, Van Gelderen P, Deng HX, Quezado MM, Danielian LE, et al. Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 tesla MRI and pathology. PLoS One. 2012;7(4):e35241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oba H, Araki T, Ohtomo K, Monzawa S, Uchiyama G, Koizumi K, et al. Amyotrophic lateral sclerosis: T2 shortening in motor cortex at MR imaging. Radiology. 1993;189(3):843–6. [DOI] [PubMed] [Google Scholar]
- 31.Paganoni S, Alshikho MJ, Zurcher NR, Cernasov P, Babu S, Loggia ML, et al. Imaging of glia activation in people with primary lateral sclerosis. Neuroimage Clin. 2018;17:347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eisen A, Shytbel W, Murphy K, Hoirch M. Cortical magnetic stimulation in amyotrophic lateral sclerosis. Muscle Nerve. 1990;13(2):146–51. [DOI] [PubMed] [Google Scholar]
- 33.Huynh W, Dharmadasa T, Vucic S, Kiernan MC. Functional Biomarkers for Amyotrophic Lateral Sclerosis. Front Neurol. 2018;9:1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geevasinga N, Menon P, Sue CM, Kumar KR, Ng K, Yiannikas C, et al. Cortical excitability changes distinguish the motor neuron disease phenotypes from hereditary spastic paraplegia. Eur J Neurol. 2015;22(5):826–31, e57–8. [DOI] [PubMed] [Google Scholar]
- 35.Magistris MR, Rosler KM. The triple stimulation technique to study corticospinal conduction. Suppl Clin Neurophysiol. 2003;56:24–32. [DOI] [PubMed] [Google Scholar]
- 36.Magistris MR, Rosler KM, Truffert A, Landis T, Hess CW. A clinical study of motor evoked potentials using a triple stimulation technique. Brain. 1999;122 (Pt 2):265–79. [DOI] [PubMed] [Google Scholar]
- 37.Moise N, Wood D, Cheung YKK, Duan N, Onge TS, Duer-Hefele J, et al. Patient preferences for personalized (N-of-1) trials: a conjoint analysis. J Clin Epidemiol. 2018;102:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woodcock J, LaVange LM. Master Protocols to Study Multiple Therapies, Multiple Diseases, or Both. N Engl J Med. 2017;377(1):62–70. [DOI] [PubMed] [Google Scholar]
- 39.Simon R Critical Review of Umbrella, Basket, and Platform Designs for Oncology Clinical Trials. Clin Pharmacol Ther. 2017;102(6):934–41. [DOI] [PubMed] [Google Scholar]
- 40.Bateman RJ, Benzinger TL, Berry S, Clifford DB, Duggan C, Fagan AM, et al. The DIAN-TU Next Generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimers Dement. 2017;13(1):8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alexander BM, Ba S, Berger MS, Berry DA, Cavenee WK, Chang SM, et al. Adaptive Global Innovative Learning Environment for Glioblastoma: GBM AGILE. Clin Cancer Res. 2018;24(4):737–43. [DOI] [PubMed] [Google Scholar]
- 42.Berry SM, Connor JT, Lewis RJ. The platform trial: an efficient strategy for evaluating multiple treatments. JAMA. 2015;313(16):1619–20. [DOI] [PubMed] [Google Scholar]
- 43.Coalition APT. Adaptive platform trials: definition, design, conduct and reporting considerations. Nat Rev Drug Discov. 2019;18(10):797–807. [DOI] [PubMed] [Google Scholar]
- 44.Janos T Sean M Healey & AMG Center for ALS at Mass General receives “May Proceed” notice for three drugs in first ALS platform trial 2020. [Available from: https://www.massgeneral.org/neurology/als/news/2020-01-22-Healey-ALS-platform-trial-may-proceed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.