

Summary
Cyclic GMP–AMP synthase (cGAS) recognition of cytosolic DNA is critical for the immune response to cancer and pathogen infection. Here we discover that cGAS-DNA phase separation is required to resist negative regulation and allow efficient sensing of immunostimulatory DNA. We map the molecular determinants of cGAS condensate formation and demonstrate that phase separation functions to limit activity of the cytosolic exonuclease TREX1. Mechanistically, phase separation forms a selective environment that suppresses TREX1 catalytic function and restricts DNA degradation to an outer shell at the droplet periphery. We identify a TREX1 mutation associated with the severe autoimmune disease Aicardi-Goutières syndrome that increases penetration of TREX1 into the repressive droplet interior and specifically impairs degradation of phase-separated DNA. Our results define a critical function of cGAS-DNA phase separation and reveal a molecular mechanism that balances cytosolic DNA degradation and innate immune activation.
Graphical Abstract
eTOC Blurb
cGAS-DNA phase separation is required to enable DNA sensing in the presence of active negative regulators in cells. Dysfunction of phase separation is an additional mechanism to explain TREX1-dependent autoimmune disease.
Introduction
Cytosolic DNA sensing is critical for detection of pathogen replication and immune recognition of DNA damage (Ablasser and Chen, 2019). The cellular enzyme cyclic GMP–AMP synthase (cGAS) functions as a direct DNA sensor and controls a major mechanism cells use to detect cytosolic DNA (Sun et al., 2013). Following DNA recognition, cGAS catalyzes synthesis of 2′–5′, 3′–5′ cyclic GMP–AMP (2′3′-cGAMP) that binds to the receptor Stimulator of Interferon Genes (STING) and activates downstream Type I interferon and NF-κB signaling (Ablasser et al., 2013; Diner et al., 2013; Gao et al., 2013; Zhang et al., 2013). Inappropriate cGAS-STING activation is a direct cause of severe autoimmunity (Liu et al., 2014; Rice et al., 2007), and loss of expression of cGAS and STING is associated with oncogenesis (Konno et al., 2018). The importance of balancing cellular tolerance and sensing of cytosolic DNA demonstrates the critical role of the molecular processes that regulate cGAS-STING activation.
cGAS activation requires direct recognition of double-stranded DNA and assembly of a higher-order cGAS–DNA complex. In the minimal functional assembly, two molecules of cGAS embrace two segments of double-stranded DNA resulting in a cGAS conformational change and initiation of 2′3′-cGAMP synthesis (Civril et al., 2013; Gao et al., 2013; Li et al., 2013; Zhang et al., 2014; Zhou et al., 2018). Although the minimal cGAS–DNA complex requires only ~15–20 bp DNA segments, longer >45 bp DNA allows cooperative assembly of additional ordered cGAS-DNA interactions and is required for robust cGAS activation in cells (Andreeva et al., 2017). Structures of the human cGAS–DNA complex reveal species-specific substitutions in the DNA binding surfaces that further regulate cGAS–DNA complex assembly and demonstrate that higher-order cGAS-DNA interactions are a key point of regulation controlling cytosolic DNA sensing (Xie et al., 2019; Zhou et al., 2018).
In addition to well-defined structural contacts between cGAS and DNA, non-specific cGAS protein-DNA and protein-protein interactions drive formation of phase-separated liquid droplets that coalesce hundreds of molecules of cGAS with immunostimulatory DNA (Du and Chen, 2018; Xie et al., 2019). cGAS-DNA phase-separated droplets exhibit liquid-like properties and occur readily in vitro under physiological conditions and in cells as visible puncta following DNA stimulation (Du and Chen, 2018). Similar to the role of phase separation in diverse signal transduction pathways (Shin and Brangwynne, 2017), cGAS-DNA liquid-liquid phase separation has been hypothesized to be important for concentrating cGAS with DNA and reaction components in the crowded intracellular environment. However, it remains unknown if cGAS-DNA phase separation is important for directly controlling 2′3′-cGAMP synthesis or for an alternative regulatory interaction that occurs in cells.
Here we discover that cGAS-DNA phase separation is critical to resist negative regulation and enable efficient DNA sensing. We systematically map the determinants of cGAS-DNA phase separation and define regulation of condensate formation as a process independent of 2′3′-cGAMP synthesis. cGAS phase separation is dispensable for 2′3′-cGAMP synthesis in vitro and does not directly control enzyme activation, but cGAS mutants defective in phase separation exhibit a drastically decreased ability to sense cytosolic DNA in the cellular cytosol. Strikingly, we show that phase separation is required to resist DNA degradation and protect immunostimulatory DNA from the cytosolic exonuclease TREX1. Using time-lapse imaging, we define a mechanism where TREX1 activity is potently suppressed in the cGAS-DNA phase-separated environment and DNA degradation is limited to the outer periphery of cGAS liquid droplets. We identify TREX1 mutations associated with the severe autoimmune disease Aicardi-Goutières syndrome that drive mislocalization of TREX1 into the repressive droplet interior and explain how specific loss of the ability to degrade phase-separated DNA can contribute to autoimmunity. Together our results establish cGAS-DNA phase separation as a molecular mechanism that balances the opposing forces of DNA-dependent immune activation and suppression in the mammalian cell cytosol.
Results
Human-specific substitutions control cGAS-DNA phase separation
Previous analysis demonstrated that cGAS recognition of cytosolic DNA results in formation of phase-separated cGAS-DNA liquid droplets (Du and Chen, 2018; Xie et al., 2019). To further define the molecular basis of cGAS-DNA phase separation, we reconstituted interactions in vitro and systematically analyzed the features of cGAS required for condensate formation. Confirming previous findings (Du and Chen, 2018; Xie et al., 2019), in the presence of 100-bp DNA, human cGAS (hcGAS) rapidly forms ~1–20 μm spherical droplets that co-label with both fluorescently-labeled cGAS and labeled DNA (Figure 1A, Video S1) and at certain conditions exhibit all hallmarks of liquid-liquid phase separation (Figures 1B and S1). The hcGAS protein is composed of a disordered N-terminus appended to a C-terminal nucleotidyltransferase (NTase) domain (Figure 1A). Both the hcGAS N-terminal and NTase domains independently interact with DNA (Figure S2A) (Tao et al., 2017) and both regions of hcGAS are necessary for cGAS-DNA phase separation (Du and Chen, 2018; Xie et al., 2019). We observed that full-length hcGAS-DNA phase separation is robust and occurs at a relative saturation concentration of ~50 nM under physiological conditions (Figures 1C, S1F,G). The hcGAS N-terminal and NTase fragments each retain the ability to phase separate in the presence of DNA, but condensate formation with these partial cGAS fragments requires 10–600-fold higher saturation concentrations (Figures 1A,C and S1F,G). Although the hcGAS NTase domain phase separates in the absence of the N-terminus, the condensates are not dynamic and lack the fluid recovery observed with full-length hcGAS or the N-terminal disordered domain alone (Figure 1B). These results support that efficient hcGAS-DNA phase separation requires cooperative protein–DNA interactions with both the N-terminal and C-terminal cGAS domains and reveal a critical role for the disordered N-terminus in ensuring droplet fluidity.
Figure 1. Molecular mechanism of cGAS-DNA phase separation.

(A) Top, schematic of the domain architecture of human cGAS (hcGAS). Bottom, fluorescence microscopy of DNA-induced phase separation of hcGAS proteins with bright field (BF) images. Recombinant hcGAS (10 μM), hcGASNterm (80 μM), and hcGASCterm (20 μM) were incubated with 100 bp dsDNA (10 μM) in buffer containing 250 mM (hcGAS), 150 mM (hcGASNterm), or 250 mM salt (hcGASCterm). Scale bar, 10 μm.
(B) FRAP analysis of cGAS-DNA phase-separated condensates. Time 0 indicates the time of photobleaching. Data represent the mean ± SEM of 7 droplets in 3 independent experiments.
(C) Analysis of relative saturation concentrations of cGAS proteins by turbidity assay. A series of concentrations of protein with 100 bp dsDNA (equal amounts) were mixed at 150 mM salt, and the absorbance of 340 nm was used as the readout of turbidity. The relative saturation concentrations are indicated with red arrows. All data are expressed as the mean ± SEM of more than 5 independent experiments. Statistical significance was calculated with a two-tailed T test, **p = <0.01, *p = <0.1.
(D) Schematic representing the phase behaviors and saturation concentrations of hcGAS and mouse cGAS (mcGAS) proteins determined by fluorescence intensity as in Figures S1F,G and S2C,D.
(E) Chimera experiments mapping the molecular determinant of enhanced human cGAS phase separation to two loops hcGAS N389–C405 and E422–S434. Left, schematic of cGAS constructs. Amino acid numbers are colored magenta for hcGAS and blue for mcGAS. Right, fluorescence microscopy images analyzing phase separation. cGAS chimeras (10 μM) were incubated with 100 bp dsDNA (10 μM) at 150 mM salt. Scale bar, 10 μm.
(F) Structure of the hcGAS–DNA complex (derived from combining PDB 6CT9 and additional DNA from PDB 6EDB) and schematic highlight of the mapped hcGAS loop sequences required for enhancement of cGAS–DNA phase separation.
(G) Cartoon model of the molecular basis of cGAS-DNA phase separation.
See also Figures S1, S2, and Video S1.
The cGAS gene is rapidly evolving in mammals and human-specific cGAS substitutions play a key role in controlling DNA ligand specificity (Zhou et al., 2018). To determine if species-specific substitutions also control cGAS-DNA phase separation, we directly compared the ability of hcGAS and mouse cGAS (mcGAS) to form condensates in the presence of DNA. mcGAS exhibits a lower ability to drive phase separation and requires an ~15-fold higher saturation concentration to form condensates (Figures 1D, S2B). Similar to hcGAS, the mcGAS N-terminal domain alone is sufficient to mediate dynamic condensate formation (Figures 1D, S2C,D). In contrast, the C-terminal NTase domain of mcGAS is unable to induce phase separation and fails to exhibit the robust condensate formation observed with hcGAS (Figures 1D, S2C,D). These results suggest that adaptations in the hcGAS NTase domain control enhancement of cGAS-DNA condensate formation.
We next applied a chimera approach to map the genetic determinants of enhanced hcGAS-DNA phase separation. Through iterative rounds of chimera design and phase separation analysis we identified substitutions within two loops N389–C405 and E422–S434 that together explain the enhanced ability of hcGAS to form condensates in the presence of DNA (Figure 1E). The first loop hcGAS N389–C405 contains the conserved H (X5) CC (X6) C Zn-Ribbon motif that forms part of the DNA A-site binding interface and mediates cGAS-cGAS dimerization (Figure 1F) (Civril et al., 2013; Kranzusch et al., 2013; Li et al., 2013). Analysis of the hcGAS–DNA structure demonstrates that a human-specific substitution E402 in this loop participates in the dimeric interface and likely stabilizes protein-protein interactions (Figure S2E,F) (Li et al., 2013; Zhou et al., 2018). Consistent with previous reports (Hooy et al., 2018; Xie et al., 2019), we observed that hcGAS preferentially forms a higher-order dimer-like species even in the absence of DNA that is not observed with mcGAS and other vertebrate homologs (Figure S2G). An E402A mutation reverts hcGAS to monomer-like migration and specifically reduces hcGAS-DNA condensate formation confirming a specific role for this substitution in enhanced phase separation (Figure S2H). The second loop identified in our chimera analysis is hcGAS E422–S434 located on the opposite protein face in a recently identified tertiary DNA binding surface designated the DNA C-site (Figures 1F, S2E) (Xie et al., 2019). An mcGAS chimera (Chi 3.3) containing both the hcGAS N389–C405 and E422–S434 loops acquires the ability for robust condensate formation demonstrating that these two loops control the major difference between human and mouse cGAS-DNA phase separation (Figures 1E). Together, these data reveal that human-specific substitutions regulate protein-protein interactions and protein-DNA interactions to enhance phase separation (Figure 1G).
cGAS phase separation does not directly control 2′3′-cGAMP synthesis in vitro but is required for immune activation in cells
cGAS-DNA phase separation has been implicated as a step required for robust enzyme activation and synthesis of the second messenger 2′3′-cGAMP (Du and Chen, 2018). Our results demonstrating that specific adaptations in hcGAS enhance phase separation are therefore surprising as hcGAS is known to catalyze a significantly lower rate of 2′3′-cGAMP synthesis compared to mcGAS (Zhou et al., 2018). We therefore next characterized a panel of diverse vertebrate cGAS homologs to systematically analyze the relationship between condensate formation and 2′3′-cGAMP synthesis. Each cGAS homolog, including distantly related zebrafish and xenopus cGAS proteins, forms condensates and synthesizes 2′3′-cGAMP in the presence of DNA demonstrating that condensate formation is a highly conserved feature of vertebrate cGAS function (Figures 2A, S3A,B). Likewise, each cGAS homolog retains the key unstructured N-terminus and domain organization features required for minimal hcGAS-DNA phase separation (Figures 1G, 2B and S2E). Although nearly all cGAS homologs are capable of forming spherical liquid droplets under certain conditions, analysis over a range of salt conditions reveals that species-specific differences result in considerable variation in the extent and strength of cGAS-DNA condensate formation (Figure 2A). We observed no direct relationship between condensate formation and overall level of 2′3′-cGAMP synthesis (Figures 2C,D, S3A,B).
Figure 2. cGAS phase separation does not directly control 2′3′-cGAMP synthesis in vitro but is required for immune activation in cells.

(A) Fluorescence microscopy images of DNA-induced phase separation of cGAS homologs. Phase separation was induced as in Figure 1A with recombinant cGAS homologs and 100 bp dsDNA in buffer with varying salt concentration. Scale bars, 10 μm.
(B) Schematic of cGAS domain architecture along with the predicted isoelectric point (pI) and amino acid composition of the N-terminal regions.
(C) Relative saturation concentrations of each cGAS homolog required for phase separation determined by turbidity assay.
(D) In vitro 2′3′-cGAMP synthesis activity of each cGAS homolog (see STAR Methods). Data were normalized to hcGAS as 100% and represent the mean ± SEM of 3 independent experiments.
(E) Fluorescence microscopy images of DNA-induced phase separation of hcGAS phase separation mutants. Phase separation was induced as in Figure 1A. Scale bars, 10 μm.
(F) Analysis of hcGAS and phase separation mutant enzyme kinetics. cGAS protein enzyme activity was measured as function of varying ATP concentration, and 2′3′-cGAMP synthesis was quantified and fit according to the Michaelis-Menten kinetics. Data represent the mean ± SEM in 3 independent experiments.
(G) Schematic of analysis of cGAS activity in cells (see STAR Methods).
(H,I) ELISA analysis of 2′3′-cGAMP production and quantification of immunoblot analysis of IRF3 phosphorylation in MCF10A cGAS−/− cells reconstituted with hcGAS or hcGAS phase separation mutant alleles and transfected with plasmid DNA. Data are the mean ± SD of 3 experiments.
See also Figure S3.
To confirm that cGAS-DNA phase separation and enzymatic activity are functionally distinct properties, we measured the ability of hcGAS chimeras to catalyze 2′3′-cGAMP synthesis. hcGAS chimera 3.2 retains the ability to catalyze near wild-type levels of 2′3′-cGAMP synthesis in spite of exhibiting a dramatic reduction in condensate formation (Figure 2E,F). We further engineered hcGAS Chi 3.2 to include a double mutation in the hcGAS DNA A site K187N/L195R that relaxes DNA-length specificity and increases the rate of 2′3′-cGAMP synthesis (Zhou et al., 2018) and observed that this construct exhibits a >3-fold increase in the rate of 2′3′-cGAMP synthesis but no change in overall condensate formation (Figures 2E,F and S3C). Notably, in spite of loss of efficient cGAS-DNA condensate formation, hcGAS Chi 3.2 K187N/L195R exhibits an enhanced catalytic efficiency that exceeds wildtype hcGAS demonstrating that the features of cGAS-DNA phase separation and 2′3′-cGAMP synthesis are distinct and can be genetically uncoupled.
We next took advantage of the hcGAS Chi 3.2 mutant specifically defective in condensate formation to assess the importance of phase separation in cytosolic DNA sensing in cells. We reconstituted MCF10A cGAS−/− cells with wildtype or mutant hcGAS alleles and stimulated immune-sensing with plasmid DNA transfection (Figures 2G and S3D). Compared to cells expressing the wildtype hcGAS allele, cells expressing the hcGAS NTase domain alone or hcGAS Chi 3.2 are defective in 2′3′-cGAMP production and IRF3 phosphorylation (Figure 2H,I). This defect can be partially relieved with the enhancing K187N/L195R mutations, demonstrating a balance between the requirement of phase separation in cells and intrinsic rate of 2′3′-cGAMP synthesis. Together, these results establish a mechanism where the conserved process of cGAS-DNA phase separation is not specifically required for enhanced 2′3′-cGAMP synthesis in vitro but is essential to sense cytosolic DNA in the cellular cytosolic environment.
cGAS phase separation is critical for resistance to TREX1 exonuclease activity
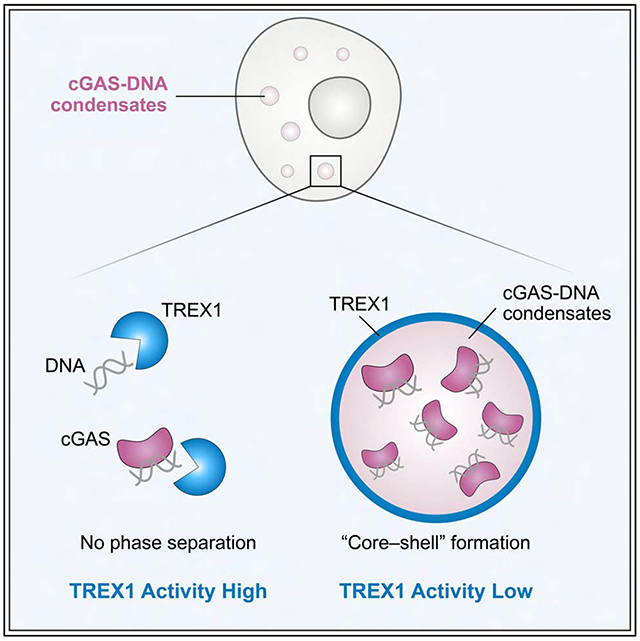
Our results demonstrate that cGAS-DNA condensate formation does not directly control 2′3′-cGAMP synthesis and that conservation of cGAS-DNA phase separation must therefore be critical for an alternative process regulating immune activation. In the cell cytosol, a major negative regulator of DNA sensing is constitutive activity of the DNA exonuclease TREX1 (Gao et al., 2015; Gray et al., 2015). TREX1 localizes to the endoplasmic reticulum in cells via a C-terminal membrane-association sequence, but TREX1 has also been detected freely in the cell cytosol (Chowdhury et al., 2006; Stetson et al., 2008). We therefore hypothesized that phase separation may protect the cGAS–DNA complex and enable sensing in the presence of active cytosolic DNA degradation. To investigate if a physical interaction exists between cGAS-DNA complexes and TREX1 in cells we transfected cells with Cy3-labeled plasmid DNA and tracked the localization of cGAS and TREX1. Following DNA transfection, endogenous cGAS forms puncta that co-localize with Cy3-labeled DNA and are consistent with observations made previously in a variety of cell types (Figure 3A) (Liu et al., 2018; Volkman et al., 2019). In agreement with our hypothesis, GFP-TREX1 also co-localizes with cGAS-DNA puncta in the cytosol suggesting active interaction between the cGAS-DNA complex and negative regulators of DNA immune sensing (Figure 3A).
Figure 3. cGAS phase separation resists DNA degradation by exonuclease TREX1.

(A) Imaging of co-localization of cGAS/DNA/TREX1 in the cell cytosol (see STAR Methods). cGAS staining indicates that cGAS predominantly localizes in either the cytosol (#1) or nucleus (#2). Scale bar, 5 μm.
(B) Left, phase diagram of purified hcGAS under various protein and DNA concentrations at 150 mM salt, where grey dots indicate no phase separation, and magenta dots indicate observable condensate formation (see also C). Right, quantification of phase separation with 1 μM DNA and increasing concentration of hcGAS. Data represent the percent area of the image occupied by condensates relative to the maximal phase separation observed with 10 μM hcGAS and are plotted as the mean ± SEM of 17 images from 3 independent experiments.
(C) cGAS phase separation protects DNA from TREX1 degradation. Left, in vitro analysis of DNA degradation by full-length TREX1 (TREX1FL) in the presence of varying degrees of cGAS-DNA phase separation. Phase separation was induced with increasing cGAS concentration as shown in (B) in the presence of 1 μM 100-bp dsDNA, and DNA degradation was measured by incubating with purified TREX1FL and resolving the remaining DNA on an agarose gel. Right, quantification of remaining DNA. Data represent the mean ± SEM of 5 independent experiments.
(D) Analysis of the TREX1 resistance by individual cGAS domains. Phase separation was induced with 1 μM DNA and titrations of N-term and C-term cGAS at indicated concentrations. DNA degradation was then initiated by adding TREX1 (M1–K242) at 0.1 μM. Remaining DNA was quantified as in (C). Data are represented as the mean ± SEM of 3 independent experiments.
(E) Analysis of TREX1 resistance by cGAS homologs. Phase separation was induced with 1 μM DNA and a titration of cGAS homolog protein as indicated. DNA degradation was measured as in (C). Data represent the mean ± SEM of 3 independent experiments.
(F) Inhibition of cGAS-DNA phase separation reduces TREX1 resistance. DNA degradation by TREX1 was measured in the presence of hcGAS or hcGAS mutants. Experiments were performed and data were quantified as in (C). Data are represented as the mean ± SEM of 4 independent experiments.
(G) Analysis of species-specific TREX1 resistance by cGAS-DNA phase separation. Human and mouse TREX1 (hTREX1 and mTREX1) DNase activity was assessed in the absence or presence of cGAS phase separation as indicated. Remaining DNA was resolved on an agarose gel and quantified. Data represent the mean ± SEM of 4 independent experiments.
See also Figure S4.
We next measured the in vitro stability of hcGAS-DNA condensates in the presence of TREX1 (Figure S4A,B). Under conditions of no or low phase separation, 100 bp DNA is sensitive to TREX1 exonuclease activity and is rapidly degraded (points P1 and P2 in Figure 3B,C). Remarkably, under conditions of robust hcGAS-DNA phase separation we observed clear DNA stabilization with TREX1 no longer capable of efficiently degrading 100 bp DNA (points P3 and P4 in Figure 3B,C). We observed that hcGAS-DNA phase separation is reduced when the ratio of cGAS to DNA exceeds ~100× (point P5 in Figures 3B and S4C,D), consistent with a reentrant phase transition as previously observed with RNA-binding proteins and RNA driven phase separation (Alshareedah et al., 2020; Banerjee et al., 2017; Milin and Deniz, 2018). Reduced cGAS phase separation by reentrant phase transition corresponds with a loss in the ability to protect DNA from TREX1 degradation (point P5 in Figure 3C), further demonstrating a strict correlation between the degree of cGAS phase separation and the ability to protect DNA from degradation (Figures 3B,C and S4C,D).
Strong resistance to TREX1 requires both the hcGAS N-terminal disordered domain and the C-terminal NTase domain (Figure 3D). hcGAS-DNA phase separation protects DNA from both full-length TREX1 and a minimal TREX1 M1–K242 exonuclease domain fragment, indicating that the TREX1 C-terminus does not strongly influence interactions with cGAS-DNA condensates (Figures 3C,D and S4B). In agreement with the enhanced ability of hcGAS to form condensates, TREX1 resistance occurs at a lower hcGAS enzyme concentration than cGAS homologs that exhibit weaker condensate formation (Figure 3E). Mouse cGAS is known to exhibit a high affinity for DNA and forms a stable “ladder-like” complex (Andreeva et al., 2017). However, consistent with weak mcGAS-DNA condensate formation (Figures 1D and 2A), mcGAS has a comparatively weak ability to protect from TREX1 DNase activity (Figure 3E). Condensate formation is therefore required to resist TREX1 DNase activity and stable cGAS-DNA binding is insufficient. Additionally, mutations to hcGAS that weaken overall phase separation reduce the ability of hcGAS to protect DNA from TREX1-mediated degradation (Figure 3F). We confirmed that human and mouse TREX1 exhibit similar rates of DNase activity in vitro and that resistance is not related to species-specific differences in TREX1 activity (Figures 3G, S4B). To understand the specificity of TREX1 resistance, we tested a panel of six model nuclease enzymes and observed a general overall ability of hcGAS-DNA phase separation to resist nuclease activity (Figure S4E,F). However, hcGAS-DNA phase separated condensates remain exquisitely sensitive to T7 exonuclease degradation demonstrating that TREX1 resistance by cGAS-DNA phase separation is not a universal feature of all nuclease enzymes. Together, these results reveal that a specific property of cGAS-DNA phase separation is resistance to TREX1-mediated DNA degradation.
cGAS-DNA phase separation creates a selective environment that restricts TREX1 diffusion
To determine the mechanism of TREX1 resistance, we used microscopy and fluorescently labeled components to track the fate of cGAS-DNA liquid droplets over time. As hcGAS forms liquid-like droplets in cells (Du and Chen, 2018), we focused our microscopy experiments on conditions at ~250 mM total salt where we observe robust cGAS-DNA liquid droplet formation (Figures 2A and Figure S5A). Under these conditions, TREX1 rapidly degrades all detectable naked DNA within minutes while cGAS-DNA droplets strongly resist TREX1 DNase activity and protect ~25% of DNA even after 2 h of incubation (Figures 4A and S5B). In agreement, >25% of the total phase-separated DNA signal remains intact throughout 2 h of imaging confirming cGAS-DNA liquid-droplet formation protects DNA from TREX1 degradation (Figures 4B and S5C,D; Video S2). Time-lapse imaging reveals TREX1 readily interacts with phase-separated DNA and co-localizes to all cGAS-DNA droplets (Figure 4B; Video S2). Remarkably, microscopy images from early time-points reveal that TREX1 localizes exclusively to the outer droplet periphery and forms a visible shell around the inner phase separated compartment (Figure 4B). Consistent with degradation occurring exclusively from the exterior, active TREX1 catalyzes a slow decrease in cGAS-DNA droplet size that proceeds steadily throughout imaging with no apparent loss in signal occurring from within the droplet center. To confirm our in vitro observations, we followed the interaction between TREX1 and immunostimulatory DNA in cells using time-lapse imaging and observed that puncta containing immunostimulatory DNA are degraded with remarkably slow kinetics (Figure 4C; Video S3). Although technical limitations preclude simultaneous time-lapse imaging of cGAS, TREX1 and labeled DNA in cells, these results are consistent with a strong ability of hcGAS-DNA phase separated condensates to resist DNA degradation.
Figure 4. cGAS phase separation restricts TREX1 DNA degradation to an outer shell at the droplet periphery.

(A) Comparison of TREX1 DNA degradation activity with (black) and without (magenta) cGAS-DNA phase separation at 250 mM salt. Time 0 indicates addition of 5 mM Mg2+ to activate TREX1. Data represent the mean ± SEM of 3 independent experiments.
(B) Time-lapse imaging of TREX1 degradation of cGAS-DNA droplets in vitro. Droplet formation was induced by hcGAS (10 μM), 100-bp DNA (10 μM) and TREX1 (1 μM) as indicated. Concentrations of cGAS and TREX1 are based on the cellular cGAS concentration being 10-fold higher than TREX1 (Hein et al., 2015). Time 0 indicates addition of 5 mM MgCl2 to activate TREX1. Scale bar, 10 μm.
(C) Time-lapse imaging of TREX1 degradation of immunostimulatory DNA (ISD) puncta in cells (see STAR Methods). Time 0 represents the start of imaging 2 h after initial Cy5-ISD transfection. Scale bar, 5 μm.
(D) Fluorescence microscopy images showing a shell-like formation of TREX1 around cGAS-DNA droplets at the early stage of degradation. cGAS-DNA-TREX1 droplet formation was induced as in (B) without adding MgCl2. The droplet labeled with a dotted line was selected for further line-scanning analysis in (F). Scale bar, 10 μm.
(E,F) Three-dimensional reconstruction and line-scanning analysis of cGAS-DNA-TREX1 droplets in (D). Corresponding fluorescence intensity along the dotted line is shown to the right.
(G) Fluorescence microscopy images showing cGAS-DNA droplets inhibit TREX1 access but not cGAS incorporation. cGAS-DNA droplet formation was induced as illustrated above. Fluorescently labeled components as indicated were subsequently added and incubated for additional time as indicated. Scale bar, 10 μm.
(H) cGAS-DNA droplet formation selectively resists TREX1 incorporation. hcGAS-DNA droplet formation was induced using non-labeled hcGAS and Cy3-labeled DNA, followed by simultaneously adding 1 μM AF647-labeled TREX1 and 1 μM AF488-labeled hcGAS. Images were collected after 1 h and fluorescence intensity along the dotted line is shown to the right. All imaging data are representative of at least 3 independent experiments.
We next withheld Mg2+ from the in vitro imaging system to prevent TREX1 DNA degradation and allow imaging of the earliest events of TREX1 interaction with cGAS-DNA droplets (Figure S5C). In the absence of catalytic activity, TREX1 forms a clear outer layer around cGAS-DNA phase-separated droplets that persists >2 h without entry into the center (Figures 4D, S5E). 3D reconstruction and line-scanning analysis confirms absence of TREX1 signal in the droplet interior and formation of a TREX1 shell that encases phase-separated DNA (Figure 4E,F). To determine the specificity of TREX1 exclusion from cGAS-DNA phase-separated droplets we tested AF488- and AF647-labeled TREX1 and confirmed that exclusion is not due to specific fluorescent labeling (Figure 4G). Additionally, we observed the same exclusion phenotype with the human paralog TREX2 (Figure S5F). We next preformed droplets with Cy3-DNA and unlabeled hcGAS, and subsequently introduced labeled hcGAS, TREX1, or ATP for imaging. AF488-labeled hcGAS rapidly incorporates throughout the entire preformed cGAS-DNA phase-separated droplet while TREX1 localization remains restricted to the droplet exterior (Figure 4G). Similar to full-length hcGAS, both the N- and C-terminal fragments of hcGAS and AF647-labeled ATP efficiently permeate to the center of cGAS-DNA droplets (Figure 4G). Finally, we simultaneously introduced labeled AF488-hcGAS and AF647-TREX1 to preformed cGAS-DNA droplets. cGAS successfully incorporates into droplets while TREX1 remains restricted to the droplet exterior demonstrating that the mechanism restricting incorporation is selective to TREX1 (Figures 4H and S5G).
Restriction of TREX1 to the cGAS-DNA liquid droplet exterior suggests that a physical mechanism limits the ability of TREX1 to permeate into the droplet center. To understand this process, we extended time-lapse imaging and observed that TREX1 incorporation into the droplet interior only occurs following an extensive increase in time (>4 h) or TREX1 protein concentration (>6 μM) (Figures 5A,B and S5H,I). We therefore hypothesized that TREX1 diffusion itself is limited inside hcGAS-DNA droplets. To test this hypothesis, we incubated cGAS-DNA droplets with TREX1 for 4 h to allow TREX1 to complete incorporation throughout the droplet and then used FRAP analysis to measure diffusion and recovery of each individual droplet component. Strikingly, fluorescent cGAS and DNA signals each rapidly recover while the TREX1 signal remains absent (Figures 5C and S5J). These results demonstrate that the phase-separated environment of cGAS-DNA droplets selectively restricts entry and diffusion of TREX1 while remaining permissive to additional cGAS incorporation.
Figure 5. Phase separation is a direct suppressor of TREX1 exonuclease activity.

(A) Time-lapse imaging of slower partitioning of TREX1 into cGAS-DNA droplets compared to cGAS. cGAS-DNA droplets were preformed as indicated, and the incorporation rates of TREX1 and cGAS were tracked using fluorescently labeled components. Scale bar, 10 μm.
(B) Line-scanning analysis of TREX1 incorporation demonstrates that TREX1 migration to the center of cGAS-DNA droplets requires extended incubation times (top) or elevated TREX1 protein levels (bottom).
(C) Analysis of the fluidity of TREX1, cGAS and DNA in cGAS-DNA droplets. cGAS-DNA droplets were incubated for 4 h to allow complete incorporation of TREX1 and then analyzed by FRAP to measure component fluidity. Time 0 indicates the time of photo bleaching. Plots are generated from 12 droplets and data represent the mean ± SEM of 3 independent experiments.
(D) Schematic of engineered cGAS-TREX1 fusion constructs developed to permit rapid incorporation of TREX1 into cGAS-DNA droplets (Left). Right, fluorescence microscopy images of construct incorporation into preformed cGAS-DNA droplets as in Figure 4G. Scale bar, 10 μm.
(E) Entry into cGAS-DNA droplets restricts TREX1 nuclease activity. Following induction of cGAS-DNA phase separation, DNA degradation was initiated with addition of TREX1 or cGASNterm–TREX1 and remaining DNA was quantified by agarose gel analysis. Data represent the mean ± SEM of 3 independent experiments.
(F) Fluorescence microscopy images of TREX1 interactions with cGAS-DNA droplets formed with linear dsDNA, closed circular dsDNA, or ssDNA. Scale bar, 10 μm.
(G) Fluorescence microscopy images showing dsDNA-induced phase separation of TREX1. TREX1-dsDNA droplet formation was induced by TREX1 (20 μM) and 100-bp dsDNA (10 μM) at various salt concentrations as indicated. Scale bar, 10 μm.
(H) Schematic of hypothetical TREX1-dsDNA interactions that drive liquid-liquid phase separation.
(I) Left, phase diagram of purified TREX1 under various protein and dsDNA concentrations at 150 mM salt with no MgCl2. Dots indicate conditions where a single phase (grey) or two phases (orange) are present. Right, quantification of TREX1-dsDNA phase separation with 1 μM dsDNA and increasing concentration of TREX1. Data correspond to the percent area of the image occupied by droplets and were quantified relative to maximal phase separation observed with 100 μM TREX1. Data represent the mean ± SEM of 7 images in 3 independent experiments.
(J) TREX1-dsDNA phase separation inhibits TREX1 exonuclease activity. TREX1-dsDNA phase separation was induced with 100-bp dsDNA and increasing TREX1 concentration as shown in (I) in the presence of 5 mM MgCl2. Reactions were inactivated and DNA was quantified by agarose gel analysis. Data represent the mean ± SEM of 3 independent experiments.
See also Figures S5, S6, and Video S4.
Phase separation is a direct suppressor of TREX1 exonuclease activity
To understand if access to DNA or suppression of TREX1 activity itself explains resistance to DNA degradation we next tested the impact of forcing TREX1 localization within the cGAS-DNA droplet interior. We created engineered cGAS–TREX fusion constructs and observed that fusion of the unstructured cGAS N-terminal domain to TREX1 (cGASNterm–TREX1) permits rapid droplet entry (Figure 5D). In spite of disruption of the selective barrier restricting TREX1 entry, cGAS-DNA droplets maintain resistance to TREX1-mediated degradation (Figure 5E). Although both wildtype TREX1 and cGASNterm–TREX1 efficiently degrade unprotected DNA in solution (Figure S6A), the ability of the cGASNterm–TREX1 construct to degrade phase-separated DNA in complex with cGAS is reduced compared to the wildtype TREX1 (Figures 5E and S6B). We confirmed these findings using time-lapse imaging demonstrating that once fully incorporated into the phase separated environment TREX1 remains unable to degrade DNA (Figure S6C). These results indicate that TREX1 is not capable of degrading DNA while within the interior of the cGAS-DNA phase-separated environment.
The inability of TREX1 to degrade DNA once inside cGAS-DNA droplets suggests that phase separation suppresses TREX1 enzymatic activity. The TREX1 exonuclease domain functions as an obligate dimer and interacts with DNA through recognition of the 3′ ends of double-stranded or single-stranded DNA substrates (de Silva et al., 2007; Grieves et al., 2015). We formed cGAS-DNA droplets using linear dsDNA, single-stranded DNA, or closed circular dsDNA with no exposed 3′ ends (Figures 5F and S6D) and observed that TREX1 can only interact with cGAS-dsDNA droplets formed with linear dsDNA (Figure 5F). Although TREX1 can degrade both ssDNA and dsDNA in solution, the requirement of multiple exposed 3′ ends for interaction with phase-separated cGAS-DNA suggests that TREX1 recruitment involves multivalent interactions and an independent phase separation-like property. In agreement, we observed that TREX1 alone can undergo a phase separation transition in vitro in the presence of dsDNA to form dynamic liquid-like droplets (Figure 5G,H; Video S4). However, formation of TREX1-DNA phase separation requires atypically low salt conditions or elevated protein concentrations that exceed the physiological ~5 nM concentration of TREX1 in cells (Figures 5G-I and S6E-G) (Hein et al., 2015). Similar to a model previously proposed with hnRNPA1a incorporation into FUS liquid droplets (Wang et al., 2018), our results suggest a mechanism where TREX1 is a factor that can participate in cGAS-DNA phase-separated interactions but is unable to independently initiate droplet formation under normal cellular conditions. However, independent formation of TREX1-DNA liquid droplets allowed us to next directly test the impact of phase separation on TREX1 activity in the absence of cGAS. The onset of TREX1 phase separation at ~10–25 μM TREX1 protein results in specific loss of TREX1 dsDNA DNase activity with >50% dsDNA remaining at 50 μM TREX1 (Figure 5I,J). In contrast, TREX1 ssDNA DNase activity remains robust even at 50 μM TREX1 protein where no obvious TREX1 condensate formation occurs (Figure S6H,I). At the highest tested protein concentration (100 μM) TREX1 is able to form liquid droplets in the presence of ssDNA. Under this condition we began to observe inhibition of ssDNA DNase activity, suggesting that phase separation can also regulate TREX1 ssDNA DNase activity but only at extreme protein concentrations far removed from the ability of phase separation to regulate TREX1 dsDNA DNase activity (Figure S6H,I). Together, these results reveal a strict relationship where phase separation directly inhibits TREX1 enzymatic function and establish a model where cGAS-DNA phase separation creates a selective environment that suppresses TREX1 to shield immunostimulatory DNA from nuclease degradation.
A human TREX1 disease-causing mutation alters interactions with cGAS-DNA droplets
Mutations in TREX1 that impair control of cGAS activation are one of the most frequent causes of Aicardi-Goutières syndrome and systemic lupus erythematosus (Rice et al., 2015; Yan, 2017; Gao et al., 2015; Gray et al., 2015). Direct loss of DNA exonuclease activity explains most TREX1 disease-causing mutations, but the molecular basis of disease for TREX1 mutations that do not disable catalytic function has remained enigmatic (Orebaugh et al., 2013). We therefore tested TREX1 disease-associated mutations outside the nuclease active site to determine if they may alter interactions with cGAS-DNA droplets. Strikingly, imaging analysis demonstrates that the disease variant TREX1 E198K exhibits a marked change in phase separation behavior (Figure 6; Video S5). TREX1 E198K requires a lower protein concentration to begin interaction with the cGAS-DNA droplet periphery and TREX1 E198K readily permeates into the internal suppressive environment at the droplet center (Figure 6A). Additionally, the E198K mutation enables TREX1 incorporation into cGAS-DNA droplets formed with ssDNA (Figures 6B and S7A), indicating removal of the requirement for multivalent 3′ end interactions and alteration to the physical rules that normally control TREX1 interactions with phase-separated cGAS-DNA.
Figure 6. The disease mutation TREX1 E198K alters interactions with cGAS-DNA droplets.

(A) Fluorescence microscopy images of incorporation of TREX1 and TREX1 E198K into cGAS-DNA droplets. cGAS-dsDNA droplet formation was induced as in Figure 4G, and TREX1-incorporation was analyzed with increasing concentration of AF488-labeled TREX1 (top) and TREX1 E198K (bottom). Scale bar, 10 μm.
(B) Time-lapse images of incorporation of TREX1 into cGAS-ssDNA droplets. cGAS-ssDNA droplets were formed as in Figure 5F, and AF488-labeled TREX1 (top) or TREX1 E198K (bottom) was added to the preformed ssDNA droplets. Scale bar, 10 μm.
(C) 1.8 Å crystal structure of the mouse TREX1E198K dimer with zoom-in cutaways of the locations of residues E198 and K66 in wildtype mTREX1 (top, PDB: 3MXJ) and mTREX1 E198K (bottom). The 2FO–FC electron density map is contoured at 1.0 σ for TREX1 and 0.7 σ for TREX1E198K
(D) The crystal structure of TREX1E198K reveals extensive remodeling of surface electrostatic potential. Surface electrostatic potentials of TREX1 and TREX1 E198K (blue positive, red negative) with E198, K66, and E198K positions highlighted with dashed outlines.
(E) Left, fluorescence microscopy images of TREX1-dsDNA (top) and TREX1E198K-dsDNA (bottom) phase separation with various salt concentrations. Scale bar, 10 μm. Right, quantification of phase separation by the percent area of the image occupied by droplets. Data are represented as the mean ± SEM of 9 images in 3 independent experiments.
(F) FRAP analysis of TREX1-dsDNA (black) and TREX1E198K-dsDNA (orange) droplets formed by mixing 100-bp dsDNA and unlabeled TREX1 or TREX1E198K protein for 3 h. Time 0 indicates the time of photobleaching pulse. Plots are generated from 6 droplets and data represent the mean ± SEM of 3 independent experiments.
(G,H) In vitro analysis and quantification of exonuclease activity of TREX1 mutation E198K in the absence or presence of cGAS phase separation. cGAS-DNA phase separation was induced as in Figure 5E, and DNA degradation initiated by adding TREX1 and TREX1 E198K. Remaining DNA was resolved on an agarose gel and quantified. Data represent the mean ± SEM of 3 independent experiments.
To understand the molecular basis of how disease-associated mutations can alter TREX1 phase separation interactions we determined a 1.8 Å crystal structure of mouse TREX1 E198K (Figure 6C and Table 1). In previous TREX1 structures, E198 forms a salt-bridge with K66 that spans the homodimer interface (de Silva et al., 2007; Huang et al., 2018). The TREX1 E198K structure reveals remodeling of TREX1 surface electrostatics due to loss of all hydrogen-bond contacts at this portion of the interface (Figure 6C,D). The TREX1 E198K mutation only remodels surface residues at the dimer interface and we confirmed that the E198K mutation has no impact on TREX1 dimerization in solution (Figure S7B). In agreement with a direct change to TREX1 surface electrostatics impacting phase separation behavior, TREX1 E198K readily initiates formation of phase-separated DNA droplets with increased fluidity at normal physiological cellular conditions (Figures 6E,F and S7C,D; Video S5). Comparing the activity of TREX1 E198K on normal DNA or phase-separated cGAS-DNA condensates reveals that the disease-causing mutation impairs TREX1 activity only in the context of DNA phase separation (Figures 6G and S7E,F). TREX1 E198K is dramatically impaired in its ability to degrade phase-separated cGAS-DNA with the majority of DNA signal remaining after the wildtype enzyme has completed elimination of >95% of the cGAS-protected phase-separated DNA (Figure 6H). Together, these results confirm the importance of cGAS-DNA phase separation controlling TREX1 activity and reveal altered TREX1-cGAS phase separation interactions as an additional mechanism to explain autoimmune disease.
Table 1.
Crystallographic Statistics
| TREX1E198K | |
|---|---|
| Data Collection | |
| Resolution (Å)a | 46.08–1.80 (1.84–1.80) |
| Wavelength (Å) | 0.9792 |
| Space group | P 21 21 21 |
| Unit cell: a, b, c (Å) | 65.58, 85.34, 99.47 |
| Unit cell: α, β, γ (°) | 90.0, 90.0, 90.0 |
| Molecules per ASU | 1 |
| No. reflections: total | 710239 |
| No. reflections: unique | 52490 (3071) |
| Completeness (%)a | 99.9 (99.4) |
| Multiplicitya | 13.5 (13.1) |
| I/σIa | 8.3 (1.3) |
| CC(1/2)1 (%)a | 99.6 (69.4) |
| Rpim2 (%)a | 4.4 (49.3) |
| Refinement | |
| Resolution (Å) | 46.08–1.80 |
| No. free reflections | 1998 |
| R-factor / R-free | 17.77 / 20.35 |
| Bond distance (RMS Å) | 0.010 |
| Bond angles (RMS °) | 1.07 |
| Structure/Stereochemistry | |
| No. atoms: protein | 3352 |
| No. atoms: water | 483 |
| Average B-factor: protein | 33.89 |
| Average B-factor: water | 44.38 |
| Ramachandran plot: favored | 99.29% |
| Ramachandran plot: allowed | 0.71% |
| Ramachandran plot: outliers | 0% |
| Rotamer outliers: | 0% |
| MolProbity3 score | 1.31 |
| Protein Data Bank ID | 6W10 |
Highest resolution shell values in parenthesis
cGAS phase separation resists suppression by multiple negative regulators of cytosolic immune-sensing
To determine if TREX1 nuclease activity is alone sufficient to explain the cellular role of cGAS-DNA phase separation, we deleted TREX1 in cells and tested the impact of cGAS phase separation-altering mutations. In the absence of TREX1, cells expressing cGAS and cGAS mutants exhibit a generally elevated level of 2′3′-cGAMP synthesis and IRF3 phosphorylation (Figures 2H,I, 7A and S3D, S7G). However, hcGAS Chi 3.2 mutants defective in phase separation remain weaker at activating downstream signaling compared to wildtype hcGAS suggesting that phase separation may be required to resist other forms of negative regulation (Figure 7A). In addition to TREX1, many factors have been reported as negative regulators of cGAS activation (Boyer et al., 2020; Cao et al., 2020; Guey et al., 2020; Kujirai et al., 2020; Ma et al., 2020; Michalski et al., 2020; Pathare et al., 2020; Shannon et al., 2018; Volkman et al., 2019; Zhao et al., 2020; Zierhut et al., 2019) including barrier-to-autointegration factor 1 (BAF) which inhibits cGAS to prevent inappropriate innate immune signaling. In agreement with previously reported results (Guey et al., 2020), purified human BAF inhibits hcGAS 2′3′-cGAMP synthesis in vitro in a dose-dependent manner (Figures 7B,C and S7H,I). We imaged interaction between BAF and phase separated cGAS-DNA in vitro and observed that cGAS-DNA droplets restrict localization of BAF to the outer periphery where it forms a shell-like appearance identical to TREX1 (Figure 7D,E). Line-scanning analysis confirms absence of BAF signal in the droplet interior demonstrating the ability of cGAS-DNA phase separation to selectively filter negative regulators beyond TREX1 (Figures 7E and S7J). Together, our data suggest a model where cGAS-DNA phase separation functions as a general mechanism to resist suppression by negative regulators of innate immune activation (Figure 7F).
Figure 7. cGAS phase separation resists immune suppression by multiple negative regulators.

(A) ELISA analysis of 2′3′-cGAMP production in MCF10A cGAS−/− TREX1−/− cells reconstituted with hcGAS or hcGAS mutant alleles and transfected with plasmid DNA. Data are the mean ± SD of 3 experiments.
(B,C) In vitro analysis of BAF-dependent inhibition of cGAS 2′3′-cGAMP synthesis. Purified hcGAS enzyme was stimulated with 100-bp DNA in reactions supplemented with an increasing concentration of BAF and 2′3′-cGAMP production was analyzed and quantified as in Figure S1B. Data represent the mean ± SEM of 4 independent experiments.
(D) Fluorescence microscopy images (left) and line-scanning analysis (right) showing that BAF is excluded to an outer shell at the cGAS-DNA droplet periphery. cGAS-DNA droplet formation was induced as in Figure 4D. Scale bar, 10 μm. Fluorescence intensity along the dotted lines was analyzed (right). Plots are generated from 3 droplets and data represent the mean ± SD.
(E) Fluorescence microscopy images of dosage-dependent incorporation of BAF and ATP into cGAS-DNA droplets. cGAS-dsDNA droplet formation was induced as in Figure 5A, and BAF/ATP incorporation was analyzed with increasing concentration of AF647-labeled BAF (top) or AF647-labeled ATP (bottom). Scale bar, 10 μm.
(F) Model of the role of cGAS-DNA phase separation in resisting negative regulation. DNA-induced cGAS phase separation creates a selective environment that suppresses entry of negative regulators and allows sensing of immunostimulatory DNA.
See also Figure S7.
Discussion
Our results reveal a critical role for cGAS-DNA phase separation in balancing the opposing forces of DNA recognition and DNA degradation in innate immune sensing. We demonstrate that condensate formation protects DNA from nuclease degradation and directly inhibits the enzymatic activity of the major cytosolic DNA exonuclease TREX1. cGAS mutations that weaken condensate formation in vitro severely attenuate immune activation in cells, further confirming the importance of phase separation and nuclease resistance in cytosolic DNA sensing (Du and Chen, 2018; Xie et al., 2019). Mechanistically, we demonstrate that cGAS-DNA phase separation functions as a selective filter. cGAS and reaction components are permitted to rapidly enter and diffuse throughout the liquid environment, while TREX1 fluidity and exonuclease activity is potently suppressed in the droplet interior (Figures 4 and 5). This physical process restricts TREX1 DNA degradation activity to an outer shell at the cGAS-DNA droplet periphery and allows immunostimulatory DNA to resist rapid degradation.
cGAS-DNA phase separation is regulated independently of cGAS enzymatic activity and is not directly required to control 2′3′-cGAMP synthesis. Using comparison of divergent cGAS homologs, we find that phase separation is highly conserved throughout vertebrate cGAS evolution and is specifically enhanced in hcGAS (Figures 2, S1-3). Although the ability of phase separated droplets to recruit cGAS and concentrate reaction components likely contributes to 2′3′-cGAMP synthesis in cells (Figure 4) (Du and Chen, 2018), our findings support that a major function of cGAS-DNA phase separation is to protect the activated complex from negative regulation and create a compartment where immune sensing can occur. The relative balance between TREX1 resistance and cGAS enzymatic rate may explain why some species like mouse cGAS that weakly form phase-separated condensates instead compensate with a significantly higher rate of 2′3′-cGAMP synthesis (Figures 1 and 2) (Zhou et al., 2018). Recently, structural and cell biology insights have demonstrated that formation of stable higher-order assemblies is a key step common to many immune sensing pathways including activation of toll-like receptors, RIG-I-like receptors and inflammasome signaling (Ablasser and Hur, 2020; Kagan et al., 2014). Similar to the ability of cGAS-DNA condensates to resist TREX1 nuclease activity, organization of other supramolecular assemblies in innate immunity may also play an important role in stabilizing immunostimulatory ligands from degradation and counteracting negative regulation.
Building on the initial discovery of cGAS-DNA phase separation (Du and Chen, 2018), our findings further establish that cGAS-DNA condensates are the key active state responsible for cytosolic DNA sensing. The TREX1 mutation E198K associated with disease only exhibits a biochemical phenotype with cGAS-DNA phase-separated condensates, demonstrating the importance of specifically assessing regulation of cGAS in the context of phase separation (Figure 6). In addition to TREX1, cGAS-DNA phase separation also restricts BAF to the outer shell of droplets revealing that this process is a general mechanism balancing the effects of negative regulators of cGAS activation in the intracellular environment (Figure 7). Additional protein co-factors PQBP1 and G3BP1 have been identified in cellular experiments as critical for regulating cGAS activity (Liu et al., 2019; Shannon et al., 2018; Yoh et al., 2015) but these proteins are dispensable for cGAS-DNA complex formation and 2′3′-cGAMP synthesis in vitro. PQBP1 and G3BP1 each contain low-complexity disordered sequences and may therefore be involved in cellular regulation of cGAS-DNA condensate formation. Likewise, specific targeting of cGAS-DNA condensates may be an important consideration to explain the cellular potency of promising small molecule cGAS inhibitors (Hall et al., 2017; Vincent et al., 2017). The discovery of cGAS-DNA phase separation as a selective filter controlling recruitment and activity of TREX1 provides an important framework to explain regulation of cytosolic DNA sensing and reveals a mechanism that enables the cellular balance between tolerance and innate immune activation.
Limitations
An important open question is to understand how interactions between cGAS-DNA condensates and negative regulators occur in real-time within the complex environment of the cell cytosol. Using purified components, we show that localization of the negative regulators TREX1 and BAF is restricted to a ring-like zone that forms at the outer periphery of cGAS-DNA droplets similar to ring-like localization observed in cells for TDP-43 (Figures 4D-H and 7D,E) (Yu et al., 2020). High-resolution imaging and live cell microscopy are required to extend these findings and further define interactions between TREX1 and cGAS-DNA puncta during active DNA sensing. Additionally, a major proportion of TREX1 in cells occurs associated with the ER membrane and recent work demonstrates that TREX1 ER-association is critical to enable localization to ruptured micronuclei and control of the cGAS-dependent immune response to chromosomal instability (Mohr et al. 2020). Mutations that remove the TREX1 C-terminal membrane-association sequence cause autoimmune disease (Richards et al., 2007; Yan, 2017), indicating that membrane localization is a further cellular complexity to consider in understanding TREX1 interactions with cGAS-DNA condensates. Defining the role of mutations in disrupting normal interactions of cellular proteins in a phase separated environment remains a new challenge and opportunity to understand the cellular basis of disease (Tsang et al., 2020).
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Philip J. Kranzusch (philip_kranzusch@dfci.harvard.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Coordinates of the TREX1 E198K structure have been deposited in the RCSB Protein Data Bank under accession number 6W10.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Escherichia coli strains
Recombinant cGAS, TREX1 and BAF proteins were expressed in E. coli BL21-RIL DE3 (Agilent) bacteria harboring a pRARE2 tRNA plasmid. Transformations and starter cultures were grown in 30 mL of MDG media (25 mM Na2HPO4, 25 mM KH2PO4, 50 mM NH4Cl, 5 mM Na2SO4, 2 mM MgSO4, 0.5% glucose, 0.25% aspartic acid, 100 mg mL−1 ampicillin, 34 mg mL−1 chloramphenicol, and trace metals) overnight at 37°C, and used to seed 1–4 L M9ZB media cultures (47.8 mM Na2HPO4, 22 mM KH2PO4, 18.7 mM NH4Cl, 85.6 mM NaCl, 2 mM MgSO4, 0.5% glycerol, 1% Cas-amino acids, 100 mg mL−1 ampicillin, 34 mg mL−1 chloramphenicol, and trace metals) grown at 37°C until OD600 of 1.5–2.5. Bacterial cultures were cooled on ice for 20 min, and then recombinant protein expression was induced by supplementation with 0.5 mM IPTG. Cultures were further incubated at 16°C with shaking at 230 RPM for ~16 h before harvest.
Mammalian Cell Lines
MCF10A cells (a gift from Maria Jasin, MSKCC) were cultured in a 1:1 mixture of DMEM/F12 media supplemented with 5% horse serum (Thermo Fisher Scientific), 20 ng mL−1 human EGF (Sigma-Aldrich), 0.5 mg mL−1 hydrocortisone (Sigma-Aldrich), 100 ng mL−1 cholera toxin (Sigma-Aldrich) and 10 μg mL−1 recombinant human insulin (Sigma-Aldrich). HEK293 cells were purchased from American Type Culture Collection (ATCC) and grown in DMEM supplemented with 10% FBS. All media was supplemented with 1% penicillin-streptomycin. Unless otherwise noted, all media and supplements were supplied by the MSKCC core facility.
METHOD DETAILS
Protein Expression and Purification
The DNA sequences of recombinant proteins were cloned into a custom pET16 vector for expression of a 6×His-SUMO2 fusion protein in E. coli BL21-RIL DE3 bacteria (Agilent) co-transformed with a pRARE2 tRNA plasmid. Starter cultures of E. coli were grown in MDG media, subsequently cultured in ~2 L of M9ZB media, and induced with IPTG for large-scale protein expression as previously described (Zhou et al., 2018). Bacteria cultures were pelleted, flash-frozen in liquid nitrogen, and stored at −80°C until purification.
Protein purification was performed as previously described (Zhou et al., 2018; 2019). Briefly, bacterial pellets were re-suspended in lysis buffer (20 mM HEPES-KOH pH 7.5, 400 mM NaCl, 10% glycerol, 30 mM imidazole, 1 mM DTT) and lysed by sonication. The initial purification was performed using Ni-NTA (Qiagen) affinity chromatography. Protein eluted from Ni-NTA was supplemented with ~250 μg of human SENP2 protease to remove the SUMO2 solubility tag and dialyzed in dialysis buffer (20 mM HEPES-KOH pH 7.5, 300 mM NaCl, 1 mM DTT) at 4°C for ~14 h. Untagged protein was further purified using Heparin HP ion-exchange (GE Healthcare) and eluted with a gradient of 300–1000 mM NaCl. Target protein was then further purified with size-exclusion chromatography using a 16/600 Superdex S75 or S200 column (GE Healthcare) equilibrated with protein storage buffer (20 mM HEPES-KOH pH 7.5, 250 mM KCl, 1 mM TCEP). The final recombinant protein was concentrated to ~10 mg mL−1, flash-frozen in liquid nitrogen, and stored as aliquots at −80°C for further usage.
cGAS chimeras were purified as described above, except that after Ni-NTA purification they were dialyzed using storage buffer (20 mM HEPES-KOH pH 7.5, 250 mM KCl, 1 mM TCEP) without tag removal. Following dialysis, aliquots were flash-frozen in liquid nitrogen and stored at −80°C for further usage. TREX1 and TREX1 variants were purified as cGAS, except minor changes to buffers used for dialysis (20 mM HEPES-KOH pH 7.5, 150 mM NaCl, 1 mM DTT) and ion-exchange (20 mM HEPES-KOH pH 7.5, 1 mM DTT, and a gradient of 150–1000 mM NaCl for elution).
BAF was expressed and purified with denaturation and protein-refolding. Briefly, the DNA sequence of full-length human BAF was cloned into a pET16 vector for expression of a 6×His-SUMO2 fusion protein as indicated above. Following protein expression, the bacterial pellet was re-suspended in lysis buffer (20 mM HEPES-KOH pH 7.5, 400 mM NaCl, 10% glycerol, 30 mM imidazole, 1 mM DTT) and lysed by sonication. The lysate was centrifuged for 30 min, 47,850 × g at 4°C, and the pellet fraction containing BAF was then solubilized in denaturing buffer (20 mM HEPES-KOH pH 7.5, 400 mM NaCl, 10% glycerol, 30 mM imidazole, 1 mM DTT, 6 M Gu-HCl) and agitated at 4°C for ~1 h. The solubilized solution was then centrifuged for 30 min, 47,850 × g at 4°C, and the supernatant fraction containing BAF was collected for purification. Initial purification was performed using standard Ni-NTA (Qiagen) affinity chromatography and 6×His-SUMO2 tagged BAF was eluted with buffer (20 mM HEPES-KOH pH 7.5, 400 mM NaCl, 10% glycerol, 300 mM imidazole, 1 mM DTT, 6 M Gu-HCl). Tagged BAF was refolded during dialysis against 20 mM HEPES-KOH pH 7.5, 150 mM NaCl, 1 mM DTT, 0.01% NP-40 at 4°C for ~14 h. Following incubation, the 6×His-SUMO2 solubility tag was removed by adding human SENP2 protease and dialyzing against fresh dialysis buffer at 4°C for ~14 h. BAF was further purified using a combination of Heparin HP and Q ion-exchange and eluted with a gradient of 300–1000 mM NaCl. An additional round of Ni-NTA affinity chromatography was used to remove minor contamination of the 6×His-SUMO2 tag. Final BAF protein was stored in 20 mM HEPES-KOH pH 7.5, 250 mM KCl, 1 mM TCEP at ~7 mg mL−1, flash-frozen in liquid nitrogen, and stored as aliquots at −80°C for further usage.
In vitro cGAS 2′3′-cGAMP Synthesis Assays
In vitro cGAS 2′3′-cGAMP synthesis was performed as previously described with minor modifications (Zhou et al., 2018). Briefly, 1 μM 100-bp dsDNA (Du and Chen, 2018) was incubated with cGAS or cGAS variants at different protein concentrations as indicated in the figure legends in a 20 μL reaction containing 50 mM Tris-HCl pH 7.5, 35 mM KCl, 10 mM Mg(OAc)2, 1 mM DTT, 25 μM ATP/GTP each, and [α-32P] ATP (~1 μCi; Perkin Elmer) at 37°C for 30 min. Reactions were terminated by heating at 95°C for 3 min and immediately placing on ice for 3 min. The unreacted NTPs were further hydrolyzed by treating with alkaline phosphatase (~4 U; New England Biolabs) at 37°C for 30 min. 1 μL of each reaction was spotted on a PEI-Cellulose F thin-layer chromatography plate (EMD Biosciences) and separated by thin-layer chromatography using a running buffer of 1.5 M KH2PO4 (pH 3.8). 2′3′-cGAMP product formation was monitored by phosphorimaging with a Typhoon Trio Variable Mode Imager system (GE Healthcare) and quantified using ImageQuant TL v8.2.0 (GE Healthcare). The relative activity of cGAS enzymes was measured as the ratio of radiolabeled 2′3′-cGAMP to the total radiolabeled product signal.
In vitro Electrophoretic Mobility Shift Assay (EMSA)
In vitro measurement of cGAS–DNA complex formation by EMSA was performed as previously described with minor modifications (Zhou et al., 2018). Briefly, 1 μM 100-bp dsDNA was incubated with cGAS or cGAS variants at a gradient of protein concentrations (0.5, 1, 2 μM) in a 20 μL reaction containing 20 mM HEPES-NaOH pH 7.8, 75 mM KCl, 1 mM DTT at 4°C for 20 min. The resulting reactions were subsequently separated on a 2% agarose gel at 4°C using 0.5 × TB buffer (45 mM Tris, 45 mM boric acid) as a running buffer. After electrophoresis, the agarose gel was stained in 0.5 × TB buffer supplemented with 10 μg mL−1 ethidium bromide and complex formation was visualized with a ChemiDoc MP Imaging System (Bio-Rad).
Measurement of the Relative Saturation Concentrations by Turbidity Assay and Fluorescence Intensity Analysis
To determine saturation concentrations in vitro, cGAS proteins and DNA mixtures were prepared at indicated protein concentrations with the same DNA concentrations using a reaction buffer of 20 mM Tris-HCl (pH 7.5), 15 mM NaCl, 135 mM KCl, 1 mg mL−1 BSA, and 1 mM TCEP. Phase separation was induced at 25°C for 30 min to fully equilibrate samples and reactions were then analyzed by measuring either turbidity or fluorescence intensity. For turbidity measurements, light scattering was quantified as the absorbance at 340 nm at 25°C using a DeNovix DS-11+ UV-Vis spectrophotometer with a 10 mm optical path length. The relative saturation concentrations were determined by a two-tailed T test of the adjacent values across each pair of points in the protein/DNA titration. For fluorescence intensity analysis, microscopy images were acquired at 25°C using a Leica TCS SP5 X (Leica Microsystems) mounted on an inverted microscope (DMI6000; Leica Microsystems) with an oil immersion 63×/numerical aperture 1.4 objective lens (HCX PL APO; Leica Microsystems). For each field of view, phase separation was quantified with FIJI (Schindelin et al., 2012) by measuring total fluorescence intensity for reach field of view. The relative saturation concentrations were determined by a two-tailed T test of the adjacent values across each pair of points in the protein/DNA titration.
In vitro TREX1 DNA Degradation Assays
For in vitro analysis of TREX1 exonuclease activity, 1 μM 100-bp dsDNA was incubated with TREX1 or TREX1 variants at 25°C in a 20 μL reaction containing 20 mM Tris-HCl pH 7.5, 15 mM NaCl, 135 mM KCl, 5 mM MgCl2, and 1 mg mL−1 BSA as specified in the figure legends. Reactions were terminated by adding 10 mM EDTA and ~0.5 U proteinase K (GoldBio) and incubating at 55°C for 30 min. Reactions were separated on a 4% agarose gel (containing 10 μg mL−1 ethidium bromide) using 0.5 × TB buffer (45 mM Tris, 45 mM boric acid) as a running buffer and DNA was visualized by ChemiDoc MP Imaging System and quantified using FIJI (Schindelin et al., 2012). In Figure 5J reactions were inactivated by adding SDS (0.2% final) and EDTA (10 mM final) and then heat-inactivating at 75°C for 15 min prior to remaining DNA isolation by standard PCR clean-up. The relative activity of TREX1 was measured as the ratio of the remaining DNA compared to a negative-control reaction without TREX1. TREX1 DNA degradation in the context of cGAS phase separation was performed as above, except that the cGAS-DNA droplets were preformed by mixing cGAS and DNA for 30 min and then TREX1 was added to the cGAS-DNA phase separation reactions and incubated for analysis of DNA degradation.
Mammalian Cell Culture
For lentiviral transduction, GFP-hcGAS, GFP-hcGASCterm, GFP-hcGAS Chi 3.2FL, GFP-hcGAS Chi 3.2FL K187N/L195R and GFP-TREX1 open reading frames were cloned into pLenti-CMV-GFP-blast (Addgene #17445). Constructs were co-transfected together with lentiviral packaging plasmids psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) into HEK293 packaging cells using calcium phosphate precipitation. Cell supernatants containing lentivirus were filtered, mixed 1:1 with target cell media and supplemented with 4 μg mL−1 polybrene. Successfully transduced cells were selected using 5 μg mL−1 blasticidin (Fisher) and transduced cells were then further enriched by two rounds of flow sorting for the GFP+ cell population.
hcGAS and TREX1 knockout cells were prepared using CRISPR/Cas9 editing and multiple sgRNAs designed to introduce a large deletion in the target gene. sgRNAs were cloned into pU6-(BbsI)_CBh-Cas9-T2A-mCherry (Addgene #64324) and 5 μg of the resulting plasmids were co-transfected into MCF10A cells by nucleofection (Lonza). Successfully transfected cells were isolated by flow cytometry (mCherry+) and subcloned into 96-well plates. Subclones were screened for successful gene deletion by immunoblotting, and biallelic targeting was verified by sequencing of TOPO-cloned PCR products.
hcGAS and TREX1 double knockout cells were prepared by knocking out cGAS in TREX1 knockout cells using the protocol described above.
CRISPR target sequences:
hcGAS #1: 5′-GTTCGGCCCCGCCAGGAAGT-3′
hcGAS #2: 5′-GGCCCCCATTCTCGTACGGA-3′
hTREX1 #1: 5′-TCAACGCTTCGATGACAACC-3′
hTREX1 #2: 5′-GCATCTACACCCGCCTGTAC-3′
hTREX1 #3: 5′-CCACTGGAACAACCAACCTA-3′
Analysis of cGAS Activity in Cells
1.5 × 106 of MCF10A cells were plated into 10-cm dishes, and 24 h later cells were stimulated by transfection with 4 μg of pMax cloning plasmid (Lonza) using Fugene HD transfection reagent (Promega) per manufacturer’s instructions. 24 h after transfection cells were harvested, pelleted, and stored at −80°C. To quantify 2′3′-cGAMP levels, 2 × 106 cells were resuspended in 120 μL lysis buffer (20 mM Tris-HCl pH 7.7, 100 mM NaCl, 10 mM NaF, 20 mM β-glycerophosphate, 5 mM MgCl2, 0.1% Triton X-100, 5% glycerol) and lysed with a 28 ½ gauge needle. Lysates were incubated on ice for 30 min, centrifuged at 16,000 × g, 4°C for 10 min and 2′3′-cGAMP levels were quantified using the 2′3′-cGAMP ELISA Kit (Arbor Assays, #K067-H5) according to the manufacturer’s instructions.
Immunoblotting
MCF10A cells were lysed in RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS), supplemented with phosphatase inhibitors (10 mM NaF, 20 mM β-glycerophosphate) and protease inhibitor (Thermo Scientific #A32965). Total proteins were prepared, and protein concentration was determined using a BCA protein assay (Thermo Scientific #23227). 20 μg total protein was loaded in each lane for SDS-PAGE analysis. After SDS-PAGE, proteins were transferred to membranes, and probed with specific antibodies. In brief, primary antibodies were diluted in blocking buffer (LI-COR) supplemented with 0.2% Tween and incubated with membranes overnight at 4°C. Secondary antibodies (goat anti-rabbit and goat anti-mouse Alexa Fluor Plus 800 and 680; Invitrogen) were used at 1:20,000 dilutions in blocking buffer supplemented with 0.2% Tween. Fluorescence was measured using an infrared imaging scanner (Odyssey; LI-COR).
Antibody information:
Anti-IRF3 (phosphor S386) (Abcam #ab76493); 1:1000 dilution.
Anti-hcGAS (Cell signaling technology #15102); 1:500 dilution.
Anti-β-actin (Abcam #ab8224 or Abcam #ab8227); 1:3000 dilution.
Anti-GFP (Santa cruz biotechnology #9996); 1:1000 dilution.
Live-Cell Imaging
Cells were plated onto 35-mm glass bottom dishes (Cellvis) 48 h before imaging and cells were stimulated by transfection of Cy5-labeled immunostimulatory DNA. Synthetic Cy5-labeled DNA was purified by HPLC (IDT) and consisted of the following duplexed 45 bp sequence: TACAGATCTACTAGTGATCTATGACTGATCTGTACATGATCTACA, and transfected into MCF10A TREX1 knockout cells stably expressing GFP-TREX1 by Fugene HD (Promega) according to the manufacturer’s instructions. 1 h before imaging media was replaced with fresh medium. Live-cell imaging was performed at 37°C and 5% CO2 using a Nikon Eclipse Ti2-E equipped with a CSU-W1 spinning disk with Borealis microadapter, Perfect Focus 4, motorized turret and encoded stage, polycarbonate thermal box, 5 line laser launch [405 (100 mw), 445 (45 mw), 488 (100 mw), 561 (80 mw), 640 (75 mw)], PRIME 95B Monochrome Digital Camera, and environmental enclosure (Tokai Hit) and CI Plan Apo Lambda 60x 1.40 NA. Images were acquired using NIS-Elements Advanced Research Software on a Dual Xeon Imaging workstation. Maximum intensity projection of z-stacks and adjustment of brightness and contrast were performed using FIJI software.
Co-localization analysis of cGAS, DNA, and TREX1 in cells
MCF10A TREX1 KO cells stably expressing GFP-TREX1 were plated onto 35 mm glass bottom dishes and transfected with Cy3-labeled plasmid DNA (Mirus; MIR7905). 2 h after transfection, cells were rinsed in TBS and fixed for 10 min in 2% paraformaldehyde. Cells were then permeabilized by incubation in TBS with 0.05% saponin for 5 min and incubated in blocking buffer (1 mg mL−1 BSA, 3% goat serum, 0.05% saponin, 1 mM EDTA in TBS) for 1 h. anti-cGAS (CST; 15102) and anti-GFP (Invitrogen; A-11120) primary antibodies diluted in blocking buffer were added for 2 h. After 4× washes with TBS and 0.05% saponin, cells were incubated with goat anti-mouse IgG Alexa Fluor 488 (Invitrogen; A11001) and goat anti-rabbit IgG Alexa Fluor 647 (Invitrogen; A21245) secondary antibodies. Following incubation with secondary antibodies, cells were washed with TBS with 0.05% saponin and DNA was stained with Hoechst33342 at 1 pg mL−1 for 10 min. Imaging was performed using the Nikon Eclipse Ti2-E confocal imaging system described above.
Protein Labeling
Purified proteins (cGAS, cGASNterm, cGASCterm, TREX1, cGASNterm–TREX1, TREX1E198K, BAF) were labeled with Alexa Fluor 488 and/or Alexa Fluor 647 carboxylic acid (succinimidyl ester) (Thermo Fisher Scientific) according to manufacturer’s protocols. Typically, the purified protein was labeled with the ester dye with a molar ratio of 1:5 at 4°C for 4 h. The labeling products were then dialyzed in dialysis buffer (20 mM HEPES-KOH pH 7.5, 250 mM KCl, 1 mM DTT) at 4°C overnight to remove free dye. Fluorescently-labeled proteins were further purified with a PD-10 desalting column (GE Healthcare) and elution with storage buffer (20 mM HEPES-KOH pH 7.5, 250 mM KCl, 1 mM TCEP). The labeled proteins were concentrated to ~5 mg mL−1, flash-frozen in liquid nitrogen, and stored at −80°C for microscopy imaging.
In vitro Phase Separation Assays and Image Collection
In vitro phase-separated droplet formation was analyzed by imaging reactions in 384-well non-binding microplates (Greiner Bio-One). Phase separation was induced by mixing protein and DNA in buffer (20 mM Tris-HCl pH 7.5, 1 mg mL−1 BSA, 1 mM TCEP) with various salt concentrations at 25°C in a total reaction volume of 20 μL. Each imaging experiment was performed using only 1 μM fluorescently-labeled protein or DNA with the remaining concentration consisting of unlabeled protein or DNA components to limit the impact of fluorescent labeling (e.g. 1 μM fluorescently-labeled cGAS with 9 μM unlabeled cGAS and 1 μM fluorescently-labeled DNA with 9 μM unlabeled DNA). The details of incubation time and specific protein, DNA, and salt concentrations are indicated in each figure legend and the corresponding figure schematics. In each case, total salt is comprised of ~50 mM KCl from purified protein and the remaining amount is from NaCl in the reaction buffer. The salt concentration indicated in the figure legends reflects the total salt of KCl and NaCl. To test the TREX1 interactions with cGAS-DNA droplets formed with linear dsDNA, closed circular dsDNA, or ssDNA (Figure 5F), linear dsDNA droplets were induced by cGAS (10 μM) and 100-bp dsDNA (10 μM, containing 1 μM Cy3-labeled DNA) at 250 mM salt; closed circular dsDNA droplets were induced by N-terminal cGAS (11 μM, containing 1 μM AF488-labeled cGASNterm) and 9.3-kb plasmid DNA (0.035 μM) at 150 mM salt; ssDNA droplets were formed by cGAS (10 μM) and 100-nt ssDNA (10 μM, containing 1 μM Cy3-labeled ssDNA) at 150 mM salt. Microscopy images were acquired at 25°C using a Leica TCS SP5 X (Leica Microsystems) mounted on an inverted microscope (DMI6000; Leica Microsystems) with an oil immersion 63x/numerical aperture 1.4 objective lens (HCX PL APO; Leica Microsystems). The formation of phase separation was measured as the percentage of the imaging area occupied by fluorescently-labeled components. In brief, unprocessed images from fluorescence microscopy imaging were analyzed with FIJI. Images were converted to 16-bit form and the thresholds were determined using the built-in image thresholder (typically using the default method). The droplet areas were then calculated by FIJI particle analyzer algorithm (smallest detected size of droplets was set to 1 μm2).
For time-lapse microscopy, droplet formation was induced as indicated in figure legends, and fluorescence images were taken at an interval of 10 s with an inverted fluorescence microscope (Leica SP5 X; Leica Microsystems). Fluorescence images of AF488, Cy3, and AF647 were acquired with excitation at 488 nm (emission at 500–530 nm), 550 nm (emission at 560–590 nm), and 650 nm (emission at 660–700 nm), respectively. For 3D reconstruction, confocal microscopy images were acquired with a z-interval of 0.25 μm, and a total of 118 images along the z-axis were recorded.
Fluorescence Recovery After Photobleaching (FRAP)
FRAP analysis was performed using fluorescently-labeled components (AF488-cGAS, AF647-TREX1, AF488-TREX1, or Cy3-DNA) and microscopy images were collected using a confocal microscope (Leica TCS SP5 X; Leica Microsystems). For FRAP analysis of cGAS-DNA and TREX1-DNA phase separation, photobleaching of the Cy3 signal in selected droplets was performed with 100% maximum laser power of a 550 nm laser. The recovery was immediately recorded at an interval of 2 s for ~200 s. For cGAS-DNA-TREX1 droplets, photobleaching of AF488, Cy3, and AF647 signals in selected droplets was carried out by the combination of a 488 laser (70% maximum laser power), a 550 nm laser (56% maximum laser power), and a 650 nm laser (30% maximum laser power) and FRAP recovery was recorded at an interval of 2 s for ~1000 s. Where indicated, experiments were fit according to one-phase association non-linear regression.
Crystallization and Structure Determination
For crystallization, purified mTREX1 E198K (residues 1–242) was diluted to ~14 mg mL−1 in a final buffer consisting of 20 mM HEPES-KOH pH 7.5, 73 mM KCl, 1 mM TCEP. Optimized mTREX1 E198K crystals were grown in hanging-drop format using 15-well EasyXtal trays (Qiagen) in 2 μL drops containing a 1:1 mixture of protein solution and reservoir solution (0.1 M NH4OAc, 17.5% PEG 3350). Crystals completed growth after 1 day at 18°C and were cryo-protected with NVH oil (Cargille) prior to freezing in liquid nitrogen.
X-ray diffraction data were collected at the Northeastern Collaborative Access Team beamline 24-ID-E (P30 GM124165) and used an Eiger detector (S10OD021527) and the Argonne National Laboratory Advanced Photon Source (DE-AC02-06CH11357). Data were processed with XDS and AIMLESS (Kabsch, 2010) using the SSRL autoxds script (A. Gonzalez, Stanford SSRL). The crystal of mTREX1 E198K indexed according to the orthorhombic spacegroup P 21 21 21 and included one copy of a mTREX1 E198K dimer in the asymmetric unit. Phases were then determined with molecular replacement using Phaser-MR in PHENIX (Adams et al., 2010) and the apo TREX1 structure (PDB: 3MXJ) as a search model. Model building of the final 1.8 Å structure was completed with Coot (Emsley and Cowtan, 2004) and PHENIX (See details of collection and refinement statistics in Table 1) (Chen et al., 2010; Karplus and Diederichs, 2012; Weiss, 2001).
QUANTIFICATION AND STATISTICAL ANALYSIS
Information on statistical analysis and replicates for each experiment can be found in the figure legends and outlined in the corresponding methods details section. Statistical analyses were performed using Graphpad Prism Version 9.0. Data are plotted with error bars representing the standard error of the mean (SEM) or standard deviation of the mean (SD).
Supplementary Material
Video S1. Time-lapse imaging of cGAS-DNA phase separation, related to Figure 1
cGAS-DNA liquid droplet formation was induced by mixing hcGAS (10 μM, containing 1 μM AF488-labeled cGAS) and 100-bp dsDNA (10 μM, containing 1 μM Cy3-labeled DNA) at 250 mM salt (total NaCl and KCl) at 25°C for 30 min. Time-lapse imaging of cGAS-DNA phase separation was recorded for an additional 76 min. Left, AF488-labeled cGAS. Middle, Cy3-labeled DNA. Right, merge of AF488 and Cy3 channels.
Video S2. Time-lapse movie of TREX1 degradation cGAS-DNA droplets, related to Figure 4
cGAS-DNA droplets were formed by mixing hcGAS (10 μM, containing 1 μM AF488-labeled cGAS) and 100-bp dsDNA (10 μM, containing 1 μM Cy3-labeled DNA) at 250 mM salt (total NaCl and KCl) at 25°C for 30 min, and then AF647-labeled TREX1 (1 μM final) was added to the preformed cGAS-DNA droplets. Droplets were incubated for 60 min to allow TREX1 interaction and then supplemented with MgCl2 (5 mM final) to activate TREX1 and begin imaging for 60 min. Top left, AF488 labeled cGAS. Top right, Cy3-labeled DNA. Bottom left, AF647-labeled TREX1. Bottom right, merge of AF488, Cy3 and AF647 channels.
Video S3. Time-lapse movie of TREX1 degradation of immunostimulatory DNA (ISD) puncta in cells, related to Figure 4
MCF10A TREX1−/− cells stably expressing GFP-TREX1 (green) were transfected with Cy5-labeled ISD (red). After 2 h, imaging was started, and DNA degradation was tracked over time. Scale bar, 5 μm.
Video S4. Time-lapse movie of TREX1-dsDNA phase separation, related to Figure 5
TREX1-dsDNA liquid droplet formation was induced by mixing TREX1 (20 μM, containing 1 μM AF647-labeled TREX1) and 100-bp dsDNA (10 μM, containing 1 μM Cy3-labeled DNA) at 125 mM salt (total NaCl and KCl) and 25°C and images were recorded for 1 h. Left, AF647-labeled TREX1. Middle, Cy3-labeled dsDNA. Right, merge of AF647 and Cy3 channels.
Video S5. Time-lapse movie of TREX1-dsDNA and TREX1E198K-dsDNA phase separation, related to Figure 6
TREX1-dsDNA and TREX1E198K-dsDNA liquid droplet formation was induced by mixing TREX1 or TREX1E198K (20 μM each) with 100-bp dsDNA (10 μM, containing 1 μM Cy3-labeled DNA) at 150 mM salt (total NaCl and KCl) at 25°C. The time-lapse movie was subsequently recorded for 1 h. Left, Cy3 labeled TREX1-dsDNA droplets. Right, Cy3 labeled TREX1E198K-dsDNA droplets.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-IRF3 (phosphor S386) (Clone EPR2346) | Abcam | Cat# ab76493; RRID: AB_1523836 |
| Rabbit monoclonal anti-cGAS (clone D1D3G) | Cell Signaling Technology | Cat# 15102; RRID: AB_2732795 |
| Mouse monoclonal anti-beta Actin (Clone mAbcam 8224) | Abcam | Cat# ab8224; RRID: AB_449644 |
| Rabbit polyclonal anti-beta Actin | Abcam | Cat# ab8227; RRID: AB_2305186 |
| Mouse monoclonal anti-GFP (B-2) | Santa Cruz Biotechnology | Cat# sc-9996; RRID: AB_627695 |
| Mouse monoclonal anti-GFP (clone 3E6) | Invitrogen | Cat# A-11120; RRID: AB_221568 |
| Bacterial Strains | ||
| E. coli BL21-RIL DE3 | Agilent | 230245 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ni-NTA Agarose | Qiagen | 30250 |
| HiTrap Heparin HP Column | GE Healthcare | 17040703 |
| HiTrap Q HP Column | GE Healthcare | 17115401 |
| HiLoad 16/600 Superdex 75 pg Column | GE Healthcare | 28989333 |
| HiLoad 16/600 Superdex 200 pg Column | GE Healthcare | 28989335 |
| [α-32P] ATP | Perkin Elmer | BLU003H250UC |
| Alkaline phosphatase | New England Biolabs | M0290L |
| PEI-Cellulose F TLC plate | EMD Biosciences | EM1.05579.0001 |
| ATP, GTP | New England Biolabs | N0450S |
| HEPES | VWR | 97061-824 |
| Tris base | VWR | 97062-416 |
| Guanidinium hydrochloride | VWR | 97061-350 |
| NP-40 | VWR | 80601-882 |
| Imidazole | VWR | 97065-016 |
| Tris[−2carboxyethyl] phosphine hydrochloride (TCEP) | GoldBio | TCEP50 |
| Dithiothreitol (DTT) | GoldBio | DTT25 |
| Bovine serum albumin (BSA) | New England Biolabs | B9000S |
| Ammonium acetate | VWR | EM-AX1222-5 |
| PEG-3350 | Sigma-Aldrich | 202444 |
| NVH oil | Hampton Research | HR3-617 |
| Alexa Fluor™ 488 NHS Ester (Succinimidyl Ester) | Thermo Fisher | A20000 |
| Alexa Fluor™ 647 NHS Ester (Succinimidyl Ester) | Thermo Fisher | A20006 |
| Alexa Fluor™ 647 ATP | Thermo Fisher | A22362 |
| Disposable PD 10 Desalting Columns | GE Healthcare | 17-0851-01 |
| Proteinase K | GoldBio | P-480-100 |
| Critical Commercial Assays | ||
| 2′3′-cGAMP ELISA Kit | Arbor Assays | K067-H5 |
| Deposited Data | ||
| Mouse TREX1 E198K structure | This paper | PDB: 6W10 |
| Experimental Models: Cell Lines | ||
| Human: MCF10A cells | Laboratory of Maria Jasin (MSKCC) | N/A |
| Human: HEK293 cells | ATCC | CRL-1573 |
| Oligonucleotides | ||
| 100 bp dsDNA sense: 5′- ACATCTAGTACATGTCTAGTCAGTATCTA GTGATTATCTAGACATACATCTAGTACAT GTCTAGTCAGTATCTAGTGATTATCTAGA CATGGACTCATCC -3′ | Integrated DNA Technologies | N/A |
| 100 bp dsDNA antisense: 5′- GGATGAGTCCATGTCTAGATAATCACTA GATACTGACTAGACATGTACTAGATGTAT GTCTAGATAATCACTAGATACTGACTAGA CATGTACTAGATGT -3′ | Integrated DNA Technologies | N/A |
| CRISPR targeting sequence cGAS #1: 5′- GTTCGGCCCCGCCAGGAAGT -3′ | This paper | N/A |
| CRISPR targeting sequence cGAS #2: 5′- GGCCCCCATTCTCGTACGGA -3′ | This paper | N/A |
| CRISPR targeting sequence TREX1 #1: 5′- TCAACGCTTCGATGACAACC -3′ | This paper | N/A |
| CRISPR targeting sequence TREX1 #2: 5′- GCATCTACACCCGCCTGTAC -3′ | This paper | N/A |
| CRISPR targeting sequence TREX1 #3: 5′- CCACTGGAACAACCAACCTA -3′ | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pET16-6×His-SUMO2 | Zhou et al., 2018 | N/A |
| Plasmid: pLenti-CMV-GFP-blast | Addgene | 17445 |
| Plasmid: psPAX2 | Addgene | 12260 |
| Plasmid: pMD2.G | Addgene | 12259 |
| Plasmid: pU6-(BbsI)_CBh-Cas9-T2A-mCherry | Addgene | 64324 |
| Software and Algorithms | ||
| Phenix 1.18.2-3874 | Adams et al., 2010 | https://www.phenix-online.org/ |
| Coot 0.8.9.2 | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Pymol v2.3.3 | Schrödinger, LLC | https://pymol.org/ |
| Prism 9.0.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/index.html |
| ImageQuant TL v8.2.0 | GE Healthcare | https://www.gelifesciences.com/en/us/shop/protein-analysis/molecular-imaging-for-proteins/imaging-software/imagequant-tl-8-1-p-00110?current=29000605 |
Highlights.
cGAS-DNA phase separation is required for 2′3′-cGAMP synthesis in cells but not in vitro
Phase separation sequesters cGAS and limits access of negative regulators TREX1 and BAF
Human-specific substitutions enhance cGAS-DNA phase separation and TREX1 resistance
Phase separation provides a new mechanism to explain TREX1-dependent autoimmune disease
Acknowledgements
The authors are grateful to L. Wang, A. Lee, B. Morehouse, K. Chat, and members of the Kranzusch laboratory for helpful discussion, L. Ding and the DFCI Light Microscopy Core Facility for assistance with data collection and B. Lowey for assistance with SEC-MALS. The work was funded by grants to P.J.K. from the Richard and Susan Smith Family Foundation, Charles H. Hood Foundation, V Foundation, Concern Foundation, a Cancer Research Institute CLIP grant, and Parker Institute for Cancer Immunotherapy; grants to J.M. from the National Cancer Institute (R00CA212290), the Geoffrey Beene Cancer Research Center, and a MSKCC core grant (P30-CA008748); W.Z. is supported as a Benacerraf Fellow in Immunology and through a Charles A. King Trust Postdoctoral Fellowship.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ablasser A, and Chen ZJ (2019). cGAS in action: Expanding roles in immunity and inflammation. Science 363, eaat8657. [DOI] [PubMed] [Google Scholar]
- Ablasser A, and Hur S (2020). Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat. Immunol 21, 17–29. [DOI] [PubMed] [Google Scholar]
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner K-P, Ludwig J, and Hornung V (2013). cGAS produces a 2′–5′–linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshareedah I, Moosa MM, Raju M, Potoyan DA, and Banerjee PR (2020). Phase transition of RNA-protein complexes into ordered hollow condensates. Proceedings of the National Academy of Sciences 117, 15650–15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva L, Hiller B, Kostrewa D, Lässig C, de Oliveira Mann CC, Jan Drexler D, Maiser A, Gaidt M, Leonhardt H, Hornung V, et al. (2017). cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 549, 394–398. [DOI] [PubMed] [Google Scholar]
- Banerjee PR, Milin AN, Moosa MM, Onuchic PL, and Deniz AA (2017). Reentrant Phase Transition Drives Dynamic Substructure Formation in Ribonucleoprotein Droplets. Angew. Chem. Int. Ed. Engl 56, 11354–11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer JA, Spangler CJ, Strauss JD, Cesmat AP, Liu P, McGinty RK, and Zhang Q (2020). Structural basis of nucleosome-dependent cGAS inhibition. Science 370, 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brucet M, Querol Audí J, Serra M, Ramirez-Espain X, Bertlik K, Ruiz L, Lloberas J, Macias MJ, Fita I, and Celada A (2007). Structure of the dimeric exonuclease TREX1 in complex with DNA displays a proline-rich binding site for WW Domains. J. Biol. Chem 282, 14547–14557. [DOI] [PubMed] [Google Scholar]
- Cao D, Han X, Fan X, Xu R-M, and Zhang X (2020). Structural basis for nucleosome-mediated inhibition of cGAS activity. Cell Res. 30, 1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung J-S, Demple B, Perrino FW, and Lieberman J (2006). The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell 23, 133–142. [DOI] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, and Hopfner K-P (2013). Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva U, Choudhury S, Bailey SL, Harvey S, Perrino FW, and Hollis T (2007). The crystal structure of TREX1 explains the 3' nucleotide specificity and reveals a polyproline II helix for protein partnering. J. Biol. Chem 282, 10537–10543. [DOI] [PubMed] [Google Scholar]
- Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, and Vance RE (2013). The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 3, 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, and Chen ZJ (2018). DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Gao D, Li T, Li X-D, Chen X, Li Q-Z, Wight-Carter M, and Chen ZJ (2015). Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proceedings of the National Academy of Sciences 112, E5699–E5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, et al. (2013). Cyclic [G(2',5')pA(3',5')p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Treuting PM, Woodward JJ, and Stetson DB (2015). Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutières Syndrome. J. Immunol 195, 1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieves JL, Fye JM, Harvey S, Grayson JM, Hollis T, and Perrino FW (2015). Exonuclease TREX1 degrades double-stranded DNA to prevent spontaneous lupus-like inflammatory disease. Proceedings of the National Academy of Sciences 112, 5117–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guey B, Wischnewski M, Decout A, Makasheva K, Kaynak M, Sakar MS, Fierz B, and Ablasser A (2020). BAF restricts cGAS on nuclear DNA to prevent innate immune activation. Science 369, 823–828. [DOI] [PubMed] [Google Scholar]
- Hall J, Brault A, Vincent F, Weng S, Wang H, Dumlao D, Aulabaugh A, Aivazian D, Castro D, Chen M, et al. (2017). Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS ONE 12, e0184843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, et al. (2015). A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163, 712–723. [DOI] [PubMed] [Google Scholar]
- Hooy RM, and Sohn JS (2018). The allosteric activation of cGAS underpins its dynamic signaling landscape. eLife 7, e39984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K-W, Liu T-C, Liang R-Y, Chu L-Y, Cheng H-L, Chu J-W, and Hsiao Y-Y (2018). Structural basis for overhang excision and terminal unwinding of DNA duplexes by TREX1. PLoS Biol. 16, e2005653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). XDS. Acta Crystallogr. D Biol. Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan JC, Magupalli VG, and Wu H (2014). SMOCs: supramolecular organizing centres that control innate immunity. Nat. Rev. Immunol 14, 821–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus PA, and Diederichs K (2012). Linking crystallographic model and data quality. Science 336, 1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H, Yamauchi S, Berglund A, Putney RM, Mulé JJ, and Barber GN (2018). Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 37, 2037–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee AS-Y, Berger JM, and Doudna JA (2013). Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 3, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujirai T, Zierhut C, Takizawa Y, Kim R, Negishi L, Uruma N, Hirai S, Funabiki H, and Kurumizaka H (2020). Structural basis for the inhibition of cGAS by nucleosomes. Science 370, 455–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, and Li P (2013). Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39, 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, Jiang Y, Fei Y, Zhu C, Tan R, et al. (2018). Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563, 131–136. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. (2014). Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med 371, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z-S, Cai H, Xue W, Wang M, Xia T, Li W-J, Xing J-Q, Zhao M, Huang Y-J, Chen S, et al. (2019). G3BP1 promotes DNA binding and activation of cGAS. Nat. Immunol 20, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Qian W, Bambouskova M, Collins PL, Porter SI, Byrum AK, Zhang R, Artyomov M, Oltz EM, Mosammaparast N, et al. (2020). Barrier-to-Autointegration Factor 1 Protects against a Basal cGAS-STING Response. MBio 11, e00136–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski S, de Oliveira Mann CC, Stafford CA, Witte G, Bartho J, Lammens K, Hornung V, and Hopfner K-P (2020). Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 587, 678–682. [DOI] [PubMed] [Google Scholar]
- Milin AN, and Deniz AA (2018). Reentrant Phase Transitions and Non-Equilibrium Dynamics in Membraneless Organelles. Biochemistry 57, 2470–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr L, Toufektchan E, von Morgen P, Chu K, Kapoor A, and Maciejowski J (2020). ER-directed TREX1 limits cGAS activation at micronuclei. Mol. Cell, 10.1016/j.molcel.2020.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orebaugh CD, Fye JM, Harvey S, Hollis T, Wilkinson JC, and Perrino FW (2013). The TREX1 C-terminal region controls cellular localization through ubiquitination. J. Biol. Chem 288, 28881–28892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathare GR, Decout A, Glück S, Cavadini S, Makasheva K, Hovius R, Kempf G, Weiss J, Kozicka Z, Guey B, et al. (2020). Structural mechanism of cGAS inhibition by the nucleosome. Nature 587, 668–672. [DOI] [PubMed] [Google Scholar]
- Rice GI, Rodero MP, and Crow YJ (2015). Human disease phenotypes associated with mutations in TREX1. J. Clin. Immunol 35, 235–243. [DOI] [PubMed] [Google Scholar]
- Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, et al. (2007). Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am. J. Hum. Genet 81, 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards A, van den Maagdenberg AMJM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-LaBarca M-L, Terwindt GM, Kasai Y, et al. (2007). C-terminal truncations in human 3′–5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet 39, 1068–1070. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon JL, Murphy MS, Kantheti U, Burnett JM, Hahn MG, Dorrity TJ, Bacas CJ, Mattice EB, Corpuz KD, and Barker BR (2018). Polyglutamine binding protein 1 (PQBP1) inhibits innate immune responses to cytosolic DNA. Mol. Immunol 99, 182–190. [DOI] [PubMed] [Google Scholar]
- Shin Y, and Brangwynne CP (2017). Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382. [DOI] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, and Medzhitov R (2008). Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Zhang X-W, Jin J, Du X-X, Lian T, Yang J, Zhou X, Jiang Z, and Su X-D (2017). Nonspecific DNA Binding of cGAS N Terminus Promotes cGAS Activation. J. Immunol 198, 3627–3636. [DOI] [PubMed] [Google Scholar]
- Tsang B, Pritišanac I, Scherer SW, Moses AM, and Forman-Kay JD (2020). Phase Separation as a Missing Mechanism for Interpretation of Disease Mutations. Cell 183, 1742–1756. [DOI] [PubMed] [Google Scholar]
- Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, Okamoto R, Imaeda T, Aida J, Rothamel K, et al. (2017). Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun 8, 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman HE, Cambier S, Gray EE, and Stetson DB (2019). Tight nuclear tethering of cGAS is essential for preventing autoreactivity. eLife 8, e47491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Choi J-M, Holehouse AS, Lee HO, Zhang X, Jahnel M, Maharana S, Lemaitre R, Pozniakovsky A, Drechsel D, et al. (2018). A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell 174, 688–699.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MS (2001). Global indicators of X-ray data quality. J. Appl. Cryst 34, 130–135. [Google Scholar]
- Xie W, Lama L, Adura C, Tomita D, Glickman JF, Tuschl T, and Patel DJ (2019). Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proceedings of the National Academy of Sciences 116, 11946–11955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N (2017). Immune Diseases Associated with TREX1 and STING Dysfunction. J. Interferon Cytokine Res 37, 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh SM, Schneider M, Seifried J, Soonthornvacharin S, Akleh RE, Olivieri KC, De Jesus PD, Ruan C, de Castro E, Ruiz PA, et al. (2015). PQBP1 Is a Proximal Sensor of the cGAS-Dependent Innate Response to HIV-1. Cell 161, 1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lu S, Gasior K, Singh D, Vazquez-Sanchez S, Tapia O, Toprani D, Beccari MS, Yates JR, Da Cruz S, et al. (2020). HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science 17, eabb4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51, 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wu J, Du F, Xu H, Sun L, Chen Z, Brautigam CA, Zhang X, and Chen ZJ (2014). The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 6, 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Xu P, Rowlett CM, Jing T, Shinde O, Lei Y, West AP, Liu WR, and Li P (2020). The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature 587, 673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Whiteley AT, and Kranzusch PJ (2019). Analysis of human cGAS activity and structure. Meth. Enzymol 625, 13–40. [DOI] [PubMed] [Google Scholar]
- Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, Gray NS, Mekalanos JJ, and Kranzusch PJ (2018). Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 174, 300–311.e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierhut C, Yamaguchi N, Paredes M, Luo J-D, Carroll T, and Funabiki H (2019). The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 178, 302–315.e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Time-lapse imaging of cGAS-DNA phase separation, related to Figure 1
cGAS-DNA liquid droplet formation was induced by mixing hcGAS (10 μM, containing 1 μM AF488-labeled cGAS) and 100-bp dsDNA (10 μM, containing 1 μM Cy3-labeled DNA) at 250 mM salt (total NaCl and KCl) at 25°C for 30 min. Time-lapse imaging of cGAS-DNA phase separation was recorded for an additional 76 min. Left, AF488-labeled cGAS. Middle, Cy3-labeled DNA. Right, merge of AF488 and Cy3 channels.
Video S2. Time-lapse movie of TREX1 degradation cGAS-DNA droplets, related to Figure 4
cGAS-DNA droplets were formed by mixing hcGAS (10 μM, containing 1 μM AF488-labeled cGAS) and 100-bp dsDNA (10 μM, containing 1 μM Cy3-labeled DNA) at 250 mM salt (total NaCl and KCl) at 25°C for 30 min, and then AF647-labeled TREX1 (1 μM final) was added to the preformed cGAS-DNA droplets. Droplets were incubated for 60 min to allow TREX1 interaction and then supplemented with MgCl2 (5 mM final) to activate TREX1 and begin imaging for 60 min. Top left, AF488 labeled cGAS. Top right, Cy3-labeled DNA. Bottom left, AF647-labeled TREX1. Bottom right, merge of AF488, Cy3 and AF647 channels.
Video S3. Time-lapse movie of TREX1 degradation of immunostimulatory DNA (ISD) puncta in cells, related to Figure 4
MCF10A TREX1−/− cells stably expressing GFP-TREX1 (green) were transfected with Cy5-labeled ISD (red). After 2 h, imaging was started, and DNA degradation was tracked over time. Scale bar, 5 μm.
Video S4. Time-lapse movie of TREX1-dsDNA phase separation, related to Figure 5
TREX1-dsDNA liquid droplet formation was induced by mixing TREX1 (20 μM, containing 1 μM AF647-labeled TREX1) and 100-bp dsDNA (10 μM, containing 1 μM Cy3-labeled DNA) at 125 mM salt (total NaCl and KCl) and 25°C and images were recorded for 1 h. Left, AF647-labeled TREX1. Middle, Cy3-labeled dsDNA. Right, merge of AF647 and Cy3 channels.
Video S5. Time-lapse movie of TREX1-dsDNA and TREX1E198K-dsDNA phase separation, related to Figure 6
TREX1-dsDNA and TREX1E198K-dsDNA liquid droplet formation was induced by mixing TREX1 or TREX1E198K (20 μM each) with 100-bp dsDNA (10 μM, containing 1 μM Cy3-labeled DNA) at 150 mM salt (total NaCl and KCl) at 25°C. The time-lapse movie was subsequently recorded for 1 h. Left, Cy3 labeled TREX1-dsDNA droplets. Right, Cy3 labeled TREX1E198K-dsDNA droplets.
Data Availability Statement
Coordinates of the TREX1 E198K structure have been deposited in the RCSB Protein Data Bank under accession number 6W10.