

Abstract
A dual catalytic system for cross-electrophile coupling reactions between aryl halides and alkyl halides that features a Ni catalyst, a Co cocatalyst, and a mild homogeneous reductant is described. Mechanistic studies indicate that the Ni catalyst activates the aryl halide, while the Co cocatalyst activates the alkyl halide. This allows the system to be rationally optimized for a variety of substrate classes by simply modifying the loadings of the Ni and Co catalysts based on the reaction product profile. For example, the coupling of aryl bromides and aryl iodides with alkyl bromides, alkyl iodides, and benzyl chlorides is demonstrated using the same Ni and Co catalysts under similar reaction conditions but with different optimal catalyst loadings in each case. Our system is tolerant of numerous functional groups and is capable of coupling heteroaryl halides, di-ortho-substituted aryl halides, pharmaceutically relevant druglike aryl halides, and a diverse range of alkyl halides. Additionally, the dual catalytic platform facilitates a series of selective one-pot three-component cross-electrophile coupling reactions of bromo(iodo)arenes with two distinct alkyl halides. This demonstrates the unique level of control that the platform provides and enables the rapid generation of molecular complexity. The system can be readily utilized for a wide range of applications as all reaction components are commercially available, the reaction is scalable, and toxic amide-based solvents are not required. It is anticipated that this strategy, as well as the underlying mechanistic framework, will be generalizable to other cross-electrophile coupling reactions.
Keywords: cross-electrophile coupling, nickel, medicinal chemistry, synthetic methods, mechanism
Graphical Abstract

INTRODUCTION
Cross-electrophile coupling in which two electrophiles are coupled in the presence of a reducing agent is a powerful method for C–C bond formation that complements, extends, and provides orthogonal reactivity to traditional cross-coupling reactions.1 In particular, nickel-catalyzed cross-electrophile coupling reactions that generate new C(sp2)–C(sp3) bonds have received significant attention over the last decade due to the prevalence of these linkages in natural products and pharmaceuticals and the limitations of the current synthetic methods to form these bonds (Figure 1a).2 Despite the widespread interest, there is still only a limited amount of knowledge about key elementary reactions in the proposed mechanism of these reactions (Figure 1b).3 As a result, reaction development has relied on the empirical screening of a wide range of reaction parameters.4 Using this strategy, it is often unclear how individual elementary reactions are affected by changes in reaction conditions, which can result in reaction development that is often specific to a limited range of substrates and involves the use of inscrutable additives.5 This is especially the case when coupling more complex, pharmaceutically relevant substrates.6 An increased understanding of the mechanism of cross-electrophile coupling reactions could result in the development of more practical systems with broader substrate scopes.
Figure 1.
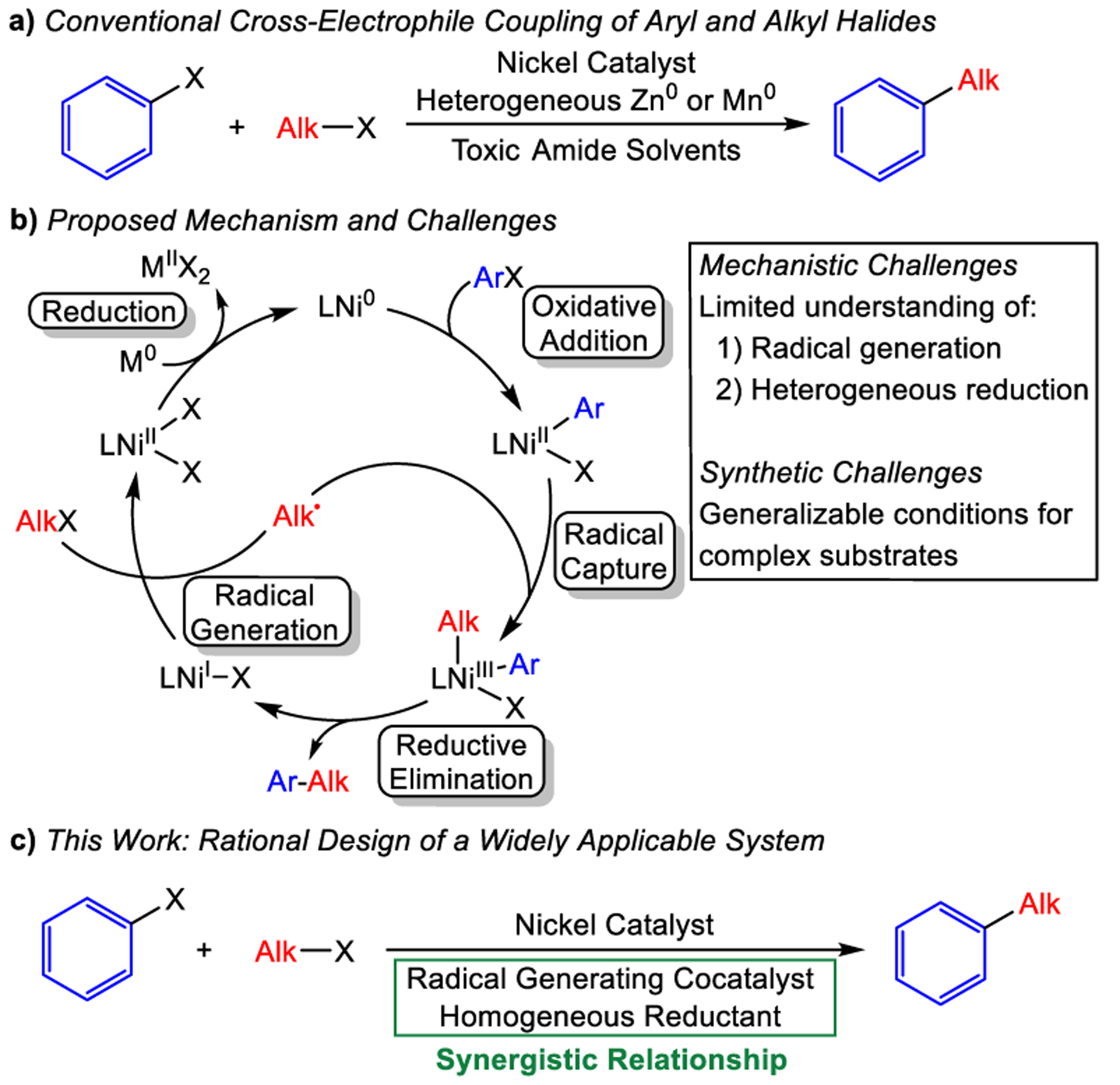
(a) General depiction and (b) mechanism of conventional nickel-catalyzed cross-electrophile coupling reactions using a heterogeneous reductant.3a (c) Cross-electrophile coupling reactions described in this work with a dual catalyst system and a homogeneous reductant.
There are two areas where greater understanding of the mechanism of nickel-catalyzed cross-electrophile coupling reactions between alkyl and aryl halides could provide opportunities for improved transformations. The first is related to the pathway by which the alkyl halide is activated to generate a transient radical. In the proposed mechanism, the alkyl halide is proposed to be activated through a radical chain process initiated by an on-cycle NiI halide complex (Figure 1b), which presents two complications for reaction optimization.3a First, it is unclear how NiI halide species are initially formed as in the current mechanism, an alkyl radical is required to generate the NiI halide species, which in turn, is needed to generate an alkyl radical.7,8 Second, the NiI halide that generates a radical is in the same catalytic cycle as the intermediate that captures the radical, LNiII(Ar)X (Figure 1b). As a result, the rate of radical generation cannot be independently tuned relative to the rate of radical capture, which lowers our ability to control the relative rates of the key steps during catalysis. An appealing solution to circumvent both of these challenges is to employ a cocatalyst that has the sole role of independently generating a radical from the alkyl halide. The use of cocatalysts to generate alkyl radicals has already been implemented by several groups using substrates including epoxides2i,j,ak and alkyl (pseudo)halides.9 However, expanding upon this methodology could enable rational control of catalytic systems, leading to more widely applicable reaction conditions.10
The second area where greater mechanistic understanding could provide opportunities for improved transformations is related to the reduction of catalytic intermediates (Figure 1b). Typically, heterogeneous reductants such as Zn0 and Mn0 are utilized in nickel-catalyzed reductive couplings, but our understanding of the mechanism of electron transfer from a solid-state reductant to a solution-state catalyst is limited.11 As a result, it is unclear how changes to the solvent and ancillary ligand, as well as the introduction of additives, impact electron transfer events, which makes it difficult to predictably control the rate of reduction in cross-electrophile coupling reactions. Heterogeneous reductants also present practical limitations as reaction success can vary with scale due to mass transfer issues and irreproducible kinetics, and they are often only effective when utilized in highly toxic amide-based solvents.12 Furthermore, they raise major challenges for the potential application of cross-electrophile coupling reactions in flow chemistry or automated chemical synthesis.13 To this end, homogeneous organic electron donors (OEDs), which have more well-defined mechanisms of electron transfer, are an attractive alternative to heterogeneous reductants that could help to address these limitations and expand the generalizability of cross-electrophile coupling.14 To date, a limited number of C(sp2)–C(sp3) cross-electrophile couplings have been reported that utilize homogeneous OEDs (vide infra);15,9a,16 however, the utility of these reactions is limited, especially in comparison to systems that utilize heterogeneous reductants. Furthermore, the potential benefits of using OEDs in tandem with a radical generating cocatalyst have not been evaluated.
Here, we leverage the combined effects of a weak homogeneous reductant and a radical generating cocatalyst in Ni-catalyzed cross-electrophile coupling to report one of the most active and operationally simple systems for the coupling of aryl and alkyl halides (Figure 1c). We demonstrate that the product profile encodes mechanistic details about the fate of the LNiII(Ar)X intermediate, and by varying the ratio of the nickel and cobalt catalysts, we can control whether this intermediate traps a radical, which leads to productive catalysis, or undergoes deleterious off-cycle processes. As a result, a wide range of aryl halides can be coupled with alkyl halides in high yield, including challenging substrates such as di-ortho-substituted arenes and heteroarenes. The unique control that our strategy offers is highlighted through a series of novel one-pot three-component couplings of bromo(iodo)arenes with two distinct alkyl electrophiles. Furthermore, we demonstrate the utility of our dual catalytic platform in complex molecule synthesis by performing reactions using medicinally relevant substrates. From a practical perspective, our system allows challenging cross-electrophile coupling reactions to be performed using commercially available starting materials, including a homogeneous reductant, which provides advantages in terms of both scope and practicality compared to reactions with heterogeneous reductants. Additionally, from a mechanistic perspective, we present a dual catalytic strategy that may be broadly translatable to a variety of reductive transformations.
RESULTS AND DISCUSSION
Development of a Dual Catalytic System for Cross-Electrophile Coupling.
Complexes of the type LNiII(Ar)X (X = Cl, Br, or I), which arise from oxidative addition of an aryl halide to a coordinatively unsaturated Ni0 species, are proposed to be key intermediates in cross-electrophile coupling because they are likely the catalyst resting state and are responsible for capturing alkyl radicals (Figures 1b and 2).3a In an ideal cross-electrophile coupling reaction, the LNiII(Ar)X intermediate would be stable and the rate at which reactive radicals are generated would be optimized relative to the concentration of LNiII(Ar)X to facilitate effective radical capture. In current systems for cross-electrophile coupling, the rate of alkyl radical generation by a NiI halide cannot be tuned independently of the concentration of LNiII(Ar)X because they are connected (vide supra). Additionally, under the reaction conditions typically utilized, LNiII(Ar)X complexes are unstable and can undergo two deleterious side reactions: (i) bimolecular decomposition via a process involving ligand rearrangement between two molecules of LNiII(Ar)X to form LNiIIX2 and LNiII(Ar)2, which in turn undergoes reductive elimination to generate a biaryl and a Ni0 species (Figure 2)3d,17 or (ii) direct reduction (depending on the reaction conditions and the reductant) to highly unstable LNiI(Ar) species, which typically decompose to give coordinatively unsaturated Ni0 species as well as aryl and biaryl products (Figure 2).3c,d,11b,18 We hypothesized that we could solve these challenges and improve cross-electrophile coupling reactions by developing a strategy in which: (i) a well-defined cocatalyst is used to generate a radical from an alkyl electrophile so that the concentration of LNiII(Ar)X can be controlled relative to the concentration of alkyl radicals, (ii) reactions are performed at low nickel loadings so that radical capture by LNiII(Ar)X is favored over bimolecular decomposition, and (iii) a reductant that is weaker than Zn0 or Mn0, but still able to reduce LNiIIX2, is used so that the reductive decomposition of LNiII(Ar)X is minimized.
Figure 2.
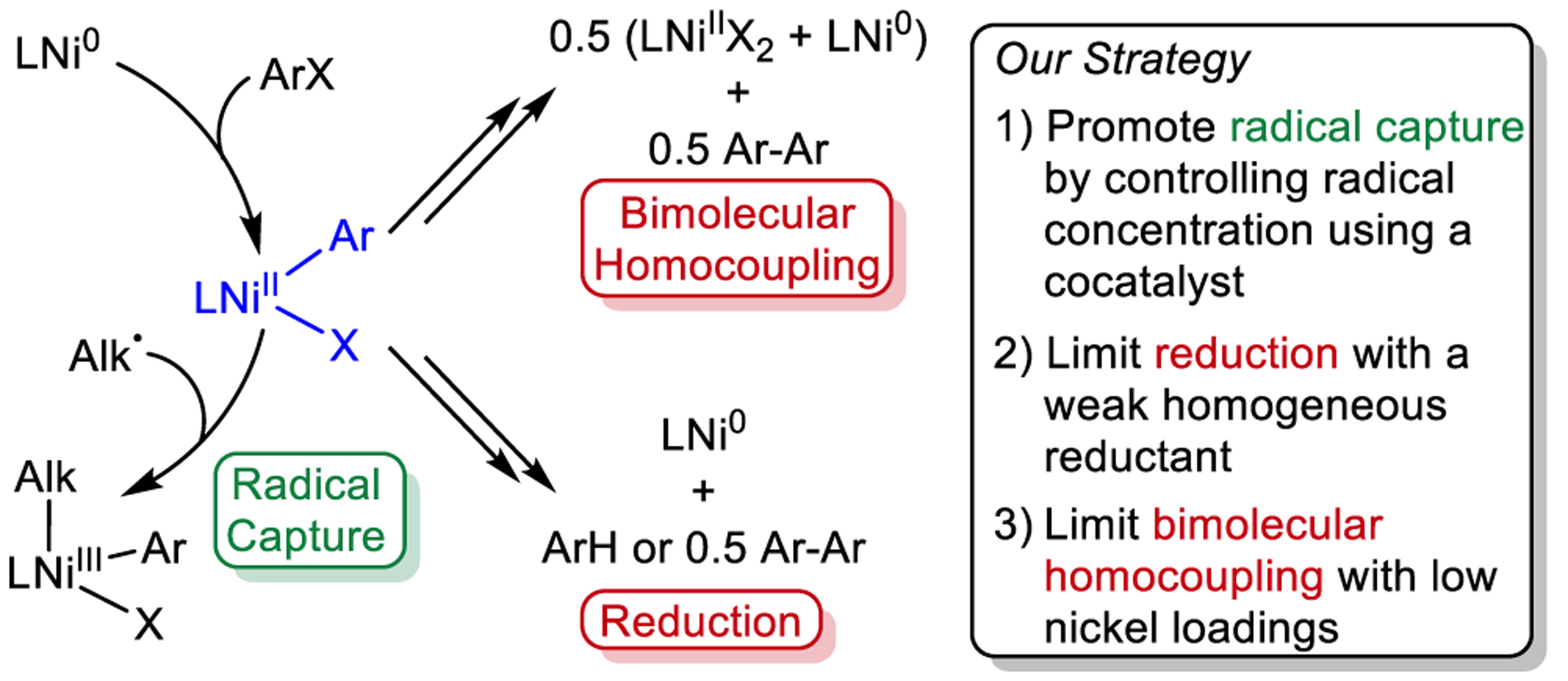
Potential reactions of LNiII(Ar)X in catalysis and an envisioned strategy for system development.
A recent example of a cross-electrophile coupling reaction using a weaker reductant than Zn0 was described by Weix and co-workers.15a They demonstrated that (dtbbpy)NiIIBr2 (dtbbpy = 4,4′-ditertbutyl-2,2′-bipyridine) is an effective precatalyst for the coupling of a limited range of alkyl halide electrophiles, such as benzyl chlorides, with aryl iodides using tetrakis(dimethylamino)ethylene (TDAE), a homogeneous OED with E° = −0.57 V vs NHE,11b as the reductant (Scheme 1).19 Apart from being a weaker reductant than Mn0 (E° = −1.19 V vs NHE)11b or Zn0 (E° = −0.86 V vs NHE),11b TDAE also has a practical advantage because it can facilitate cross-electrophile coupling reactions in nonamide solvents, which is often not possible with heterogeneous reductants.15a In preliminary experiments, we examined the stability of the model catalytic intermediate (dtbbpy)NiII(o-tol)I in a range of solvents both in the presence and absence of TDAE (see Supporting Information). In weakly or nonpolar solvents, such as 1,4-dioxane or toluene, no reaction is observed between TDAE and (dtbbpy)NiII(o-tol)I over days at room temperature. In contrast, a reaction between TDAE and (dtbbpy)NiII(o-tol)I can be observed over minutes to hours in more polar solvents, such as acetonitrile (see Supporting Information for further discussion). These results suggest that a weakly polar solvent, such as 1,4-dioxane, which stabilizes the key LNiII(Ar)X intermediate, could be beneficial for reactions with nonactivated alkyl halides where the rate of radical generation is slower.
Scheme 1.
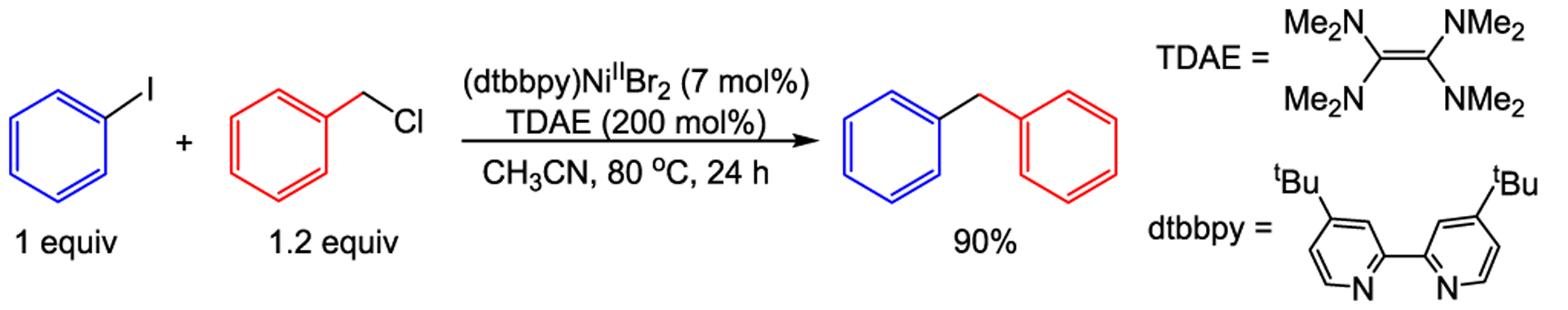
Cross-Electrophile Coupling of Aryl Iodides with Benzyl Chlorides Using TDAE Reported by Weix et al15a
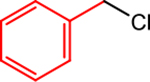
Under the conditions reported by Weix et al.,15a we explored a cross-electrophile coupling reaction between phenyl iodide and benzyl chloride using 1,4-dioxane as the solvent and TDAE as the reductant. In the absence of a radical generating cocatalyst, the reaction produced diphenylmethane in 40% yield (Table 1, Entry 1). Notably, significant quantities of benzyl chloride were still present at the end of the reaction, suggesting that radical generation is relatively slow. Remarkably, in the presence of 0.1 mol % of the air-stable cobalt cocatalyst, CoII(Pc) (Pc = phthalocyanine), which has been reported by the Weix and Reisman groups to generate radicals from benzyl electrophiles under reductive coupling conditions,9b,e the yield increased to 90% (Entry 3). High yields were also obtained when the CoII(Pc) loading was increased to 1 mol % (Entries 4–6), but the yield began to diminish as the CoII(Pc) loading was increased above 2.5 mol % (Entries 7 and 8). The identity of the byproducts and the starting materials that remained at the end of the reaction changed as the loading of CoII(Pc) was varied (Table 1). At low loadings of CoII(Pc), unreacted benzyl chloride was observed and biphenyl was formed in appreciable quantities (Entries 1 and 2). In contrast, at high CoII(Pc) loadings, no unreacted benzyl chloride was detected, only small amounts of biphenyl were formed, and significant amounts of phenyl iodide remained unreacted (Entries 7 and 8). Mechanistically, these observations align with our hypotheses and suggest that the main roles of the cobalt catalyst are to activate the alkyl electrophile and generate an alkyl radical and the main roles of the nickel catalyst are to activate the aryl electrophile, capture the alkyl radical, and facilitate C–C bond formation. We note that nickel is capable of productively engaging benzyl chloride in the absence of CoII(Pc) (Table 1, Entry 1); however, it is likely that the cobalt catalyst more readily activates benzyl chloride compared to nickel alone when efficient catalysis is observed (Entries 3–6). Additionally, more challenging substrates, such as unactivated primary alkyl bromides (vide infra), were not productively engaged by nickel under similar reaction conditions (see Supporting Information).
Table 1.
 | ||||||
|---|---|---|---|---|---|---|
| entry | CoII(Pc) (X mol %) | product (%) | unreacted ArI (%) | biphenyl (%) | unreacted BnCl (%)c | catalytic regime |
| 1 | 0 | 40 | 3 | 21 | 28 | 1 |
| 2 | 0.01 | 77 | <1 | 6 | 16 | 1 |
| 3 | 0.1 | 90 | 5 | 2 | 4 | 2 |
| 4 | 0.25 | 87 | 4 | 2 | <1 | 2 |
| 5 | 0.5 | 96 | 3 | 1 | <1 | 2 |
| 6 | 1 | 90 | 4 | 2 | <1 | 2 |
| 7 | 2.5 | 75 | 14 | 1 | <1 | 3 |
| 8 | 5 | 62 | 21 | 1 | <1 | 3 |
Reaction conditions: iodobenzene (0.0625 mmol), benzyl chloride (0.075 mmol), (dtbbpy)NiIIBr2(0.0044 mmol), and TDAE (0.075 mmol) in 1,4-dioxane (0.5 mL) at 80 °C for 24 h.
Yields are reported as the average of two trials and were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
Yield of recovered benzyl chloride reported relative to iodobenzene loading.
To explore the proposed interplay between nickel and cobalt in catalysis, we performed a stoichiometric reaction between (dtbbpy)NiII(o-tol)I and two equivalents of benzyl chloride in the presence of excess TDAE and catalytic amounts of CoII(Pc) in 1,4-dioxane (Table 2). This resulted in the generation of the diarylmethane cross-product, (o-tolyl)-(phenyl)methane, in 76% yield (Table 2, Entry 1). No product formation, however, was observed without CoII(Pc) in either the presence or absence of TDAE (Entries 2 and 3), consistent with our hypothesis that cobalt primarily activates the alkyl electrophile. Furthermore, the use of stoichiometric CoII(Pc) without TDAE also yielded no cross-product (Entry 4), suggesting that the activation of alkyl electrophiles occurs at a reduced cobalt center. In agreement with this proposal, the reduction potential of TDAE2+/0 is more negative than that of CoII(Pc)0/1−.20 Low-valent cobalt complexes similar to CoI(Pc)− are known to undergo oxidative addition with alkyl halides through an SN2 mechanism to form CoIII(Pc)(Alk) species.21 In turn, these high valent CoIII complexes can undergo homolysis of the CoIII–Alk bond, which produces an alkyl radical and regenerates CoII(Pc).22 Further support for the proposal that CoII(Pc) is capable of generating alkyl radicals in the presence of TDAE was obtained by performing an analogous radical trapping experiment using 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as the radical acceptor instead of (dtbbpy)NiII(o-tol)I.23 In a similar fashion to our experiment with (dtbbpy)NiII(o-tol)I, trapping of the benzyl radical by TEMPO is only observed in the presence of excess TDAE and catalytic amounts of CoII(Pc) (see Supporting Information). Altogether, these experiments provide evidence for the cobalt-mediated generation of free radicals from an alkyl electrophile and subsequent radical capture by LNiII(Ar)X species in catalysis.
Table 2.
Stoichiometric Reaction of (dtbbpy)NiII(o-tol)I with Benzyl Chloride under Various Reaction Conditionsab
 | ||
|---|---|---|
| entry | deviation from conditions | yield (%) |
| 1 | none | 76 |
| 2 | No CoII(Pc) | <1 |
| 3 | No CoII(Pc) & no TDAE | <1 |
| 4 | 100 mol % CoII(Pc) & no TDAE | <1 |
Reaction conditions: (dtbbpy)NiII(o-tol)I (0.0132 mmol), benzyl chloride (0.0264 mmol), CoII(Pc) (0.00185 mmol), and TDAE (0.0264 mmol) in 1,4-dioxane (1.5 mL) at room temperature for 1 h.
Yields are reported as the average of two trials and were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
On the basis of our experimental results, we propose a mechanism containing two cycles for the coupling of phenyl iodide and benzyl chloride catalyzed by (dtbbpy)NiIIBr2 and CoII(Pc) (Figure 3). Initially, the (dtbbpy)NiIIBr2 precatalyst is reduced by TDAE to generate a catalytically active Ni0 species and the two-electron oxidized form of TDAE, which likely precipitates out of solution as [TDAE2+][Br−]2. The Ni0 species undergoes oxidative addition with an aryl halide to form a (dtbbpy)NiII(Ar)X intermediate. Subsequently, the (dtbbpy)NiII(Ar)X intermediate captures an alkyl radical, which is liberated upon the homolysis of a CoIII(Pc)(Alk) species. The CoIII(Pc)(Alk) species is generated in an independent catalytic cycle through initial reduction of CoII(Pc) to form an anionic [CoI(Pc)]− complex, which can react with an alkyl halide via an SN2 mechanism. Following radical capture by (dtbbpy)NiII(Ar)X, a putative (dtbbpy)NiIII(Ar)(Alk)X species is produced, which rapidly reductively eliminates at the NiIII center to liberate the product and form a (dtbbpy)NiIX species. Finally, we propose that the (dtbbpy)NiIX species is reduced by TDAE to regenerate Ni0, closing the catalytic cycle. Further mechanistic work to explore all the potential roles of NiI species is ongoing.
Figure 3.
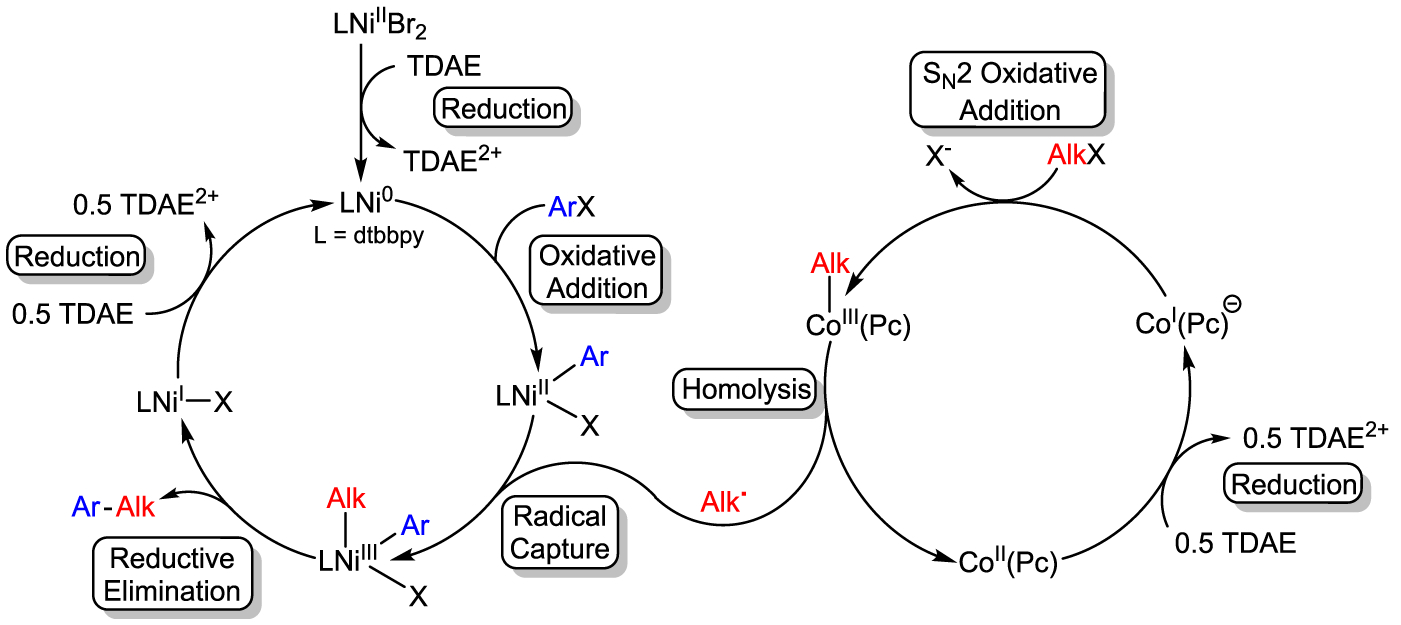
Proposed mechanism for the cross-electrophile coupling of aryl and alkyl halides in the presence of CoII(Pc).
Our mechanism and preliminary catalytic results are consistent with the presence of three distinct regimes, which are related to the relative loadings of (dtbbpy)NiIIBr2 and CoII(Pc). These regimes can be identified by analyzing the byproducts and the identity of any unreacted substrates. Regime 1 occurs when the rate of radical formation is slow relative to the rate of decomposition of (dtbbpy)NiII(Ar)X. This results in the presence of unreacted alkyl electrophile when all of the aryl electrophile has been consumed. Additionally, a significant amount of biphenyl is generated from the decomposition of (dtbbpy)NiII(Ar)X.14d Regime 2 occurs when the rate of alkyl radical formation is optimal relative to the formation of (dtbbpy)NiII(Ar)X. High yields of the coupled product are observed when the concentrations of radical and (dtbbpy)NiII(Ar)X are matched, so neither decomposes before the trapping event. Regime 3 occurs when alkyl radical formation is faster than the generation of (dtbbpy)NiII(Ar)X. In this case, the alkyl radical decomposes before it can be trapped by nickel. As a result, after all of the alkyl electrophile is consumed, the aryl electrophile is still present. This proposal is supported by the data in Table 1, which is separated into the three possible regimes using dashed lines as the CoII(Pc) loading is altered with a fixed loading of (dtbbpy)NiIIBr2. The same trends are also obtained when the loading of (dtbbpy)NiIIBr2 is varied at a fixed loading of CoII(Pc) (see Supporting Information). Practically, these observations suggest that reaction yields may be rationally optimized by tuning only two variables, the loadings of (dtbbpy)NiIIBr2 and CoII (Pc), based on the byproducts and recovered starting materials observed.
Using our approach for the cross-electrophile coupling of aryl iodides with benzyl chlorides as a guide, we applied our strategy of using a dual catalytic system with a homogeneous OED to the coupling of unactivated aryl and alkyl bromides. To date, these synthetically important substrates have typically proven incompatible with homogeneous reductants. Remarkably, in an analogous fashion to our results with benzyl chlorides (vide supra), we were able to optimize the coupling of 4-tert-butyl-bromobenzene with 1-bromo-3-phenylpropane by fixing the loading of CoII(Pc) at 2.5 mol % and altering the loading of (dtbbpy)NiIIBr2 based on the byproducts (Table 3). Using 1 mol % of (dtbbpy)NiIIBr2 and 2.5 mol % CoII(Pc), we obtained a yield of 84% in 1,4-dioxane (Table 3, Entry 3). Notably, no product was observed under these conditions in the absence of CoII(Pc), demonstrating the importance of the cobalt cocatalyst (see Supporting Information). Furthermore, we are also able to couple iodobenzene with 1-iodo-3-phenylpropane in 97% yield under similar conditions after optimizing the loadings of nickel and cobalt (Scheme 2 and see Supporting Information). These results demonstrate that we are able to perform cross-electrophile coupling reactions with relatively unactivated bromo-electrophiles under nearly the same conditions we used to couple aryl iodides with benzyl chlorides and alkyl iodides, with the only difference being the loadings of the nickel and cobalt catalysts. This generalizability of reaction conditions across aryl iodides and bromides, benzyl chlorides, and alkyl bromides and iodides is uncommon for cross-electrophile coupling reaction conditions, which are often specific to a more narrow range of substrates (vide supra). Furthermore, in our dual catalytic system, the role of each reagent is understood and there is no need for commonly utilized additives, such as alkali halide salts or pyridines. As a result, our system provides a method for simple, rational optimization through modulation of catalyst loadings, which, as we show in the following sections, can be broadly applied to a wide range of challenging and novel substrates.
Table 3.
Cross-Electrophile Coupling of 4-tert-Butyl-Bromobenzene with 1-Bromo-3-Phenylpropane with Varying Amounts of (dtbbpy)NiIIBr2a,b
 | ||||||
|---|---|---|---|---|---|---|
| entry | (dtbbpy)NiIIBr2 (X mol %) | product (%) | unreacted ArBr (%) | biphenyl (%) | unreacted AlkBr (%)c | catalytic regime |
| 1d | 0.1 | 55 | 29 | <1 | 3 | 3 |
| 2d | 0.5 | 68 | 22 | <1 | 6 | 3 |
| 3 | 1 | 84 | 6 | <1 | <1 | 2 |
| 4e | 2.5 | 66 | 4 | 9 | 10 | 1 |
| 5f | 5 | 53 | <1 | 12 | 30 | 1 |
Reaction conditions: 1-bromo-4-tertbutylbenzene (0.0625 mmol), 1-bromo-3-phenylpropane (0.075 mmol), CoII(Pc) (0.0016 mmol), and TDAE (0.075 mmol) in 1,4-dioxane (0.5 mL) at 80 °C for 24 h.
Yields are reported as the average of two trials and were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
Yield of recovered 1-bromo-3-phenylpropane reported relative to 4-tert-butyl-bromobenzene loading.
Reaction run for 48 h.
Reaction run for 12 h.
Reaction run for 4 h.
Scheme 2.
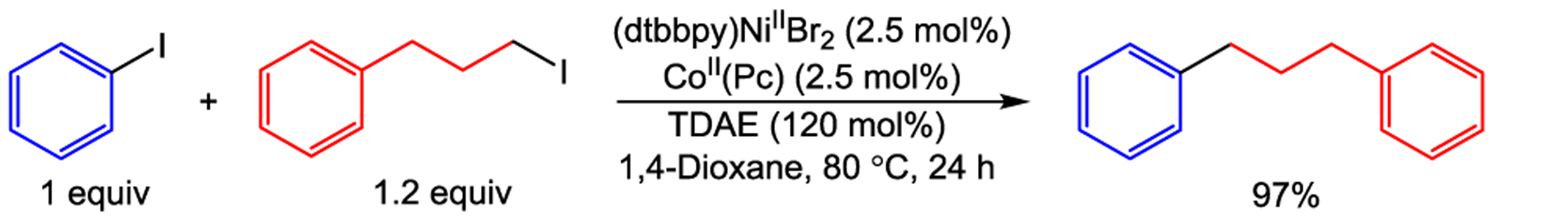
Cross-Electrophile Coupling of Iodobenzene with 1-Iodo-3-Phenylpropane
Substrate Scope for Cross-Electrophile Coupling of Simple Aryl and Alkyl Bromides.
Given our development of a system for the cross-electrophile coupling of aryl and alkyl bromides using a homogeneous reductant, we optimized the reaction conditions (see Supporting Information) and evaluated the substrate scope of the method (Figure 4). Aryl electrophiles were primarily coupled with 1-bromo-3-phenyl-propane although in some cases, N-(3-bromopropyl)-phthalimide was used as the alkyl electrophile because the polar functional group assists with isolation. To show the generality and simplicity of our optimization protocol, we optimized each substrate to a yield greater than 75% by proton nuclear magnetic resonance (1H NMR) spectroscopy by modulating catalyst loadings according to the observed product profile (see Supporting Information).24 Notably, the optimized conditions for each substrate deviate only slightly from our standard reaction conditions, indicating the ease by which high yields can be obtained. Furthermore, good yields can be obtained over more than an order of magnitude variation in CoII(Pc) or (dtbbpy)NiIIBr2 loadings (Tables 1 and 3). This suggests that a wide range of substrates may be successfully coupled under a standard set of conditions even without performing the simple catalyst loading optimization. In a similar fashion to conventional cross-electrophile coupling reactions using heterogeneous reductants2b,4 our homogeneous dual catalytic system has a broad substrate scope and excellent functional group tolerance. For example, using 1 mol % (dtbbpy)NiIIBr2 and 2.5 mol % CoII(Pc), we are able to couple bromobenzene (4a) with N-(1-bromopropyl)phthalimide as well as electron-rich 4-bromoanisole (4b) and electron-deficient 4-bromotriflurobenzene (4c) with 1-bromo-3-phenylpropane in good yields. Additionally, under analogous conditions, aryl halides with a variety of different functional groups, such as 4-bromobenzonitrile (4d), 4-bromophenyl methyl sulfone (4e), 4-bromobenzaldehyde (4f), 4-bromoacetophenone (4g), methyl 4-bromobenzoate (4h), and 4-bromo-N-methylbenzamide (4i), can also be coupled with 1-bromo-3-phenylpropane, in good yields. While these types of aryl halides are commonly utilized as substrates in cross-electrophile coupling reactions, it is noteworthy that our system requires only a 1 mol % loading of the nickel catalyst, whereas most previously reported reactions require higher loadings.1e Interestingly, when 1-bromo-4-chlorobenzene (4j) is used as a substrate with 2 mol % (dtbbpy)NiIIBr2 and 2.5 mol % CoII(Pc), our system is selective for coupling the aryl bromide, which offers opportunities for orthogonal reactivity with traditional cross-coupling reactions.1h Our dual catalytic system is also able to couple substrates with acidic groups, such as 4-bromophenethyl alcohol (4k) and 5-bromoindole (4l) using just 1 mol % of (dtbbpy)NiIIBr2. Furthermore, the reaction between 4-iodoaniline (4m) and 1-iodo-3-phenylpropane using 2 mol % (dtbbpy)NiIIBr2 and 2.5 mol % CoII(Pc) generated the desired product in 80% yield. However, the coupling of 4-bromoaniline with 1-bromo-3-phenylpropane resulted in a lower yield (44%, see Supporting Information). In this case, we propose that more reactive iodo-substituted substrates are required because the rates of deleterious side reactions between the acidic group and the catalysts are more competitive when bromo-substituted substrates are utilized.
Figure 4.
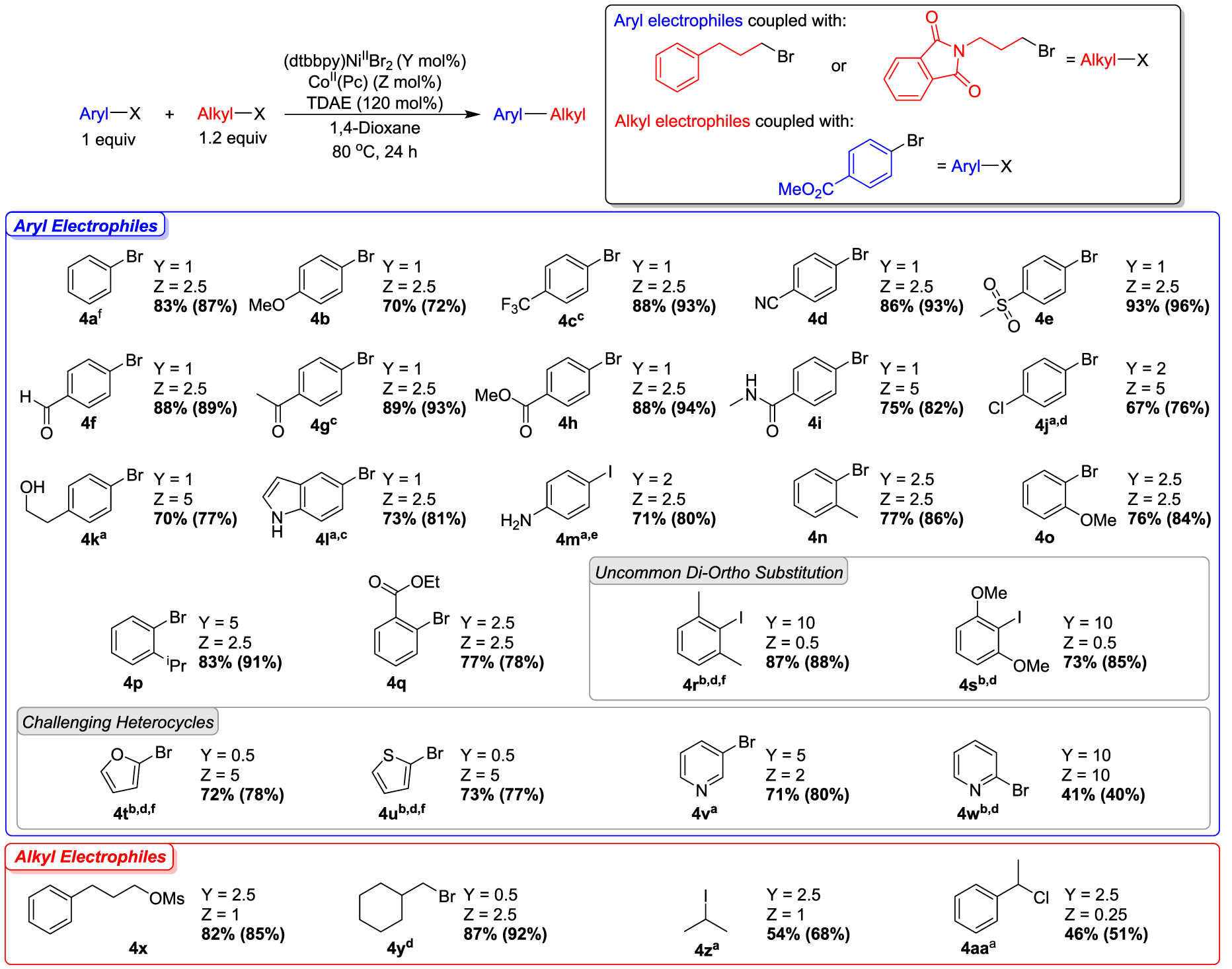
Substrate scope for dual-catalyzed cross-electrophile coupling between aryl halides and alkyl halides or pseudohalides. Values outside of parentheses are isolated yields and values inside of parentheses are NMR yields, which were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard. a1.6 equiv. of alkyl substrate, 140 mol % TDAE. b2.0 equiv. of alkyl substrate, 160 mol % TDAE. c36 h. d48 h. e1-iodo-3-phenylpropane used as an alkyl substrate. fN-(3-bromopropyl)phthalimide used as an alkyl substrate instead of 1-bromo-3-phenylpropane.
A major advantage of our system for cross-electrophile coupling is its compatibility with sterically bulky aryl halides. For example, the ortho-substituted aryl bromides 2-bromotoluene (4n), 2-bromoanisole (4o), 2-bromocumene (4p), and ethyl 2-bromobenzoate (4q) can all be coupled with 1-bromo-3-phenylpropane using 2.5–5 mol % of (dtbbpy)NiIIBr2 and 2.5 mol % of CoII(Pc). In particular, the coupling of 4q is significant as Weix et al. previously reported that it was difficult to couple aryl halides with bulky ortho-directing groups.4 Even more significantly, our system is able to couple the 2,6-disubstituted aryl halides 2-iodo-1,3-dimethylbenzene (4r) and 2-iodo-1,3-dimethoxybenzene (4s) for which there is virtually no precedent in the cross-electrophile coupling literature.4,25 For these substrates, aryl iodides are required instead of aryl bromides along with a 10 mol % loading of (dtbbpy)NiIIBr2. This is likely due to the difficulty associated with oxidative addition for these sterically bulky systems and tuning of the ancillary ligand on the nickel catalyst may enable these reactions to be performed with aryl bromide congeners at lower catalyst loadings. Additionally, from a mechanistic perspective, ortho-substitution of aryl ligands is known to inhibit the decomposition of LNiII(Ar)X intermediates via bimolecular homocoupling, relative to complexes with aryl ligands that do not have ortho-substitution (Figure 2).26 This should be advantageous for promoting productive radical capture at LNiII(Ar)X intermediates. In agreement with this proposal, catalysis with ortho-substituted aryl substrates can be performed with high efficiency (regime 2, above) using a wider relative range of nickel to cobalt loadings compared to analogous reactions without ortho-substitution on the aryl substrate (see Supporting Information).
Heteroaryl halides are important substrates because heteroaromatic groups are common structures in medicinal chemistry.27 Traditionally, it has proven difficult to use 2-halofurans and 2-halothiophenes as substrates in cross-electrophile coupling reactions, especially when there is no substitution in the 5-position.2l,14j Our dual catalytic system can couple 2-bromofuran (4t) and 2-bromothiophene (4u) in high yields using just 0.5 mol % of (dtbbpy)NiIIBr2, although 5 mol % of the CoII(Pc) cocatalyst is required. Pyridyl halides are another highly challenging class of substrates in cross-electrophile coupling reactions. As a result, complex ligands and various additives are commonly required to facilitate their coupling.4,6a In contrast, we can couple 3-bromopyridine (4v) with 1-bromo-3-phenylpropane in 71% yield simply by modulating the catalyst loadings to 5 mol % (dtbbpy)NiIIBr2 and 2 mol % CoII(Pc), demonstrating the broad generalizability of our dual catalytic system. 2-bromopyridine (4w) is a more difficult substrate and only a 41% yield is observed using 10 mol % of (dtbbpy)NiIIBr2 and 10 mol % of CoII(Pc).
Different types of alkyl electrophiles that are compatible with our dual catalytic system were examined using the same optimization strategy that was utilized for exploring the scope of the aryl electrophile. Benzyl chloride can be coupled with phenyl iodide (vide supra) and methyl 4-bromobenzoate in excellent yields (see Supporting Information), but the reaction requires high loadings of (dtbbpy)NiIIBr2 (5–7 mol %) relative to CoII(Pc) (0.5–1 mol %), consistent with the facile radical generation from the highly reactive alkyl electrophile. In addition to unactivated primary alkyl iodides (vide supra) and bromides (4a–4w), the primary alkyl mesylate, 3-phenylpropyl methanesulfonate (4x), is compatible with our conditions and can be coupled with methyl 4-bromobenzoate in a high yield using 2.5 mol % (dtbbpy)NiIIBr2 and 1 mol % CoII(Pc). This result is notable because alkyl mesylates can be readily generated in situ from the corresponding alcohols,9b,28 which are abundant and diverse building blocks that are commonly used in pharmaceutical research.9c However, our system is limited to substrates that can be activated via an SN2 mechanism, consistent with the proposed mechanism (Figure 3). Accordingly, substrates with some steric bulk at the α-carbon of alkyl bromides, such as (bromomethyl)cyclohexane (4y), can be coupled with methyl 4-bromobenzoate using 0.5 mol % (dtbbpy)NiIIBr2 and 2.5 mol % CoII(Pc), but no product is generated when either neopentyl bromide or iodide is used as a substrate (see Supporting Information). Similarly, branched secondary alkyl halides such as iodides (4z) and benzyl chlorides (4aa) can be coupled in moderate yields, but branched secondary alkyl bromides and iodocyclohexane are unreactive (see Supporting Information). In a subsequent section, we explore the types of functionalized alkyl bromides and iodides and benzyl chlorides that are compatible with our system.
Cross-electrophile coupling reactions between vinyl and alkyl electrophiles have been reported under related conditions to those reported for reactions between aryl and alkyl electrophiles using a nickel catalyst with a heterogeneous reductant.2b,q,ai Our dual catalytic method is also compatible with vinyl electrophiles as demonstrated by the coupling of 1-bromo-2-methyl-1-propene and N-(3-bromopropyl)-phthalimide in 74% yield, using 1 mol % (dtbbpy)NiIIBr2 and 2.5 mol % CoII(Pc) (Scheme 3).
Scheme 3.
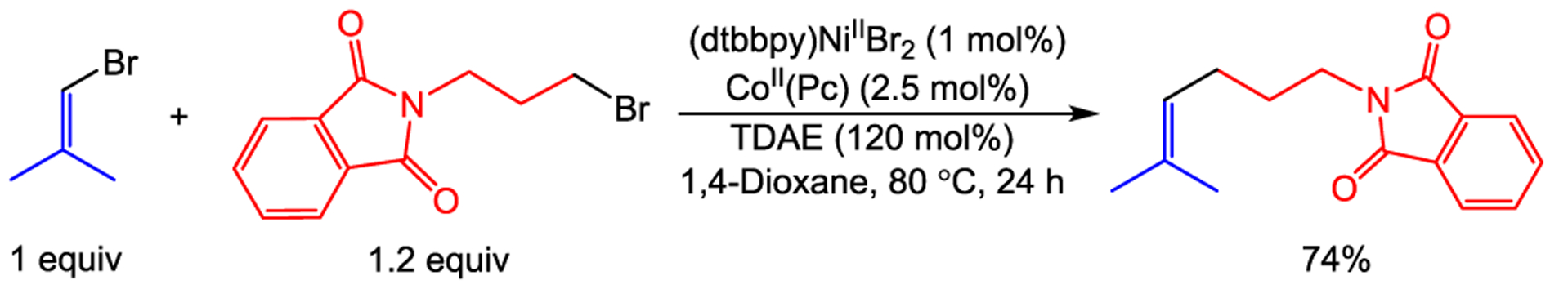
Cross-Electrophile Coupling of 1-Bromo-2-Methyl-1-Propene with N-(3-bromopropyl)Phthalimide
Overall, these results demonstrate that our dual catalytic platform can not only enable the cross-electrophile coupling of many substrates using lower catalyst loadings than those typically reported but can also couple a range of challenging substrates, such as heteroaryl halides and di-ortho-substituted aryl halides, simply by rationally changing the relative loading of the two catalysts. We show in the next sections the unique control that our system offers, which provides new opportunities for the discovery of novel transformations and the functionalization of complex molecules.
Potential Applications of the Dual Catalytic System for Cross-Electrophile Coupling.
Three-Component Coupling Reactions.
Reactions that enable the modification of simple aryl rings in a modular fashion are valuable for the creation of diverse libraries of compounds, which often facilitate the discovery of lead structures in medicinal chemistry.29 To this end, readily accessible dihaloarenes are promising starting materials that can be used to directly and efficiently construct widely diverse structures.30 Although there are currently numerous methods for the sequential introduction of aryl groups into dihaloarenes via standard C(sp2)–C(sp2) cross-coupling reactions,29−31 reports that demonstrate the sequential introduction of alkyl groups are limited32 due, in part, to the difficulties associated with C(sp2)–C(sp3) bond formation. We hypothesized that our dual catalytic system is uniquely suited for the dialkylation of bromo(iodo)arenes through consecutive cross-electrophile coupling reactions for three reasons: (i) it is able to efficiently couple a range of highly activated and unactivated aryl and alkyl substrates under a general set of conditions; (ii) TDAE is utilized in only stoichiometric quantities, which means that after initial coupling at the more activated iodide site of the bromo-(iodo)arene, the bromide site should remain unreacted for use in a subsequent coupling reaction. This advantage is unique to our system compared to conventional cross-electrophile coupling reactions, which generally use superstoichiometric quantities of heterogeneous Zn0 or Mn0 reductants; and (iii) we observed regioselective activation of the aryl bromide in the dihaloarene substrate 1-bromo-4-chlorobenzene (4j), indicating that preferential reactivity at one site in a disubstituted aryl halide is possible.
We performed an initial reaction between 1-bromo-4-iodo-2-methoxybenzene and benzyl chloride using 5 mol % (dtbbpy)NiIIBr2, 0.5 mol % CoII(Pc), and 120 mol % TDAE. The reaction was highly selective for the iodide position and produced a monoalkylated bromoarene in 93% yield (see Supporting Information). We then performed the same reaction in the presence of a second alkyl electrophile 1-bromo-3-phenylpropane and 260 mol % TDAE (see Scheme 4 and Supporting Information for full reaction optimization). In this case, a 76% yield of the desired bis-alkylated product was observed, where the aryl iodide had presumably initially coupled with benzyl chloride, and subsequently, the less reactive aryl and alkyl bromides had coupled. We suggest that there are two requirements for achieving such high selectivity in one step: (i) a highly activated alkyl electrophile, such as a benzyl chloride, must be employed, which reacts preferentially with the Co catalyst over a second, less activated alkyl electrophile, such as a primary alkyl bromide. (ii) The optimal catalyst loading of nickel and cobalt for the initial coupling must overlap with the optimal catalyst loadings for the second coupling. Analysis of the optimized reaction conditions for coupling alkyl electrophiles in single-component cross-electrophile coupling reactions (vide supra) indicates there are only a limited number of substrates that meet the requirements for use in a one-pot single-step reaction.
Scheme 4.
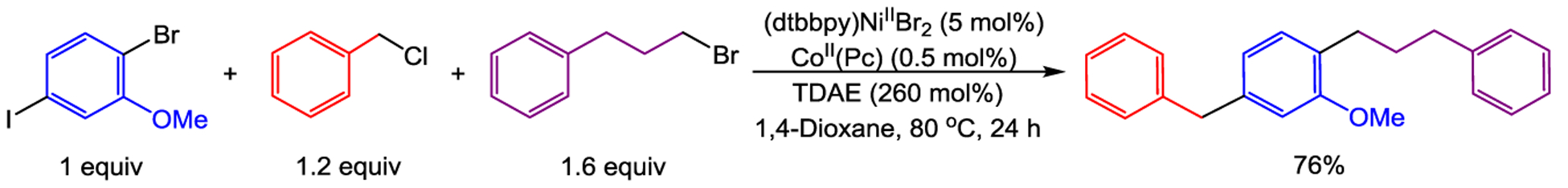
Single-Step Three-Component Cross-Electrophile Coupling of 1-Bromo-4-Iodo-2-Methoxybenzene with Benzyl Chloride and 1-Bromo-3-Phenylpropane
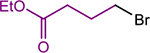
To overcome the limitations of our single-step three-component coupling reaction, we explored the possibility of a one-pot three-component cross-electrophile coupling reaction involving the sequential addition of two alkyl halides (Table 4). Initially, upon completion of the reaction between 1-bromo-4-iodo-2-methoxybenzene and benzyl chloride using 5 mol % (dtbbpy)NiIIBr2 and 0.5 mol % CoII(Pc) to form a monoalkylated bromoarene product, we added ethyl 4-bromobutyrate and TDAE and continued the reaction for another 24 h at 80 °C. Remarkably, the in situ-generated bromoarene product underwent a second cross-electrophile coupling reaction with the alkyl bromide in the same pot, without the need to add additional amounts of either catalyst (Table 4, Entry 1 and see Supporting Information for optimization). Across the two steps, the isolated yield of the bis-alkylated product was 82%. This result suggests that there is no significant catalyst death either during or upon completion of initial alkylation.
Table 4.
 | ||||||
|---|---|---|---|---|---|---|
| Entry |  |
 |
 |
Y1, Z1 (mol%) | Y2, Z2 (mol%) | Yield (%) |
| 1 | 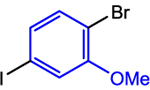 |
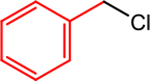 |
 |
5, 0.5 | 0, 0 | 82 (88) |
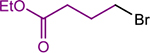
| 2b | 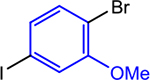 |
 |
 |
1, 2.5 | 4, 0 | 84 (91) |
| 3 | 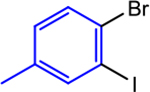 |
 |
 |
5, 0.5 | 0, 0 | 91 (95) |
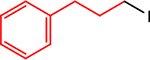
| 4 | 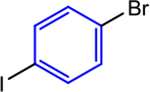 |
 |
 |
2.5, 0.1 | 0, 5 | 70 (78) |
Yields outside of parentheses are isolated yields and yields inside of parentheses are NMR yields, which were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
1.1 equivalents of 1-iodo-3-phenylpropane and 110 mol % TDAE were used in initial coupling.
Using the sequential addition strategy, we extended our one-pot three-component coupling reaction beyond combinations of highly activated and weakly activated alkyl halides. For example, a primary alkyl iodide, such as 1-iodo-3-phenylpropane, and a primary alkyl bromide, such as ethyl 4-bromobutyrate, were sequentially coupled with 1-bromo-4-iodo-2-methoxybenzene in 84% yield using 1 mol % (dtbbpy)NiIIBr2 and 2.5 mol % CoII(Pc) for the initial coupling of the organic iodides followed by the addition of 4 mol % (dtbbpy)NiIIBr2, TDAE, and alkyl bromide to accomplish the second coupling (Entry 2). Although the nickel catalyst was shown to remain active after the initial coupling, an additional 4 mol % of (dtbbpy)NiIIBr2 was added for the second coupling because the optimal conditions for this reaction utilize 5 mol % (dtbbpy)NiIIBr2 and 2.5 mol % CoII(Pc). The three-component reaction is also compatible with an ortho-substituted bromo(iodo)arene. Specifically, 1-bromo-2-iodo-4-methylbenzene can be coupled with benzyl chloride and ethyl 4-bromobenzoate in 91% yield in a two-step one-pot process (Entry 3). In this case, 5 mol % (dtbbpy)NiIIBr2 and 0.5 mol % CoII(Pc) are the optimal catalyst loadings for both the first reaction and the second reaction. Thus, only ethyl 4-bromobenzoate and TDAE need to be added after the first coupling to facilitate the second coupling. Unsubstituted bromo(iodo)arenes can also be used to perform sequential coupling reactions. For instance, 1-bromo-4-iodobenzene was coupled with benzyl chloride and ethyl 4-bromobenzoate in 70% yield using 2.5 mol % (dtbbpy)NiIIBr2 and 0.1 mol % CoII(Pc) for the initial coupling of the benzyl chloride followed by the introduction of an additional 5 mol % CoII(Pc), TDAE, and alkyl bromide to accomplish the second coupling (Entry 4). The four examples presented here serve as proof-of-principle that our dual catalytic platform can be used for the rapid construction of multiple C(sp2)–C(sp3) bonds using readily available bromo(iodo)arene and alkyl halide starting materials. Our method is a significant advancement over the existing methodology for dialkylation of bromo-(iodo)arenes, which cannot be performed in one-pot and requires the use of preformed organometallic nucleophiles that are not commercially available and generally require multistep syntheses.32b As we demonstrate in the next section, our findings should be compatible with medicinally relevant systems as our new catalytic system can promote couplings using complex substrates.
Coupling with Medicinally Relevant Substrates.
Despite the significant attention that C(sp2)–C(sp3) cross-electrophile coupling has received over the past decade, methods to couple complex, medicinally relevant substrates are limited or only applicable to a narrow set of substrates.2aa,6b,c,14e The major challenge associated with using druglike aryl halides in transition metal-catalyzed transformations is that these substrates contain functional groups that can either bind to the catalyst and sequester it in undesired equilibria or directly cause catalyst decomposition. Consequently, the concentration of catalytic intermediates is perturbed in an unpredictable fashion, which complicates reaction optimization. Given the high value of compounds containing alkylated arene groups in the development and study of pharmaceuticals, a robust method to form C(sp2)–C(sp3) linkages that is compatible with a range of medicinally relevant substrates would be valuable for drug discovery.33
We hypothesized that our dual catalytic platform would be well-suited to overcoming the challenges of working with complex molecules because of its functional group tolerance (Figure 4) and the improved control offered by the ability to independently control the rates of activation of the aryl and alkyl halides. To this end, we tested the compatibility of our reaction conditions with aryl halides from the MSD Aryl Halide Informer Library as these compounds were at one time intermediates in drug discovery programs, and therefore, represent relevant chemical space for medicinal chemisty.34 For all substrates, we rationally optimized the reaction by varying the loading of (dtbbpy)NiIIBr2 and CoII(Pc) (vide supra) without changing other reaction parameters, such as solvent, temperature, or the identity of the ancillary ligand.35 We also did not add additives to the reaction. As a result, it was straightforward to perform the reaction optimization using standard high-throughput experimentation (HTE) techniques (see Supporting Information).36 Aryl halides 5a–5h were successfully coupled in moderate to high yields (42–84% yield by 1H NMR spectroscopy) with 1-bromo-3-phenylpropane (Figure 5). The range of functional groups present in these aryl halides highlights the power of our method. For example, successful reactions were observed in the presence of esters, amides, sulfones, alcohols, triazoles, thiophenes, pyridines, and both free and protected amines among many other functional groups. Notably, the challenging, di-ortho substituted aryl halide, 5i, was coupled in a lower, but still medicinally useful, yield (22% by 1H NMR spectroscopy) with 1-iodo-3-phenylpropane. Additionally, we isolated the product from the reaction of 5a with 1-bromo-3-phenylpropane in a good yield (72%) as proof-of-principle that our method will enable the generation of compounds for drug discovery.
Figure 5.
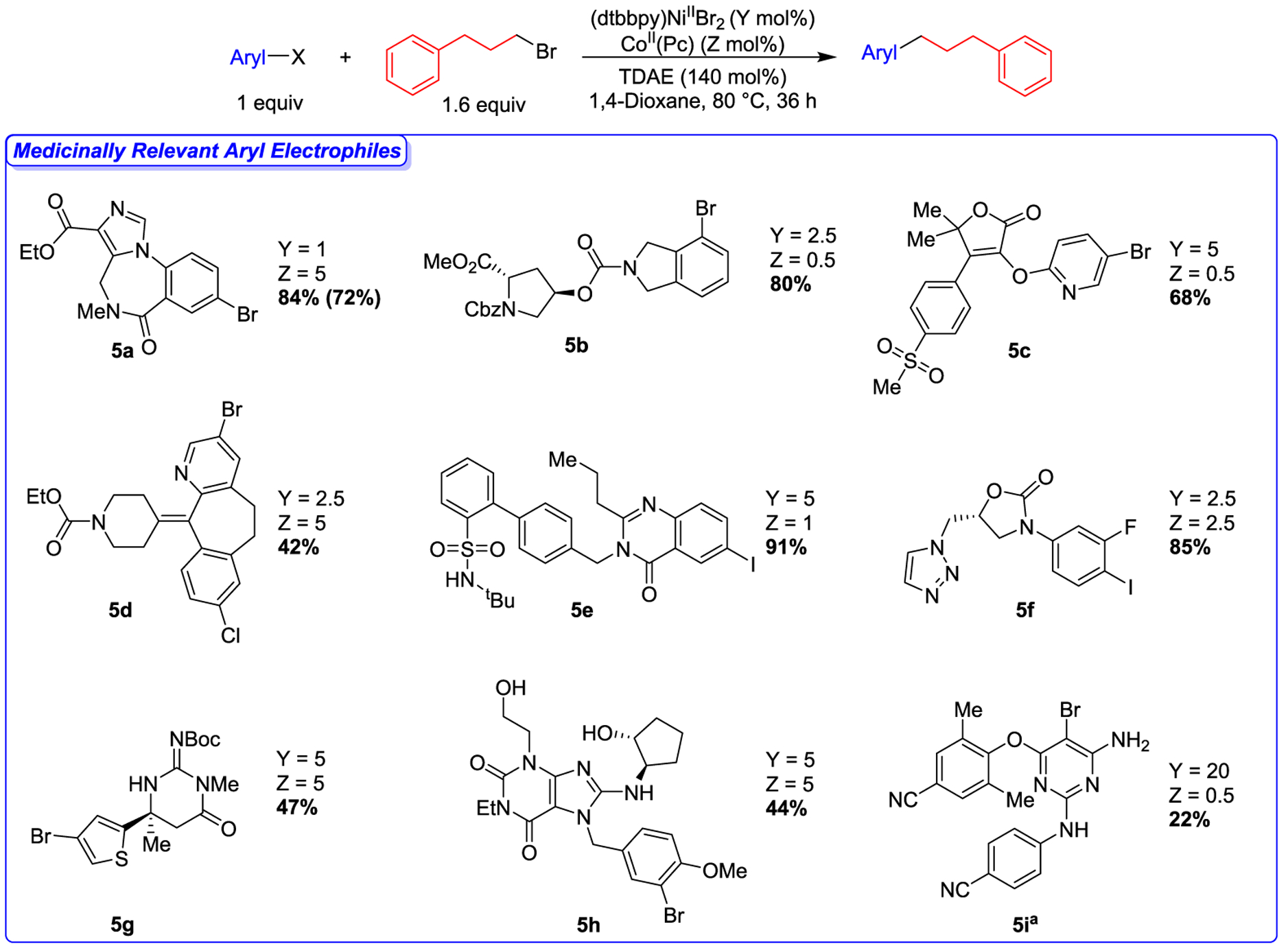
Substrate scope for dual-catalyzed cross-electrophile coupling between MSD Aryl Halide Informer Library and 1-bromo-3-phenylpropane. Values outside of parentheses are NMR yields, which were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard, and values inside of parentheses are isolated yields on 0.10 mmol scale. a2 equivalents of 1-iodo-3-phenylpropane alkyl substrate, 160 mol % TDAE, 48 h.
To further investigate the potential applicability of our reaction conditions to C(sp2)–C(sp3) bond formation in molecules relevant to drug discovery, we performed a parallel library synthesis via late-stage diversification of aryl halide 5f, a precursor to oxazolidinone antibacterials, with different alkyl halides (Figure 6).37 First, we optimized the loadings of (dtbbpy)NiIIBr2 and CoII(Pc) for the reactions of 5f with benzyl chloride, 1-iodo-3-phenylpropane, and 1-bromo-3-phenylpropane (see Supporting Information). We then used the optimized conditions for each class of alkyl halide to evaluate the ability of our system to couple a series of functionalized benzyl chlorides and alkyl bromides and iodides with 5f using HTE techniques (see Supporting Information). For example, all primary alkyl bromides used in the experiment were coupled under the optimal conditions determined for the coupling of 5f with 1-bromo-3-phenylpropane (6r). For each reaction, the amount of 5f transformed to the product is reported as a ratio of desired product to all other known byproducts, as determined by UV–vis spectroscopy (see Supporting Information). We validated this method by determining the 1H NMR yields for the reactions of 6a, 6i, and 6m with 5f and showing that they agreed within 10% of the conversion values obtained using UV–vis spectroscopy (Figure 6).
Figure 6.
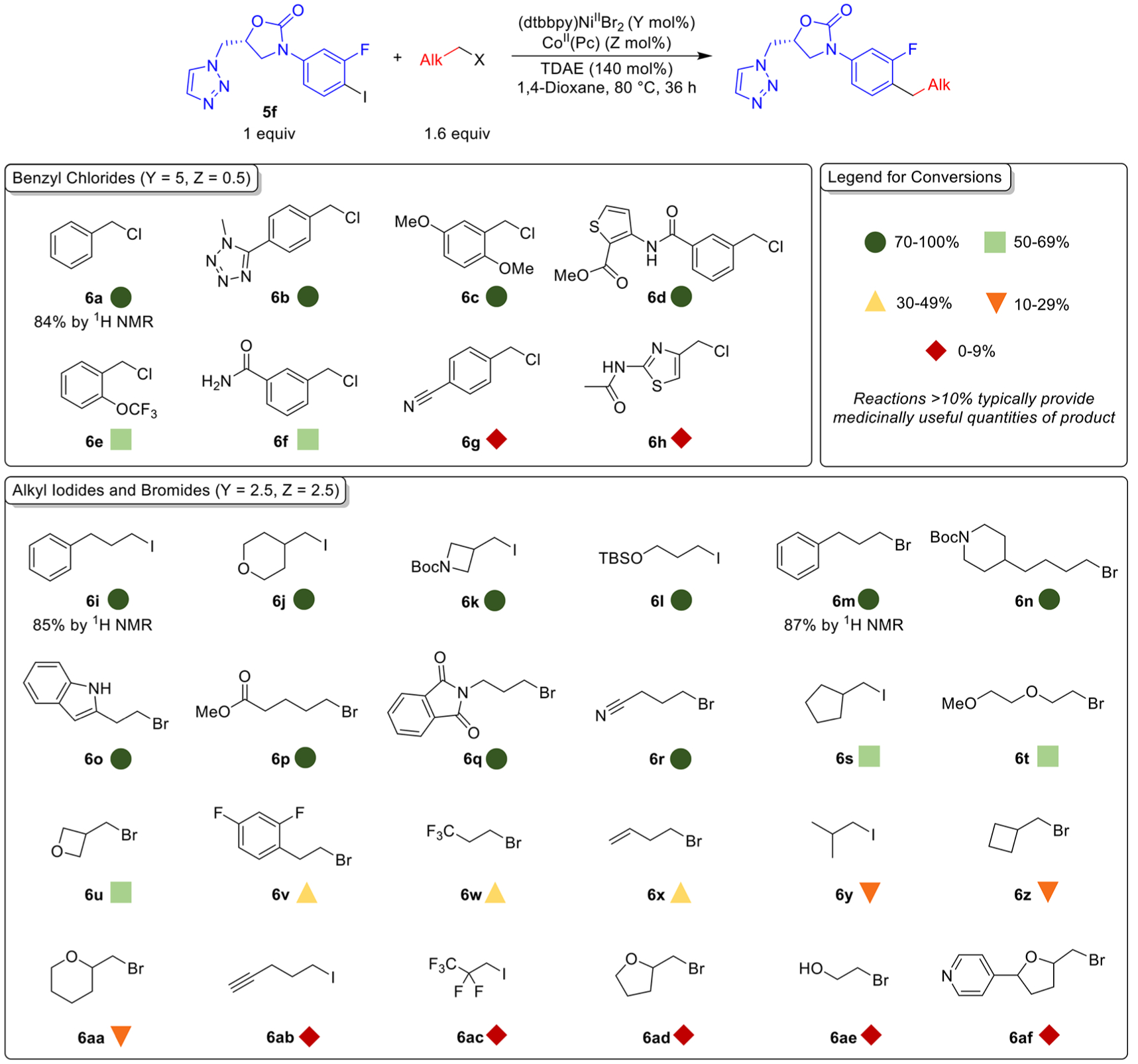
Dual-catalyzed cross-electrophile coupling reactions between 5f and a series of benzyl chlorides, alkyl iodides, and alkyl bromides. Values are reported as the conversion to product relative to all known species derived from 5f determined by UV–vis spectroscopy (see Supporting Information for details). NMR yields were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
Using our strategy, we observed that 25 out of 32 products were formed in greater than 10% conversion, an overall 78% success rate. A range of functionalized primary benzyl chloride electrophiles could be coupled using 5 mol % (dtbbpy)NiIIBr2 and 0.5 mol % CoII(Pc). Notably, substrates containing a tetrazole (6b) or thiophene (6d) ring, or a protic amide substituent (6f) were all successfully coupled. Furthermore, various primary alkyl iodide and bromide electrophiles could be coupled using 2.5 mol % (dtbbpy)NiIIBr2 and 2.5 mol % CoII(Pc). Notably, challenging substrates containing an unprotected indole (6o) and terminal alkenes (6x), which are susceptible to Giese-type additions in related reactions,16c,d generated product. Additionally, heterocyclic rings, which are prevalent in medicinal chemistry, such as azetidine (6k), piperidine (6n), and cyclic ethers (6j, 6u, and 6aa) are compatible with our method.38 Overall, this experiment demonstrates that our reaction conditions can be generally applied across a series of alkyl halide substrates for parallel library synthesis using a complex aryl halide and HTE methods. It also shows that in an analogous fashion to the aryl halide substrate, our methodology can tolerate a large number of functional groups on the alkyl halide substrate.
Finally, to assess if our method is amenable to scale up, we coupled 5f with 1-iodo-3-phenylpropane on a 3.0 mmol scale using the linearly scaled conditions from the reaction on a 0.03 mmol scale. In the 0.03 mmol scale reaction, a 96:4 ratio of product to starting material was observed by analysis of the crude reaction mixture by 19F NMR spectroscopy. In the 3 mmol scale reaction, a 95:5 ratio of product to starting material was observed by analysis of the crude reaction mixture by 19F NMR spectroscopy, demonstrating high reproducibility of the reaction on a large scale (see Supporting Information). From this mixture, we were able to isolate the product in 64% yield (Scheme 5). Our system presents two advantages over conventional C(sp2)–C(sp3) cross-electrophile coupling reactions that are specific to large-scale reactions: (i) it is compatible with a wide range of nontoxic solvents, including several green solvents such as 2-methyltetrahydrofuran, isopropyl acetate, and methyl ethyl ketone (see Supporting Information), which assists in finding conditions where substrates are fully soluble and reduces environmental impact, and (ii) it utilizes a homogeneous reductant as opposed to a heterogeneous reductant, which should provide more reproducible kinetic profiles.
Scheme 5.
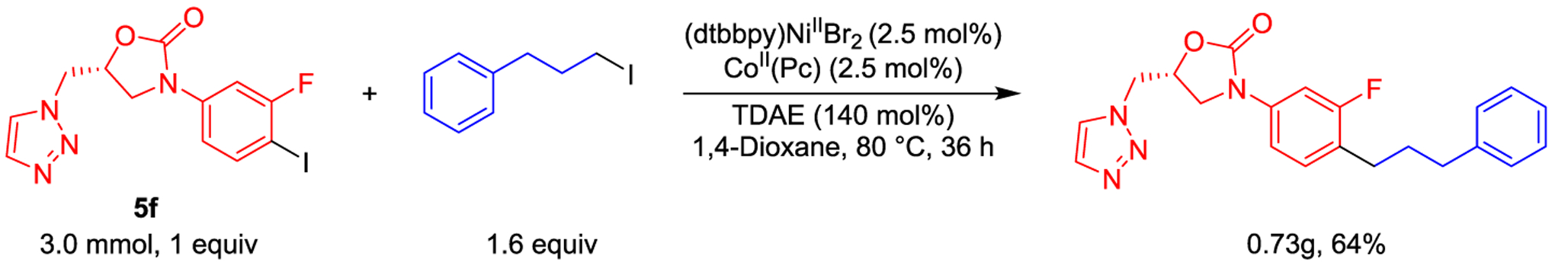
3.0 mmol Scale Cross-Electrophile Coupling of 5f with 1-Iodo-3-Phenylpropane
CONCLUSIONS
We have developed a novel system for C(sp2)–C(sp3) cross-electrophile coupling reactions between aryl halides and alkyl halides. Our system uses a nickel and cobalt dual catalytic platform in tandem with a relatively weak homogenous reductant, TDAE, to provide a high level of control over cross-electrophile coupling reactions. Mechanistic studies suggest that the cobalt catalyst selectively activates the alkyl electrophile to generate an alkyl radical, while the nickel catalyst primarily activates the aryl electrophile and captures the alkyl radical, ultimately leading to the formation of a new C(sp2)–C(sp3) bond. The method allows for reactions to be rationally optimized by modulating the relative loadings of the two catalysts based on the reaction byproducts. Our dual catalyst platform is able to efficiently couple a wide range of substrates with excellent functional group tolerance. Notably, we can couple heteroaryl substrates, such as 2-bromothiophene, 2-bromofuran, and 2-bromopyridine, and di-ortho-substituted aryl iodides, which are rarely compatible with methods for C(sp2)–C(sp3) cross-electrophile coupling. We further demonstrate the unique control that our methodology provides through a series of novel one-pot three-component dialkylations of bromo(iodo)arenes. Finally, we show the utility of our strategy to medicinal chemistry in two ways: (i) through the successful coupling of a range of druglike aryl halides and functionally diverse alkyl halides using HTE and (ii) through a larger scale reaction involving a druglike aryl halide. Overall, our mechanistic work has enabled the discovery of a system for C(sp2)–C(sp3) cross-electrophile coupling that has a broad scope and is applicable to both synthetic and medicinal chemistry. Furthermore, the underlying principles behind our dual catalytic strategy can likely be used to improve a number of existing transformations and facilitate the discovery of new reactions.
Supplementary Material
ACKNOWLEDGMENTS
N.H. acknowledges support from the NIHGMS under Award Number R01GM120162. We thank Professor Jon Ellman and Dr. Louis-Charles Campeau for valuable discussions. We also thank Sr. Scientist Scott Borges, May Ann Desaca, and Sr. Scientist Tao Meng for support in conducting purification, Assoc. Scientist Ming Wang for assistance with high-throughput analytical sample processing, and Dr. Fabian Menges for help with mass spectrometry measurements.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c03237.
General methods and instrumentation methods, procedure for cross-electrophile coupling of aryl and alkyl halides, reactivity of (dtbbpy)NiII(o-tol)I with TDAE, varying (dtbbpy)NiIIBr2 loading in dual-catalyzed cross-electrophile coupling of iodobenzene with benzyl chloride, stoichiometric C(sp2)–C(sp3) bond formation with (dtbbpy)NiII(o-tol)I, radical trapping experiments with TEMPO, optimization of cross-electrophile coupling of phenyl iodide with 1-iodo-3-phenylpropane, optimization of concentration, solvent screen, and temperature, effect of aryl halide ortho-substitution on optimization of catalyst loadings, additional reactions for two-component and three-component cross-electrophile couplings, optimization of single-step one-pot three-component coupling, procedure for optimization of two-step one-pot three-component coupling for 1H NMR yields, high-throughput experimentation for optimization of druglike aryl halides, additional reactions for druglike aryl halide cross-electrophile coupling, parallel library synthesis using substrate 5f, procedure and general information of 3 mmol scale reaction of 5f with 1-iodo-3-phenylpropane, isolation procedures and characterization of products of two-component cross-electrophile coupling, two-step one-pot three-component coupling, and two-component cross-electrophile coupling with druglike aryl halides, procedure for 1H NMR yields of products, NMR spectra of isolated products, and ultraperformance liquid chromatography traces from HTE experiments (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acscatal.0c03237
The authors declare no competing financial interest.
Contributor Information
Mycah R. Uehling, Merck & Co., Inc., Discovery Chemistry, HTE and Lead Discovery Capabilities, Kenilworth, New Jersey 07033, United States
Susan L. Zultanski, Merck & Co., Inc., Department of Process Research and Development, Rahway, New Jersey 07065, United States
REFERENCES
- (1).(a) Everson DA; Weix DJ Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem 2014, 79, 4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Knappke CEI; Grupe S; Gärtner D; Corpet M; Gosmini C; Jacobi von Wangelin A Reductive Cross-Coupling Reactions Between Two Electrophiles. Chem. – Eur. J 2014, 20, 6828–6842. [DOI] [PubMed] [Google Scholar]; (c) Moragas T; Correa A; Martin R Metal-Catalyzed Reductive Coupling Reactions of Organic Halides with Carbonyl-Type Compounds. Chem. – Eur. J 2014, 20, 8242–8258. [DOI] [PubMed] [Google Scholar]; (d) Gu J; Wang X; Xue W; Gong H Nickel-Catalyzed Reductive Coupling of Alkyl Halides with Other Electrophiles: Concept and Mechanistic Considerations. Org. Chem. Front 2015, 2, 1411–1421. [Google Scholar]; (e) Weix DJ Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc. Chem. Res 2015, 48, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Richmond E; Moran J Recent Advances in Nickel Catalysis Enabled by Stoichiometric Metallic Reducing Agents. Synthesis 2018, 50, 499–513. [Google Scholar]; (g) Tortajada A; Juliá-Hernández F; Börjesson M; Moragas T; Martin R Transition-Metal-Catalyzed Carboxylation Reactions with Carbon Dioxide. Angew. Chem. Int. Ed 2018, 57, 15948–15982. [DOI] [PubMed] [Google Scholar]; (h) Campeau L-C; Hazari N Cross-Coupling and Related Reactions: Connecting Past Success to the Development of New Reactions for the Future. Organometallics 2019, 38, 3–35. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Poremba KE; Dibrell SE; Reisman SE Nickel-Catalyzed Enantioselective Reductive Cross-Coupling Reactions. ACS Catal. 2020, 8237–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2). For leading references, see.; (a) Everson DA; Shrestha R; Weix DJ Nickel-Catalyzed Reductive Cross-Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc 2010, 132, 920–921. [DOI] [PubMed] [Google Scholar]; (b) Everson DA; Jones BA; Weix DJ Replacing Conventional Carbon Nucleophiles with Electrophiles: Nickel-Catalyzed Reductive Alkylation of Aryl Bromides and Chlorides. J. Am. Chem. Soc 2012, 134, 6146–6159. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wotal AC; Weix DJ Synthesis of Functionalized Dialkyl Ketones from Carboxylic Acid Derivatives and Alkyl Halides. Org. Lett 2012, 14, 1476–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cherney AH; Kadunce NT; Reisman SE Catalytic Asymmetric Reductive Acyl Cross-Coupling: Synthesis of Enantioenriched Acyclic α,α-Disubstituted Ketones. J. Am. Chem. Soc 2013, 135, 7442–7445. [DOI] [PubMed] [Google Scholar]; (e) Cherney AH; Reisman SE Nickel-Catalyzed Asymmetric Reductive Cross-Coupling Between Vinyl and Benzyl Electrophiles. J. Am. Chem. Soc 2014, 136, 14365–14368. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Molander GA; Traister KM; O’Neill BT Reductive Cross-Coupling of Nonaromatic, Heterocyclic Bromides with Aryl and Heteroaryl Bromides. J. Org. Chem 2014, 79, 5771–5780. [DOI] [PubMed] [Google Scholar]; (g) Molander GA; Wisniewski SR; Traister KM Reductive Cross-Coupling of 3-Bromo-2,1-borazaronaphthalenes with Alkyl Iodides. Org. Lett 2014, 16, 3692–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhao C; Jia X; Wang X; Gong H Ni-Catalyzed Reductive Coupling of Alkyl Acids with Unactivated Tertiary Alkyl and Glycosyl Halides. J. Am. Chem. Soc 2014, 136, 17645–17651. [DOI] [PubMed] [Google Scholar]; (i) Zhao Y; Weix DJ Nickel-Catalyzed Regiodivergent Opening of Epoxides with Aryl Halides: Co-Catalysis Controls Regioselectivity. J. Am. Chem. Soc 2014, 136, 48–51. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Zhao Y; Weix DJ Enantioselective Cross-Coupling of meso-Epoxides with Aryl Halides. J. Am. Chem. Soc 2015, 137, 3237–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Arendt KM; Doyle AG Dialkyl Ether Formation by Nickel-Catalyzed Cross-Coupling of Acetals and Aryl Iodides. Angew. Chem. Int. Ed 2015, 54, 9876–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Kadunce NT; Reisman SE Nickel-Catalyzed Asymmetric Reductive Cross-Coupling between Heteroaryl Iodides and α-Chloronitriles. J. Am. Chem. Soc 2015, 137, 10480–10483. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Molander GA; Traister KM; O’Neill BT Engaging Nonaromatic, Heterocyclic Tosylates in Reductive Cross-Coupling with Aryl and Heteroaryl Bromides. J. Org. Chem 2015, 80, 2907–2911. [DOI] [PubMed] [Google Scholar]; (n) Wang X; Wang S; Xue W; Gong H Nickel-Catalyzed Reductive Coupling of Aryl Bromides with Tertiary Alkyl Halides. J. Am. Chem. Soc 2015, 137, 11562–11565. [DOI] [PubMed] [Google Scholar]; (o) Hu L; Liu X; Liao X Nickel-Catalyzed Methylation of Aryl Halides with Deuterated Methyl Iodide. Angew. Chem. Int. Ed 2016, 55, 9743–9747. [DOI] [PubMed] [Google Scholar]; (p) Huihui KMM; Caputo JA; Melchor Z; Olivares AM; Spiewak AM; Johnson KA; DiBenedetto TA; Kim S; Ackerman LKG; Weix DJ Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides. J. Am. Chem. Soc 2016, 138, 5016–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Johnson KA; Biswas S; Weix DJ Cross-Electrophile Coupling of Vinyl Halides with Alkyl Halides. Chem. – Eur. J 2016, 22, 7399–7402. [DOI] [PMC free article] [PubMed] [Google Scholar]; (r) Konev MO; Hanna LE; Jarvo ER Intra- and Intermolecular Nickel-Catalyzed Reductive Cross-Electrophile Coupling Reactions of Benzylic Esters with Aryl Halides. Angew. Chem. Int. Ed 2016, 55, 6730–6733. [DOI] [PubMed] [Google Scholar]; (s) Liu J; Ren Q; Zhang X; Gong H Preparation of Vinyl Arenes by Nickel-Catalyzed Reductive Coupling of Aryl Halides with Vinyl Bromides. Angew. Chem. Int. Ed 2016, 55, 15544–15548. [DOI] [PubMed] [Google Scholar]; (t) Chen F; Chen K; Zhang Y; He Y; Wang Y-M; Zhu S Remote Migratory Cross-Electrophile Coupling and Olefin Hydroarylation Reactions Enabled by in Situ Generation of NiH. J. Am. Chem. Soc 2017, 139, 13929–13935. [DOI] [PubMed] [Google Scholar]; (u) He Y; Cai Y; Zhu S Mild and Regioselective Benzylic C–H Functionalization: Ni-Catalyzed Reductive Arylation of Remote and Proximal Olefins. J. Am. Chem. Soc 2017, 139, 1061–1064. [DOI] [PubMed] [Google Scholar]; (v) Poremba KE; Kadunce NT; Suzuki N; Cherney AH; Reisman SE Nickel-Catalyzed Asymmetric Reductive Cross-Coupling To Access 1,1-Diarylalkanes. J. Am. Chem. Soc 2017, 139, 5684–5687. [DOI] [PMC free article] [PubMed] [Google Scholar]; (w) Woods BP; Orlandi M; Huang C-Y; Sigman MS; Doyle AG Nickel-Catalyzed Enantioselective Reductive Cross-Coupling of Styrenyl Aziridines. J. Am. Chem. Soc 2017, 139, 5688–5691. [DOI] [PubMed] [Google Scholar]; (x) Schneider P; Schneider G Privileged Structures Revisited. Angew. Chem. Int. Ed 2017, 56, 7971–7974. [DOI] [PMC free article] [PubMed] [Google Scholar]; (y) Peng L; Li Y; Li Y; Wang W; Pang H; Yin G Ligand-Controlled Nickel-Catalyzed Reductive Relay Cross-Coupling of Alkyl Bromides and Aryl Bromides. ACS Catal 2018, 8, 310–313. [Google Scholar]; (z) Wang X; Ma G; Peng Y; Pitsch CE; Moll BJ; Ly TD; Wang X; Gong H Ni-Catalyzed Reductive Coupling of Electron-Rich Aryl Iodides with Tertiary Alkyl Halides. J. Am. Chem. Soc 2018, 140, 14490–14497. [DOI] [PubMed] [Google Scholar]; (aa) Xu C; Guo W-H; He X; Guo Y-L; Zhang X-Y; Zhang X Difluoromethylation of (hetero)aryl Chlorides with Chlorodifluoromethane Catalyzed by Nickel. Nature Commun 2018, 9, 1170. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ab) Liao J; Basch CH; Hoerrner ME; Talley MR; Boscoe BP; Tucker JW; Garnsey MR; Watson MP Deaminative Reductive Cross-Electrophile Couplings of Alkylpyridinium Salts and Aryl Bromides. Org. Lett 2019, 21, 2941–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ac) Ni S; Li C-X; Mao Y; Han J; Wang Y; Yan H; Pan Y Ni-Catalyzed Deaminative Cross-Electrophile Coupling of Katritzky Salts with Halides via C–N Bond Activation. Sci. Adv 2019, 5, 9516. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ad) Ni S; Padial NM; Kingston C; Vantourout JC; Schmitt DC; Edwards JT; Kruszyk MM; Merchant RR; Mykhailiuk PK; Sanchez BB; Yang S; Perry MA; Gallego GM; Mousseau JJ; Collins MR; Cherney RJ; Lebed PS; Chen JS; Qin T; Baran PS A Radical Approach to Anionic Chemistry: Synthesis of Ketones, Alcohols, and Amines. J. Am. Chem. Soc 2019, 141, 6726–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ae) Tian Z-X; Qiao J-B; Xu G-L; Pang X; Qi L; Ma W-Y; Zhao Z-Z; Duan J; Du Y-F; Su P; Liu X-Y; Shu X-Z Highly Enantioselective Cross-Electrophile Aryl-Alkenylation of Unactivated Alkenes. J. Am. Chem. Soc 2019, 141, 7637–7643. [DOI] [PubMed] [Google Scholar]; (af) Wang J; Cary BP; Beyer PD; Gellman SH; Weix DJ Ketones from Nickel-Catalyzed Decarboxylative, Non-Symmetric Cross-Electrophile Coupling of Carboxylic Acid Esters. Angew. Chem. Int. Ed 2019, 58, 12081–12085. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ag) Ye Y; Chen H; Sessler JL; Gong H Zn-Mediated Fragmentation of Tertiary Alkyl Oxalates Enabling Formation of Alkylated and Arylated Quaternary Carbon Centers. J. Am. Chem. Soc 2019, 141, 820–824. [DOI] [PubMed] [Google Scholar]; (ah) Yue H; Zhu C; Shen L; Geng Q; Hock KJ; Yuan T; Cavallo L; Rueping M Nickel-Catalyzed C–N Bond Activation: Activated Primary Amines as Alkylating Reagents in Reductive Cross-Coupling. Chem. Sci 2019, 10, 4430–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ai) Duan J; Du Y-F; Pang X; Shu X-Z Ni-Catalyzed Cross-Electrophile Coupling Between Vinyl/Aryl and Alkyl Sulfonates: Synthesis of Cycloalkenes and Modification of Peptides. Chem. Sci 2019, 10, 8706–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]; (aj) Kim S; Goldfogel MJ; Gilbert MM; Weix DJ Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Chlorides with Primary Alkyl Chlorides. J. Am. Chem. Soc 2020, 142, 9902–9907. [DOI] [PMC free article] [PubMed] [Google Scholar]; (ak) Parasram M; Shields BJ; Ahmad O; Knauber T; Doyle AG Regioselective Cross-Electrophile Coupling of Epoxides and (Hetero)aryl Iodides via Ni/Ti/Photoredox Catalysis. ACS Catal 2020, 10, 5821–5827. [DOI] [PMC free article] [PubMed] [Google Scholar]; (al) Wang J; Hoerrner ME; Watson MP; Weix DJ Nickel-Catalyzed Synthesis of Dialkyl Ketones from the Coupling of N-Alkyl Pyridinium Salts with Activated Carboxylic Acids. Angew. Chem. Int. Ed 2020, 59, 13484–13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Biswas S; Weix DJ Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Diccianni JB; Katigbak J; Hu C; Diao T Mechanistic Characterization of (Xantphos)Ni(I)-Mediated Alkyl Bromide Activation: Oxidative Addition, Electron Transfer, or Halogen-Atom Abstraction. J. Am. Chem. Soc 2019, 141, 1788–1796. [DOI] [PubMed] [Google Scholar]; (c) Lin Q; Diao T Mechanism of Ni-Catalyzed Reductive 1,2-Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc 2019, 141, 17937–17948. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mohadjer Beromi M; Brudvig GW; Hazari N; Lant HMC; Mercado BQ Synthesis and Reactivity of Paramagnetic Nickel Polypyridyl Complexes Relevant to C(sp2)–C(sp3) Coupling Reactions. Angew. Chem. Int. Ed 2019, 58, 6094–6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hansen EC; Pedro DJ; Wotal AC; Gower NJ; Nelson JD; Caron S; Weix DJ New Ligands for Nickel Catalysis From Diverse Pharmaceutical Heterocycle Libraries. Nature Chem. 2016, 8, 1126–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Isbrandt ES; Sullivan RJ; Newman SG High Throughput Strategies for the Discovery and Optimization of Catalytic Reactions. Angew. Chem. Int. Ed 2019, 58, 7180–7191. [DOI] [PubMed] [Google Scholar]
- (6).(a) Hansen EC; Li C; Yang S; Pedro D; Weix DJ Coupling of Challenging Heteroaryl Halides with Alkyl Halides via Nickel-Catalyzed Cross-Electrophile Coupling. J. Org. Chem 2017, 82, 7085–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hughes JME; Fier PS Desulfonylative Arylation of Redox-Active Alkyl Sulfones with Aryl Bromides. Org. Lett 2019, 21, 5650–5654. [DOI] [PubMed] [Google Scholar]; (c) Mennie KM; Vara BA; Levi SM Reductive sp3–sp2 Coupling Reactions Enable Late-Stage Modification of Pharmaceuticals. Org. Lett 2020, 22, 556–559. [DOI] [PubMed] [Google Scholar]
- (7). In reference 3a, Weix and co-workers discuss different pathways for the generation of an alkyl radical in cross-electrophile coupling prior to the formation of a NiI species. They postulated that LNiII(Ar)X could be involved in radical generation but were unable to conclusively verify this hypothesis.
- (8). In traditional cross-coupling reactions, there are examples of the generation of alkyl radicals through the reaction of a NiII species with an alkyl halide, see.; (a) Schley ND; Fu GC Nickel-Catalyzed Negishi Arylations of Propargylic Bromides: A Mechanistic Investigation. J. Am. Chem. Soc 2014, 136, 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yin H; Fu GC Mechanistic Investigation of Enantioconvergent Kumada Reactions of Racemic α-Bromoketones Catalyzed by a Nickel/Bis(oxazoline) Complex. J. Am. Chem. Soc 2019, 141, 15433–15440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Takai K; Nitta K; Fujimura O; Utimoto K Preparation of Alkylchromium Reagents by Reduction of Alkyl Halides with Chromium(II) Chloride Under Cobalt Catalysis. J. Org. Chem 1989, 54, 4732–4734. [Google Scholar]; (b) Ackerman LKG; Anka-Lufford LL; Naodovic M; Weix DJ Cobalt Co-Catalysis for Cross-Electrophile Coupling: Diarylmethanes From Benzyl Mesylates and Aryl Halides. Chem. Sci 2015, 6, 1115–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Komeyama K; Ohata R; Kiguchi S; Osaka I Highly Nucleophilic Vitamin B12-Assisted Nickel-Catalysed Reductive Coupling of Aryl Halides and Non-Activated Alkyl Tosylates. Chem. Commun 2017, 53, 6401–6404. [DOI] [PubMed] [Google Scholar]; (d) Komeyama K; Yamahata Y; Osaka I Nickel and Nucleophilic Cobalt-Catalyzed Trideuteriomethylation of Aryl Halides Using Trideuteriomethyl p-Toluenesulfonate. Org. Lett 2018, 20, 4375–4378. [DOI] [PubMed] [Google Scholar]; (e) Hofstra JL; Cherney AH; Ordner CM; Reisman SE Synthesis of Enantioenriched Allylic Silanes via Nickel-Catalyzed Reductive Cross-Coupling. J. Am. Chem. Soc 2018, 140, 139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Komeyama K; Michiyuki T; Osaka I Nickel/Cobalt-Catalyzed C(sp3)–C(sp3) Cross-Coupling of Alkyl Halides with Alkyl Tosylates. ACS Catal 2019, 9, 9285–9291. [Google Scholar]; (g) Ociepa M; Wierzba AJ; Turkowska J; Gryko D Polarity-Reversal Strategy for the Functionalization of Electrophilic Strained Molecules via Light-Driven Cobalt Catalysis. J. Am. Chem. Soc 2020, 142, 5355–5361. [DOI] [PubMed] [Google Scholar]
- (10). Cocatalytic generation of a radical in tandem with nickel catalysis has also been used in reactions other than cross-electrophile coupling. For selected examples, see.; (a) Tellis JC; Primer DN; Molander GA Single-Electron Transmetalation in Organoboron Cross-Coupling by Photoredox/Nickel Dual Catalysis. Science 2014, 345, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Merging Photoredox with Nickel Catalysis: Coupling of α-carboxyl sp3-Carbons with Aryl Halides. Science 2014, 345, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Green SA; Matos JLM; Yagi A; Shenvi RA Branch-Selective Hydroarylation: Iodoarene–Olefin Cross-Coupling. J. Am. Chem. Soc 2016, 138, 12779–12782. [DOI] [PubMed] [Google Scholar]; (d) Perry IB; Brewer TF; Sarver PJ; Schultz DM; DiRocco DA; MacMillan DWC Direct Arylation of Strong Aliphatic C–H Bonds. Nature 2018, 560, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Green SA; Huffman TR; McCourt RO; van der Puyl V; Shenvi RA Hydroalkylation of Olefins To Form Quaternary Carbons. J. Am. Chem. Soc 2019, 141, 7709–7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Iyoda M; Sakaitan M; Otsuka H; Oda M Reductive Coupling of Benzyl Halides Using Nickel(0)-Complex Generated In Situ in the Presence of the Tetraethylammonium Iodide, A Simple and Convenient Synthesis of BiBenzyls. Chem. Lett 1985, 14, 127–130. [Google Scholar]; (b) Charboneau DJ; Brudvig GW; Hazari N; Lant HMC; Saydjari AK Development of an Improved System for the Carboxylation of Aryl Halides through Mechanistic Studies. ACS Catal. 2019, 9, 3228–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huang L; Ackerman LKG; Kang K; Parsons AM; Weix DJ LiCl-Accelerated Multimetallic Cross-Coupling of Aryl Chlorides with Aryl Triflates. J. Am. Chem. Soc 2019, 141, 10978–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Jessop PG Searching for Green Solvents. Green Chem. 2011, 13, 1391–1398. [Google Scholar]; (b) Diorazio LJ; Hose DRJ; Adlington NK Toward a More Holistic Framework for Solvent Selection. Org. Proc. Res. Devel 2016, 20, 760–773. [Google Scholar]; (c) Prat D; Wells A; Hayler J; Sneddon H; McElroy CR; Abou-Shehada S; Dunn PJ CHEM21 Selection Guide of Classical- and Less Classical-Solvents. Green Chem. 2016, 18, 288–296. [Google Scholar]; (d) Yin J; Maguire CK; Yasuda N; Brunskill APJ; Klapars A Impact of Lead Impurities in Zinc Dust on the Selective Reduction of a Dibromoimidazole Derivative. Org. Proc. Res. Devel 2017, 21, 94–97. [Google Scholar]; (e) Nimmagadda SK; Korapati S; Dasgupta D; Malik NA; Vinodini A; Gangu AS; Kalidindi S; Maity P; Bondigela SS; Venu A; Gallagher WP; Aytar S; González-Bobes F; Vaidyanathan R Development and Execution of an Ni(II)-Catalyzed Reductive Cross-Coupling of Substituted 2-Chloropyridine and Ethyl 3-Chloropropanoate. Org. Proc. Res. Devel 2020, 24, 1141–1148. [Google Scholar]
- (13).(a) Hartman RL Managing Solids in Microreactors for the Upstream Continuous Processing of Fine Chemicals. Org. Proc. Res. Devel 2012, 16, 870–887. [Google Scholar]; (b) Godfrey AG; Masquelin T; Hemmerle H A Remote-Controlled Adaptive Medchem Lab: An Innovative Approach to Enable Drug Discovery in the 21st Century. Drug Discovery Today 2013, 18, 795–802. [DOI] [PubMed] [Google Scholar]; (c) Li J; Ballmer SG; Gillis EP; Fujii S; Schmidt MJ; Palazzolo AM; Lehmann JW; Morehouse GF; Burke MD Synthesis of Many Different Types of Organic Small Molecules Using One Automated Process. Science 2015, 347, 1221–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Buitrago Santanilla A; Regalado EL; Pereira T; Shevlin M; Bateman K; Campeau L-C; Schneeweis J; Berritt S; Shi Z-C; Nantermet P; Liu Y; Helmy R; Welch CJ; Vachal P; Davies IW; Cernak T; Dreher SD Nanomole-Scale High-Throughput Chemistry for the Synthesis of Complex Molecules. Science 2015, 347, 49–53. [DOI] [PubMed] [Google Scholar]; (e) Plutschack MB; Pieber B; Gilmore K; Seeberger PH The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev 2017, 117, 11796–11893. [DOI] [PubMed] [Google Scholar]; (f) Jensen KF Flow Chemistry–Microreaction Technology Comes of Age. AIChE J. 2017, 63, 858–869. [Google Scholar]; (g) Perera D; Tucker JW; Brahmbhatt S; Helal CJ; Chong A; Farrell W; Richardson P; Sach NW A Platform for Automated Nanomole-Scale Reaction Screening and Micromole-Scale Synthesis in Flow. Science 2018, 359, 429–434. [DOI] [PubMed] [Google Scholar]
- (14). Systems that utilize light, via a photocatalyst, or electricity, in place of a heterogeneous reductant have been reported. While these methods address some limitations associated with heterogeneous reductants, they have different sets of practical challenges. For selected systems, see.; (a) Duan Z; Li W; Lei A Nickel-Catalyzed Reductive Cross-Coupling of Aryl Bromides with Alkyl Bromides: Et3N as the Terminal Reductant. Org. Lett 2016, 18, 4012–4015. [DOI] [PubMed] [Google Scholar]; (b) Zhang P; Le CC; MacMillan DWC Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc 2016, 138, 8084–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Perkins RJ; Pedro DJ; Hansen EC Electrochemical Nickel Catalysis for sp2-sp3 Cross-Electrophile Coupling Reactions of Unactivated Alkyl Halides. Org. Lett 2017, 19, 3755–3758. [DOI] [PubMed] [Google Scholar]; (d) Li H; Breen CP; Seo H; Jamison TF; Fang Y-Q; Bio MM Ni-Catalyzed Electrochemical Decarboxylative C–C Couplings in Batch and Continuous Flow. Org. Lett 2018, 20, 1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Bacauanu V; Cardinal S; Yamauchi M; Kondo M; Fernández DF; Remy R; MacMillan DWC Metallaphotoredox Difluoromethylation of Aryl Bromides. Angew. Chem. Int. Ed 2018, 57, 12543–12548. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) DeLano TJ; Reisman SE Enantioselective Electroreductive Coupling of Alkenyl and Benzyl Halides via Nickel Catalysis. ACS Catal. 2019, 9, 6751–6754. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Koyanagi T; Herath A; Chong A; Ratnikov M; Valiere A; Chang J; Molteni V; Loren J One-Pot Electrochemical Nickel-Catalyzed Decarboxylative sp2–sp3 Cross-Coupling. Org. Lett 2019, 21, 816–820. [DOI] [PubMed] [Google Scholar]; (h) Martin-Montero R; Yatham VR; Yin H; Davies J; Martin R Ni-Catalyzed Reductive Deaminative Arylation at sp3 Carbon Centers. Org. Lett 2019, 21, 2947–2951. [DOI] [PubMed] [Google Scholar]; (i) Perkins RJ; Hughes AJ; Weix DJ; Hansen EC Metal-Reductant-Free Electrochemical Nickel-Catalyzed Couplings of Aryl and Alkyl Bromides in Acetonitrile. Org. Proc. Res. Devel 2019, 23, 1746–1751. [Google Scholar]; (j) Yi J; Badir SO; Kammer LM; Ribagorda M; Molander GA Deaminative Reductive Arylation Enabled by Nickel/Photoredox Dual Catalysis. Org. Lett 2019, 21, 3346–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Chen TQ; MacMillan DWC A Metallaphotoredox Strategy for the Cross-Electrophile Coupling of α-Chloro Carbonyls with Aryl Halides. Angew. Chem. Int. Ed 2019, 58, 14584–14588. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Brill ZG; Ritts CB; Mansoor UF; Sciammetta N Continuous Flow Enables Metallaphotoredox Catalysis in a Medicinal Chemistry Setting: Accelerated Optimization and Library Execution of a Reductive Coupling between Benzylic Chlorides and Aryl Bromides. Org. Lett 2020, 22, 410–416. [DOI] [PubMed] [Google Scholar]; (m) Jiao K-J; Liu D; Ma H-X; Qiu H; Fang P; Mei T-S Nickel-Catalyzed Electrochemical Reductive Relay Cross-Coupling of Alkyl Halides to Aryl Halides. Angew. Chem. Int. Ed 2020, 59, 6520–6524. [DOI] [PubMed] [Google Scholar]; (n) Truesdell BL; Hamby TB; Sevov CS General C(sp2)–C(sp3) Cross-Electrophile Coupling Reactions Enabled by Over-charge Protection of Homogeneous Electrocatalysts. J. Am. Chem. Soc 2020, 142, 5884–5893. [DOI] [PubMed] [Google Scholar]; (o) Xu B; Troian-Gautier L; Dykstra R; Martin RT; Gutierrez O; Tambar UK Photocatalyzed Diastereoselective Isomerization of Cinnamyl Chlorides to Cyclopropanes. J. Am. Chem. Soc 2020, 142, 6206–6215. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Sakai HA; Liu W; Le CC; MacMillan DWC Cross-Electrophile Coupling of Unactivated Alkyl Chlorides. J. Am. Chem. Soc 2020, 142, 11691–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Anka-Lufford LL; Huihui KMM; Gower NJ; Ackerman LKG; Weix DJ Nickel-Catalyzed Cross-Electrophile Coupling with Organic Reductants in Non-Amide Solvents. Chem. – Eur. J 2016, 22, 11564–11567. [DOI] [PubMed] [Google Scholar]; (b) Suzuki N; Hofstra JL; Poremba KE; Reisman SE Nickel-Catalyzed Enantioselective Cross-Coupling of N-Hydroxyphthalimide Esters with Vinyl Bromides. Org. Lett 2017, 19, 2150–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16). For other Ni-catalyzed transformations that use homogeneous reductants, see reference 11b and.; (a) Kuroboshi M; Tanaka M; Kishimoto S; Goto K; Mochizuki M; Tanaka H Tetrakis-(dimethylamino)ethylene (TDAE) as a Potent Organic Electron Source: Alkenylation of Aldehydes Using an Ni/Cr/TDAE Redox System. Tetrahedron Lett. 2000, 41, 81–84. [Google Scholar]; (b) Lv L; Qiu Z; Li J; Liu M; Li C-J N2H4 as Traceless Mediator for Homo- and Cross-Aryl Coupling. Nature Commun. 2018, 9, 4739. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) García-Domínguez A; Li Z; Nevado C Nickel-Catalyzed Reductive Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc 2017, 139, 6835–6838. [DOI] [PubMed] [Google Scholar]; (d) Shu W; García-Domínguez A; Quirós MT; Mondal R; Cárdenas DJ; Nevado C Ni-Catalyzed Reductive Dicarbofunctionalization of Nonactivated Alkenes: Scope and Mechanistic Insights. J. Am. Chem. Soc 2019, 141, 13812–13821. [DOI] [PubMed] [Google Scholar]; (e) Wei X; Shu W; García-Domínguez A; Merino E; Nevado C Asymmetric Ni-Catalyzed Radical Relayed Reductive Coupling. J. Am. Chem. Soc 2020, 142, 13515–13522. [DOI] [PubMed] [Google Scholar]
- (17).(a) Tsou TT; Kochi JK Mechanism of Biaryl Synthesis with Nickel Complexes. J. Am. Chem. Soc 1979, 101, 7547–7560. [Google Scholar]; (b) Osakada K; Yamamoto T Transmetallation of Alkynyl and Aryl Complexes of Group 10 Transition Metals. Coord. Chem. Rev 2000, 198, 379–399. [Google Scholar]
- (18).(a) Mohadjer Beromi M; Nova A; Balcells D; Brasacchio AM; Brudvig GW; Guard LM; Hazari N; Vinyard DJ Mechanistic Study of An Improved Ni Precatalyst for Suzuki–Miyaura Reactions of Aryl Sulfamates: Understanding the Role of Ni(I) Species. J. Am. Chem. Soc 2017, 139, 922–936. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mohadjer Beromi M; Banerjee G; Brudvig GW; Hazari N; Mercado BQ Nickel (I) Aryl species: Synthesis, Properties, and Catalytic Activity. ACS Catal 2018, 8, 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19). In reference 15a, a limited number of reactions were also performed involving alkyl iodides and aryl bromides.
- (20).(a) Mho S.-i.; Ortiz B; Doddapaneni N; Park SM Electrochemical and Spectroelectrochemical Studies on Metallophthalocyanine-Oxygen Interactions in Nonaqueous Solutions. J. Electrochem. Soc 1995, 142, 1047. [Google Scholar]; (b) Eberle B; Hübner O; Ziesak A; Kaifer E; Himmel H-J What Makes a Strong Organic Electron Donor (or Acceptor)? Chem. – Eur. J 2015, 21, 8578–8590. [DOI] [PubMed] [Google Scholar]
- (21).Alonso F; Beletskaya IP; Yus M Metal-Mediated Reductive Hydrodehalogenation of Organic Halides. Chem. Rev 2002, 102, 4009–4092. [DOI] [PubMed] [Google Scholar]
- (22).Demarteau J; Debuigne A; Detrembleur C Organocobalt Complexes as Sources of Carbon-Centered Radicals for Organic and Polymer Chemistries. Chem. Rev 2019, 119, 6906–6955. [DOI] [PubMed] [Google Scholar]
- (23). For a previous example where TEMPO has been used to trap alkyl radicals from CoIII–Alk species see:; Ng FTT; Rempel GL; Mancuso C; Halpern J Decomposition of α-phenethylbis-(dimethylglyoximato)cobalt(III) Complexes. Influence of Electronic and Steric Factors on the Kinetics and Thermodynamics of Cobalt-Carbon Bond Dissociation. Organometallics 1990, 9, 2762–2772. [Google Scholar]
- (24). An alternative method for optimizing reactions involves utilizing additional equivalents of the alkyl electrophile. See the Supporting Information for further details.
- (25).Yin H; Sheng J; Zhang K-F; Zhang Z-Q; Bian K-J; Wang X-S Nickel-Catalyzed Monofluoromethylation of (Hetero)aryl Bromides via Reductive Cross-Coupling. Chem. Commun 2019, 55, 7635–7638. [DOI] [PubMed] [Google Scholar]
- (26).Mohadjer Beromi M; Banerjee G; Brudvig GW; Charboneau DJ; Hazari N; Lant HMC; Mercado BQ Modifications to the Aryl Group of dppf-Ligated Ni σ-Aryl Precatalysts: Impact on Speciation and Catalytic Activity in Suzuki-Miyaura Coupling Reactions. Organometallics 2018, 37, 3943–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Roughley SD; Jordan AM The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- (28).Do H-Q; Chandrashekar ERR; Fu GC Nickel/Bis(oxazoline)-Catalyzed Asymmetric Negishi Arylations of Racemic Secondary Benzylic Electrophiles to Generate Enantioenriched 1,1-Diarylalkanes. J. Am. Chem. Soc 2013, 135, 16288–16291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mendel M; Kalvet I; Hupperich D; Magnin G; Schoenebeck F Site-Selective, Modular Diversification of Polyhalogenated Aryl Fluorosulfates (ArOSO2F) Enabled by an Air-Stable PdI Dimer. Angew. Chem. Int. Ed 2020, 59, 2115–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kalvet I; Magnin G; Schoenebeck F Rapid Room-Temperature, Chemoselective C-C Coupling of Poly(pseudo)-halogenated Arenes Enabled by Palladium(I) Catalysis in Air. Angew. Chem. Int. Ed 2017, 56, 1581–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).(a) He L-Y; Schulz-Senft M; Thiedemann B; Linshoeft J; Gates PJ; Staubitz A Nucleophile-Selective Cross-Coupling Reactions with Vinyl and Alkynyl Bromides on a Dinucleophilic Aromatic Substrate. Eur. J. Org. Chem 2015, 2015, 2498–2502. [Google Scholar]; (b) Fyfe JWB; Fazakerley NJ; Watson AJB Chemoselective Suzuki–Miyaura Cross-Coupling via Kinetic Transmetallation. Angew. Chem. Int. Ed 2017, 56, 1249–1253. [DOI] [PubMed] [Google Scholar]
- (32).(a) Yamashita Y; Tellis JC; Molander GA Protecting Group-Free, Selective Cross-Coupling of Alkyltrifluoroborates with Borylated Aryl Bromides via Photoredox/Nickel Dual Catalysis. Proc. Natl. Acad. Sci 2015, 112, 12026–12029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lin K; Wiles RJ; Kelly CB; Davies GHM; Molander GA Haloselective Cross-Coupling via Ni/Photoredox Dual Catalysis. ACS Catal 2017, 7, 5129–5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).(a) Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]; (b) Lovering F Escape from Flatland 2: Complexity and Promiscuity. Med. Chem. Comm 2013, 4, 515–519. [Google Scholar]
- (34).Kutchukian PS; Dropinski JF; Dykstra KD; Li B; DiRocco DA; Streckfuss EC; Campeau L-C; Cernak T; Vachal P; Davies IW; Krska SW; Dreher SD Chemistry Informer Libraries: A Chemoinformatics Enabled Approach to Evaluate and Advance Synthetic Methods. Chem. Sci 2016, 7, 2604–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35). The concentration of the aryl halide was reduced to 0.1 M when working with medicinally relevant substrates due to their 135reduced solubility in 1,4-dioxane.
- (36).Krska SW; DiRocco DA; Dreher SD; Shevlin M The Evolution of Chemical High-Throughput Experimentation To Address Challenging Problems in Pharmaceutical Synthesis. Acc. Chem. Res 2017, 50, 2976–2985. [DOI] [PubMed] [Google Scholar]
- (37).(a) Reck F; Zhou F; Eyermann CJ; Kern G; Carcanague D; Ioannidis G; Illingworth R; Poon G; Gravestock MB Novel Substituted (Pyridin-3-yl)phenyloxazolidinones: Antibacterial Agents with Reduced Activity against Monoamine Oxidase A and Increased Solubility. J. Med. Chem 2007, 50, 4868–4881. [DOI] [PubMed] [Google Scholar]; (b) Komine T; Kojima A; Asahina Y; Saito T; Takano H; Shibue T; Fukuda Y Synthesis and Structure–Activity Relationship Studies of Highly Potent Novel Oxazolidinone Antibacterials. J. Med. Chem 2008, 51, 6558–6562. [DOI] [PubMed] [Google Scholar]
- (38).Taylor RD; MacCoss M; Lawson ADG Rings in Drugs. J. Med. Chem 2014, 57, 5845–5859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.