

Summary:
Interleukin-12 (IL-12) and IL-23 are heterodimeric cytokines produced by antigen presenting cells to regulate the activation and differentiation of lymphocytes and which share IL-12Rβ1 as a receptor signaling subunit. We present a crystal structure of the quaternary IL-23(IL-23p19/p40)/IL-23R/IL-12Rβ1 complex, together with cryo-EM maps of the complete IL-12(IL-12p35/p40)/IL-12Rβ2/IL-12Rβ1 and IL-23 receptor complexes, which reveal ‘non-canonical’ topologies whereby IL-12Rβ1 directly engages the common p40 subunit. We targeted the shared IL-12Rβ1/p40 interface to design a panel of IL-12 partial agonists that preserved interferon gamma (IFNγ) induction by CD8+ T cells, but impaired cytokine production from natural killer (NK) cells in vitro. These cell-biased properties were recapitulated in vivo, where IL-12 partial agonists elicited anti-tumor immunity to MC-38 murine adenocarcinoma absent the NK cell-mediated toxicity seen with wild-type IL-12. Thus, the structural mechanism of receptor sharing used by IL-12 family cytokines provides a protein interface blueprint for tuning this cytokine axis for therapeutics.
eTOC blurb:
Structures of the IL-12 and IL-23 receptor complexes guide design of T cell biased IL-12 agonists with reduced cytokine pleiotropy to support anti-tumor immunity without inducing toxicity.
Graphical Abstract
Introduction:
The use of shared signaling receptors is a pervasive mechanism used by cytokines to regulate pleiotropic, often redundant aspects of physiology through multimerization of cell surface receptors and induction of the JAK-STAT signaling pathway (Murray, 2007). The principal shared receptor subunits: gp130, common gamma (γc), common beta (βc) and IL-10 receptor beta (IL-10Rβ), among others, are shared between multiple cytokines (Wang et al., 2009). The interleukin-12 (IL-12) family cytokines, IL-12 and IL-23, share the IL-12 receptor beta 1 subunit (IL-12Rβ1) and are produced by antigen presenting cells in response to pathogen associated molecular patterns (Vignali and Kuchroo, 2012).
IL-12 was the first heterodimeric cytokine identified and consists of a four-helix bundle α subunit, IL-12p35, and a β subunit, p40, with homology to type I cytokine receptors (Kobayashi et al., 1989). IL-12 signals through a receptor complex of IL-12Rβ1 and IL-12Rβ2 expressed on NK cells and T cells (Figure 1A) (Chua et al., 1994; Presky et al., 1996). Dimerization of the IL-12 receptor induces activation of receptor-associated Janus Kinase (JAK) molecules, JAK2 and Tyk2, which trans-phosphorylate one another as well as tyrosine residues in the intracellular domain of IL-12Rβ2 which serve as docking sites for the SH2 containing signal transducer and activator of transcription 4 (STAT4) (Yao et al., 1999). Receptor-associated STAT4 proteins are then phosphorylated before translocating to the nucleus where they promote the expression of interferon gamma (IFNγ) and the polarization of CD4+ T cells towards a T helper 1 (Th1) phenotype (Trinchieri, 2003). Given the similarities between immunity to intracellular pathogens and cancer, therapeutic approaches that stimulate Th1 responses, either indirectly through selection of vaccine adjuvants and epitopes, or directly, through administration of IL-12, have been explored in the context of cancer immunotherapy (Berraondo et al., 2018; Lasek et al., 2014).
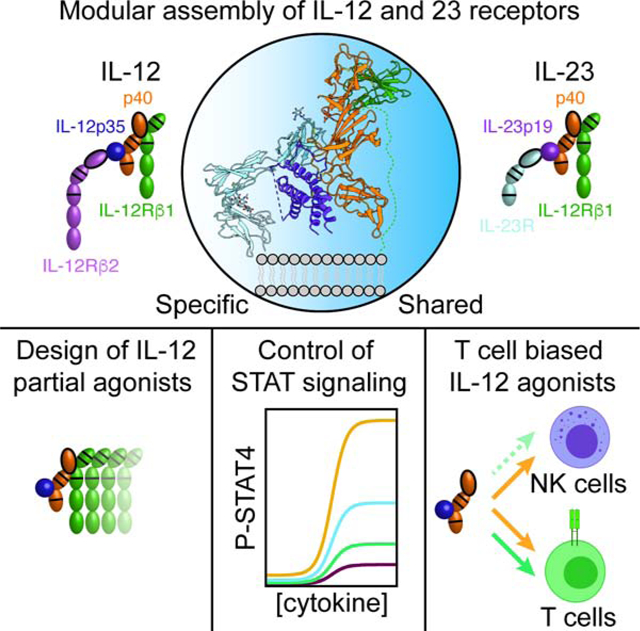
Figure 1. Crystal structures of IL-12Rβ1 and the quaternary IL-23 receptor complex.
(A) Schematic of IL-12 and IL-23 cytokine composition and receptor usage. Fibronectin-type three (FNIII) domains are shown as ovals. Immunoglobin (Ig) domains are shown as ovals with black outline. Cytokine-binding homology region (CHR) consisting of tandem FNIII domains are shown with a double line in the upper domain and a single line in the lower domain. Four-helix bundle components are represented as circles.
(B) Crystal structure of the N-terminal D1–D2 domains of IL-12Rβ1.
(C) Cartoon representation of the quaternary IL-23 receptor complex as viewed from the side. IL-23R is shown in light blue, IL-23p19 in violet, p40 in orange, and IL-12Rβ1 in green.
(D) Quaternary IL-23 receptor complex as viewed from the top looking down towards the membrane.
(E) View of the site 1 interaction between IL-23p19 and p40 showing the intermolecular disulfide bond formed between Cys73 in IL-23p19 and Cys199 in p40.
(F) View of the site 2 interaction between p40 and IL-12Rβ1 focusing on the positively charged FG loop of p40 D2 interacting with negatively charged residues in IL-12Rβ1.
(G) View of the site 3 interaction between IL-23R and IL-23p19 highlighting Trp156 in IL-23p19 packing against Gly116 in IL-23R and the N-terminal extension of IL-23R making contacts with the top of helix D in IL-23p19.
In pre-clinical models, IL-12 administration has been shown to promote tumor clearance (Ardolino et al., 2014; Brunda et al., 1995; Brunda et al., 1993; Momin et al., 2019); however, translating these findings to human immunotherapy has been challenging due to toxicity associated with IL-12 administration (Cohen, 1995; Leonard et al., 1997). In mice, IL-12 toxicity is mediated by NK cells when co-administered with IL-2 (Carson et al., 1999), IL-15 (Biber et al., 2002; Carson et al., 1999) or IL-18 (Carson et al., 2000), suggesting that narrowing the pleotropic actions of IL-12 may be beneficial in reducing toxicity.
IL-12 shares its p40 subunit with another four-helix bundle, IL-23p19, to form the cytokine IL-23 (Oppmann et al., 2000). IL-23 signals through a receptor complex formed by IL-12Rβ1 and IL-23 receptor (IL-23R) (Figure 1A) (Parham et al., 2002). As a shared receptor for IL-12 and IL-23, IL-12Rβ1 is expressed on T cells, NK cells, and monocytes (Uhlen et al., 2015) while IL-23R is found on γδ T cells (Liang et al., 2013) and CD4+ T cells (Yeste et al., 2014). Despite shared use of IL-12Rβ1, IL-12 and IL-23 have distinct phenotypic effects. In CD4+ T cells, IL-23 signaling promotes phosphorylation of STAT3 and stabilization of the IL-17-producing Th17 lineage (Zhou et al., 2007). While Th17 cells and IL-23 signaling play an important role in the immune response against extracellular pathogens, aberrant Th17 activity has been associated with multiple autoimmune conditions (Littman and Rudensky, 2010). Indeed, genetic deficiency in IL-23p19 protects mice against experimental autoimmune encephalomyelitis (Thakker et al., 2007) and colitis (Yen et al., 2006). Clinically, antagonist antibodies targeting IL-23 have been approved for the treatment of moderate to severe plaque psoriasis, psoriatic arthritis, Crohn’s disease and ulcerative colitis; however, many of these antibodies block both IL-12 and IL-23 signaling (Chan et al., 2018; Kopp et al., 2015; Koutruba et al., 2010). Thus, a deeper understanding of the basis of IL-12Rβ1 sharing could enable better separation of the divergent functions of IL-12 and IL-23.
Currently, it is unclear how these closely related cytokines engage their shared receptor subunits. A crystal structure of IL-23R bound to IL-23 showed the N-terminal Ig domain of IL-23R engages IL-23 in an extended conformation (Bloch et al., 2018), analogous to the ‘site 3’ paradigm established by the interaction between IL-6 and gp130 (Boulanger et al., 2003b; Chow et al., 2001). However, blocking antibodies which bind the shared p40 subunit neutralize signaling of both IL-12 and IL-23 (Benson et al., 2011), suggesting that the assembly of IL-12 and IL-23 receptors may be unique compared to the gp130 model.
Here we report a 2.0Å resolution crystal structure of IL-12Rβ1, a 3.4Å resolution crystal structure of the IL-23(IL-23p19/p40)/IL-23R/IL-12Rβ1 receptor complex, as well as cryo-EM structures of complete IL-12 and IL-23 receptor extracellular domain (ECD) complexes. Together, these structures reveal a modular mechanism of IL-12 and IL-23 receptor assembly which is unique among the cytokine superfamily. We targeted the ‘shared’ IL-12Rβ1/p40 interaction site to design a series of IL-12 partial agonists that preferentially supported T cell activation with reduced activity on NK cells. IL-12 partial agonists exhibited similar cell-type biases in vivo and promoted anti-tumor responses in the MC-38 murine colorectal cancer model without inducing toxicity associated with wild-type IL-12 administration. Thus, the unique mechanism of IL-12Rβ1 sharing provides a blueprint to engineer functionally selective agonists.
Results:
Crystal structures of IL-12Rβ1 and the quaternary IL-23 receptor complex
IL-23(IL-23p19/p40) signals through a receptor complex composed of IL-23R and IL-12Rβ1 (Figure 1A). The ECD of IL-12Rβ1 consists of 5 fibronectin type III (FNIII) domains of which the N-terminal D1–D2 domains mediate binding to IL-23 (Schroder et al., 2015). To determine the structural basis for IL-23 receptor assembly, we crystalized a complex of IL-12Rβ1 D1–D2 with IL-23 and the IL-23R ectodomain. The quaternary complex diffracted to 3.4 Å resolution (Table S1) and we determined a structure of a partial complex by molecular replacement using the previously published IL-23R ternary (IL-23p19/p40/IL-23R) complex (Bloch et al., 2018). Additional electron density for IL-12Rβ1 was observed but there was no published IL-12Rβ1 structure to use as a search model. Thus, we determined the structure of the human IL-12Rβ1 D1–D2 domains to a resolution of 2.0 Å by multi-wavelength anomalous dispersion (MAD) (Figure 1B, Table S1). We then used this structure as a search model and were able to place the IL12Rβ1 D1 domain in the electron density of the quaternary complex. The D2 domain was not visible, likely due to interdomain flexibility (Figure 1C–D, Figure S1A–B, Table S1).
The quaternary IL-23 receptor complex exhibits a modular architecture in which IL-23 serves as a bridge connecting IL-23R with IL-12Rβ1 to initiate intracellular JAK2/Tyk2 trans-phosphorylation (Figure 1C–G). The tip of IL-23p19 and the side of p40 collectively engage the N-terminal Ig domain of IL-23R in a classical “site 3” interaction as previously observed (Bloch et al., 2018) (Figure 1G). Similar to other IL-6 family cytokines, this interaction is anchored by an aromatic residue at the tip of IL-23p19 (Trp156) which packs against the inner beta sheet of IL-23R D1 Ig domain (Boulanger et al., 2003b). Alignment of the two IL-23R bound structures on IL-23p19 shows that IL-23R is rotated by 18° in our structure compared to the previously reported partial receptor complex (Figure S1C, Table S2).
The shared receptor, IL-12Rβ1 binds to the “back” of p40, at the intersection between the N-terminal D1 Ig and D2 fibronectin domains of p40. IL-12Rβ1 binds p40 in a single, 1532 Å2 interface that is characterized by a high degree of charge complementarity between the interacting proteins. The base of the interface is formed by a contiguous, positively charged loop in p40 (His216, Lys217, and Lys219) which interacts with a negatively charged patch in IL-12Rβ1 made up of Glu28, Asp58 and Asp101 (Figure 1F). Above these charge-charge interactions sits a hydrophobic strip on p40 formed by the aromatic residues, Trp37 and Phe82, which is ringed by polar residues in IL-12Rβ1 (Gln102, Ser106, Tyr109, Gln132, and Tyr134) that make hydrogen bonding interactions with side chain and main chain atoms in p40 (Table S2). Interestingly, the IL-12Rβ1 binding site on p40 appears to be a hotspot for protein-protein interactions as both the clinical antibodies, Ustekinumab and Briakinumab, along with the nanobody chaperone, Nb22E11, bind to this site (Figure S2) (Bloch et al., 2018; Desmyter et al., 2017; Luo et al., 2010).
The mechanism of IL-23 receptor assembly is distinct from IL-6 family cytokines
The IL-23p19/p40 heterodimer bears a strong resemblance to the IL-6Rα/IL-6 binary ‘site 1’ complex, leading to the conjecture that IL-23 would use a similar receptor assembly mechanism as seen in IL-6 and other gp130-family cytokines (Figure 2A–B) (Boulanger et al., 2003a; Boulanger et al., 2003b; Lupardus and Garcia, 2008; Yoon et al., 2000). IL-6 has formed the paradigm for assembly of ‘tall’ cytokine receptors such as gp130, LIFR, and OSM-R, which contain additional membrane-proximal FNIII domains compared to other ‘short’ cytokine receptors, such as growth hormone receptor (GHR) and IL-2 receptors. This assembly model has been correctly applied to signaling complexes for ciliary neurotrophic factor (CNTF) (Skiniotis et al., 2008) and leukemia inhibitory factor (LIF) (Boulanger et al., 2003a; Huyton et al., 2007). In the IL-6 paradigm, the four-helix bundle of IL-6 binds to IL-6Rα and gp130 through opposing helical faces, sites 1 and 2, respectively, while engaging a second gp130 molecule at the tip of IL-6 (site 3) to generate a full, hexameric complex (Figure 2C) (Boulanger et al., 2003b).
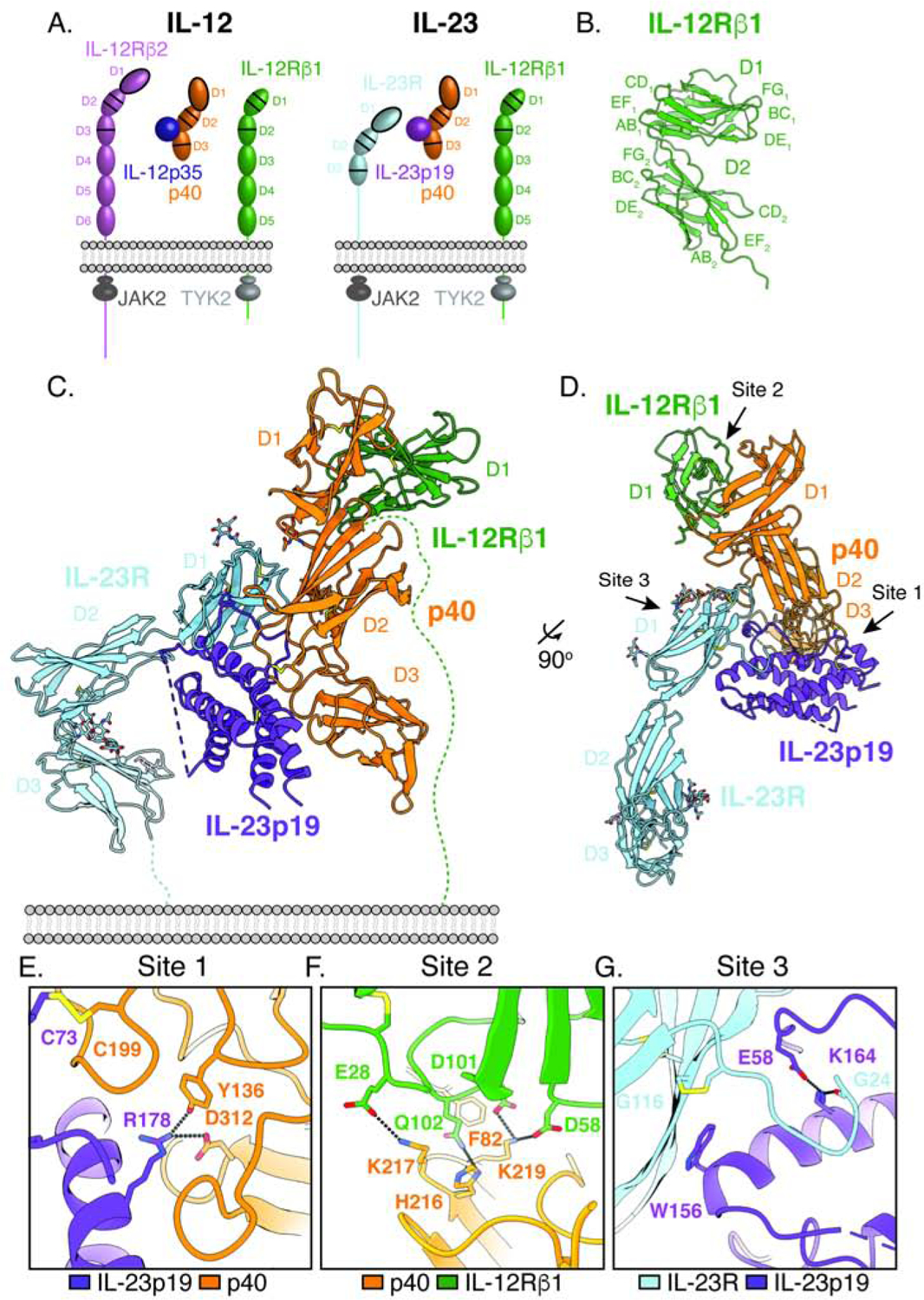
Figure 2. The mechanism of IL-23 receptor assembly is distinct from IL-6 family cytokines.
(A–B) Structural homology between the IL-23 (PDB: 3DUH) and the ‘site 1’ IL-6Rα/IL-6 complex modeled using crystal structures of the hexameric IL-6 receptor complex (PDB: 1P9M) and IL-6Rα D1–D3 (PDB: 1N26).
(C) Top-down view of the hexameric IL-6 receptor complex (PDB: 1P9M). Arrows indicate the site 1, 2, 3 interactions between IL-6 and receptor subunits.
(D) Top-down view of the quaternary IL-23 receptor complex. Arrows indicate the site 1 and site 3 interactions, analogous to those in the IL-6 complex. IL-23 does not use a canonical site 2 interface on IL-23p19; instead, IL-12Rβ1 binds p40 at a site distal to IL-23p19.
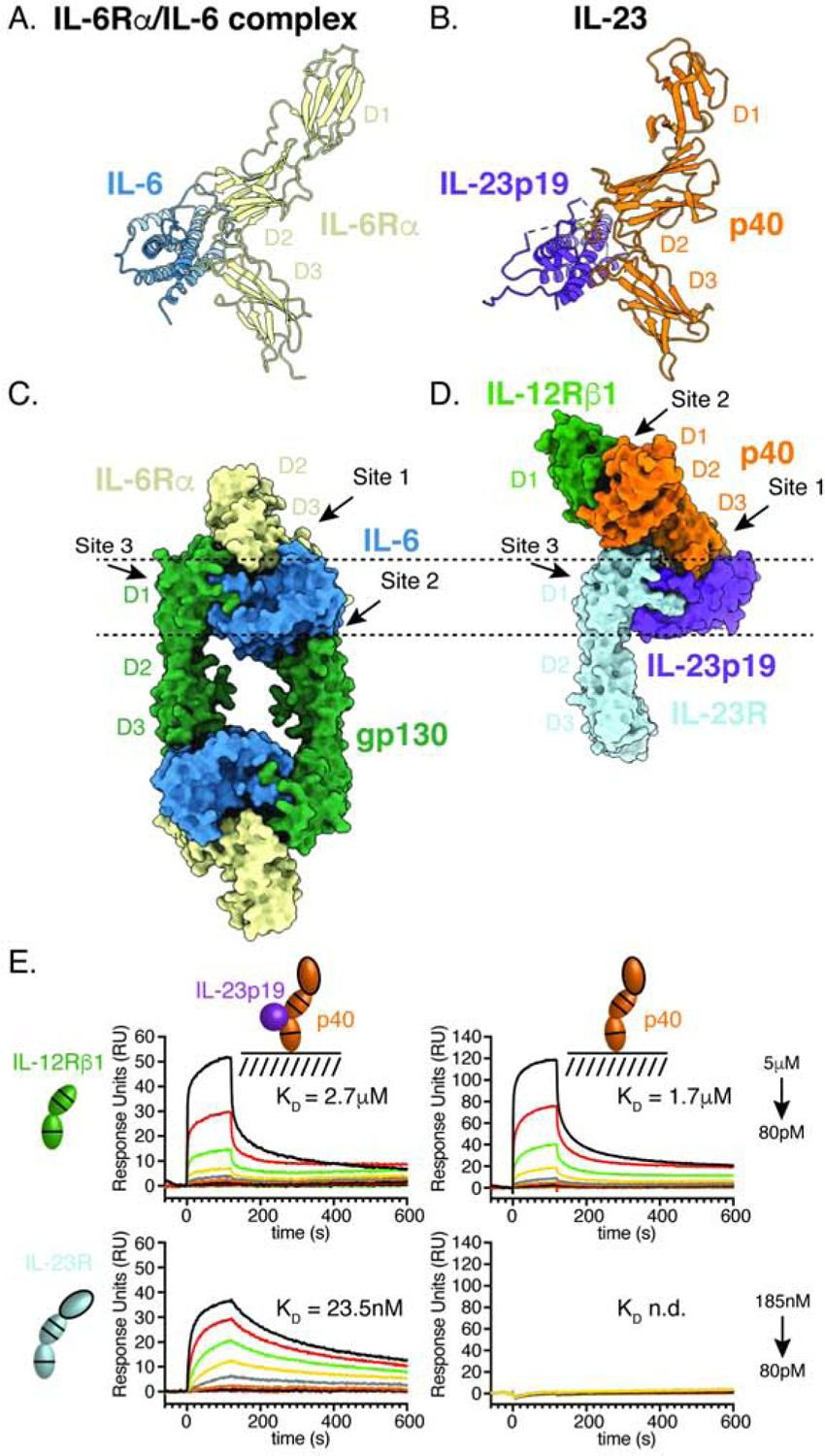
(E) IL-23p19 is required for IL-23R binding but is dispensable for IL-12Rβ1 recruitment. Surface plasmon resonance (SPR) sensorgrams showing binding of receptor subunits to immobilized IL-23 (left column) or p40 (right column). Receptor extracellular domains were flowed over the sensor chip at 3-fold dilution from 185nM to 80pM (IL-23R) or 5μM to 80pM (IL-12Rβ1). Dissociation constants (KD) for IL-23R (top row) and IL-12Rβ1 (bottom row) were determined using a steady state affinity model. For pairs in which no interaction was observed, KD is listed as n.d. for not determined.
Analogous to IL-6, IL-23 consists of a site 1 interaction between IL-23p19 and p40. IL-23R binds IL-23 via a site 3 interaction between the N-terminal Ig domain of IL-23R and the tip of IL-23p19. However, IL-23 breaks with the IL-6 paradigm in that IL-12Rβ1 is predicted to bind to IL-23p19 opposite p40 at the site analogous to IL-6/gp130 “site 2”. Instead, IL-12Rβ1 binds directly to p40, having no contact with the helical cytokine p19 (Figure 2D). This interaction predicts that p40 should bind IL-12Rβ1 independently of IL-23p19. Consistent with this, we find that IL-12Rβ1 binds both IL-23 and p40 with similar affinity and thus is not affected by the presence of the helical IL-23p19 subunit (Figure 2E). In contrast, IL-23R binding requires the presence of IL-23p19, as predicted by the site 3 interaction observed in the crystal structure.
The modular interaction between p40 and IL-12Rβ1 represents a striking deviation from the typical mechanism of receptor sharing employed by other cytokines. Rather than interacting with multiple cytokines through a cross-reactive interface, IL-12Rβ1 binds the p40 subunit using highly specific interactions characterized by charge complementarity and hydrogen bonding.
Affinity maturation of IL-12Rβ1 facilitates comparison of IL-12 and IL-23 receptor complexes by cryo-EM
We wished to determine if the unusual assembly mechanism we observed for IL-23 extended to the IL-12 receptor complex. Since cryo-EM studies are carried out at relatively dilute protein concentrations at which low affinity complexes dissociate, we ‘affinity-matured’ IL-12Rβ1 binding to IL-23 using an error prone yeast display library in order to stabilize the receptor complexes for structural studies by cryo-EM (Figure 3A). Although the selections were performed using IL-23, enriched clones displayed heightened binding to both IL-12 and IL-23, consistent with IL-12Rβ1 binding to the shared p40 subunit of both cytokines (Figure 3B). Sequencing of the enriched population revealed a pair of affinity enhancing mutations in IL-12Rβ1 (Tyr109Ser Gln132Leu, referred to hereafter as affi-IL-12Rβ1), which map to the p40 binding interface, thereby providing independent validation of the interaction observed in the IL-23 receptor crystal structure (Figure 3C–D).
Figure 3. Affinity maturation of IL-12Rβ1 facilitates structural characterization of IL-12 and IL-23 receptor complexes by cryo-EM.
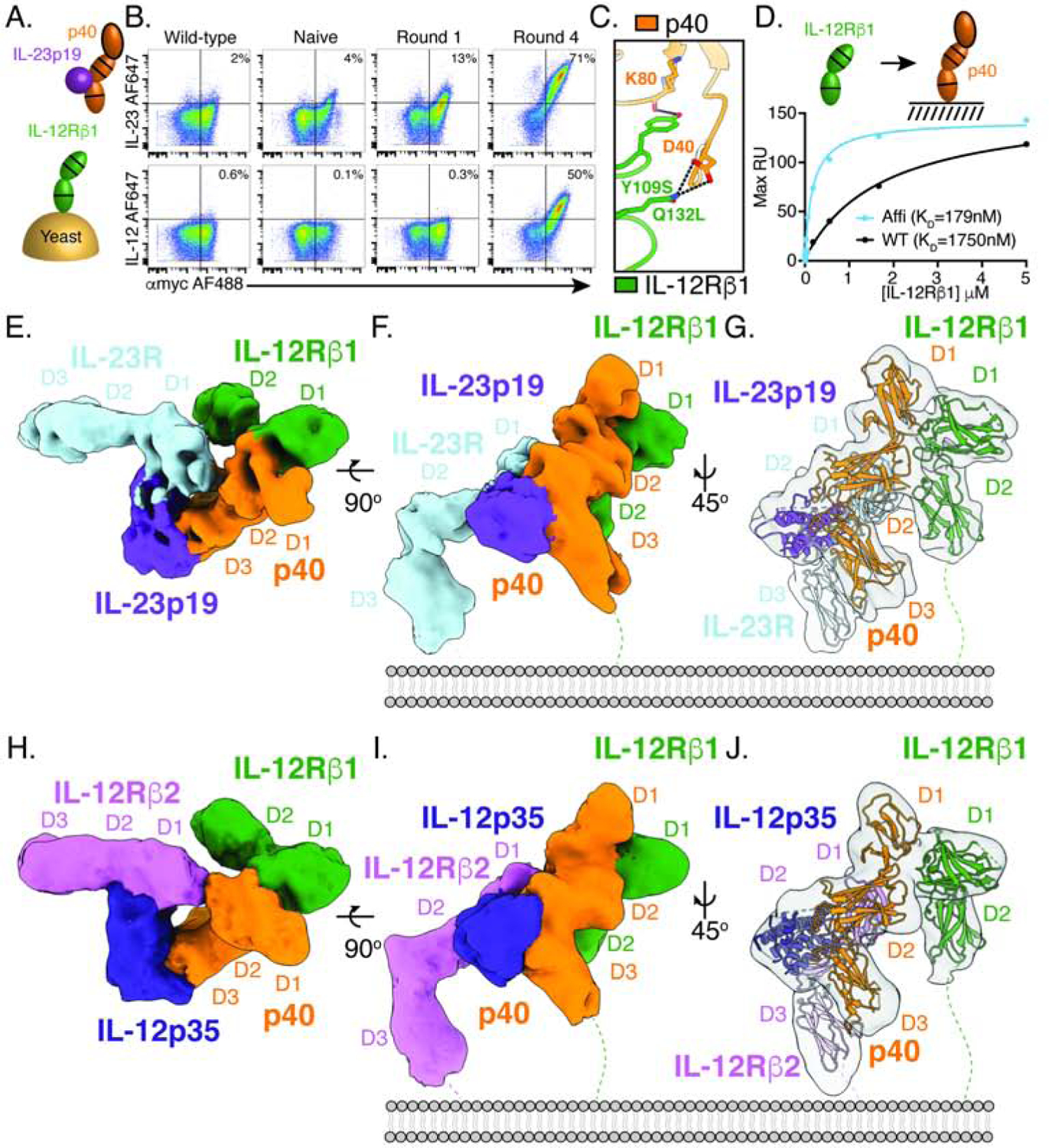
(A) Schematic of IL-12Rβ1 affinity maturation strategy. The D1–D2 domains of IL-12Rβ1 were expressed as a fusion with N-terminal Aga2 and an error prone library was generated. Yeast were selected with biotinylated IL-23, increasing the stringency of selection each round.
(B) Affinity maturation of IL-12Rβ1 for IL-23 enhances binding to IL-23 and IL-12. Flow cytometry plots showing binding of 10nM IL-23 (top) or 10nM IL-12 (bottom) to IL-12Rβ1 expressing yeast. Yeast were stained with αmyc-AF488 (x-axis) as a measure of IL-12Rβ1 surface expression and biotinylated cytokine followed by streptavidin-AF647 (y-axis) as a measure of cytokine binding. The left-hand column shows isogenic yeast expressing wild-type IL-12Rβ1 and subsequent columns show the error prone library prior to selection (naïve) or at different rounds, as indicated.
(C) Affinity enhancing mutations map to the interface between IL-12Rβ1 and p40. Interface view of IL-12Rβ1 binding to p40 with labels indicating convergent mutations identified in the error prone selections (Tyr109Ser Gln132Leu).
(D) IL-12Rβ1 mutations enhance p40 binding in solution. Surface plasmon resonance (SPR) curves showing binding of IL-12Rβ1 D1–D2 to immobilized p40. Dissociation constants (KD) were determined using a steady state affinity model. RU, response units.
(E–F) Cryo-EM map of the IL-23 receptor complex as viewed from the top (E) and side
(F). Individual subunits are colored as in Figure 1.
(G) Cartoon model showing individual components IL-23R (PDB: 5MZV), IL-23 (PDB: 3DUH), and IL-12Rβ1 (reported here) fit to the map density.
(H–I) Cryo-EM map of the IL-12 receptor complex as viewed from the top (H) and side
(I). Individual subunits are colored as follows: IL-12Rβ2 in magenta, IL-12p35 in blue, p40 in orange, and IL-12Rβ1 in green.
(J) Cartoon model showing IL-12Rβ2 (no reported crystal structure, using IL-23R PDB: 5MZV in model), IL-12 (PDB: 1F45), and IL-12Rβ1 (reported here) fit to the map density.
See also Figure S3.
For structural analysis, IL-23 was complexed with the ectodomain of IL-23R and the full extracellular domain of IL-12Rβ1 (D1–D5) containing affinity-enhancing mutations. The complex was purified by size exclusion chromatography and vitrified on grids for single-particle cryo-EM analysis. Particle projections from 2D averages with well-defined features for the complex were selected for 3D classification, thereby identifying a 3D class that was refined to a resolution of 8.0 Å (figure S3A–D, Table S3). Despite the low resolution of the maps, individual subunits can be clearly delineated. The overall assembly is consistent with the binding mode observed in the crystal structure in which receptors IL-23R and IL-12Rβ1 engage the IL-23p19 and p40 cytokine components, respectively (Figure 3E–G). While both the D1–D2 domains of IL-12Rβ1 are observed in the cryo-EM maps, poor density is seen for IL-12Rβ1 D3–D5, indicating that this region is flexible.
To visualize the IL-12 receptor complex, we applied a similar approach, complexing IL-12, IL-12Rβ2 D1–D3, and affi-IL-12Rβ1 D1–D5. This yielded a structure of the IL-12 receptor complex at a resolution of 10.0 Å (Figure S3E–H, Table S3). The overall architecture of the IL-12 receptor complex is similar to the arrangement seen in the IL-23 crystal structure and cryo-EM maps. IL-12Rβ2 engages IL-12 via an N-terminal Ig domain similar to the interaction between IL-23R and IL-23p19 while IL-12Rβ1 binds the back of p40 (Figure 3H–J). In both IL-12 and IL-23, the shared p40 subunit directly binds the common IL-12Rβ1, indicating a convergent function of p40 in mediating the assembly of IL-12 and IL-23 signaling complexes.
p40 acts as a common regulator of IL-12 and IL-23 signaling
Despite use of the shared p40 subunit, IL-12 and IL-23 play non-redundant roles in the immune system. Comparison of the cryo-EM maps indicate that functional differences between IL-12 and IL-23 are likely the result of their unique four-helix bundle components, IL-12p35 and IL-23p19, specifically engaging private receptors, IL-12Rβ2 and IL-23R, which leads to STAT4 and STAT3 activation, respectively. To determine if the four-helix bundle components of IL-12 and IL-23 mediate distinct receptor interactions, we measured binding of IL-12Rβ2 and IL-23R to IL-12 and IL-23 by SPR. We found that these ‘private receptors’ mediate highly specific interactions: IL-12Rβ2 binds specifically to IL-12, but not IL-23, and IL-23R binds to IL-23 but not IL-12 (Figure 4A).
Figure 4. P40 acts as a common regulator of IL-12 and IL-23 signaling.
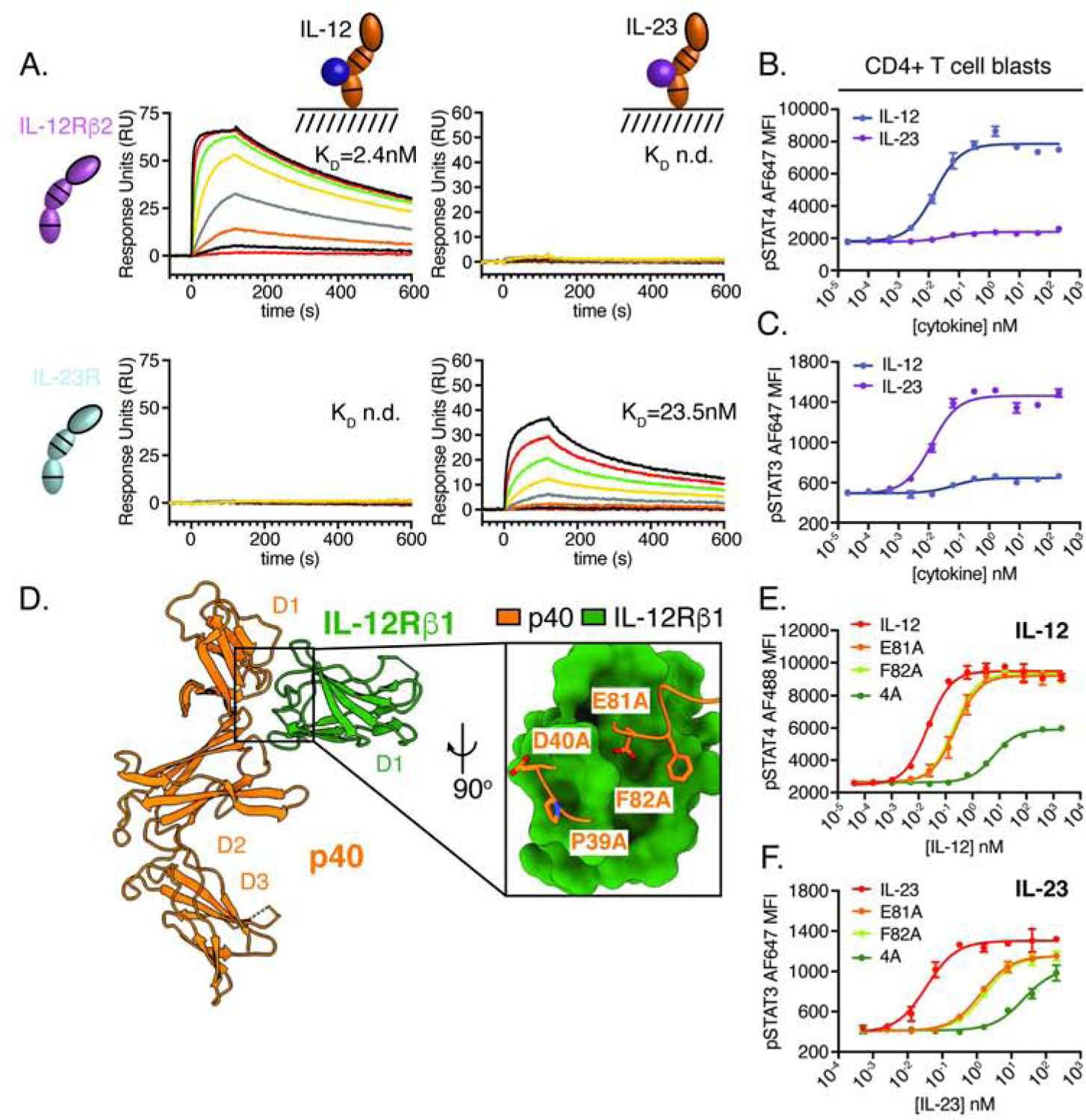
(A) IL-12p35 and IL-23p19 mediate specific interactions with receptor extracellular domains. Surface plasmon resonance (SPR) sensorgrams showing binding of IL-12Rβ2 (top row) and IL-23R (bottom row) to immobilized IL-12 (first column) and IL-23 (second column). Receptor extracellular domains were flowed over the sensor chip at 3-fold dilution from 185nM to 80 pM. Dissociation constants (KD) were determined using a steady state affinity model or listed as n.d. for not determined when no interaction was observed. The interaction between IL-23R and IL-23 originally shown in Figure 2E is reproduced here to highlight the specificity of the interaction.
(B–C) IL-12 and IL-23 induce distinct patterns of STAT phosphorylation in CD4+ T cell blasts. Human peripheral blood mononuclear cells were activated with 2.5μg/mL αCD3, 5μg/mL αCD28, and 100IU/mL IL-2 for 2 days prior to being rested overnight, stained to identify CD4+ T cells, stimulated for 20 minutes with IL-12 or IL-23 and analyzed by phospho-flow cytometry.
(D) Snapshot of the quaternary IL-23 receptor complex highlighting the interaction between IL-12Rβ1 and p40. A panel of p40 muteins (Glu81Ala, Phe82Ala and 4A: Pro39Ala/Asp40Ala/Glu81Ala/Phe82Ala) were co-expressed with IL-12p35 or IL-23p19 to generate IL-12 and IL-23 variants for analysis of pSTAT signaling. (E) Phospho-flow cytometry of pSTAT4 in human CD4+ T cell blasts following stimulation with IL-12 variants for 20 minutes. (F) Phospho-flow cytometry of pSTAT3 in human CD4+ T cell blasts following stimulation with IL-23 variants for 20 minutes. MFI, mean fluorescence intensity. Phospho-STAT data were expressed as mean ± standard deviation of two biological replicates and are representative of two or more independent experiments.
This extracellular receptor specificity is relayed to distinct patterns of intracellular signaling where IL-12 led to phosphorylation of STAT4 with minimal activation of STAT3 while IL-23 led to phosphorylation of STAT3 without inducing STAT4 phosphorylation in primary human CD4+ T cells (Figure 4B–C). The specificity of IL-12 and IL-23 signaling suggests that p40 plays similar roles in both complexes to recruit IL-12Rβ1/Tyk2 and initiate STAT signaling specified by phospho-tyrosine motifs in IL-12Rβ2 and IL-23R.
Based on the shared role of the p40/IL-12Rβ1 interaction in both the IL-12 and IL-23 receptor complexes, we targeted this interface to control STAT4 signaling in the context of IL-12, and STAT3 signaling in the context of IL-23, by modulating the efficiency of IL-12Rβ1 recruitment. We created a series of IL-12 and IL-23 variants by introducing alanine substitutions in two loops of p40 D1 that mediate interactions with IL-12Rβ1 (Figure 4D). We found that individual alanine mutations (Glu81Ala and Phe82Ala) reduced the potency of IL-12 and IL-23, as indicated by an increase in the half-maximal dose (effective concentration 50, EC50) for pSTAT4 and pSTAT3. This effect was exaggerated by multiple alanine mutations (4A: Pro39Ala/Asp40Ala/Glu81Ala/Phe82Ala) which had decreased maximal STAT phosphorylation and increased the EC50 relative to wild-type cytokines (Figure 4E–F). The magnitude of these effects were larger for IL-23, likely due to the ~10 fold difference in affinity between IL-12 and IL-23 for IL-12Rβ2 and IL-23R, respectively. The ability of a single interface on p40 to modulate pSTAT3 and pSTAT4 suggest that p40 can serve as a fulcrum to control both IL-12 and IL-23 signaling.
IL-12 partial agonists preferentially support T cell function with reduced activity on NK cells based on differences in IL-12Rβ1 expression
IL-12 has shown promise as a cancer therapy by potentiating both T cells and NK cells, however, systemic administration of IL-12 leads to toxicity associated with heightened IFNγ production (Cohen, 1995; Leonard et al., 1997). Biasing IL-12 to preferentially activate T cells with reduced NK cell IFNγ induction may decrease toxicity by focusing the actions of IL-12 on antigen-specific T cells. An important difference in IL-12 signaling between T cells and NK cells is that antigen stimulation through the T cell receptor (TCR) enhances IL-12 sensitivity through upregulation of IL-12 receptor subunits (Szabo et al., 1997). Using p40 as a FACS staining reagent to assess IL-12Rβ1 surface expression, we found low levels of IL-12Rβ1 on NK cells and CD8+ T cells, however, stimulation of CD8+ T cells (blasts) led to upregulation of IL-12Rβ1 (Figure 5A, Figure S4A).
Figure 5. IL-12 partial agonists preferentially support T cell function with reduced activity on NK cells based on differences in IL-12Rβ1 expression.
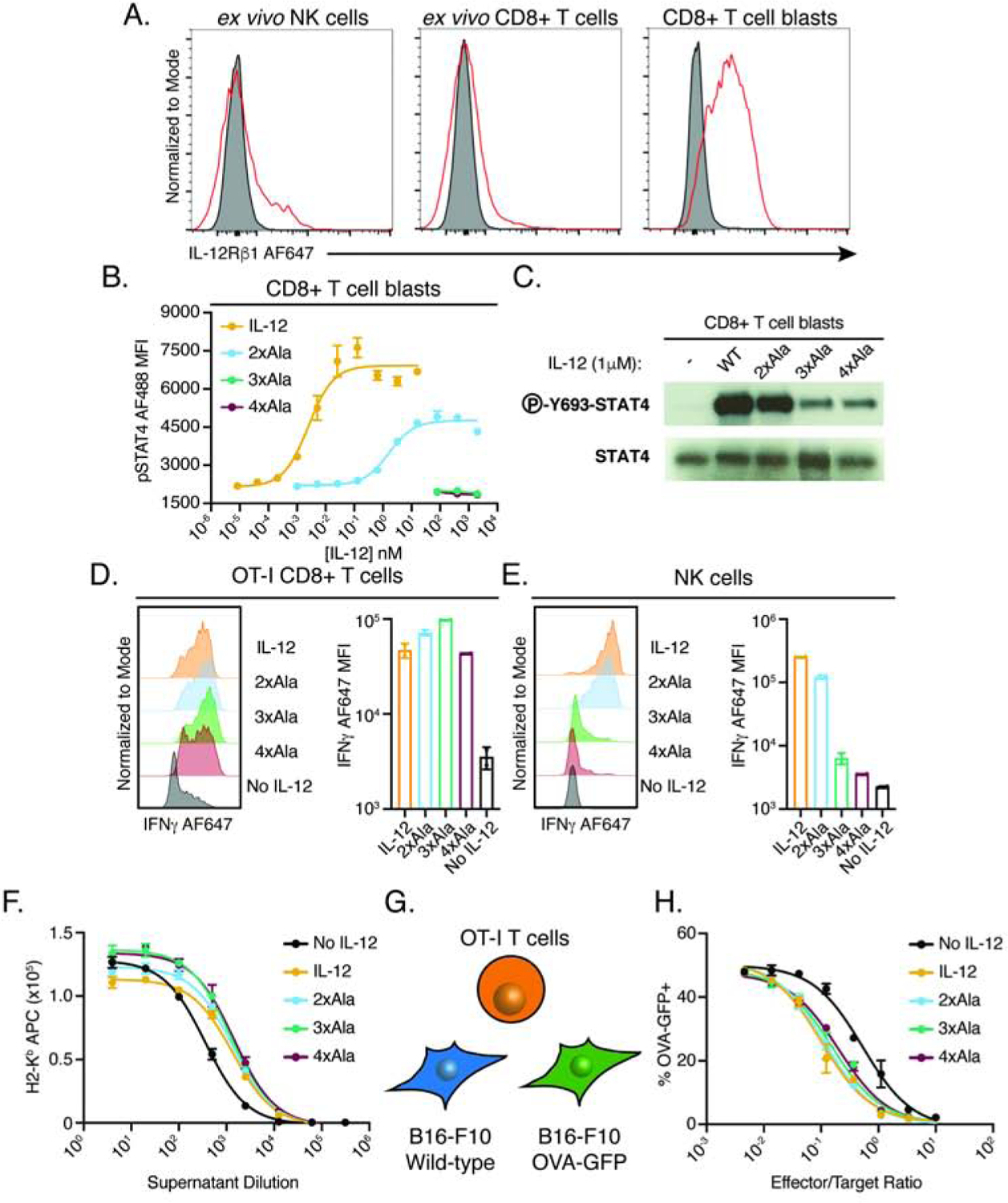
(A) Cell-type and activation-state dependent expression of IL-12Rβ1. Flow cytometry plots showing IL-12Rβ1 expression levels measured by mouse p40 tetramer staining of murine NK cells (CD3-NK1.1+) or CD8+ T cells (CD3+CD8+). Red line indicates 200nM tetramer staining, gray population represents streptavidin staining alone. Single cell suspension of spleen and lymph nodes from female C57BL/6J mice were stained with p40 tetramer either directly (ex vivo) or following 2-day stimulation with 2.5μg/mL αCD3, 5μg/mL αCD28, and 100IU/mL IL-2 (blasts). For gating, see Figure S4A.
(B–C) p40 mutations modulate IL-12 signaling in CD8+ T cell blasts. (B) Phospho-flow cytometry of STAT4 signaling in CD8+ T cell blasts following 20-minute incubation with IL-12 or partial agonists (2xAla: Glu81Ala/Phe82Ala, 3xAla: Glu81Ala/Phe82Ala/Lys106Ala, 4xAla: Glu81Ala/Phe82Ala/Lys106Ala/Lys217Ala). (C) Western blot of STAT4 phosphorylation in whole cell lysate of CD8+ T cell blasts stimulated with IL-12 partial agonists for 20 minutes.
(D) IL-12 partial agonists promote IFNγ production by antigen-specific CD8+ T cells. Representative histograms (left) and quantification (right) of intracellular IFNγ in OT-I CD8+ T cells (CD3+CD8+) following 48-hour stimulation with 1μg/mL OVA (257–264), 0.5μg/mL αCD28, 100IU/mL IL-2, and 1μM IL-12 variants. See also Figure S4C–F.
(E) IL-12 partial agonists display attenuated IFNγ induction in NK cell. Purified NK cells were stimulated with 50ng/mL IL-18 and 1μM IL-12 variants for 48 hours. See also Figure S5C–E.
(F) Supernatants from IL-12 and partial agonist stimulated OT-I effectors more potently upregulate MHC-I on B16-F10 melanoma cells. Supernatants from OT-I effectors generated with or without IL-12 agonists were added to B16-F10 overnight and MHC-I expression was read out by surface staining for H-2Kb.
(G–H) IL-12 partial agonists support antigen-specific tumor cell killing. (G) Schematic of the specific killing assay. A 1:1 mixture of wild-type cells and OVA-GFP expressing B16-F10 cells were incubated with varying ratios of OT-I effectors for 36 hours and the frequency of OVA-GFP+ cells was read-out to measure antigen-specific killing. (H) Dose response curves showing specific killing of OT-I effectors generated with or without IL-12 agonists.
Dose response curves and bar graphs show mean ± standard deviation of two biological replicates and are representative of two or more independent experiments. MFI, mean fluorescence intensity.
Based on our structural finding that p40 mediates recruitment of IL-12Rβ1 in order to initiate heterodimerization with IL-12Rβ2 or IL-23R, we reasoned that reducing the affinity of p40 for IL-12Rβ1 may more severely impair signaling on NK cells which have reduced levels of IL-12Rβ1 expression relative to antigen-experienced T cells. To test this, we designed a series of alanine mutations in murine p40 that are predicted to disrupt binding to IL-12Rβ1 based on sequence homology with human p40 (Figure S4B). To characterize mouse IL-12 variants, we tested signaling on CD8+ T cell blasts and, as predicted, mutations in p40 at the IL-12Rβ1 binding interface increase the EC50 and reduce maximal STAT4 phosphorylation (Figure 5B). IL-12 with two alanine substitutions (2xAla: Glu81Ala/Phe82Ala) reduced STAT4 phosphorylation to roughly 50% of wildtype while IL-12 with three (3xAla: Glu81Ala/Phe82Ala/Lys106Ala) or four (4xAla: Glu81Ala/Phe82Ala/Lys106Ala/Lys217Ala) alanine substitutions did not induce measurable STAT4 phosphorylation by flow cytometry, however, we are able to observe residual pSTAT4 by western blot (Figure 5C). Functionally, these variants act as IL-12 partial agonists by reducing the maximal signaling output at saturating concentrations of cytokine.
A well-documented effect of IL-12 signaling in both T cells and NK cells is the induction of IFNγ. To determine the capacity of IL-12 partial agonists to promote IFNγ production in antigen-specific CD8+ T cells, we activated ovalbumin(OVA)-specific OT-I T cell (Hogquist et al., 1994) for 48 hours in the presence of IL-12 variants. IL-12 along with the 2xAla, 3xAla, and 4xAla variants led to upregulation of IFNγ as measured by intracellular cytokine stain (Figure 5D, Figure S4C–E) and supernatant ELISA (Figure S4F). Similar effects were observed in CD4+ T cells where TCR stimulation led to upregulation of IL-12Rβ1 and IL-12 partial agonists supported Th1 polarization of naïve CD4+ T cells as measured by IFNγ stain, although to a lesser extent than wild-type IL-12 (Figure S4G–I). Thus, IL-12 partial agonists support IFNγ induction in both CD4+ and CD8+ T cells.
To assess the ability of IL-12 variants to stimulate NK cells, we first confirmed that IL-12 partial agonists modulate STAT4 signaling in NK cells. Similar to the effects in CD8+ T cell blasts, we find that mutations in p40 at the IL-12Rβ1 binding site reduce STAT4 phosphorylation in NK cells (Figure S5A). Unlike T cells which require stimulation through the T cell receptor to respond to IL-12, NK cells produce IFNγ in response to IL-12 in combination with the IL-1 family cytokine, IL-18 (Figure S5B). IL-12 and IL-18 stimulation induced robust IFNγ expression, a response that was attenuated with 3xAla and 4xAla variants as measured by intracellular cytokine stain (Figure 5E, Figure S5C–D) and supernatant ELISA (Figure S5E). IL-12 and IL-18 also promoted upregulation of Ifng at the transcript level following 8-hour stimulation, an effect that was reduced with 3xAla/IL-18 stimulation (Figure S5F); however, under these conditions, we did not observe induction of Tigit by IL-12 (Figure S5G). Previously, the γc family cytokines IL-2 and IL-15 have been shown to modulate the activity of NK cells and lead to upregulation of IL-12 receptor components (Lehmann et al., 2001; Oka et al., 2020; Romee et al., 2012). Consistent with these reports, we find that pre-activation of NK cells with IL-2 led to a slight upregulation of IL-12Rβ1 (Figure S5H). Addition of IL-2 to NK cell cultures increased IFNγ production above IL-18 alone; however, IL-2 did not synergize with 3xAla and 4xAla to enhance IFNγ induction above IL-2/IL-18 (Figure S5I).
To determine if human IL-12 partial agonists were capable of eliciting cell-type specific responses, we assessed IL-12Rβ1 expression and IFNγ production in human peripheral blood mononuclear cells (PBMCs). Similar to our findings in mouse, TCR stimulation enhanced IL-12Rβ1 expression in CD8+ T cells above that of non-activated T cells and NK cells (Figure S6A–B). We generated analogous IL-12 muteins and tested pSTAT4 signaling in CD8+ T cell blasts (Figure S6C–D). Human IL-12 partial agonists preferentially supported induction of IFNγ by CD8+ T cells relative to NK cells (Figure S6E–F). These findings indicate that upregulation of IL-12Rβ1 is a conserved mechanism used by T cells to enhance sensitivity to IL-12 signaling and that IL-12 partial agonists are capable of biasing signaling towards T cells in both human and mouse.
IL-12 partial agonists promote antigen-specific tumor killing
In CD8+ T cells, IL-12 acts to potentiate antigen-specific killing of tumors and virally infected cells through upregulation of cytotoxic factors, such as granzyme B, and secretion of inflammatory cytokines including IFNγ (Aste-Amezaga et al., 1994; Rubinstein et al., 2015). A well described role of IFNγ is the upregulation of class I major histocompatibility complex (MHC-I), which can render transformed cells sensitive to T cell surveillance (Zhou, 2009). To determine if supernatants from OT-I effector T cells were capable of driving MHC-I upregulation, we assessed MHC-I surface expression on B16-F10 murine melanoma cells by antibody stain following incubation with supernatants from OT-I effectors. Supernatants from IL-12 and partial agonist cultures more potently induced MHC-I expression than supernatant generated in the absence of IL-12, consistent with elevated IFNγ levels observed by intracellular stain and ELISA (Figure 5F).
Our finding that IL-12 partial agonists promote IFNγ production and subsequent upregulation of MHC-I on tumor cell lines led us to examine the capacity of IL-12 partial agonists to potentiate tumor cell killing. To measure antigen-specific CD8+ T cell killing, we transduced B16-F10 cells with a plasmid containing OVA along with a GFP marker (OVA-GFP) and added these cells 1:1 with wild-type B16-F10 cells. This mixture of B16-F10 cells was then incubated with OT-I effectors and the frequency of OVA-GFP expressing cells was used as a read-out of antigen-specific tumor cell killing (Figure 5G). OT-I effectors generated in the presence of IL-12 or partial agonists were able to kill OVA-expressing tumor cells at a lower effector to target ratio, indicating increased potency of antitumor response (Figure 5H). Together, these data indicate that IL-12 partial agonists with reduced affinity for IL-12Rβ1 promote IFNγ production and tumor cell killing by antigen-specific CD8+ T cells with reduced activity on NK cells.
IL-12 partial agonists support antigen-specific T cell response with reduced NK cell activation in vivo
To test whether IL-12 partial agonists elicit cell-type specific responses in vivo, we adoptively transferred OT-I CD8+ T cells into congenic recipients and immunized with OVA (257–264) in Incomplete Freud’s Adjuvant (OVA-IFA) followed by daily cytokine administration for 5 days (Figure 6A). For in vivo studies, we expressed IL-12 and partial agonists in mammalian cells (Expi293F) and confirmed that mammalian-expressed IL-12 partial agonists retain cell-type bias in vitro, as seen for the baculovirus-expressed material used previously (Figure S7A–F).
Figure 6. IL-12 partial agonists elicit cell-type specific responses in vivo.
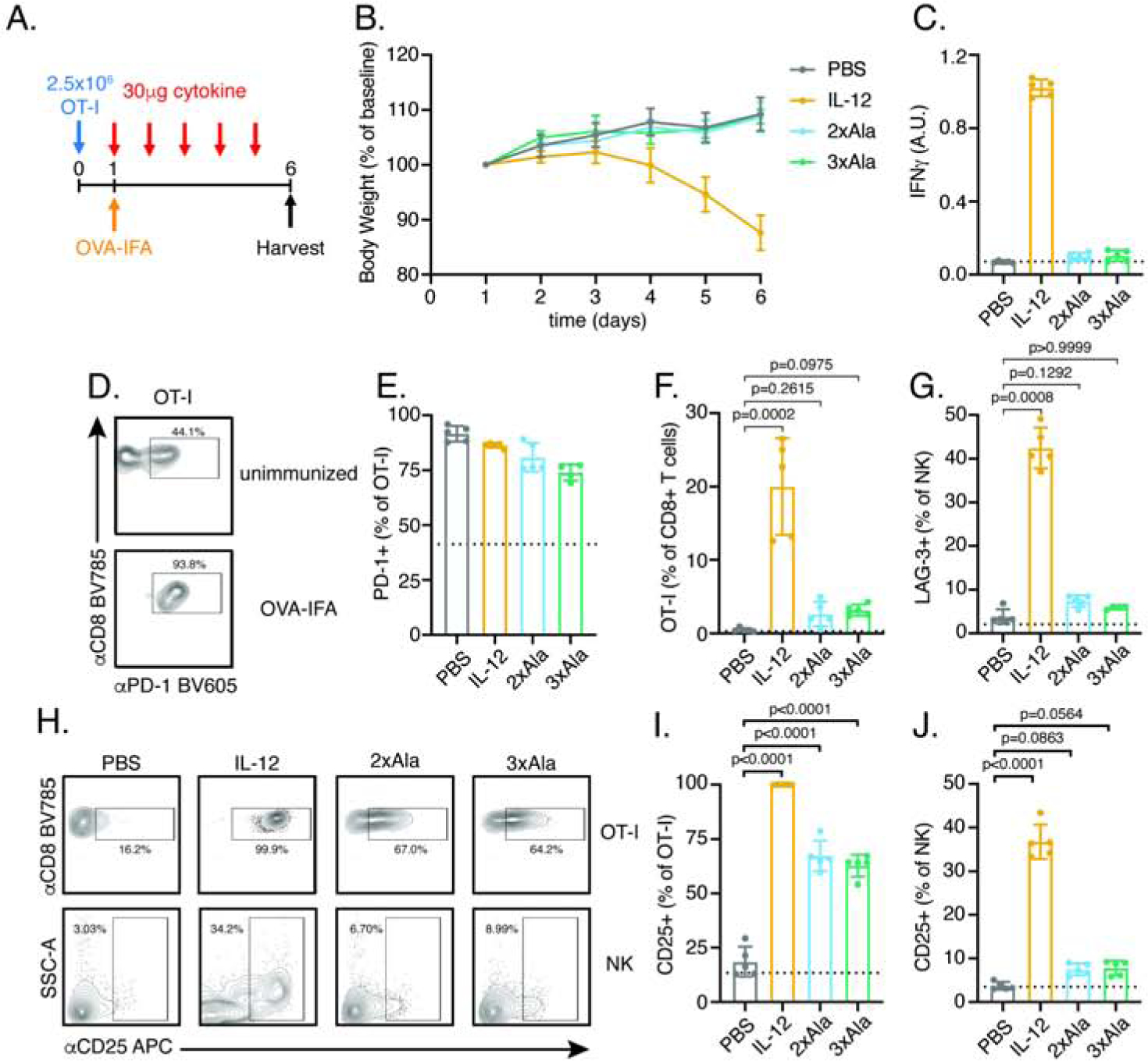
(A) Schematic of experimental design. CD8+ T cells from an OT-I TCR transgenic mouse (Thy1.2) were transferred to congenic recipient mice (Thy1.1) on day 0. The following day, mice were immunized subcutaneously with OVA (257–264) in Incomplete Freund’s Adjuvant (IFA) and daily interperitoneally injection of 30μg cytokine by was begun. Following 5 days of cytokine treatment, mice were euthanized for analysis of serum IFNγ by ELISA and cell-type profiling in draining lymph nodes by flow cytometry.
(B) IL-12 but not partial agonists result in weight loss. Mouse weight was monitored daily and normalized to body weight on day 1 prior to initiation of cytokine treatment.
(C) IL-12 but not partial agonists elevate systemic IFNγ as measured by serum ELISA on day 6. Dashed line represent measurement from unimmunized mice in this and subsequent panels.
(D–E) Immunization increases the frequency of PD-1+ OT-I T cell independent of cytokine treatment. (D) Representative FACS plots showing PD-1 expression in OT-I+ T cells identified as CD3+CD8+Thy1.2+. (E) Quantification of PD-1+ cells as a frequency of OT-I+ T cells.
(F) IL-12 but not partial agonists expand OT-I T cells. OT-T cells were identified as Thy1.2+ and expressed as a frequency of total CD8+ T cells. Data were analyzed by Kruskal-Wallis test with Dunn’s multiple comparisons.
(G) IL-12 but not partial agonists increase the frequency of LAG-3+ NK cells. Data were analyzed by Kruskal-Wallis test with Dunn’s multiple comparisons. See also Figure S7H.
(H–J) IL-12 partial agonists preferentially increase the frequency of CD25+ expressing OT-I T cells with reduced activity on NK cells relative to IL-12. (H) Representative FACS plots showing CD25 expression in OT-I T cells (top) and NK cells (bottom). (I) Quantification of CD25+ OT-I T cells. (J) Quantification of CD25+ NK cells. Data were analyzed by one-way ANOVA with Tukey’s multiple comparisons.
Data are expressed as mean ± standard deviation of n=5mice/group and are representative of two independent experiments. For gating, see Figure S7G.
Treatment with IL-12 but not 2xAla and 3xAla induced weight loss and elevated levels of IFNγ in serum (Figure 6B–C). To assess the impact of immunization on T cell activation, we monitored expression of the inhibitory receptor, PD-1, on OT-I T cells. Immunization increased the frequency of PD-1+ OT-I T cells independent of cytokine treatment, indicating activation of adoptively transferred cells (Figure 6D–E, Figure S7G). The effects of immunization were potentiated by IL-12 which increased the frequency of OT-I T cells in the draining lymph node, an effect not seen with partial agonists (Figure 6F). Within NK cells, IL-12 but not partial agonists increased a population of activated NK cells as measured by high side scatter (SSC) and expression of the inhibitory receptor, LAG-3 (Figure 6G, Figure S7H).
Previously the IL-2Rα chain, CD25, has been described as a marker of activated T cells (Au-Yeung et al., 2017) and NK cells (Leong et al., 2014). IL-12 strongly upregulated CD25 expression on both OT-I T cells and NK cells while the 2xAla and 3xAla partial agonists led to intermediate upregulation of CD25 on OT-I T cells without increasing expression on NK cells (Figure 6H–J). Interestingly, while the 2xAla variant did not exhibit as significant T/NK cell bias as the 3xAla variant in vitro (Figure S7E–F), it shows comparably strong T/NK cell bias to 3xAla in vivo, highlighting that the therapeutic window will likely be quantitatively different in vitro versus in vivo. These results indicate that IL-12 partial agonists support intermediate levels of T cell activation with reduced NK cell stimulation and toxicity in vivo.
IL-12 partial agonists support anti-tumor immunity with reduced toxicity relative to IL-12
Based on in vitro characterization and in vivo cell profiling, IL-12 partial agonists may be capable of supporting anti-tumor T cell immunity without systemic toxicity by biasing the activity of IL-12 towards antigen-specific T cells and away from NK cells. To determine the capacity of IL-12 partial agonists to provide therapeutic benefit in vivo, we performed tumor studies using the colon adenocarcinoma MC-38, which has been shown to be responsive to IL-12 (Corbett et al., 1975; Nastala et al., 1994). Mice were engrafted with MC-38 for 1 week prior to initiation of daily cytokine treatment for 7 days (Figure 7A). IL-12 administration, either at 1μg or 30μg, resulted in profound toxicity as measured by weight loss (Figure 7B), elevated serum IFNγ (Figure 7C) and reduced mobility (Figure 7D–E). All mice administered 30μg of IL-12 succumbed to lethal toxicity between days 13 and 15. As a result, we were unable to measure mobility of these mice on day 16. In contrast, the 2xAla and 3xAla partial agonists were well tolerated and did not induce toxicity in tumor-bearing mice. Although additional IL-12 responsive cell types including intraepithelial type 1 innate lymphoid cells (Fuchs et al., 2013) and mucosal associated invariant T (MAIT) cells (Wallington et al., 2018) are present in vivo, we find that IL-12 partial agonists with reduced activity on NK cells do not induce systemic toxicity consistent with the requirement for NK cells in IL-12 mediated toxicity (Biber et al., 2002; Carson et al., 2000; Carson et al., 1999).
Figure 7. IL-12 partial agonists support MC-38 anti-tumor response without inducing IL-12 associated toxicity.
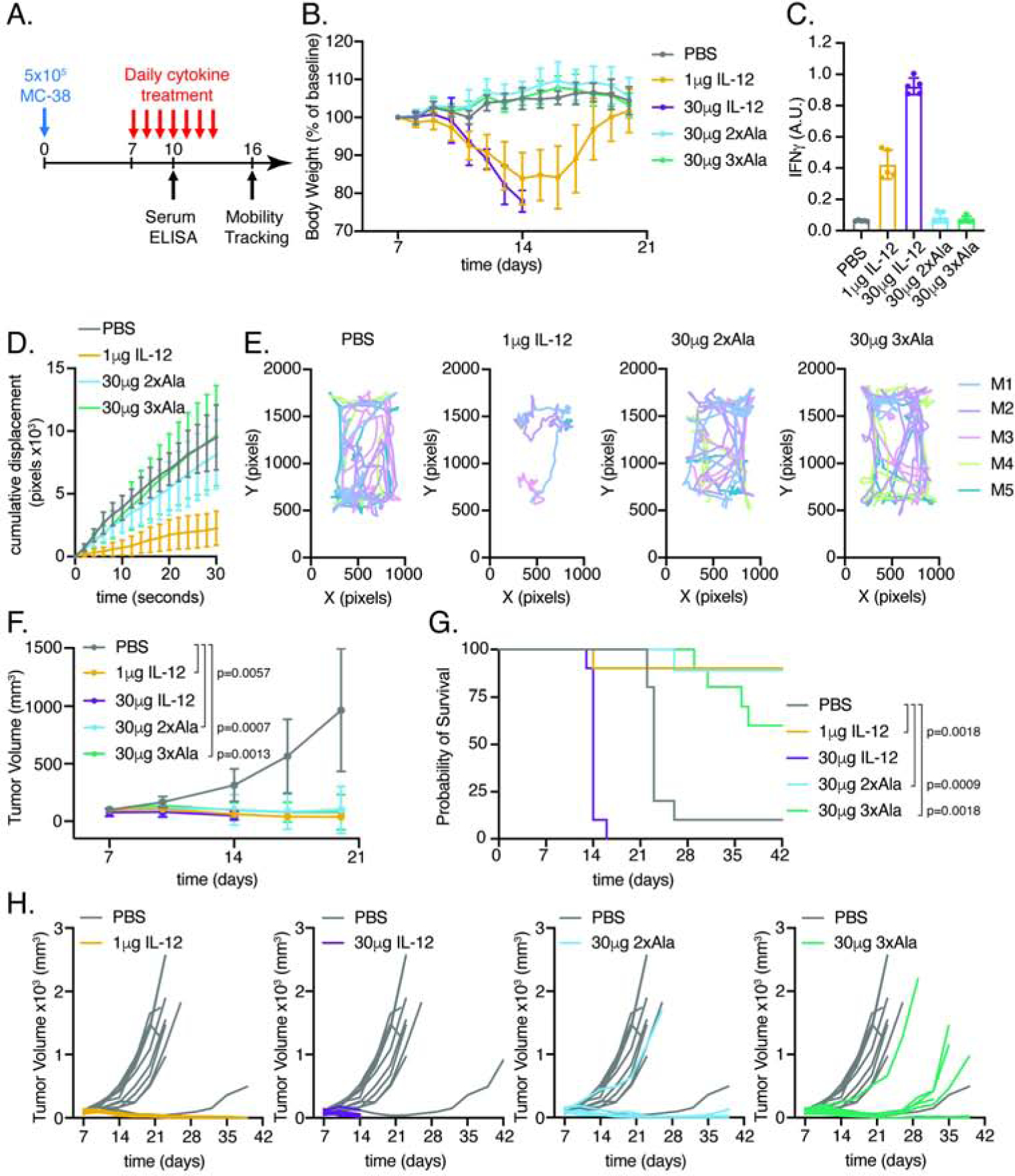
(A) Schematic of experimental design. Mice were implanted with 5×105 MC-38 cells in Matrigel on day 0. Beginning on day 7, mice were administered daily injections of PBS (n=10), 1μg IL-12 (n=10), 30μg IL-12 (n=9), 30μg 2xAla (n=9), or 30μg 3xAla (n=10) as indicated.
(B) IL-12, but not partial agonists, induces weight loss in tumor-bearing mice. Body weights were normalized to day 7 prior to cytokine treatment. Mice administered 30μg dose of IL-12 succumbed to cytokine toxicity between days 13 and 15.
(C) IL-12, but not partial agonists, enhances systemic IFNγ. Serum IFNγ ELISA on day 10, n=5mice/group.
(D) IL-12, but not partial agonists, reduce mobility. Cumulative displacement of MC-38 bearing mice following cytokine treatment. Quantitation of 30 second videos captured on day 16. Cumulative displacement was calculated as the sum of ΔX and ΔY over time. Data are shown as mean ± standard deviation of n=5 mice/group.
(E) Traces showing XY position of mice in a 30 second video.
(F) IL-12 and partial agonists attenuate MC-38 tumor growth. Tumor volumes were compared on day 20 by Kruskal-Wallis test with Dunn’s multiple comparisons.
(G) IL-12 and partial agonists extend survival of MC-38 bearing mice. Kaplan-Meier curves of mice treated with PBS or IL-12 variants. P values from log-rank test were corrected for multiple comparisons using the Holm-Šídák method.
(H) Individual tumor growth curves of MC-38 bearing mice. Growth curves for PBS-treated mice are shown in gray for comparison with cytokine-treated mice in color.
Data are expressed as mean ± standard deviation and are representative of two independent experiments.
Both IL-12 and partial agonists attenuated tumor growth and prolonged survival relative to treatment with PBS (Figure 7F–H). However, the 2xAla and 3xAla partial agonists did so without inducing systemic toxicity observed with IL-12 administration. These results provide additional in vivo support for our hypothesis that the biased agonists, designed based on the structure of the IL-12Rβ1 shared interface, have the capacity to decouple T cell from NK cell activation, significantly reducing IL-12 pleiotropy.
Discussion
A key feature of cytokine signaling is the use of shared receptors that act as common signal transducing subunits for multiple cytokines Unlike other shared receptors which interact with multiple four-helix bundle cytokines through cross-reactive interfaces (Wang et al., 2009), we find that IL-12Rβ1 binds directly to the shared p40 subunit of IL-12 and IL-23 which is an Ig-fold protein reminiscent of cytokine receptor ECDs, rather than the IL-23p19 four-helical bundle typically engaged by cytokine receptors. The nature of this interaction is completely unique amongst the known cytokine receptor complexes.
How this ‘non-canonical’ mechanism of receptor assembly emerged is unclear. It is worth noting that p40 shares homology with the single pass transmembrane cytokine receptors, IL-6Rα, IL-11Rα, and ciliary neurotrophic factor receptor (CNTFR). These Ig Super Family (IgSF) proteins share a similar domain architecture with an N-terminal Ig domain followed by a cytokine-binding homology region formed by tandem fibronectin domains. Despite structural similarity between IL-6Rα/IL-6 and IL-23, the mechanism of receptor assembly differs as IL-6 interacts with both IL-6Rα and gp130 receptor subunits through sites on the four-helix bundle cytokine (sites 1, 2, and 3), while IL-23 engages IL-23R and IL-12Rβ1 through two distinct sites on IL-23p19 and p40. Thus, IL-12 and IL-23 have evolved in such a way to dispense with the typical site 2 interaction and have created a new interaction with the N-terminal Ig domain of p40. Interestingly, IL-6Rα, IL-11Rα, and CNTFR also contain an N-terminal Ig domain; however, this domain is not required for assembly of the canonical IL-6 receptor complex (Boulanger et al., 2003b). What role, if any, this domain serves is currently unclear.
In addition to IL-12 and IL-23, a more recently identified set of heterodimeric cytokines have been characterized which utilize the secreted factor, Epstein Bar Virus Induced 3 (Ebi3). Similar to p40, Ebi3 contains tandem fibronectin domains and has been shown to pair with four-helix bundles IL-27p28 (IL-27), IL-12p35 (IL-35), and IL-23p19 (IL-39) to form a second set of heterodimeric cytokines that signal through the shared gp130 receptor (Collison et al., 2012; Collison et al., 2007; Pflanz et al., 2004; Pflanz et al., 2002; Wang et al., 2016). However, unlike p40, Ebi3 lacks an N-terminal Ig domain. Our finding that p40 engages IL-12Rβ1 via its N-terminal Ig-domain suggests that the mechanism of sharing for Ebi3-containing cytokines is likely different than that of IL-12 and IL-23. Instead, we suggest that these cytokines may assemble receptor complexes in a manner similar to the site1/2/3 paradigm of IL-6/CNTF/LIF, etc.
The modular receptor assembly mechanism used by IL-12 and IL-23 suggests that expression of cytokine components alone should antagonize signaling by binding to receptors but failing to induce dimerization. Indeed, expression of either IL-12p35 (Dambuza et al., 2017) or IL-12p40 (Mattner et al., 1993; Mondal et al., 2020) in isolation has been reported to antagonize IL-12 signaling. Consistent with these reports, blocking antibodies which bind p40 inhibit signaling of both IL-12 and IL-23 (Benson et al., 2011). Comparison of p40 binding antibodies and nanobodies with the IL-12Rβ1 complex reported here explain the basis for their antagonism and suggest that p40 targeted antagonists will be unable to confer cytokine specific activity as they directly compete with IL-12Rβ1 for cytokine binding (Figure S2). Instead, cytokine-specific antagonists must target the specific interaction between the helical subunits of the cytokines and IL-12Rβ2 or IL-23R. A second generation of IL-23 antagonists targeting IL-23p19 are in clinical trials or have been approved for use in plaque psoriasis (Chan et al., 2018; Kopp et al., 2015). Unlike p40 binding antagonists, these therapies limit antibody blockade to IL-23 while preserving IL-12 signaling and as a result may limit unwanted side-effects of p40 blockade (Koutruba et al., 2010). Increased specificity of IL-23 blockade may reduce off-target effects, however, this strategy blocks both pathogenic as well as protective effects of IL-23. Future work will focus on generating IL-23 partial agonists that preserve immune function at barrier sites without potentiating autoimmune conditions.
In contrast to IL-23, therapeutic modulation of IL-12 signaling has focused on potentiation of IL-12 signaling in the context of cancer immunotherapy. Current therapeutic approaches have attempted to limit toxicity associated with IL-12 administration by targeting IL-12 to tumors through antibody fusion (Pasche et al., 2012), collagen-binding (Momin et al., 2019), transfer of IL-12 expressing cells (Pegram et al., 2012; Zitvogel et al., 1995) or introduction of IL-12 coding nucleic acid (Fewell et al., 2009; Tamura et al., 2001). Broadly, these strategies rely on regulating the spatial distribution of cytokine. This is in contrast with the approach used here in which we narrowed the pleiotropic effects of IL-12 by leveraging differences in receptor expression across cell types and activation states. By limiting the activity of IL-12 to T cells, IL-12 partial agonists elicit anti-tumor activity without inducing systemic toxicity associated with IL-12 administration. As toxicity has been a major limitation to the clinical implementation of IL-12 (Cohen, 1995; Leonard et al., 1997), IL-12 partial agonists with reduced cytokine pleiotropy may provide a mechanism to enhance the therapeutic utility of IL-12. Collectively, our results reveal new strategies to design IL-12 and IL-23 agonists and antagonists with improved therapeutic potential.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, K. Christopher Garcia (kcgarcia@stanford.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code availability
Structure factors and coordinates have been deposited in the Protein Data Bank with identification numbers PDB: 6WDP (IL-12Rβ1) and PDB: 6WDQ (IL-23 receptor complex). Cryo-EM maps have been deposited to Electron Microscopy Data Bank (EMDB) under accession ID EMD-21646 (IL-23 receptor complex) and EMD-21645 (IL-12 receptor complex).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian cell lines and culture conditions
For structural studies, protein was produced in N-acetylglucosaminyltransferase I (GnTI) deficient HEK293S suspension cells (Reeves et al., 2002) maintained in FreeStyle 293 Expression Media (Gibco) with 1% fetal bovine serum (FBS, Fisher Scientific) and GlutaMAX (Gibco). For cell signaling assays and in vivo studies, proteins were expressed in Expi293F cells (Gibco) maintained in Expi293 Expression Media (Gibco). HEK293S and Expi293 cells are of female human origin and were cultured at 37°C with 5% CO2 and gentle agitation.
For in vitro tumor cell killing, B16-F10 cells (ATCC) derived from male murine pulmonary melanoma were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco) supplemented with 10% FBS (Fisher Scientific), GlutaMAX (Gibco), and penicillin-streptomycin (Gibco). For in vivo tumor studies, MC-38 (Kerafast) derived from female murine colon adenocarcinoma were maintained in DMEM (Gibco) supplemented with 10% FBS, MEM non-essential amino acids (Gibco), sodium pyruvate (Gibco), 10mM HEPES (Gibco), and penicillin-streptomycin (Gibco). Tumor cells were grown at 37°C with 5% CO2. Cell lines were not authenticated but were checked regularly for morphology and viability.
Insect cell lines and culture conditions
For recombinant protein expression, baculovirus was produced in Spodoptera frugiperda (Sf9) ovarian cells (ATCC) maintained in Sf-900 III medium (Gibco) with 10% FBS (Fisher Scientific) and GlutaMAX (Gibco). Protein was expressed by baculoviral infection of Trichoplusia ni (T. ni) ovarian cells (Expression Systems) maintained in ESF 921 Insect Cell Culture Medium (Expression Systems). Insect cells were grown at 27°C with ambient CO2 and gentle agitation.
Yeast strains and culture conditions
EBY100 yeast (ATCC) were cultured at 30°C with shaking in YPD media (Sigma) and selected in media containing glucose and casamino acids (SDCAA): 1L deionize water with 20g dextrose (Sigma), 6.7g yeast nitrogen base (RPI), 5g Bacto casamino acids (Gibco), 10.4g sodium citrate (Sigma), and 6.4g citric acid monohydrate (Sigma), pH 4.5. Expression was induced at 20°C with shaking in galactose containing media (SGCAA) which resembles SDCAA but includes 20g galactose (Sigma) instead of glucose.
Human primary cells and culture conditions
Peripheral mononuclear cells (PBMCs) from healthy human donors were obtained from Stanford Blood Bank and cultured in complete RPMI: RPMI 1640-glutaMAX (Gibco) supplemented with 10% FBS (Fisher Scientific), 50μM 2-mercaptoethanol (βME, Sigma), MEM non-essential amino acids (Gibco), sodium pyruvate (Gibco), 15mM HEPES (Gibco), and penicillin-streptomycin (Gibco) at 37°C with 5% CO2.
Animal primary cells and culture conditions
Primary mouse lymphocytes were isolated from secondary lymphoid organs of female mice and cultured in complete RPMI at 37°C with 5% CO2.
In vivo animal studies
Female C57BL/6J, C57BL/6-Tg(TcraTcrb)1100Mjb/j and B6.PL-Thy1a/CyJ age 8–12 weeks were obtained from Jackson Labs or bred in house. Mice were maintained on a 12-hour light-dark cycle at Stanford University. All mouse experiments were conducted according to protocols approved by the Institutional Animal Care and Use Committee, Administrative Panel on Laboratory Animal Care (APLAC) protocol number 32279.
METHOD DETAILS
Crystallography
For crystallographic studies, human p40 (23–328), IL-23p19 (20–189), and IL-12Rβ1 D1–D2 (25–239) were cloned into the pAcSG2 baculoviral vector with an N-terminal GP64 signal sequence. P40 and IL-12Rβ1 were expressed with a C-terminal 6xHis while IL-23p19 lacked a C-terminal tag. Human IL-23R (1–315) was cloned into the pVLAD6 BacMam vector with C-terminal 3C protease site, Protein C tag, and 6xHis. Baculovirus was produced by transfection of Sf9 insect cells with Cellfectin II (Gibco) and Sapphire Baculovirus DNA (Allele) followed by viral amplification in Sf9. For IL-23 production, T. ni cells were coinfected with p40 and IL-23p19 baculovirus in the presence of the endoplasmic reticulum mannosidase I (ERM1) inhibitor, kifunensine (Toronto Research Chemicals). 48–72 hours post infection, heterodimeric IL-23 was purified by Ni-NTA affinity chromatography followed by size exclusion chromatography (SEC) using Superdex S200 Increase column (Cytiva). Similarly, IL-12Rβ1 was expressed by baculoviral infection of T. ni cells followed by Ni-NTA purification and SEC. IL-23R was expressed by baculoviral infection of GnTI deficient HEK293S suspension cells, Ni-NTA purification and SEC.
For IL-12Rβ1 structure determination, initial crystals of native IL-12Rβ1 were grown by vapor diffusion from 1.75 M ammonium sulfate, 0.2 M NaCl, and 0.1 M sodium cacodylate pH 5.9. These crystals were then used for microseeding to grow both native and SeMet-labeled crystals using the same precipitant solution. For selenomethionine (SeMet)-labeling, IL-12Rβ1 D1–D2 (25–239) was expressed by baculovirus infection of T. ni cells in Met-free medium (Expression Systems) in the presence of SeMet (Fisher Scientific) and purified as described for native IL-12Rβ1. Data of native and SeMet-derivative crystals were collected at a temperature of 100K at beamline 8.2.2 of the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory (LBNL) and at beamline 12–2 of the Stanford Synchrotron Radiation Laboratory (SSRL), respectively. All data were processed with the XDS package (Kabsch, 2010). The structure of IL-12Rβ1 was solved by SeMet multi-wavelength anomalous diffraction (MAD) using autoSHARP (Vonrhein et al., 2007). The program COOT (Emsley et al., 2010) was used for model building. Refinement against the native dataset was carried out in PHENIX (Liebschner et al., 2019). Data collection and refinement statistics are summarized in Table S1. The structure was deposited in the RCSB protein databank with accession ID 6WDP.
For complex structural determination, IL-23 and receptor components were treated with endoglycosidase H (EndoH) and 3C protease along with carboxypeptidase A (Sigma) and B (Sigma) overnight at 4°C. SEC fractions containing all complex components were collected and concentrated to 12.4mg/mL. The complex crystalized in a solution of Peg12K, CAPSO pH9.4, 5% dioxane, 10% spermine HCl. Diffraction data was collected at 100K at SSRL beamline 12–2. Data were indexed, integrated, and scaled using XDS. The quaternary complex structure was solved by molecular replacement with the program PHASER using the partial complex of IL-23R and IL-23 (PDB: 5MZV) (Bloch et al., 2018) and D1 of IL-12Rβ1 (reported here) as search models. Due to clashes with symmetry mates, D1–D2 of IL-12Rβ1 could not be placed as a unit by molecular replacement and only D1 is modeled in the complex structure (Figure S1D). The final model was built by iterative cycles of reciprocal space refinement with PHENIX and manual rebuilding using COOT. The structure was deposited in the RCSB protein databank with accession ID 6WDQ. Crystallographic software used in the project was installed and configured by SBGrid (Morin et al., 2013).
Surface plasmon resonance
For affinity measurements, human p40 (23–328) was cloned into pD649 vector with an N-terminal HA signal peptide, C-terminal AviTag (GLNDIFEAQKIEWHE) and 6xHis. Human IL-12p35 (23–219) and IL-23p19 (28–189) were cloned into pD649 with an N-terminal HA signal peptide, Flag-tag, and TEV protease site. IL-12 (IL-12p35 and p40), IL-23 (IL-23p19 and p40) and p40 alone were expressed by transient transfection of Expi293F cells (Gibco) using ExpiFectamine 293 Transfection Kit (Gibco) according to manufacturer’s protocols. Supernatants were subject to Ni-NTA purification and incubated with soluble BirA ligase in buffer containing 0.5mM Bicine (pH 8.3), 100mM ATP, 100mM magnesium acetate, and 500μM biotin overnight at 4°C. Biotinylated proteins were then purified by SEC.
Affinity measurements were made using a BIAcore T100 instrument. Biotinylated cytokines or biotinylated frizzled 8 (Fzd8) control were immobilized on a streptavidin sensor chip (Cytiva) at low density (~100RU) and binding to recombinant receptor ECDs was measured. For BIAcore studies, human IL-23R (1–309) was cloned into the pVLAD6 BacMam vector with C-terminal 3C protease site, Protein C tag, and 6xHis. hIL-23R was produced in GnTI- HEK293S cells and purified by Ni-NTA followed by SEC. Human IL-12Rβ2 D1–D3 (24–317) was cloned into the pAcSG2 with an N-terminal GP64 signal sequence and a C-terminal 6xHis tag. IL-12Rβ2 was expressed in T. ni cells and purified by Ni-NTA followed by SEC. The D1–D2 domains of IL-12Rβ1, either wild-type or Tyr109Ser Gln132Leu (affi-IL-12Rβ1), was expressed in T. ni cells as described for crystallization.
Binding measurements were performed at 25°C with a flow rate of 50μL/min. Receptor ECDs were diluted in HBS containing 0.005% P20 (HBS-P, Cytiva) and injected over the chip for 120 seconds followed by a 500 second dissociation phase. Following each injection, the chip was regenerated with 4M MgCl2. Binding curves were exported for analysis in Prism (Graphpad Software) and steady state affinity was calculated using BIAevaluation software (Cytiva).
Affinity maturation of IL-12Rβ1
The D1–D2 domains of human IL-12Rβ1 (25–234) were cloned into the pCT302 yeast display vector with an N-terminal Aga2 followed by a 3C protease site and a C-terminal Myc-tag (Midelfort et al., 2004). EBY100 yeast (ATCC) were transformed with IL-12Rβ1 display vector, selected in media containing glucose and casamino acids (SDCAA) followed by induction with galactose containing media (SGCAA) (Chao et al., 2006). Proper folding was confirmed by αmyc AF488 staining (Cell Signaling Technologies) and IL-23 tetramer (biotinylated IL-23 complexed with streptavidin-AF647 at a 4:1 molar ratio). For IL-23 staining, human IL-23p19 (28–189) was cloned into the pAcSG2 baculoviral vector with GP64 signal peptide and co-expressed with human p40 (23–328) in pAcGP67a baculoviral vector with an N-terminal GP64 signal peptide and C-terminal BirA substrate peptide (LHHILDAQKMVWNHR) and 6xHis tag. Similarly, IL-12 was expressed by co-infection of human p40 with human IL-12p35 (22–212) in pAcSG2 with an N-terminal GP64 signal sequence. Heterodimeric IL-12 and IL-23 were purified by Ni-NTA and biotinylated in vitro. An error prone library of IL-12Rβ1 was generated using the GeneMorph II Random Mutagenesis kit (Agilent Technologies) with 0.1ng DNA/reaction, to achieve an average mutation rate of 3.3 mutations/gene. EBY100 yeast were electroporated to generate a library with a diversity of 1.09×109. The library was selected for IL-23 binding over five rounds of increasing selection stringency starting with 200nM magnetic-activated cell sorting (MACS) using Streptavidin microbeads (Miltenyi Cat#130-048-101) with LS magnetic selection column (Miltenyi Cat#130-042-401) and finishing with 1nM monomer selection by fluorescence assisted cell sorting (FACS) using a SH800S cell sorter (Sony). DNA from enriched yeast was isolated using the Zymoprep Yeast Plasmid Miniprep II Kit (Zymo Research), transformed into DH5α (Zymo Research), and sequenced by rolling circle amplification to identify convergent mutations. To track enrichment of cytokine binding, yeast expressing wild-type IL-12Rβ1 or error prone library from various rounds were stained with αmyc AF488 and 10nM cytokine (biotinylated IL-12 or IL-23) followed by 50nM streptavidin-AF647 to detect binding of biotinylated monomer. Fluorescence intensity was measured using CytoFlex flow cytometer (Beckman Coulter) and analyzed using FlowJo software (BD).
IL-12 and IL-23 receptor complex Cryo-EM
For electron microscopy, the ectodomain of human affi-IL-12Rβ1 (25–542) was cloned into the pAcGP67a baculoviral vector with an N-terminal GP64 signal peptide and C-terminal 6xHis tag. IL-23R was expressed as described for SPR measurements and complexed with T. ni expressed IL-23 (coinfection of IL-23p19 and p40, as described in Affinity maturation of IL-12Rβ1) and affi-IL-12Rβ1. The complex was purified by SEC and concentrated to 1.04mg/mL. Samples were spiked with 0.05% octyl beta glucoside and applied to a glow-discharged Quantifoil Au1.2/1.3, 200 mesh grids (SPI supplies), then plunge-froze in liquid ethane using FEI Vitrobot. Data were collected using Titan Krios at 300 kV with Gatan K3 direct detection camera. Raw images were collected as a movie, recorded at a magnification of 29,000 × corresponding to 0.85 pixel per Å. Each of these movies were recorded for 2 sec at 0.05 sec/frame with a total dose of 44 electrons/Å2 and defocus values ranging from −1.5 to −2.4 microns. The frames were motion-corrected, dose-weighted using motioncor2 (Zheng et al., 2017), and contrast transfer function (CTF) parameters were determined by CTFFIND4 (Rohou and Grigorieff, 2015). Particles were semi-automatically picked based on a template built from manually picked particles using Autopick software from Relion pipeline (Zivanov et al., 2018). Particle projections were sorted by reference-free 2D classification and 2D averages with well-defined complex features were selected for 3D classification with an initial model. The most well resolved 3D class was further refined to a resolution of 8.0 Å ,according to 0.143 “gold-standard” Fourier Shell Correlation (FSC) criteria and deposited to the Electron Microscopy Data Bank (EMDB) under accession ID EMD-21646 (Figure S3A–D). X-ray crystal structures of the individual components were fit into the map by using Chimera software to visualize the relative arrangement of the receptors. For the IL-23 model individual components of IL-23R (PDB: 5MZV), IL-23 (PDB: 3DUH), and IL-12Rβ1 (reported here) fit to the map density.
IL-12 complexes for cryo-EM were analogous to those used for IL-23. Human IL-12p35 (22–212) was cloned into pAcSG2 with an N-terminal GP64 signal sequence. Human IL-12Rβ2 (24–317) was expressed as described for SPR measurements. Heterodimeric IL-12 (coinfection of IL-12p35 and p40), affi-IL-12Rβ1 D1–D5, and IL-12Rβ2 were expressed in T. ni cells. Complex components were mixed and purified by SEC. Fractions were concentrated to 0.9mg/mL and samples were processed as described above. Data were collected and processed similar to the approach described for IL-23 with the exception that Gctf (Zhang, 2016) was used to obtain CTF parameters (Figure S3E–H). The most well resolved 3D class was further refined to a resolution of 10.0 Å, according to 0.143 “gold-standard” Fourier Shell Correlation (FSC) criteria and deposited to the Electron Microscopy Data Bank (EMDB) under accession ID EMD-21645. For the IL-12 model, IL-12Rβ2 (no reported crystal structure, using IL-23R PDB: 5MZV in model), IL-12 (PDB: 1F45), and IL-12Rβ1 (reported here) were fit to the map density.
Human T cell signaling
For human T cell signaling, IL-12 and IL-23 variants were produced in Expi293F cells (Gibco) as described for surface plasmon resonance experiments. IL-12p35 and IL-23p19 were co-transfected with p40 variants (wild type, Glu81Ala, Phe82Ala, 4A: Pro39Ala Asp40Ala Glu81Ala Phe82Ala, 2xAla: Glu81Ala Phe82Ala, 3xAla: Glu81Ala Phe82Ala Lys106Ala) and purified by Ni-NTA followed by SEC. Human peripheral mononuclear cells (PBMCs) were isolated from Stanford Blood Bank samples using SepMate-50 columns (StemCell Technologies) with Ficoll-Paque PLUS (Cytiva). Cells were diluted in sterile PBS (Gibco) with 2% FBS (Fisher Scientific) and added to SepMate-50 columns pre-loaded with 15ml Ficoll. Red blood cells were lysed using ACK lysis buffer (Gibco) for 5 min, quenched with PBS containing 2% FBS (Fisher Scientific) and resuspended at 50×106/mL in freezing media containing 90% FBS (Fisher Scientific) and 10% DMSO (Fisher Scientific). Cells were stored frozen at −80°C until ready for use. Human PBMCs were stimulated in 6 well plates coated with 2.5μg/mL αCD3 (OKT3, BioLegend) in complete RPMI supplemented with 5μg/mL αCD28 (CD28.2, BioLegend), and 100IU/mL recombinant human IL-2. Cells were cultured for 48 hours at 37°C with 5% CO2, washed once, and rested overnight in complete RPMI. For STAT signaling, cells were stained with αCD4 PacBlue (RPA-T4, BD) for CD4+ T cell signaling or αCD8 BV605 (SK1, BioLegend) for CD8+ T cells signaling and stimulated with IL-12 and IL-23 variants for 20 minutes at 37°C prior to fixation with 1.6% paraformaldehyde (Fisher Scientific) for 10 minutes at room temperature. Cells were pelleted, resuspended in methanol and stored at −20°C until analysis. Cells were washed in FACS buffer (PBS pH7.4 with 2% FBS and 2mM EDTA) and stained with antibodies against STAT4 pY693 AF488 (38/p-Stat4, BD) and STAT3 pY705 AF647 (4/P-STAT3, BD) for 1 hour at room temperature. Fluorescence intensity of pSTAT staining in CD4+ T cells was analyzed using a CytoFlex flow cytometer (Beckman Coulter).
For analysis of IL-12Rβ1 in human PBMCs, cells were stained directly (ex vivo) or activated as described above to generate T cell blasts. To identify T cells and NK cells, Fc receptors were blocked with TruStain FcX (BioLegend) and cells were stained with a phenotyping panel of αCD3 Pacific Blue (UCHT1, BioLegend), αCD4 FITC (OKT4, BioLegend), αCD8 AF750 (R&D systems), and αCD56 BV605 (HCD56, BioLegend). Human p40 tetramers were prepared by mixing 200nM streptavidin-AF647 with four-fold molar excess of biotinylated p40 expressed as described in the surface plasmon resonance section. Cells were stained for 2 hours at 4°C followed by live cell detection using propidium iodide (PI, Invitrogen). Samples were analyzed using CytoFlex flow cytometer (Beckman Coulter) followed by analysis in FlowJo (BD). CD8+ T cells were defined as liveCD3+CD8+, NK cells were defined as liveCD3-CD56+. See Figure S6B for gating.
For human CD8+ T cell IFNγ induction assay, CD8+ T cells were isolated from PBMCs by MACS using CD8+ T cell isolation kit (Miltenyi) and LS magnetic columns (Miltenyi). Purified CD8+ T cells were stimulated at 80,000 cells/well in 96-well round bottom plates in coated with 2μg/mL αCD3 (OKT3, BioLegend) in the presence of 0.5μg/mL αCD28 (CD28.2, BioLegend), and 5ng/mL human IL-2. After 48 hours, cells were pelleted and supernatant was analyzed using human IFNγ ELISA MAX Deluxe (BioLegend) with Nunc MaxiSorp ELISA plates (BioLegend).
For human NK cell IFNγ induction assays, NK cells were isolated from PBMCs by MACS using the EasySep human NK cell isolation kit (StemCell) with EasySep Magnet (StemCell). Purified NK cells were stimulated at 40,000 cells/well in 96 well round bottom plates in the presence of 100ng/mL IL-18 (R&D systems). After 48 hours, supernatant was harvested and processed as described for CD8+ T cell IFNγ induction assays.
Mouse surface staining, IL-12 signaling, IFNγ induction and tumor cell killing
For IL-12Rβ1 surface staining, mouse p40 (23–335) was cloned into pAcGP67a with N-terminal GP64 signal peptide and C-terminal AviTag and 6xHis tags. Mouse p40 is secreted as a disulfide bonded homodimer. To obtain monomeric p40, Ni-NTA purified protein was reduced with 20mM cysteine and alkylated with 40mM iodoacetamide in HEPES buffered saline (HBS) pH 8.2 followed by SEC. Monomeric p40 was biotinylated with recombinant BirA and purified by a second round of SEC.
For surface staining and cell signaling, spleen and lymph nodes from female C57BL/6J mice were isolated, red blood cells were lysed using ACK lysis buffer (Gibco) and single cell suspension was generated. For T cell blasts, single cell suspension was added to plates coated with 2.5μg/mL αCD3 (145–2c11, BioLegend) in complete RMPI with 5μg/mL αCD28 (37.51, Bio X Cell), and 100IU/mL recombinant mouse IL-2 for 48 hours at 37°C. For cell staining, ex vivo cells and T cell blasts were incubated with TruStain FcX (93, BioLegend) and stained with a phenotyping panel of αCD3 FITC (17A2, eBiosciences), αCD4 PerCP-Cy5.5 (GK1.5, BioLegend), αCD8 BV785 (53–6.7, Biolegend), and αNK1.1 eFluor450 (PK136, eBioscience). Mouse p40 tetramers were prepared by mixing 200nM streptavidin-AF647 with four-fold molar excess of biotinylated p40 and cells were stained for 2 hours at 4°C followed by live cell detection using propidium iodide (PI, Invitrogen). Samples were analyzed using CytoFlex flow cytometer (Beckman Coulter) followed by analysis in FlowJo (BD). CD8+ T cells were defined as liveCD3+CD8+, CD4+ T cells were gated as liveCD3+CD4+, and NK cells were identified as liveCD3-NK1.1+ (Figure S4A).
For in vitro studies, mouse IL-12 was expressed as a single chain with N-terminal p40 (23–335) followed by a 3xGGGS linker, 3C protease site, and IL-12p35 (23–215). Single chain mouse IL-12 was cloned into pAcGP67a with an N-terminal GP64 signal peptide and C-terminal 6xHis tag. Mouse IL-12 variants were expressed in T. ni cells and purified by Ni-NTA and SEC. For cell signaling, mouse T cell blasts were prepared as described above, rested overnight in complete RPMI, stained with αCD8 BV785 (53–6.7, Biolegend) and stimulated for 20 minutes at 37°C with IL-12 variants before fixation, permeabilization, and staining as described for human T cell signaling. For NK cell signaling, NK cells were MACS isolated from spleen and lymph nodes of female C57BL/6J mice using NK cell isolation kit (Miltenyi) and LS magnetic columns (Miltenyi). Purified NK cells were mixed with Cell Trace Violet (CTV, Invitrogen) labeled Expi293F (Gibco) carrier cells and stimulated with cytokine for 20 minutes before fixation in FixLyse buffer (BD) and permeabilization in Perm Buffer III (BD). Carrier cells were used in these assays to improve cell retention during washing and staining. Cells were stained with anti-STAT4 pY693 AF488 (38/p-Stat4, BD) and analyzed using CytoFlex flow cytometer (Beckman Coulter). NK cells were identified as CTV negative.
For western blot analysis, CD8+ T cell blasts were generated as described above. After 48-hour activation, CD8+ T cells were isolated by MACS using CD8+ T cell isolation kit (Miltenyi) and LS magnetic columns (Miltenyi). Purified CD8+ T cells were rested overnight in complete RPMI. In the morning, 1×106 cells were starved for 1 hour in RPMI without serum and stimulated for 20 minutes with 1μM cytokine. Cells were washed once in cold PBS and lysed in 100μL of Pierce IP lysis buffer (Pierce) supplemented with 1:100 Abcam phosphatase inhibitor cocktail (Abcam), 1:100 Halt phosphatase inhibitor cocktail (Thermo Scientific), 1:200 Sodium Orthovanadate (NEB) and 1:10 cOmplete protease inhibitor cocktail (Roche). Lysates were resolved on 12% SDS-PAGE (BioRad Cat#4561046) and transferred to PVDF membranes using Trans-Blot Turbo transfer kit (BioRad). Membranes were blocked for 1 hour in TBS-T (20mM Tris pH7.4, 150mM NaCl, 0.1% tween 20) containing Blotting-Grade Blocker (BioRad). Primary antibodies against phosphorylated STAT4 (Tyr693, Cell Signaling Technologies) and total STAT4 (C46B10, Cell Signaling Technologies) were detected with swine anti-rabbit HRP (Dako) and enhanced chemiluminescence (ECL) detection reagent (Cytiva). Blots were developed on Hyperfilm ECL (Cytiva)
For NK cell IFNγ induction assays, NK cells were isolated as described for pSTAT4 signaling. Purified NK cells were stimulated at 25,000 cells/well in a 96 well round bottom plate with 50ng/mL recombinant mouse IL-18 (R&D systems) and IL-12 variants for 48 hours at 37°C. In the final four hours of culture, GolgiStop (BD) was added to prevent further cytokine secretion. Cells were fixed and permeabilized using Cytofix/Cytoperm kit (BD) and stained with αIFNγ AF647 (XMG1.2, BD). Fluorescence intensity was recorded using CytoFlex flow cytometer (Beckman Coulter) and analyzed in FlowJo (BD). Supernatants were assessed for IFNγ using the ELISA MAX Deluxe Set (BioLegend) and Nunc MaxiSorp ELISA plates (BioLegend).
For transcriptional analysis, MACS isolated NK cells were stimulated with 50ng/mL IL-18 (R&D systems) and 1μM IL-12 variants for 8 hours followed by RNA isolation using RNeasy Plus Mini Kit (Qiagen). cDNA was generated using iScript Reverse Transcription Supermix for RT-qPCR (BioRad). Primer sets for Ifng (Mm.PT.58.41769240), Tigit (Mm.PT.56a.6925849), and Gapdh (Mm.PT.39a.1) were purchased from Integrated DNA Technologies (IDT). qPCR was performed using PowerTrack SYBR Green Master Mix (Applied Biosystems) and analyzed using the StepOnePlus Real-Time PCR system (Applied Biosystems). For analysis, Ct values were exported from StepOne Software (Applied Biosystems) and ΔCt was calculated for all samples relative to Gapdh control. ΔΔCt was calculated relative to unstimulated cells and fold change was expressed as 2−ΔΔCt.
For CD8+ T cell assays, splenocytes from female C57BL/6-Tg(TcraTcrb)1100Mjb/j OT-I TCR transgenic mice (Hogquist et al., 1994) were stimulated in complete RPMI containing 1μg/mL OVA (257–264, GenScript), 0.5μg/mL αCD28 (37.51, Bio X Cell), 100IU/mL IL-2, and IL-12 variants. For IFNγ induction assays, cells were stimulated for 48 hours at 80,000 cells/well in a 96 well round bottom plate. For the final four hours, GolgiStop (BD) was added to prevent further cytokine secretion. Supernatants were analyzed for IFNγ by ELISA as described for NK assays and cells were stained with αCD3 eFluor450 (17A2, eBioscience) and αCD8 BV785 (53–6.7, BioLegend) before being fixed/permeabilized using the Cytofix/Cytoperm kit (BD) and stained with αIFNγ AF647 (XMG1.2, BD). OT-I T cells were gated on CD3+CD8+ cells and αIFNγ AF647 staining was assessed using CytoFlex flow cytometer (Beckman Coulter) followed by analysis in FlowJo (BD). For gating see Figure S4C. For OT-I effectors used in in vitro tumor cell killing assay, OT-I T cells were stimulated as described above for 72 hours in 24 well plates. Cells were spun down and supernatant saved.
For MHC-I upregulation, 25,000 B16-F10 melanoma cells (ATCC) were plated on 96 flat bottom plates for 4 hours at 37°C prior to exchanging media for OT-I effector supernatants diluted in compete RPMI. After 16 hours at 37°C, media was removed and cells were detached using TrypLE (Gibco). Cells were stained with αH-2Kb APC (AF6–88.5, BioLegend) and PI to identify live cells. Data were collected on CytoFlex flow cytometer (Beckman Coulter) and analyzed in FlowJo (BD).
For antigen-specific tumor cell killing, B16-F10 cells expressing pCDH-EF1-cOVA-T2A-copGFP (Tseng et al., 2013) were cultured with wild-type B16-F10 wild type at a 1:1 ratio and 25,000 cells were plated on a 96 flat bottom plate. After 4 hours at 37°C, media was removed and OT-I effectors were added in complete RPMI for 36 hours at 37°C. Media was removed and cells were detached using TrypLE (Gibco) and stained with PI and αCD45.2 APC (104, eBioscience) prior to analysis on CytoFlex (Beckman Coulter). B16-F10 were identified as liveCD45.2- and % GFP+ was quantified as compared to no effector condition.
For Th1 polarization, naïve CD4+ T cells were isolated from spleen and lymph nodes of female C57BL/6J mice using Naïve CD4+ T cell isolation Kit (Miltenyi) with LS columns (Miltenyi). Naïve CD4+ T cells were stimulated at 80,000 cells/well in a 96-well round bottom plates coated with 2μg/mL αCD3 (145–2c11, BioLegend) in complete RMPI with 0.5μg/mL αCD28 (37.51, Bio X Cell), 1μg/mL αIL-4 (11b11, eBioscience), and 5ng/mL recombinant mouse IL-2. After 48 hours, cells were re-stimulated in 1μg/mL Ionomycin (Sigma), 50ng/mL phorbol 12-myristate 13-acetate (PMA, Sigma), and GolgiStop (BD) for 4 hours prior to analysis of IFNγ by intracellular cytokine stain as described for NK cells and OT-I T cells.
Adoptive transfer and immunization
For in vivo characterization, mouse IL-12p35 (23–215) and p40 (23–335) were cloned into pD649 vector with an N-terminal HA signal peptide. IL-12p35 also included a C-terminal 6xHis tag for Ni-NTA affinity chromatography. Mouse IL-12 and variants were expressed in Expi293F cells (Gibco) by co-transfection of plasmids containing IL-12p35 and p40. IL-12 heterodimer was purified by Ni-NTA affinity chromatography followed by SEC. Endotoxin was removed using High Capacity NoEndo Columns (Protein Ark) and levels were quantified using the LAL Chromogenic Endotoxin Quantitation Kit (Pierce). All in vivo preparations had <1U endotoxin/dose.
To assess cell-type bias in vivo, female B6.PL-Thy1a/CyJ Thy1.1 mice were seeded with 2.5×106 CD8+ T cells from female OT-I TCR transgenic mice by retro-orbital transfer. The following day, mice were immunized subcutaneously in the chest with 50μg OVA (257–264, Genscript) in Incomplete Freund’s Adjuvant (IFA, BD) and cytokine was administered daily by interperitoneally injection for five days. On day six, mice were sacrificed for analysis of serum IFNγ and cellularity in draining lymph nodes. For analysis of serum cytokine, blood was allowed to clot for 20 minutes at room temperature and samples were spun down at 2,000xg for 10 minutes. Serum was diluted and analysis of IFNγ using ELISA MAX Deluxe Set (BioLegend) and Nunc MaxiSorp ELISA plates (BioLegend). For cell profiling, a single cell suspension from draining lymph nodes was stained with TruStain FcX (93, BioLegend) and a phenotyping panel of αCD3 PerCP-Cy5.5 (145–2c11, eBioscience), αCD4 PE-Cy7 (GK1.5, eBioscience), αCD8 BV785 (53–6.7, BioLegend), αThy1.2 PE (30-H12, BioLegend), αNK1.1 eFluor450 (PK136, eBioscience), and αNKp46 FITC (29A1.4, eBioscience). For gating, see Figure S7G. To assess activation by cell surface staining, αLAG-3 AF647 (C9B7W, BioLegend), αPD-1 BV605 (29F.1A12, BioLegend), and αCD25 APC (PC61, BioLegend) were used as indicated.
MC-38 tumor studies
Female C57BL/6J mice were inoculated subcutaneously in the flank with 5×105 MC-38 (Kerafast) in Matrigel (Corning). After 7 days, tumors volumes were calculated as: v = a2b/2, where a is minor radius and b is major radius. Mice with tumors between 50–200 mm3 were randomized to groups and administered daily cytokine injections for 7 days. Measurements of serum IFNγ were performed as described for adoptive transfer and immunization on day 10 following 3 doses of cytokine. Mice were euthanized when tumor volumes reached around 2000 mm3, tumors showed profound ulceration, or when animals displayed limited locomotion and were moribund.
For mobility tracking, 30 second movies of individual mice were exported to an image series at 5 frames/second and imported into ImageJ using Fiji (Schindelin et al., 2012). Images were converted to 8-bit Grayscale and a threshold was set to identify mice (dark object on light background). Mice were tracked using MTrack2 (Klopfenstein and Vale, 2004). X, Y positions were plotted in Prism (GraphPad Software) and cumulative displacement was calculated as the sum of the absolute value of ΔX and ΔY for each time point.
QUANTIFICATION AND STATISTICAL ANALYSIS
SPR data was analyzed using BIAevaluation software (Cytiva). Flow cytometry data was analyzed using FlowJo software (BD). qPCR data was analyzed using StepOne Software (Applied Biosystems). Statistical analyses were performed using Prism v8.4 (GraphPad Software). Groups were compared by one-way ANOVA with Tukey’s multiple comparisons test if Brown-Forsythe test indicated no difference in standard deviation between groups and Kolmogorov-Smirnov test indicated the data were normally distributed. Otherwise Kruskal-Wallis test with Dunn’s correction for multiple comparisons was performed. Survival analysis was performed using Kaplan-Meier test and P values from log-rank test were corrected for multiple comparisons using the Holm-Šídák method. In vitro experiments are shown as mean ± standard deviation of duplicate wells. N values for in vivo experiments indicate the number of mice in each condition. All data are representative of two or more independent experiments. Additional information, including n values and number of replicates, can be found in figure legends.
Supplementary Material
Figure S1. IL-23 receptor crystallography. Related to Figure 1. (A–B) Quaternary complex structure with density corresponding to IL-12Rβ1 D1 domain shown with 2mFo-DFc electron density map contoured at 1.0σ. (B) Inset showing interface view of p40 binding to IL-12Rβ1.
(C) Alignment of IL-23R bound IL-23 from quaternary complex reported here and a previously published crystal structure of the partial complex (PDB: 5MZV). Structures were aligned on the IL-23p19 subunit to show the hinge-arm motion of IL-23R.
(D) Crystal packing of the quaternary IL-23 receptor complex occludes IL-12Rβ1 D2. The IL-12Rβ1 D1–D2 crystal structure was aligned to IL-12Rβ1 D1 in the quaternary complex structure. An IL-23R subunit from a neighboring asymmetric unit is shown as a ribbon trace of backbone alpha carbons with associated 2mFo-DFc electron density map contoured at 1.0σ.
Figure S2. Convergent p40 binding at the IL-12Rβ1 docking site. Related to Figure 1. (A) Crystal structure of the quaternary IL-23R complex showing the interaction between IL-12Rβ1 and p40.
(B) Ustekinumab bound to p40 (PDB: 3HMX).
(C) Briakinumab bound to p40 (PDB: 5NJD).
(D) Nanobody 22e11 bound to p40 (PDB: 4GRW).
Figure S3. Cryo-EM data processing of the IL-12 and IL-23 receptor complexes. Related to Figure 3. (A–D) Cryo-EM of the IL-23 receptor complex. (A) Representative image from data acquisition. (B) 2D class averages. (C) Data processing flow chart. (D) Fourier shell correlation (FSC) curve with dashed line at FSC=0.143.
(E–H) Cryo-EM of the IL-12 receptor complex. (E) Representative image from data acquisition. (F) 2D class averages. (G) Data processing flow chart. (H) Fourier shell correlation (FSC) curve with dashed line at FSC=0.143.
Figure S4. Characterization of mouse IL-12 T cell signaling. Related to Figure 5. (A) Gating of NK cells and T cells for p40 tetramer staining. Related to Figure 5A.
(B) Alignment of human and mouse p40. Sequences were aligned using Clustal Omega and colored by residue according to the ClustalX color scheme. Red stars indicate residues targeted for mutagenesis.
(C) OT-I CD8+ T cell gating. Related to Figure 5D.
(D) IL-12 induces dose-dependent IFNγ production in OT-I T cells. Splenocytes from OT-I TCR transgenic mice were stimulated as in Figure 5D with a titration of IL-12 and analyzed for IFNγ induction at 48 hours by intracellular cytokine stain.
(E) IL-12 agonists elicit dose dependent IFNγ expression in OT-I T cells. Splenocytes from OT-I TCR transgenic mice were stimulated as in Figure 5D and analyzed for IFNγ induction at 48 hours by intracellular cytokine stain.
(F) IL-12 agonists enhance IFNγ secretion by OT-I T cells. Analysis of IFNγ levels in supernatant of OT-I cultures containing 1μM IL-12 agonists by ELISA, related to Figure 5D.
(G) IL-12Rβ1 is upregulated on CD4+ T cell blasts. Analyzed as in Figure 5A.
(H–I) IL-12 partial agonists support Th1 polarization. Naïve CD4+ T cells were activated with 2μg/mL plate-bound αCD3 with 0.5μg/mL αCD28, 5ng/mL IL-2, and 1μg/mL αIL-4 in the presence of 1μM IL-12 partial agonists for 48 hours. In the last 4 hours of culture, cells were restimulated with PMA-ionomycin in the presence of GolgiStop and IFNγ was read-out by intracellular cytokine stain. (H) Representative gating showing identification of IFNγ+ Th1 cells. (I) Quantification of Th1 induction in response to IL-12 partial agonists.
Data are shown as mean ± standard deviation of two biological replicates and are representative to two or more independent experiments.
Figure S5. Characterization of mouse IL-12 signaling on NK cells. Related to Figure 5. (A) IL-12 partial agonists elicit reduced pSTAT4 signaling in NK cells. MACS purified NK cells were mixed with CTV loaded carrier cells and stimulating with IL-12 agonists for 20 minutes.
(B) IL-18 is required for IL-12 mediated IFNγ induction in NK cells. MACS purified NK cells were stimulated with 50ng/mL IL-18 and 1nM IL-12 as indicated.
(C) IL-12 induces dose dependent IFNγ production in NK cells. NK cells were stimulated as in Figure 5E with a titration of IL-12 and analyzed for IFNγ induction at 48 hours by intracellular cytokine stain.
(D) IL-12 agonists elicit dose dependent IFNγ expression in NK cells, related to Figure 5E.
(E) 3xAla and 4xAla IL-12 partial agonists have reduced secretion of IFNγ by NK cells relative to IL-12. Analysis of IFNγ in supernatant of NK cell cultures by ELISA, related to Figure 5E.
(F–G) Quantitative PCR (qPCR) of Ifng (F) and Tigit (G) from NK cells stimulated with 50ng/mL IL-18 and 1μM IL-12 for 8 hours. Data were normalized to unstimulated control and are shown as mean ± standard deviation of technical triplicates.
(H) IL-2 pre-activation upregulates IL-12Rβ1 on NK cells. MACS purified NK cells were stimulated with 1000IU/mL IL-2 for 48 hours and stained with 200nM p40 tetramer (red) or streptavidin control (grey) as in Figure 5A to identify IL-12Rβ1 expression levels.
(I) IL-2 enhances IFNγ induction in NK cells but does not synergize with IL-12 partial agonists. MACS purified NK cells were activated with 1000IU/mL IL-2, 50ng/mL IL-18 and 1μM IL-12 agonists for 48 hours. Dashed line indicates IFNγ staining in NK cells stimulated with IL-18 alone. Data are shown as mean ± standard deviation of two biological replicates unless otherwise stated and are representative of two or more experiments.
Figure S6. Characterization of human IL-12 partial agonists. Related to Figure 5. (A) Cell-type and activation-state dependent expression of IL-12Rβ1 in human PBMCs. Flow cytometry plots showing IL-12Rβ1 expression level measured by p40 tetramer staining of human NK cells (CD3-CD56+) or CD8+ T cells (CD3+CD8+). Red line indicates 200nM tetramer staining, gray population represents streptavidin staining alone. For T cell blasts, PBMCs were stimulated for 2 days with 2.5μg/mL αCD3, 5μg/mL αCD28, and 100IU/mL IL-2.
(B) NK cell and T cell gating scheme.
(C–D) Phospho-flow cytometry of CD8+ T cell blasts stimulated with IL-12 partial agonists for 20 minutes. (C) Dose-response curves of pSTAT4 signaling in human CD8+ T cell blasts. (D) Histograms show pSTAT4 staining at 8nM (IL-12) or 1μM (2xAla: Glu81Ala/Phe82Ala, 3xAla: Glu81Ala/Phe82Ala/Lys106Ala)
(E) IL-12 partial agonists support IFNγ secretion by CD8+ T cells. MACS isolated CD8+ T cells were stimulated with 2μg/mL αCD3, 0.5μg/mL αCD28, and 5ng/mL IL-2 with or without IL-12 agonists. After 48 hours, the supernatant was analyzed for IFNγ by ELISA. Dashed line indicates IFNγ level in the absence of IL-12.
(F) IL-12 partial agonists show attenuated IFNγ production by NK cells. MACS isolated NK cells were stimulated with 100ng/mL IL-18 with or without IL-12 agonists for 48 hours and the supernatant was assayed for IFNγ by ELISA. Conditions in which no IFNγ was detected above background are listed as n.d. for not determined
Data are expressed as mean ± standard deviation of two biological replicates and are representative of two independent experiments.
Figure S7. In vivo characterization of IL-12 agonists. Related to Figure 6. (A–B) Purification of IL-12 from expi293F cells. (A) Representative S200 size exclusion chromatography (SEC) of Ni-NTA purified murine IL-12. mAU: milli absorbance units. (B) SDS-PAGE of IL-12 following Ni-NTA affinity purification and SEC under reducing
(R) and non-reducing (NR) conditions.
(C–F) Characterization of mammalian-expressed mouse IL-12 variants. (C–D) pSTAT4 staining of CD8+ T cell blasts following a 20 minute stimulation with cytokine. Histograms show pSTAT4 staining at 8nM for IL-12 and 1μM for partial agonists. (E) Mammalian-expressed IL-12 partial agonists promote IFNγ production by antigen-specific CD8+ T cells. Representative histograms (left) and quantification (right) of intracellular IFNγ in OT-I CD8+ T cells (CD3+CD8+) following 48-hour stimulation with 1μg/mL OVA peptide (257–264), 0.5ug/mL αCD28, 100IU/mL IL-2, and 1μM IL-12 variants. For gating, see Figure S4C. (F) Mammalian-expressed IL-12 partial agonists display attenuated IFNγ induction in NK cell. Purified NK cells were stimulated with 50ng/mL IL-18 with 1μM IL-12 variants for 48 hours.
(G) Gating scheme to identify NK cells and OT-I T cells in draining lymph nodes following OVA-IFA immunization, related to Figure 6. OT-I T cells were defined as liveCD3+CD8+Thy1.2+. NK cells were defined as liveCD3-NKp46+. αNK1.1 was included in these panels; however, it is downregulated by IL-12 administration.
(H) Representative FACS plots showing LAG-3 expression in NK cells. Related to Figure 6G.
Highlights:
Crystal structure of the complete IL-23 receptor complex
Cryo-EM maps of the complete IL-12 and IL-23 receptor complexes
The p40 subunit of IL-12 and IL-23 is a common gateway for induction of STAT signaling
T cell biased IL-12 agonists elicit anti-tumor response without inducing toxicity
Acknowledgments
The authors would like to thank Amy Fan, Robert Saxton and members of the Garcia lab for helpful feedback and discussion. Technical support for cryo-EM was provided by Elizabeth Montabana. pCDH-EF1-cOVA-T2A-copGFP plasmid was a kind gift of Irving L. Weissman. K.C.G. is an investigator with the Howard Hughes Medical Institute. K.C.G. is supported by NIH R01-AI51321, The Mathers Foundation, and Ludwig Foundation. C.R.G is supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1656518. We thank the staff at Stanford Synchrotron Radiation Lightsource and Advanced Light Source for their assistance. The Advanced Light Source is a Department of Energy Office of Science User Facility under Contract No. DE-AC02-05CH11231. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). Graphical abstract was created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
K.C.G and C.R.G are co-inventors on a provisional patent based on discoveries described in this manuscript. K.C.G is the founder of Synthekine.
References
- Anderson R, Macdonald I, Corbett T, Hacking G, Lowdell MW, and Prentice HG (1997). Construction and biological characterization of an interleukin-12 fusion protein (Flexi-12): delivery to acute myeloid leukemic blasts using adeno-associated virus. Hum Gene Ther 8, 1125–1135. [DOI] [PubMed] [Google Scholar]
- Ardolino M, Azimi CS, Iannello A, Trevino TN, Horan L, Zhang L, Deng W, Ring AM, Fischer S, Garcia KC, et al. (2014). Cytokine therapy reverses NK cell anergy in MHC-deficient tumors. J Clin Invest 124, 4781–4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aste-Amezaga M, D’Andrea A, Kubin M, and Trinchieri G (1994). Cooperation of natural killer cell stimulatory factor/interleukin-12 with other stimuli in the induction of cytokines and cytotoxic cell-associated molecules in human T and NK cells. Cell Immunol 156, 480–492. [DOI] [PubMed] [Google Scholar]
- Au-Yeung BB, Smith GA, Mueller JL, Heyn CS, Jaszczak RG, Weiss A, and Zikherman J (2017). IL-2 Modulates the TCR Signaling Threshold for CD8 but Not CD4 T Cell Proliferation on a Single-Cell Level. J Immunol 198, 2445–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson JM, Sachs CW, Treacy G, Zhou H, Pendley CE, Brodmerkel CM, Shankar G, and Mascelli MA (2011). Therapeutic targeting of the IL-12/23 pathways: generation and characterization of ustekinumab. Nat Biotechnol 29, 615–624. [DOI] [PubMed] [Google Scholar]
- Berraondo P, Etxeberria I, Ponz-Sarvise M, and Melero I (2018). Revisiting Interleukin-12 as a Cancer Immunotherapy Agent. Clin Cancer Res 24, 2716–2718. [DOI] [PubMed] [Google Scholar]
- Biber JL, Jabbour S, Parihar R, Dierksheide J, Hu Y, Baumann H, Bouchard P, Caligiuri MA, and Carson W (2002). Administration of two macrophage-derived interferon-gamma-inducing factors (IL-12 and IL-15) induces a lethal systemic inflammatory response in mice that is dependent on natural killer cells but does not require interferon-gamma. Cell Immunol 216, 31–42. [DOI] [PubMed] [Google Scholar]
- Bloch Y, Bouchareychas L, Merceron R, Skladanowska K, Van den Bossche L, Detry S, Govindarajan S, Elewaut D, Haerynck F, Dullaers M, et al. (2018). Structural Activation of Pro-inflammatory Human Cytokine IL-23 by Cognate IL-23 Receptor Enables Recruitment of the Shared Receptor IL-12Rbeta1. Immunity 48, 45–58 e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger MJ, Bankovich AJ, Kortemme T, Baker D, and Garcia KC (2003a). Convergent mechanisms for recognition of divergent cytokines by the shared signaling receptor gp130. Mol Cell 12, 577–589. [DOI] [PubMed] [Google Scholar]
- Boulanger MJ, Chow DC, Brevnova EE, and Garcia KC (2003b). Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science 300, 2101–2104. [DOI] [PubMed] [Google Scholar]
- Brunda MJ, Luistro L, Hendrzak JA, Fountoulakis M, Garotta G, and Gately MK (1995). Role of interferon-gamma in mediating the antitumor efficacy of interleukin-12. J Immunother Emphasis Tumor Immunol 17, 71–77. [DOI] [PubMed] [Google Scholar]
- Brunda MJ, Luistro L, Warrier RR, Wright RB, Hubbard BR, Murphy M, Wolf SF, and Gately MK (1993). Antitumor and antimetastatic activity of interleukin 12 against murine tumors. J Exp Med 178, 1223–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson WE, Dierksheide JE, Jabbour S, Anghelina M, Bouchard P, Ku G, Yu H, Baumann H, Shah MH, Cooper MA, et al. (2000). Coadministration of interleukin-18 and interleukin-12 induces a fatal inflammatory response in mice: critical role of natural killer cell interferon-gamma production and STAT-mediated signal transduction. Blood 96, 1465–1473. [PubMed] [Google Scholar]
- Carson WE, Yu H, Dierksheide J, Pfeffer K, Bouchard P, Clark R, Durbin J, Baldwin AS, Peschon J, Johnson PR, et al. (1999). A fatal cytokine-induced systemic inflammatory response reveals a critical role for NK cells. J Immunol 162, 4943–4951. [PubMed] [Google Scholar]
- Chan TC, Hawkes JE, and Krueger JG (2018). Interleukin 23 in the skin: role in psoriasis pathogenesis and selective interleukin 23 blockade as treatment. Ther Adv Chronic Dis 9, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, and Wittrup KD (2006). Isolating and engineering human antibodies using yeast surface display. Nat Protoc 1, 755–768. [DOI] [PubMed] [Google Scholar]
- Chow D, He X, Snow AL, Rose-John S, and Garcia KC (2001). Structure of an extracellular gp130 cytokine receptor signaling complex. Science 291, 2150–2155. [DOI] [PubMed] [Google Scholar]
- Chua AO, Chizzonite R, Desai BB, Truitt TP, Nunes P, Minetti LJ, Warrier RR, Presky DH, Levine JF, Gately MK, et al. (1994). Expression cloning of a human IL-12 receptor component. A new member of the cytokine receptor superfamily with strong homology to gp130. J Immunol 153, 128–136. [PubMed] [Google Scholar]
- Cohen J (1995). IL-12 deaths: explanation and a puzzle. Science 270, 908. [DOI] [PubMed] [Google Scholar]
- Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, Satoskar AR, Garcia KC, Hunter CA, Drake CG, et al. (2012). The composition and signaling of the IL-35 receptor are unconventional. Nat Immunol 13, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, and Vignali DA (2007). The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 450, 566–569. [DOI] [PubMed] [Google Scholar]
- Corbett TH, Griswold DP Jr., Roberts BJ, Peckham JC, and Schabel FM Jr. (1975). Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res 35, 2434–2439. [PubMed] [Google Scholar]
- Dambuza IM, He C, Choi JK, Yu CR, Wang R, Mattapallil MJ, Wingfield PT, Caspi RR, and Egwuagu CE (2017). IL-12p35 induces expansion of IL-10 and IL-35-expressing regulatory B cells and ameliorates autoimmune disease. Nat Commun 8, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmyter A, Spinelli S, Boutton C, Saunders M, Blachetot C, de Haard H, Denecker G, Van Roy M, Cambillau C, and Rommelaere H (2017). Neutralization of Human Interleukin 23 by Multivalent Nanobodies Explained by the Structure of Cytokine-Nanobody Complex. Front Immunol 8, 884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukkipati A, Park HH, Waghray D, Fischer S, and Garcia KC (2008). BacMam system for high-level expression of recombinant soluble and membrane glycoproteins for structural studies. Protein Expr Purif 62, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairhead M, and Howarth M (2015). Site-specific biotinylation of purified proteins using BirA. Methods Mol Biol 1266, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewell JG, Matar MM, Rice JS, Brunhoeber E, Slobodkin G, Pence C, Worker M, Lewis DH, and Anwer K (2009). Treatment of disseminated ovarian cancer using nonviral interleukin-12 gene therapy delivered intraperitoneally. J Gene Med 11, 718–728. [DOI] [PubMed] [Google Scholar]
- Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, and Colonna M (2013). Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 38, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, and Carbone FR (1994). T cell receptor antagonist peptides induce positive selection. Cell 76, 17–27. [DOI] [PubMed] [Google Scholar]
- Huyton T, Zhang JG, Luo CS, Lou MZ, Hilton DJ, Nicola NA, and Garrett TP (2007). An unusual cytokine:Ig-domain interaction revealed in the crystal structure of leukemia inhibitory factor (LIF) in complex with the LIF receptor. Proc Natl Acad Sci U S A 104, 12737–12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010). Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfenstein DR, and Vale RD (2004). The lipid binding pleckstrin homology domain in UNC-104 kinesin is necessary for synaptic vesicle transport in Caenorhabditis elegans. Mol Biol Cell 15, 3729–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Fitz L, Ryan M, Hewick RM, Clark SC, Chan S, Loudon R, Sherman F, Perussia B, and Trinchieri G (1989). Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J Exp Med 170, 827–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp T, Riedl E, Bangert C, Bowman EP, Greisenegger E, Horowitz A, Kittler H, Blumenschein WM, McClanahan TK, Marbury T, et al. (2015). Clinical improvement in psoriasis with specific targeting of interleukin-23. Nature 521, 222–226. [DOI] [PubMed] [Google Scholar]
- Koutruba N, Emer J, and Lebwohl M (2010). Review of ustekinumab, an interleukin-12 and interleukin-23 inhibitor used for the treatment of plaque psoriasis. Ther Clin Risk Manag 6, 123–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Lasek W, Zagozdzon R, and Jakobisiak M (2014). Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother 63, 419–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann C, Zeis M, and Uharek L (2001). Activation of natural killer cells with interleukin 2 (IL-2) and IL-12 increases perforin binding and subsequent lysis of tumour cells. Br J Haematol 114, 660–665. [DOI] [PubMed] [Google Scholar]
- Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, and Ryan JL (1997). Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 90, 2541–2548. [PubMed] [Google Scholar]
- Leong JW, Chase JM, Romee R, Schneider SE, Sullivan RP, Cooper MA, and Fehniger TA (2014). Preactivation with IL-12, IL-15, and IL-18 induces CD25 and a functional high-affinity IL-2 receptor on human cytokine-induced memory-like natural killer cells. Biol Blood Marrow Transplant 20, 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Zuo A, Shao H, Born WK, O’Brien RL, Kaplan HJ, and Sun D (2013). IL-23 receptor expression on gammadelta T cells correlates with their enhancing or suppressive effects on autoreactive T cells in experimental autoimmune uveitis. J Immunol 191, 1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littman DR, and Rudensky AY (2010). Th17 and regulatory T cells in mediating and restraining inflammation. Cell 140, 845–858. [DOI] [PubMed] [Google Scholar]
- Luo J, Wu SJ, Lacy ER, Orlovsky Y, Baker A, Teplyakov A, Obmolova G, Heavner GA, Richter HT, and Benson J (2010). Structural basis for the dual recognition of IL-12 and IL-23 by ustekinumab. J Mol Biol 402, 797–812. [DOI] [PubMed] [Google Scholar]
- Lupardus PJ, and Garcia KC (2008). The structure of interleukin-23 reveals the molecular basis of p40 subunit sharing with interleukin-12. J Mol Biol 382, 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattner F, Fischer S, Guckes S, Jin S, Kaulen H, Schmitt E, Rude E, and Germann T (1993). The interleukin-12 subunit p40 specifically inhibits effects of the interleukin-12 heterodimer. Eur J Immunol 23, 2202–2208. [DOI] [PubMed] [Google Scholar]
- Midelfort KS, Hernandez HH, Lippow SM, Tidor B, Drennan CL, and Wittrup KD (2004). Substantial energetic improvement with minimal structural perturbation in a high affinity mutant antibody. J Mol Biol 343, 685–701. [DOI] [PubMed] [Google Scholar]
- Momin N, Mehta NK, Bennett NR, Ma L, Palmeri JR, Chinn MM, Lutz EA, Kang B, Irvine DJ, Spranger S, et al. (2019). Anchoring of intratumorally administered cytokines to collagen safely potentiates systemic cancer immunotherapy. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S, Kundu M, Jana M, Roy A, Rangasamy SB, Modi KK, Wallace J, Albalawi YA, Balabanov R, and Pahan K (2020). IL-12 p40 monomer is different from other IL-12 family members to selectively inhibit IL-12Rbeta1 internalization and suppress EAE. Proc Natl Acad Sci U S A 117, 21557–21567. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, and Sliz P (2013). Collaboration gets the most out of software. Elife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ (2007). The JAK-STAT signaling pathway: input and output integration. J Immunol 178, 2623–2629. [DOI] [PubMed] [Google Scholar]
- Nastala CL, Edington HD, McKinney TG, Tahara H, Nalesnik MA, Brunda MJ, Gately MK, Wolf SF, Schreiber RD, Storkus WJ, et al. (1994). Recombinant IL-12 administration induces tumor regression in association with IFN-gamma production. J Immunol 153, 1697–1706. [PubMed] [Google Scholar]
- Oka N, Markova T, Tsuzuki K, Li W, El-Darawish Y, Pencheva-Demireva M, Yamanishi K, Yamanishi H, Sakagami M, Tanaka Y, et al. (2020). IL-12 regulates the expansion, phenotype, and function of murine NK cells activated by IL-15 and IL-18. Cancer Immunol Immunother 69, 1699–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, et al. (2000). Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–725. [DOI] [PubMed] [Google Scholar]
- Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, et al. (2002). A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol 168, 5699–5708. [DOI] [PubMed] [Google Scholar]
- Pasche N, Wulhfard S, Pretto F, Carugati E, and Neri D (2012). The antibody-based delivery of interleukin-12 to the tumor neovasculature eradicates murine models of cancer in combination with paclitaxel. Clin Cancer Res 18, 4092–4103. [DOI] [PubMed] [Google Scholar]
- Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, and Brentjens RJ (2012). Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 119, 4133–4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflanz S, Hibbert L, Mattson J, Rosales R, Vaisberg E, Bazan JF, Phillips JH, McClanahan TK, de Waal Malefyt R, and Kastelein RA (2004). WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J Immunol 172, 2225–2231. [DOI] [PubMed] [Google Scholar]
- Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, Hibbert L, Churakova T, Travis M, Vaisberg E, et al. (2002). IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity 16, 779–790. [DOI] [PubMed] [Google Scholar]
- Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu CY, Gately MK, and Gubler U (1996). A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor subunits. Proc Natl Acad Sci U S A 93, 14002–14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves PJ, Callewaert N, Contreras R, and Khorana HG (2002). Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci U S A 99, 13419–13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, Cooper MA, and Fehniger TA (2012). Cytokine activation induces human memory-like NK cells. Blood 120, 4751–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein MP, Su EW, Suriano S, Cloud CA, Andrijauskaite K, Kesarwani P, Schwartz KM, Williams KM, Johnson CB, Li M, et al. (2015). Interleukin-12 enhances the function and anti-tumor activity in murine and human CD8(+) T cells. Cancer Immunol Immunother 64, 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder J, Moll JM, Baran P, Grotzinger J, Scheller J, and Floss DM (2015). Non-canonical interleukin 23 receptor complex assembly: p40 protein recruits interleukin 12 receptor beta1 via site II and induces p19/interleukin 23 receptor interaction via site III. J Biol Chem 290, 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skiniotis G, Lupardus PJ, Martick M, Walz T, and Garcia KC (2008). Structural organization of a full-length gp130/LIF-R cytokine receptor transmembrane complex. Mol Cell 31, 737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo SJ, Dighe AS, Gubler U, and Murphy KM (1997). Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med 185, 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Nishi T, Goto T, Takeshima H, Dev SB, Ushio Y, and Sakata T (2001). Intratumoral delivery of interleukin 12 expression plasmids with in vivo electroporation is effective for colon and renal cancer. Hum Gene Ther 12, 1265–1276. [DOI] [PubMed] [Google Scholar]
- Thakker P, Leach MW, Kuang W, Benoit SE, Leonard JP, and Marusic S (2007). IL-23 is critical in the induction but not in the effector phase of experimental autoimmune encephalomyelitis. J Immunol 178, 2589–2598. [DOI] [PubMed] [Google Scholar]
- Trinchieri G (2003). Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 3, 133–146. [DOI] [PubMed] [Google Scholar]
- Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, Seita J, Inlay MA, Weiskopf K, Miyanishi M, et al. (2013). Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A 110, 11103–11108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Vignali DA, and Kuchroo VK (2012). IL-12 family cytokines: immunological playmakers. Nat Immunol 13, 722–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C, Blanc E, Roversi P, and Bricogne G (2007). Automated structure solution with autoSHARP. Methods Mol Biol 364, 215–230. [DOI] [PubMed] [Google Scholar]
- Wallington JC, Williams AP, Staples KJ, and Wilkinson TMA (2018). IL-12 and IL-7 synergize to control mucosal-associated invariant T-cell cytotoxic responses to bacterial infection. J Allergy Clin Immunol 141, 2182–2195 e2186. [DOI] [PubMed] [Google Scholar]
- Wang X, Lupardus P, Laporte SL, and Garcia KC (2009). Structural biology of shared cytokine receptors. Annu Rev Immunol 27, 29–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wei Y, Xiao H, Liu X, Zhang Y, Han G, Chen G, Hou C, Ma N, Shen B, et al. (2016). A novel IL-23p19/Ebi3 (IL-39) cytokine mediates inflammation in Lupus-like mice. Eur J Immunol 46, 1343–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtowicz WM, Vielmetter J, Fernandes RA, Siepe DH, Eastman CL, Chisholm GB, Cox S, Klock H, Anderson PW, Rue SM, et al. (2020). A Human IgSF Cell-Surface Interactome Reveals a Complex Network of Protein-Protein Interactions. Cell 182, 1027–1043 e1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao BB, Niu P, Surowy CS, and Faltynek CR (1999). Direct interaction of STAT4 with the IL-12 receptor. Arch Biochem Biophys 368, 147–155. [DOI] [PubMed] [Google Scholar]
- Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, et al. (2006). IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest 116, 1310–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeste A, Mascanfroni ID, Nadeau M, Burns EJ, Tukpah AM, Santiago A, Wu C, Patel B, Kumar D, and Quintana FJ (2014). IL-21 induces IL-22 production in CD4+ T cells. Nat Commun 5, 3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon C, Johnston SC, Tang J, Stahl M, Tobin JF, and Somers WS (2000). Charged residues dominate a unique interlocking topography in the heterodimeric cytokine interleukin-12. EMBO J 19, 3530–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F (2009). Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol 28, 239–260. [DOI] [PubMed] [Google Scholar]
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, and Littman DR (2007). IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 8, 967–974. [DOI] [PubMed] [Google Scholar]
- Zitvogel L, Tahara H, Robbins PD, Storkus WJ, Clarke MR, Nalesnik MA, and Lotze MT (1995). Cancer immunotherapy of established tumors with IL-12. Effective delivery by genetically engineered fibroblasts. J Immunol 155, 1393–1403. [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. IL-23 receptor crystallography. Related to Figure 1. (A–B) Quaternary complex structure with density corresponding to IL-12Rβ1 D1 domain shown with 2mFo-DFc electron density map contoured at 1.0σ. (B) Inset showing interface view of p40 binding to IL-12Rβ1.
(C) Alignment of IL-23R bound IL-23 from quaternary complex reported here and a previously published crystal structure of the partial complex (PDB: 5MZV). Structures were aligned on the IL-23p19 subunit to show the hinge-arm motion of IL-23R.
(D) Crystal packing of the quaternary IL-23 receptor complex occludes IL-12Rβ1 D2. The IL-12Rβ1 D1–D2 crystal structure was aligned to IL-12Rβ1 D1 in the quaternary complex structure. An IL-23R subunit from a neighboring asymmetric unit is shown as a ribbon trace of backbone alpha carbons with associated 2mFo-DFc electron density map contoured at 1.0σ.
Figure S2. Convergent p40 binding at the IL-12Rβ1 docking site. Related to Figure 1. (A) Crystal structure of the quaternary IL-23R complex showing the interaction between IL-12Rβ1 and p40.
(B) Ustekinumab bound to p40 (PDB: 3HMX).
(C) Briakinumab bound to p40 (PDB: 5NJD).
(D) Nanobody 22e11 bound to p40 (PDB: 4GRW).
Figure S3. Cryo-EM data processing of the IL-12 and IL-23 receptor complexes. Related to Figure 3. (A–D) Cryo-EM of the IL-23 receptor complex. (A) Representative image from data acquisition. (B) 2D class averages. (C) Data processing flow chart. (D) Fourier shell correlation (FSC) curve with dashed line at FSC=0.143.
(E–H) Cryo-EM of the IL-12 receptor complex. (E) Representative image from data acquisition. (F) 2D class averages. (G) Data processing flow chart. (H) Fourier shell correlation (FSC) curve with dashed line at FSC=0.143.
Figure S4. Characterization of mouse IL-12 T cell signaling. Related to Figure 5. (A) Gating of NK cells and T cells for p40 tetramer staining. Related to Figure 5A.
(B) Alignment of human and mouse p40. Sequences were aligned using Clustal Omega and colored by residue according to the ClustalX color scheme. Red stars indicate residues targeted for mutagenesis.
(C) OT-I CD8+ T cell gating. Related to Figure 5D.
(D) IL-12 induces dose-dependent IFNγ production in OT-I T cells. Splenocytes from OT-I TCR transgenic mice were stimulated as in Figure 5D with a titration of IL-12 and analyzed for IFNγ induction at 48 hours by intracellular cytokine stain.
(E) IL-12 agonists elicit dose dependent IFNγ expression in OT-I T cells. Splenocytes from OT-I TCR transgenic mice were stimulated as in Figure 5D and analyzed for IFNγ induction at 48 hours by intracellular cytokine stain.
(F) IL-12 agonists enhance IFNγ secretion by OT-I T cells. Analysis of IFNγ levels in supernatant of OT-I cultures containing 1μM IL-12 agonists by ELISA, related to Figure 5D.
(G) IL-12Rβ1 is upregulated on CD4+ T cell blasts. Analyzed as in Figure 5A.
(H–I) IL-12 partial agonists support Th1 polarization. Naïve CD4+ T cells were activated with 2μg/mL plate-bound αCD3 with 0.5μg/mL αCD28, 5ng/mL IL-2, and 1μg/mL αIL-4 in the presence of 1μM IL-12 partial agonists for 48 hours. In the last 4 hours of culture, cells were restimulated with PMA-ionomycin in the presence of GolgiStop and IFNγ was read-out by intracellular cytokine stain. (H) Representative gating showing identification of IFNγ+ Th1 cells. (I) Quantification of Th1 induction in response to IL-12 partial agonists.
Data are shown as mean ± standard deviation of two biological replicates and are representative to two or more independent experiments.
Figure S5. Characterization of mouse IL-12 signaling on NK cells. Related to Figure 5. (A) IL-12 partial agonists elicit reduced pSTAT4 signaling in NK cells. MACS purified NK cells were mixed with CTV loaded carrier cells and stimulating with IL-12 agonists for 20 minutes.
(B) IL-18 is required for IL-12 mediated IFNγ induction in NK cells. MACS purified NK cells were stimulated with 50ng/mL IL-18 and 1nM IL-12 as indicated.
(C) IL-12 induces dose dependent IFNγ production in NK cells. NK cells were stimulated as in Figure 5E with a titration of IL-12 and analyzed for IFNγ induction at 48 hours by intracellular cytokine stain.
(D) IL-12 agonists elicit dose dependent IFNγ expression in NK cells, related to Figure 5E.
(E) 3xAla and 4xAla IL-12 partial agonists have reduced secretion of IFNγ by NK cells relative to IL-12. Analysis of IFNγ in supernatant of NK cell cultures by ELISA, related to Figure 5E.
(F–G) Quantitative PCR (qPCR) of Ifng (F) and Tigit (G) from NK cells stimulated with 50ng/mL IL-18 and 1μM IL-12 for 8 hours. Data were normalized to unstimulated control and are shown as mean ± standard deviation of technical triplicates.
(H) IL-2 pre-activation upregulates IL-12Rβ1 on NK cells. MACS purified NK cells were stimulated with 1000IU/mL IL-2 for 48 hours and stained with 200nM p40 tetramer (red) or streptavidin control (grey) as in Figure 5A to identify IL-12Rβ1 expression levels.
(I) IL-2 enhances IFNγ induction in NK cells but does not synergize with IL-12 partial agonists. MACS purified NK cells were activated with 1000IU/mL IL-2, 50ng/mL IL-18 and 1μM IL-12 agonists for 48 hours. Dashed line indicates IFNγ staining in NK cells stimulated with IL-18 alone. Data are shown as mean ± standard deviation of two biological replicates unless otherwise stated and are representative of two or more experiments.
Figure S6. Characterization of human IL-12 partial agonists. Related to Figure 5. (A) Cell-type and activation-state dependent expression of IL-12Rβ1 in human PBMCs. Flow cytometry plots showing IL-12Rβ1 expression level measured by p40 tetramer staining of human NK cells (CD3-CD56+) or CD8+ T cells (CD3+CD8+). Red line indicates 200nM tetramer staining, gray population represents streptavidin staining alone. For T cell blasts, PBMCs were stimulated for 2 days with 2.5μg/mL αCD3, 5μg/mL αCD28, and 100IU/mL IL-2.
(B) NK cell and T cell gating scheme.
(C–D) Phospho-flow cytometry of CD8+ T cell blasts stimulated with IL-12 partial agonists for 20 minutes. (C) Dose-response curves of pSTAT4 signaling in human CD8+ T cell blasts. (D) Histograms show pSTAT4 staining at 8nM (IL-12) or 1μM (2xAla: Glu81Ala/Phe82Ala, 3xAla: Glu81Ala/Phe82Ala/Lys106Ala)
(E) IL-12 partial agonists support IFNγ secretion by CD8+ T cells. MACS isolated CD8+ T cells were stimulated with 2μg/mL αCD3, 0.5μg/mL αCD28, and 5ng/mL IL-2 with or without IL-12 agonists. After 48 hours, the supernatant was analyzed for IFNγ by ELISA. Dashed line indicates IFNγ level in the absence of IL-12.
(F) IL-12 partial agonists show attenuated IFNγ production by NK cells. MACS isolated NK cells were stimulated with 100ng/mL IL-18 with or without IL-12 agonists for 48 hours and the supernatant was assayed for IFNγ by ELISA. Conditions in which no IFNγ was detected above background are listed as n.d. for not determined
Data are expressed as mean ± standard deviation of two biological replicates and are representative of two independent experiments.
Figure S7. In vivo characterization of IL-12 agonists. Related to Figure 6. (A–B) Purification of IL-12 from expi293F cells. (A) Representative S200 size exclusion chromatography (SEC) of Ni-NTA purified murine IL-12. mAU: milli absorbance units. (B) SDS-PAGE of IL-12 following Ni-NTA affinity purification and SEC under reducing
(R) and non-reducing (NR) conditions.
(C–F) Characterization of mammalian-expressed mouse IL-12 variants. (C–D) pSTAT4 staining of CD8+ T cell blasts following a 20 minute stimulation with cytokine. Histograms show pSTAT4 staining at 8nM for IL-12 and 1μM for partial agonists. (E) Mammalian-expressed IL-12 partial agonists promote IFNγ production by antigen-specific CD8+ T cells. Representative histograms (left) and quantification (right) of intracellular IFNγ in OT-I CD8+ T cells (CD3+CD8+) following 48-hour stimulation with 1μg/mL OVA peptide (257–264), 0.5ug/mL αCD28, 100IU/mL IL-2, and 1μM IL-12 variants. For gating, see Figure S4C. (F) Mammalian-expressed IL-12 partial agonists display attenuated IFNγ induction in NK cell. Purified NK cells were stimulated with 50ng/mL IL-18 with 1μM IL-12 variants for 48 hours.
(G) Gating scheme to identify NK cells and OT-I T cells in draining lymph nodes following OVA-IFA immunization, related to Figure 6. OT-I T cells were defined as liveCD3+CD8+Thy1.2+. NK cells were defined as liveCD3-NKp46+. αNK1.1 was included in these panels; however, it is downregulated by IL-12 administration.
(H) Representative FACS plots showing LAG-3 expression in NK cells. Related to Figure 6G.
Data Availability Statement
Structure factors and coordinates have been deposited in the Protein Data Bank with identification numbers PDB: 6WDP (IL-12Rβ1) and PDB: 6WDQ (IL-23 receptor complex). Cryo-EM maps have been deposited to Electron Microscopy Data Bank (EMDB) under accession ID EMD-21646 (IL-23 receptor complex) and EMD-21645 (IL-12 receptor complex).