

ABSTRACT
Irritable Bowel Syndrome (IBS), the most common gastrointestinal disorder, is diagnosed solely on symptoms. Potentially diagnostic alterations in the bacterial component of the gut microbiome (the bacteriome) are associated with IBS, but despite the known role of the virome (particularly bacteriophages), in shaping the gut bacteriome, few studies have investigated the virome in IBS. We performed metagenomic sequencing of fecal Virus-Like Particles (VLPs) from 55 patients with IBS and 51 control individuals. We detected significantly lower alpha diversity of viral clusters comprising both known and novel viruses (viral ‘dark matter’) in IBS and a significant difference in beta diversity compared to controls, but not between IBS symptom subtypes. The three most abundant bacteriophage clusters belonged to the Siphoviridae, Myoviridae, and Podoviridae families (Order Caudovirales). A core virome (defined as a cluster present in at least 50% of samples) of 5 and 12 viral clusters was identified in IBS and control subjects, respectively. We also identified a subset of viral clusters that showed differential abundance between IBS and controls. The virome did not co-vary significantly with the bacteriome, with IBS clinical subtype, or with Bile Acid Malabsorption status. However, differences in the virome could be related back to the bacteriome as analysis of CRISPR spacers indicated that the virome alterations were at least partially related to the alterations in the bacteriome. We found no evidence for a shift from lytic to lysogenic replication of core viral clusters, a phenomenon reported for the gut virome of patients with Inflammatory Bowel Disease. Collectively, our data show alterations in the virome of patients with IBS, regardless of clinical subtype, which may facilitate development of new microbiome-based therapeutics.
KEYWORDS: Bacteriophage, bile acid malabsorption, irritable bowel syndrome, gut microbiota, virome
Introduction
Irritable Bowel Syndrome (IBS) is the most common functional gastrointestinal disorder, affecting about 11% of the global population.1 Diagnosis of IBS is based on symptoms, including recurrent abdominal pain and changes in bowel habits using the Rome IV diagnostic criteria. Three clinical subtypes are recognized: IBS with constipation (IBS-C), IBS with diarrhea (IBS-D), and IBS with mixed bowel habits (IBS-M).2 The cause of IBS is unknown and perhaps multi-factorial, with input from genetics,3,4 psychological stress,5 anxiety and depression, disruption of gut-brain interactions,6 diet,7 low grade inflammation,8 and previous evidence of gastroenteritis in a subset of cases.9 Multiple studies have shown that the bacterial component of the microbiome (the bacteriome) is altered in IBS10–14 and that IBS-D and IBS-M can be distinguished from bile acid malabsorption (BAM), a differential diagnosis of IBS-D15 based upon a metabolomic signature.12
The gut virome, which is mainly composed of bacteriophages (phages) of the dsDNA Caudovirales order, plays a major role in shaping the composition and interactions of the bacteriome through predation and co-evolution and by facilitating horizontal gene transfer and nutrient turnover in the bacteriome.16 Extensive inter-individual variation as well as high temporal stability of individual gut viromes have been reported.17–20 Despite a key role in the gut microbial ecosystem, the virome is relatively understudied in gastrointestinal diseases in contrast to the bacteriome, although recent reports suggest disturbances of the gut virome in inflammatory bowel disease (IBD)17,21–24 and in one small study of IBS.25 The aims of the present study were, therefore, to investigate if the gut virome is altered in patients with IBS and to determine if any alterations distinguished the symptom-based clinical subtypes of IBS.
Results
Taxonomy of known and unknown phage sequences in IBS
The clinical features of the study participants which included 55 patients with IBS (based on Rome IV criteria) and 51 non-IBS controls are outlined in Table 1. We isolated and performed shotgun sequencing of Virus Like Particle (VLP) dsDNA from purified fecal samples. Reads were assembled into non-redundant contigs, filtered to retain those with viral signal (Figure S1 & S2) as described in Methods and clustered into 2,962 viral clusters (VCs), analogous to genus/sub-family taxonomic level, based on gene sharing (Bin Jang et al., 2019) with 2,233 VCs composed of more than 2 contigs (75.4%). Of note, each methodology used to identify putative viral contigs yielded many contigs unique to that approach (Figure S1). VCs composed of contigs that had less than 75% breadth of mapped read coverage (see Methods) were removed, retaining 2,957 VCs. Only 33 clusters contained a reference genome from the Viral RefSeq database (version 97), highlighting the amount of ‘viral dark matter’ present in the gut virome. Thus, putative family level taxonomy was assigned based on contig level taxonomy using the UniprotKB TrEMBL database as described in Methods and putative bacterial hosts were predicted at species level by comparing CRISPR spacers on bacterial metagenomic contigs from 69 patients in our IBS cohort12 which had bacterial metagenomic data available, to protospacers on viral contigs in VCs, although many VCs could not be assigned bacterial host(s) (Figure2; Tables S1 & S2). Using the above methodology, 36% of clusters (Table S3) and 46.9% of contigs (Table S4) remained unclassified at family level across all samples. At family level, 46.5% of cluster and 43.7% of contig abundance were accounted for by clusters and contigs that could not be classified (Tables S3 & S4, Figure S3). Unclassified VCs accounted for 42.2% and 45.2% of the family level abundance in controls and IBS respectively (Table S5).
Table 1.
Clinical features of control and IBS subjects
| Control (n = 51) | IBS (n = 55) | |
|---|---|---|
| Age range, years (mean) | 20–64 (45) | 18–66 (40) |
| Sex (male/female) | 12/39 | 14/41 |
| BMI Class, n (%) | ||
| Normal | 21 (41) | 24 (44) |
| Obese Class I | 10 (20) | 7 (13) |
| Obese Class II | 3 (6) | 4 (7) |
| Obese Class III | 1 (2) | 3 (5) |
| Overweight | 15 (29) | 16 (29) |
| Underweight | 1 (2) | 1 (2) |
| HADS: Anxiety, n (%) | ||
| Normal (0–10) | 46 (90) | 43 (78) |
| Abnormal (11–21) | 5 (10) | 12 (22) |
| HADS: Depression, n (%) | ||
| Normal (0–10) | 51 (100) | 48 (87) |
| Abnormal (11–21) | 0 (0) | 7 (13) |
| Bristol Stool Score, n (%) | ||
| Normal | 43 (84) | 15 (27) |
| Constipated | 7 (14) | 15 (27) |
| Diarrhea | 1 (2) | 25 (46) |
| IBS subtype, n (%) | ||
| IBS-C | 21 (38) | |
| IBS-D | N/A | 17 (31) |
| IBS-M | 17 (31) | |
| SeHCAT assayed, n (%) | 9 (18) | 31 (56) |
| Dietary group (FFQ), n (%) | ||
| Omnivore | 49 (96) | 52 (94) |
| Vegetarian | 1 (2) | 2 (4) |
| Pescatarian | 1 (2) | 0 (0) |
| Gluten-free | 0 (0) | 1 (2) |
| Drinks alcohol, n (%) | ||
| Current | 46 (90) | 39 (71) |
| Previous | 0 (0) | 0 (0) |
| Never | 5 (10) | 16 (29) |
| Smoker, n (%) | ||
| Current | 7* (14) | 7* (13) |
| Previous | 8 (16) | 14 (25) |
| Never | 36 (70) | 34 (62) |
* 1 subject in each group smoked e-cigarettes; N/A not applicable
Figure 2.
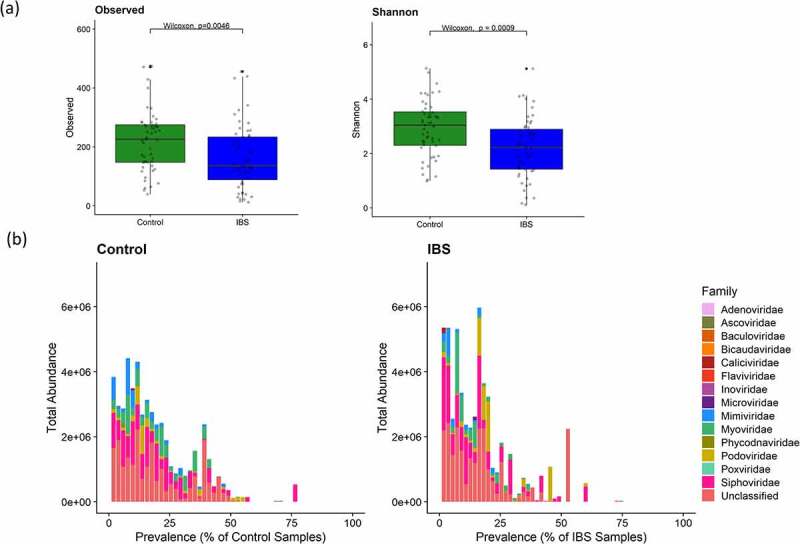
Difference in viral diversity between IBS and Controls. A) Boxplots of richness (observed) and alpha diversity (Shannon) estimates for IBS and control samples (Control: n = 51; IBS: n = 55 (IBS-C: n = 21; IBS-D: n = 17; IBS-M: n = 17) computed from viral cluster counts, showing significant differences (Wilcoxon rank-sum test p < .05) between IBS and controls. B). Barplots showing the total abundance of each viral family in control and IBS populations by prevalence in each population)
Of classified clusters, the most abundant families were those of the tailed bacteriophage viruses Siphoviridae (27.7%), Myoviridae (13%), and Podoviridae (8.9%) respectively, which are all in the Caudovirales order, in agreement with previous studies of the gut virome in healthy and disease cohorts (Table S3).17–21,24,26,27 The 4th most abundant family (6%) was the Mimiviridae family, whose members infect amoebae and protists, and whose member Mimivirus has been previously noted as a potentially dubious taxonomic classification in a study of the gut virome in ulcerative colitis.24,28 This led us to examine the confidence of the taxonomic classifications in more detail. Examination of the percentage identity and percentage coverage for each protein sequence aligned to the hit protein used for taxonomic assignment (percentage query cover) revealed a wide distribution of both scores for each family indicating that many of the classifications (Figure 1 and Figure S4) were based on low scores. While families in the Caudovirales order had high-scoring hits present, scores for most other families, including Mimiviridae were low indicating that the family-level classification was not reliable. Except for Mimiviridae, all other families with low scoring hits had very low abundances (<0.2% of total abundance at VC level and <0.6% at contig level; Tables S3 & S4, Figure 1). Ascoviridae, Iridoviridae, Marseilleviridae, Pithoviridae, and Poxviridae, all families of nucleocytoplasmic large dsDNA viruses (NCLDVs; proposed order Megavirales29) of eukaryotes had mainly low scoring hits (Figure 1) and were present in very low abundances (Figure 1; Table S1). Furthermore, Pithoviridae viruses have a diameter of 500 nm, which is larger than the 450 nm filter used to remove non-viral material in this study (Methods), and so would not be expected in our sequence data.30 Some Mimivirdae genomes are less than 450 nm and so could legitimately be present in our data. However, taxonomic assignments to families without the presence of high scoring hits or with low abundance are likely not as reliable as those for Caudovirales.
Figure 1.
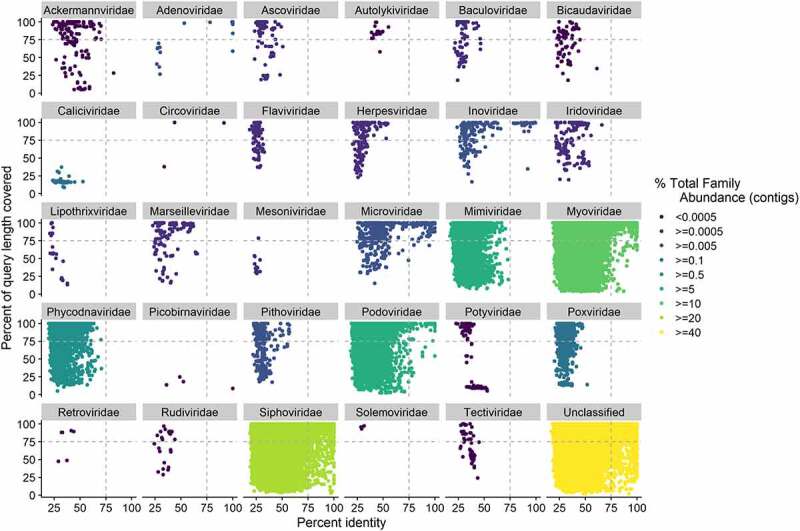
Confidence in taxonomic classifications, as measured by percentage query coverage and percent identity for protein sequences, predicted from the pooled contigs from IBS (n = 55) and control samples (n = 51), (n = 51,703 contigs). Each protein sequence was taxonomically classified using its top Diamond hit to the viral component of the UniProtKB TrEMBL database. Where the percentage of the query length covered was >100 due to gaps in the alignment, the coverage value was set to a value of 100. Family abundance was calculated by summing the abundance of all contigs assigned to that family and percentage family abundance calculated as the abundance of a family divided by the abundance of all families. Dashed lines indicate 75% identity and query coverage
The gut virome differs between IBS and controls
Alpha diversity was significantly lower in the IBS fecal virome compared with controls as measured with VCs (Figure 2a; Wilcoxon p < .05). Among the IBS clinical subtypes, the viromes of IBS-D and IBS-M were significantly less diverse than controls, based upon both observed and Shannon diversity metrics, and the IBS-D virome was significantly less diverse than IBS-C for observed VC counts (Figure S5a; Wilcoxon p < .05).
Significantly lower alpha diversity was also observed at contig level (Figure S6) in IBS and the IBS subtypes compared to controls, showing that the grouping of contigs into VCs had not masked any differences occurring at species/strain level. Visualization of the total abundance of VCs compared with the prevalence of those VCs in IBS and controls (Figure 2b) showed that VCs with lower prevalence contribute more to the total abundance in the IBS fecal phageome while there is a larger spread of the abundance values at each prevalence level in controls.
To determine if a core virome was present in IBS or control subjects, where the core virome was operationally defined as VCs present in at least 50% of individuals in a group, the prevalence of VCs in each group and across all samples was examined (Tables S6 & S7). We identified 5 and 12 core VCs in IBS and controls, respectively. Of these, four of the five VCs that were core VCs in IBS were also core VCs in the controls and six VCs were identified as core when considering all 106 subjects (Table S6). The highest prevalence was attributed to two VCs, both present in 76 samples. One was taxonomically assigned as Myoviridae and was present in 41 IBS and 35 control samples, while the other was unclassified and found in 40 IBS and 36 control subjects. All core VCs either had taxonomic assignments to families in the Caudovirales order or were unclassified but together accounted for only 6% of the total VC abundance. We could not identify a putative bacterial host for most core VCs from our analysis of CRISPR spacers with the exception of VC_1982_0, a core VC in controls only, which had some contigs with hits to Lachnospiraceae and VC_513_0, a core VC in all samples, which had some contigs with hits to Bacteroides (Table S6).
Additional examination of all pairwise distances, as measured using pairwise Bray-Curtis dissimilarity of VC abundance between samples, for samples in the IBS group, and separately, for all samples in the control group, showed that the IBS group had a greater degree of heterogeneity as defined by larger distances between samples and this difference was statistically significant between IBS and control groups (Figure S7; Wilcoxon rank-sum test p < .05). Taken together, the lower amount of core VCs and larger distances between IBS samples, demonstrate that the IBS gut virome is more individual-specific than that of controls.
Beta diversity, as measured using a PCoA of Bray-Curtis dissimilarity of VC abundance, was also significantly different between IBS and controls (PMANOVA p-value = 0.001), with the only significant split occurring at the second eigenvector (Students t-test, p-value = 0.0004) (Figure 3a). Testing beta diversity based on VCs instead of contigs accounted for more of the variance (10.7% of the total variance for VCs, see Figure 3, versus 6.4% for contigs, see Figure S8, was accounted by the first two axes) and improved the separation between IBS and controls (Figure S8a), demonstrating that VCs carry a stronger biological signal; however, stratification of IBS based on clinical subtypes was not significant at VC or contig level (PMANOVA p-value = 0.1) (Figure S5b and Figure S8b). s
Figure 3.
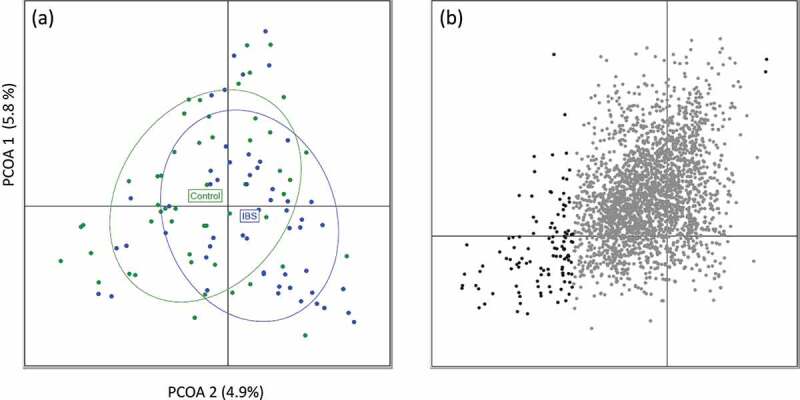
Difference in viral clusters between IBS and Controls. A) PCoA generated using the Bray-Curtis dissimilarity of viral cluster abundances (log-of abundance with pseudocount) between IBS and control samples showing a significant split between IBS and Control subjects (R2 = 0.0145, PMANOVA p-value = 0.001). The significant split was associated with the second eigenvector only (t.test p-value = 0.0004). PCoA1 is on the vertical axis, while PCoA2 is on the horizontal axis. B) Visualization of viral clusters using Canonical Correlation analysis with the Spearman methodology. Clusters significantly associated with the second eigenvector are highlighted as dark gray (n = 116). Of the 116 significant clusters, 114 are negatively associated with the second eigenvector showing a reduction in abundance and/or prevalence of these viral clusters across the PCoA2 axis which is associated with the IBS vs Control separation
Feature selection analysis using the DeSeq2 methodology on viral cluster counts, identified 10 VCs that were significantly differentially abundant between IBS and non-IBS controls, seven of which showed reduced abundance in the IBS subjects (Table S8). Of those with decreased abundance, three were unclassified at family level, one was classified as Mimiviridae and three were classified in the Caudovirales order (two as Siphoviridae and one as Podoviridae). Of those with increased abundance, one was classified as Mimiviridae, one as Podoviridae, and one as Siphoviridae. Most differentially abundant VCs were present in very few samples in either IBS or Controls (median of four samples in IBS and nine samples in controls), except for VC_1982_0. This VC, which was classified as Podoviridae, displayed increased abundance in IBS (log2FC = 3.8) and was a core VC in control samples (present in 53% of control samples and 46% of IBS samples). The DESeq2 methodology reported large log2 fold changes between groups (mean and standard deviation of absolute log2FC of 20.6 ± 9.8) but the VCs identified were often at a low prevalence in each group. A second feature-level analysis was performed that correlated VC abundance against the secondary axis of the VCs (Table S9) that were significantly associated with the IBS-to-control split after correction for multiple testing (Canonical Correlation analysis with Spearman methodology, Benjamini-Hochberg adjusted p-value <0.05). Of these, 114 showed decreased abundance in IBS. The findings from the two feature selection methodologies applied reflect the reduced abundance and loss of viral features in IBS as would be predicted by the overall decrease in gut virome alpha diversity in IBS compared with controls.
We used CRISPR protospacers to predict the bacterial hosts for the phages represented by the contigs in the ten VCs that were differentially abundant between IBS and control groups, and in the 116 VCs associated with the IBS to control split as defined above. CRISPR spacers prediction resulted in four phage contigs in one VC (VC_1832_0) with hits to spacers on two bacterial contigs and two phage contigs in another VC (VC_1982_0) with hits to two different bacterial contigs. VC_1832_0, which was unclassified at family level, had a significantly lower abundance in IBS, whereas VC_1982_0, classified as Podoviridae and a core VC in controls, had significantly higher abundance in IBS (Figure 4). Of the four contigs in VC_1832_0 contigs, three unclassified phage contigs had matches to one unclassified bacterium and one unclassified phage contig had a predicted Bacteroides host. In VC_1982_0, one contig had a predicted Lachnospiraceae host and the other had a hit to an unclassified bacterium. Both Lachnospiraceae and Bacteroides have been reported as being present at altered abundance in IBS.31,32 The remaining eight differentially abundant VCs did not yield any hits to spacers on bacterial contigs.
Figure 4.
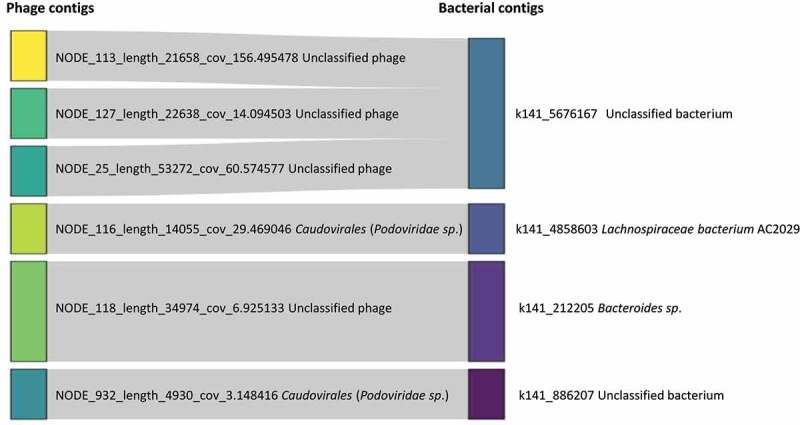
Sankey plot of putative phage contigs and their putative bacterial hosts based on CRISPR spacer sequences in VC_1832_0 and VC_1982_0, which were two of ten differentially abundant VCs between IBS and controls. To produce this plot, phage contigs in the 10 VCs were restricted to those that had a match based on CRISPR spacer sequences to a bacterial contig, resulting in 6 contigs, 4 from viral cluster, VC_1832_0 and 2 from VC_1982_0 (phage contigs beginning with NODE_116 and NODE_932). VC_1832_0 had significantly decreased abundance, and VC_1982_0 had significantly increased abundance in IBS compared with control samples, as reported by DeSeq2. For phage contigs, the putative species is indicated in brackets beside the phage family name
Analysis of bacterial hosts for the 116 VCs associated with the IBS to control split yielded 12 VCs containing contigs with hits to Prevotella (2 VCs), Faecalibacterium (4 VCs), Blautia and Ruminococcus (both associated with 1 VC), Firmicutes (3 VCs), Clostridia (I VC), and Aerobutyricum (1 VC) species in a subset of VCs with decreased abundance in IBS; however, we could not predict bacterial hosts for most contigs in these VCs (Table S9). The two VCs with hits to Prevotella were unclassified at family level but phage families Myoviridae and Siphoviridae were associated with the other bacterial hosts.
In summary, the reduced core virome, low count of gut virome species, and differential abundance observed in IBS signals a distinctive and limited virome in IBS compared with non-IBS controls.
Despite these differences, we found that the gut virome could not be used as predictive of IBS based on an XGBOOST machine learning pipeline, implemented as described previously for IBD17 with five fold cross-validations. Predictive models showed no predictive power when combined with gut virome cluster data with AUCs close to 0.5.
The gut virome does not co-vary with the bacteriome or BAM status
Bile acid malabsorption (BAM) causes diarrhea which is clinically difficult to distinguish from IBS-D.15 We recently showed that the fecal metabolome, but not the microbiome, could distinguish subjects with BAM from those with IBS.12 We first used co-inertia analysis to test for co-variance between the gut virome VCs and bacteriome for 326 bacterial species in 103 samples for which we had bacteriome information and found only limited global similarity between the gut bacteriome and virome (RV = 0.45; data not shown). Analysis using contigs instead of VCs also yielded a low RV value of 0.43 indicating that the co-variance between the VCs and the gut bacteriome of these samples was limited.
To determine if BAM status interacted with the gut virome, the BAM status of 28 patients with IBS (4 borderlines, 5 mild, 4 moderate, 12 normal, and 3 severe BAM cases) was compared with VC abundance in the fecal virome data. No significant relationship was identified (PMANOVA on Spearman distance, p-value = 0.16), although the low number of samples used was a limitation.
No evidence of conversion from lytic to lysogenic life cycle in IBS
A gut virome study of patients with IBD found evidence that the virulent phage core in healthy individuals is replaced with temperate phage in IBD patients.17 To determine if this is the case in IBS, we examined 943 VCs (31.9% of VCs) that we identified as putative temperate VCs (see Methods) for differential abundance in IBS and controls. We did not find any VCs which were differentially abundant (Wilcoxon signed-rank test, Benjamini-Hochberg adjusted p-value <0.05) indicating that there is no switch in core phage lifecycles in IBS.
Discussion
Our data show that there is a limited core virome in patients with IBS and controls. Those with IBS had reduced gut virome diversity and more individual-specific viromes compared with non-IBS controls, but there were no significant differences in diversity of the gut virome among the IBS clinical subtypes. The use of VCs helped attenuate the strong inter-individual variation observed when using strain level contigs as documented for IBD.17 It detected more sharing than seen at contig level and improved stratification between IBS and controls, especially in beta diversity analysis. Of classified VCs, families in the Caudovirales order (Myoviridae, Podoviridae, Siphoviridae) predominated in both IBS and controls, with a minimal core gut virome, as reported in other gut virome studies for diseased and healthy populations.17–22,24,26,27 The IBS gut virome is distinct from that of controls, but there is only one core VC (unclassified taxonomy) identified in IBS that was not also found as a core VC in controls.
To our knowledge, there is a single previous report on the gut virome in IBS25 which investigated the fecal virome of 25 patients with IBS and 17 controls using the subset of reads that could be classified to known viral families by alignment to the Viral RefSeq database. The larger study presented here examines both known and unknown viral sequences. We corroborated the reduction in phage alpha diversity in IBS and lack of robust differentiation among IBS subtypes reported by Ansari et al.25 However, we did not identify significant differences in families of eukaryotic viruses of the order Megavirales between patients with IBS and controls, as reported by Ansari et al.25 with the exception of two VCs assigned to the Mimiviridae family, one of which was more abundant in IBS and one with less abundance but we noted that the protein sequence hits used to classify this family had low support. All other members of the Megavirales order in our data had very low abundances and alignment scores and did not display significantly different abundances between IBS and controls. Due to the challenges of taxonomic assignment, we cannot rule out the presence of eukaryotic viruses in the gut virome as these viruses have been identified in the gut virome of both healthy subjects and those with gastroenteritis previously and have been cultured from human stools.33,34 However, they likely originate from amoebas in drinking water and a definitive role for these viruses in pathogenesis is lacking. Difficulties with taxonomic assignment of eukaryotic viruses in the gut virome have been previously reported;28 both viral genome assembly and taxonomic assignment of viruses are challenging due to the lack of a universal marker gene, high numbers of hypervariable and repeat regions,35,36 rapid mutation rates,36 horizontal transfer between viruses and bacteria and incomplete databases.35,37–40 Validated methods such as vConTACT241 can cluster contigs and assign taxonomy; in our data; however, many clusters remained unclassified with this approach highlighting the large amount of viral dark matter in the gut virome.
The core gut virome, dominated by lytic phage, seems to be replaced by temperate phage in IBD.17 To determine if a similar phenomenon is involved in IBS, we examined the differential abundance of all putative temperate VCs between IBS and controls, but we did not find any evidence of a difference and conclude that replacement of the predominant phage lifecycles is not a consistent observation in IBS.
Since residual bacterial sequences may be present in VLP datasets,42 we filtered our contigs to retain only those with viral signal, which we performed by combining the results of a comprehensive set of methodologies used in other gut virome studies.17,20,26,43 We observed low concordance across the approaches highlighting the utility of combining multiple methods. A potential limitation of our study is that novel viruses containing features that are substantially different from those cataloged in viral databases could be missed, and that bacterial contigs with loci homologous to viral features could have been erroneously included. We also attempted to identify putative bacterial hosts for phage contigs in all VCs by identifying sequence matches on phage contigs to CRISPR spacers on bacterial contigs. For the 10 VCs that were differentially abundant between IBS and controls, we could only identify hosts for contigs in two of the ten VCs, and similarly, we could only identify bacterial hosts for 12 VCs of the 116 VCs associated with the IBS to Control split. This could potentially be improved by using contigs from more samples as we only had bacterial contig information for 66% of the virome samples.
Although we did not find global co-variance of the bacteriome and virome data, this may be due to the inherent sparseness of the data which also presented challenges for feature selection and beta diversity analysis. Additionally, we did not examine the role of ssDNA or RNA viruses in this study, although RNA viruses in the gut would be expected to be mostly of dietary (i.e., plant) based origin.23,44
Use of a whole-virome-based approach in this study allowed us to identify changes in both the known and unknown components of the gut virome in IBS compared with controls. Further work is required to improve methods for taxonomic classification of viral sequences as well as for resolving phage genomes. Application of long read metagenomics to the analysis of the human gut virome using approaches such as those employed by Warwick-Dugdale and colleagues39 would improve completeness of recovered phage genomes, enable more accurate strain-level analysis, taxonomic classification, and aid exploration of virome population structure and host interactions to identify potential biomarkers and/or therapeutic targets for IBS.
Patients and methods
Study participant recruitment
Patients with IBS (Rome IV criteria) and control subjects aged 16–70 years and of the same ethnicity and geographic region were recruited to the study. Clinical subtyping of 55 patients with IBS45 included: 21 with constipation (IBS-C), 17 mixed type (IBS-M), and 17 with diarrhea (IBS-D). Exclusion criteria included the use of antibiotics within 6 weeks prior to study enrollment, other chronic illnesses including gastrointestinal diseases, severe psychiatric disease, abdominal surgery other than hernia repair or appendectomy. The inclusion/exclusion criteria for the control population were the same as for the IBS population with the exception of having to fulfill the Rome IV criteria for IBS. Gastrointestinal (GI) symptom history, psychological symptoms, diet, medical history, and medication use were collected on all participants (IBS and controls) and using the Bristol Stool Score, Hospital Anxiety and Depression Scale (HADS)46, and Food Frequency Questionnaire (FFQ).47 IBS-D and IBS-M patients reporting diarrhea as well as a subset of consenting control subjects were assessed for bile acid malabsorption by SeHCAT, a radiolabelled synthetic bile acid which is used to clinical diagnosis of BAM. Ethical approval for the study was granted by the Cork Research Ethics Committee before commencing the study and all participants provided written informed consent. Fresh fecal samples were collected from all participants and these were stored at −80°C within a few hours of the collection until processed for metagenomic sequencing of dsDNA virus-like particles (VLPs). Characteristics of the study population are presented in Table 1.
Extraction, library preparation, and shotgun sequencing of fecal VLP
We used the 1E protocol from Milani et al.48 adapted from Kleiner et al.42 to extract fecal VLP dsDNA. When compared with polyethylene glycol (PEG) and filtration methods, this protocol had the least non-viral DNA contamination and had the lowest species-specific bias. 0.5 g of stool was suspended in a sodium chloride magnesium sulfate and gelatin (SMG) buffer by vortexing and kept 1 hr on ice. The suspension was then centrifuged at 4°C for 5 min at 2500 xg. The supernatant was removed to a fresh tube and centrifuged again at 4°C for 15 min at 5000 xg. The supernatant was again transferred to a fresh tube and dithiothreitol (DTT) was added to a final concentration of 6.5 mM and incubated for 1 h at 37°C. The sample was then filtered through a 0.45 µm syringe filter and treated with 10 U DNase (Roche) for 1 hr at RT. The DNase was inactivated by heating samples for 10 min at 70°C. DNA was extracted using the Norgen Phage DNA isolation kit (Accuscience) manufacturer’s instructions. This kit isolates bacteriophage DNA from the sample using spin column chromatography without the need for phenol, chloroform, or cesium chloride. The Norgen spin column binds nucleic acids depending on ionic concentrations, thus preferentially purifying DNA while removing most RNA and proteins in the flowthrough. Briefly, lysis buffer and proteinase K were added to the sample and incubated 15 min at 55°C followed by 15 min at 65°C. Isopropanol was added before transferring the sample to the spin column, centrifuging at 6000 xg for 1 min. The spin column was washed with three rounds of wash buffer to remove any remaining impurities and the sample eluted from the column with 75 µL elution buffer. DNA concentration was quantified using Qubit High Sensitivity dsDNA kit (Life Technologies). Libraries were prepared for sequencing using the Nextera XT library prep kit (Illumina) following manufacturer’s instructions. Briefly, 1 ng of DNA was enzymatically cut, and short sequences “tagged” onto resulting fragments. Subsequently, PCR adapter and index sequences were added using the short tag sequences. Samples were run on a High Sensitivity DNA Bioanalyzer 2100 chip (Agilent) to determine the average fragment size and pool in equimolar concentrations. Tagged libraries were sent for sequencing to the commercial supplier (GATC Biotech AG, Konstanz, Germany) on a flow cell of a HiSeq 2500 sequencer, producing an average of 7,295,373 ± 1,643,253 250 bp paired-end sequences per sample.
Sequence data pre-processing
Read processing
Reads were checked using FastQC v0.11.8 and multiQC v1.7.49 Adapters were removed using Skewer v0.2.2 with adapter string ‘CTGTCTCTTATACACATCT’, minimum read length of 60 and maximum read length of 170. Low complexity reads were then removed using BBMap v38.22 BBDuk with parameters entropy = 0.5, entropywindow = 50 and entropyk = 5 to filter out reads with an average entropy less than 0.5. Reads that mapped to the human genome (ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/GCA_000001405.15_GRCh38_no_alt_analysis_set.fna.gz) were removed using BBSplit (BBMap v38.22) (https://sourceforge.net/projects/bbmap/) with default parameters resulting in an average of 6,285,899 + 1,480,880 paired-end reads with median read length of 157. The level of bacterial contamination was quantified by calculating the percentage of quality filtered reads mapped to the silva-bac-16s-id90 supplied by SortMeRNA (derived from SILVA SSU Ref NR v.119 database) using SortMeRNA version 2.1b.50
Assembly and dereplication
Quality filtered reads were assembled into contigs for each sample using metaSPAdes v3.13.051 with default k-mer sizes of 21,33,55,77,99. Contigs less than 1 kb were removed and contigs from all samples were pooled and dereplicated using BBMap v38.22 dedupe. sh with minimum percent identity of 95% keeping the longest contigs where two or more contigs had above the minimum identity threshold. This resulted in 327,267 contigs from the 106 samples.
Mapping and Abundance calculation
Reads that passed QC filtering were mapped to the dereplicated contigs using Bowtie252 v2.3.4.3 in end-to-end mapping mode. Sambamba v0.6.653 view, sort, and index functions were used to convert the Sequence Alignment/Map (SAM) file to Binary Sequence Alignment/Map (BAM), sort the BAM file, and create the BAM index file in that order. Sambamba v0.6.6 depth with threshold set to 1 (-T parameter) was used to quantify the percentage of bases in each contig that had at least 1X coverage. The abundance of each contig was calculated as the number of reads mapped to the contig (read count column in sambamba depth results). Only counts for contigs passing the step to select viral contigs were retained.
Selection of putative viral contigs
Open reading frames (ORFs) and their corresponding protein sequences were predicted for the dereplicated contigs using Prodigal v2.6.354 in metagenomic mode. HMMER hmmsearch v3.2.1 was used to search the predicted protein sequences against HMM databases of prokaryotic viral orthologous groups (pVOGs), virus orthologous groups (VOGDB) and Integrated Microbial Genomes/Virus (IMGVR) sequences with hits retained if they had a minimum full sequence E-value of 1e-5. A database of crAssphage protein sequences was compiled by downloading 22,240 protein sequences from Guerin et al.55 and 2,684 protein sequences from Yutin et al.56 producing 24,924 protein sequences. Predicted protein products from the dereplicated contigs were aligned to this database using Diamond v0.9.24 keeping only the top result for each protein that had a hit with a minimum E-value of 1e-10 and minimum identity of 80%.
To minimize any bacterial contamination, the dereplicated contigs were filtered to retain only those contigs that fulfilled one or more of the following criteria: a) be circular as predicted using ‘find_circular.py’ from VRCA57 with read length of 150 bp and minimum contig length of 1 kb b) categories 1 to 6 from VirSorter run in virome decontamination mode with the diamond aligner and default RefSeqdb (parameters: – virome, – diamond, – db 1 c) greater than 3 ORFs with HMM hits on a contig and greater than 2 HMM hits per 10 kb of that contig to the pVOG, VOG or IMG/VR database (hits had minimum E-value of 1e-5) d) contig predicted as viral or ‘unclassified’ using Kaiju v1.6.3 with the kaiju nr_euk (42 GB) database downloaded on the 5th February 2019 from http://kaiju.binf.ku.dk/server and default parameters or e) one or more protein sequences on a contig with a hit to a crAssphage protein sequence as described earlier. These steps resulted in 51,703 contigs. Contigs that had less than 75% of their bases at 1X coverage in a sample had their counts set to zero in that sample, resulting in 51,178 contigs retained for downstream analysis.
Identification of Viral Clusters (VCs)
752,654 proteins from 51,703 contigs were clustered using vConTACT2 v0.9.1141 with the PC and VC inflation index set to 1.5 and all other parameters at default. 373,129 proteins from 9,208 genomes in Viral RefSeq release 97 were downloaded and added to the pool of protein sequences input to vConTACT2 in order to facilitate taxonomic placement of the viral clusters.
Where vConTACT2 refined discordant clusters into sub-clusters, the sub-clusters were considered as independent clusters. All clusters were filtered to keep those with Benjamini-Hochberg adjusted p-values less than 0.05 and that had at least one contig from our dataset. The abundance of each cluster was calculated by summing the count associated with each contig in the cluster for each sample. This produced 2,962 clusters containing at least one contig from our data (2,233/2,962 clusters had ≥2 members, 729 clusters with only one member) but only 33 of these clusters contained a Viral RefSeq genome. Five clusters were removed as they were composed entirely of contigs that did not pass the breadth of coverage filter, resulting in 2,957 contigs encompassing a total of 11,630 contigs. The maximum number of members in any cluster was 62 (mean of four and median of two contigs in clusters).
Bacterial host prediction based on CRISPR protospacers
Bacterial metagenome contigs which were co-assembled from metagenomes from 69 fecal samples from our IBS patient cohort12 using Megahit v1.1.258 with minimum contig length set to 200, were used to search for CRISPR spacers.
CRISPR spacers were predicted on contigs ≥1kb (n = 871,531) using PILER-CR, retaining only results with E-value <1e-05, resulting in 10,502 spacer sequences from 644 contigs. The spacer sequences were aligned to the viral contigs (n = 51,703) using blastn from Blast++59 version 2.9.0+ with mode ‘blastn-short’ and E-value <1e-05. 2,993 viral contigs had hits to 2,076 spacer sequences from 644 bacterial contigs. The 644 bacterial contigs were taxonomically classified using CAT v5.0.360 with default parameters (containing diamond version 0.9.21 and using a CAT nr database downloaded from tbb.bio.uu.nl/bastiaan/CAT_prepare/CAT_prepare_20190719.tar.gz). 459 contigs were classified (71.27%). At the superkingdom level, 451 were classified as Bacteria, two as Archaea, one as Viral (k141_4221899, Inoviridae family), and five remained unclassified.
Statistical Analysis
Feature selection was performed using DESeq2 applied to the count data for the viral clusters which were prefiltered to keep clusters present in at least 10% of samples. Correction for multiple testing was applied using the Benjamini-Hochberg (BH) methodology. Significance across IBS/control stratification and IBS and BAM subgroups were performed using the Permutational MANOVA methodology from the R vegan library. Co-inertia analyses were carried out using co-inertia function from the R library ade4 to compare Metagenomic species, as defined by Metaphlan261 with Archaea and Eukaryota removed (n = 326 species), for 103 samples, with the viral contig counts for the same samples. Alpha diversity (Shannon diversity) and richness (observed counts i.e., the number of phages identified in each subject) were calculated using raw counts for contigs and VCs using the ‘estimate_richness’ function in the phyloseq v1.28.0 R package. Raw counts were scaled by taking the log10 of the count after adding a pseudocount of one in both datasets. PCA was performed on each matrix using the ‘dudi.pca’ command, coinertia was carried out using the ‘coinertia’ command and the significance of the RV value was assessed by calculating a simulated p-value using the ‘randtest’ function with 999 permutations.
Taxonomic assignment of contigs
The viral section of the UniprotKB TrEMBL database was downloaded on the 24th April 2019 from ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/taxonomic_divisions/uniprot_trembl_viruses.dat.gz. Annotation information was downloaded from https://www.uniprot.org/uniprot/?query=taxonomy%3A%22Viruses+%5B10239%5D%22+reviewed%3Ano using search term ‘taxonomy:”Viruses [10239]” AND reviewed: no’ and adding ‘Organism ID’ as a column to the table, which resulted in 3,878,757 entries. The ete3 toolkit was used in a python script to extract the official taxonomic ranks associated with each organism ID (superkingdom, kingdom, phylum, class, order, family, genus, species) and output a file with each accession number and taxonomic information. A separate script was used to extract all protein sequences from the uniprot_trembl_viruses.dat file and place them into a fasta file with accession numbers as sequence headers. Diamond v0.9.24 blastp62 was used to align the protein sequences associated with viral contigs to the protein sequences in the fasta file with parameters – max-target-seqs 1 – evalue 1e-05 and – sensitive. The diamond results were parsed to assign taxonomy to the query proteins using the file of accession numbers and taxonomic information.
Taxonomy was assigned to contigs using a voting based lowest common ancestor approach. For contigs with at least one ORF per 10 kb of length, if all protein sequences with hits to the UniprotKB TrEMBL database on that contig agreed at the species level then the contig was assigned that taxonomy. If there was a disagreement at species level but the genus level was the same, then the most prevalent species was assigned. If there was a tie in the number of disagreements at species level e.g. two protein sequences were assigned to one species and the other two to another, then only the genus level classification was kept. The same procedure was followed to assign contigs back to order level in the case of disagreements at downstream levels. Note that species-levelassignments may not be reliable as some families have very few species genomes sequenced, potentially leading to spurious species assignments.
Taxonomic assignment of viral clusters
As only 33/2,957 clusters contained a Viral RefSeq genome, taxonomy was assigned to viral clusters at family level using the family level taxonomy of the contigs that formed the clusters.
Temperate phage prediction and differential abundance
Viral contigs were assigned as belonging to putative temperate phages if they had a gene with homology to one of 28 integrase or site-specific recombinase pVOGs (E-value <1E-05, pVOGs from the list used by Clooney et al.17). Viral clusters were considered as potentially temperate if they contained at least one putative temperate contig. Differential abundance of temperate clusters was calculated using the Wilcoxon test on temperate cluster counts for IBS and Controls. Clusters with Benjamini Hochberg adjusted p-value <0.05 were considered differentially abundant.
Terminase annotation
Terminases lack bacterial homologs and so we annotated contigs that contained genes with homology to a terminase gene from a list of 72 pVOGs produced by filtering for the word ‘terminase’ in the ‘Protein Annotations’ column of http://dmk-brain.ecn.uiowa.edu/pVOGs/VOGbigtable.html.
Supplementary Material
Acknowledgments
We are grateful to the 4D pharma Cork clinical team, Caroline O’Leary, Marian O’Meara and Sinead Kelly who recruited the subjects to the study and collected the biological samples and clinical metadata. We would also like to thank gastroenterologist Dr Syed Akbar Zulquernain and Dr Elizabeth Kenny in Cork University Hospital who assisted with IBS patient recruitment. We acknowledge the input of 4D pharma Cork lab staff, Luke O’Brien, Fiona Grady, Marie McBrien and Siobhan O’Neill for sample processing for analysis. We also thank Michael Moore, Fintan Bradley, Tom Carty, Fionnuala McDonnell, Lean O’Donoghue, Pauline Beecher, Roisin Wilson, and Shane Hayes at Cork University Hospital who performed the SeHCAT assay.
Disclosure statement
IBJ, FS and PWOT are founders of 4D pharma Cork Limited which is a wholly-owned subsidiary of 4D pharma plc., a company developing live biotherapeutics for diseases including Blautix which is in clinical trials for IBS. The remaining authors disclose no conflicts.
Author contributions
IBJ, EOH, FS and PWOT conceived the study idea and design. SC, IBJ, EOH, FS and PWOT drafted and reviewed the manuscript. SC, IBJ and AD performed the bioinformatics analysis.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Lovell RM, Ford AC.. Global prevalence of and risk factors for irritable bowel syndrome: a meta-analysis. Clin Gastroenterol Hepatol. 2012; doi: 10.1016/j.cgh.2012.02.029. [DOI] [PubMed] [Google Scholar]
- 2.Lacy BE, Mearin F, Chang L, Chey WD, Lembo AJ, Simren M, Spiller R. Bowel disorders. Gastroenterology 2016; doi: 10.1053/j.gastro.2016.02.031. [DOI] [PubMed] [Google Scholar]
- 3.Beyder A, Mazzone A, Strege PR, Tester DJ, Saito YA, Bernard CE, Enders FT, Ek WE, Schmidt PT, Dlugosz A, et al. Loss-of-function of the voltage-gated sodium channel NaV1.5 (Channelopathies) in patients with irritable bowel syndrome. Gastroenterology. 2014; doi: 10.1053/j.gastro.2014.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ek WE, Reznichenko A, Ripke S, Niesler B, Zucchelli M, Rivera NV, Schmidt PT, Pedersen NL, Magnusson P, Talley NJ, et al. Exploring the genetics of irritable bowel syndrome: a GWA study in the general population and replication in multinational case-control cohorts. Gut 2015; doi: 10.1136/gutjnl-2014-307997. [DOI] [PubMed] [Google Scholar]
- 5.Qin HY, Cheng CW, Tang XD, Bian ZX. Impact of psychological stress on irritable bowel syndrome. In World J Gastroenterol. 2014; doi: 10.3748/wjg.v20.i39.14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chey WD, Kurlander J, Eswaran S. Irritable bowel syndrome: a clinical review. In JAMA - J Am Medi Assoc. 2015; doi: 10.1001/jama.2015.0954. [DOI] [PubMed] [Google Scholar]
- 7.El-Salhy M, Gundersen D. Diet in irritable bowel syndrome. Nutr J. 2015; doi: 10.1186/s12937-015-0022-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins SM. A role for the gut microbiota in IBS. Nat Rev Gastroenterol Hepatol. 2014;11:497–15. doi: 10.1038/nrgastro.2014.40. [DOI] [PubMed] [Google Scholar]
- 9.Thabane M, Marshall JK. Post-infectious irritable bowel syndrome. World J Gastroenterol. 2009;15(29):3591–3596. doi: 10.3748/wjg.15.3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carroll IM, Ringel-Kulka T, Siddle JP, Ringel Y. Alterations in composition and diversity of the intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motility. 2012; doi: 10.1111/j.1365-2982.2012.01891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casén C, Vebø HC, Sekelja M, Hegge FT, Karlsson MK, Ciemniejewska E, Dzankovic S, Frøyland C, Nestestog R, Engstrand L, et al. Deviations in human gut microbiota: a novel diagnostic test for determining dysbiosis in patients with IBS or IBD. Aliment Pharmacol Ther. 2015; doi: 10.1111/apt.13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeffery IB, Das A, O’Herlihy E, Coughlan S, Cisek K, Moore M, Bradley F, Carty T, Pradhan M, Dwibedi C, et al. Differences in fecal microbiomes and metabolomes of people with vs without irritable bowel syndrome and bile acid malabsorption. Gastroenterology 2020. doi: 10.1053/j.gastro.2019.11.301. [DOI] [PubMed] [Google Scholar]
- 13.Jeffery IB, O’Toole PW, Öhman L, Claesson MJ, Deane J, Quigley EMM, Simrén M. An irritable bowel syndrome subtype defined by species-specific alterations in faecal microbiota. Gut 2012; doi: 10.1136/gutjnl-2011-301501. [DOI] [PubMed] [Google Scholar]
- 14.Rajilić-Stojanović M, Biagi E, Heilig HGHJ, Kajander K, Kekkonen RA, Tims S, De Vos WM. Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome. Gastroenterology 2011; doi: 10.1053/j.gastro.2011.07.043. [DOI] [PubMed] [Google Scholar]
- 15.Slattery SA, Niaz O, Aziz Q, Ford AC, Farmer AD. Systematic review with meta-analysis: the prevalence of bile acid malabsorption in the irritable bowel syndrome with diarrhoea. In Alimentary Pharmacol Therapeutics. 2015; doi: 10.1111/apt.13227. [DOI] [PubMed] [Google Scholar]
- 16.Shkoporov AN, Hill C. Bacteriophages of the human gut: the “known unknown” of the microbiome. Cell Host Microbe. 2019; doi: 10.1016/j.chom.2019.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Clooney AG, Sutton TDS, Shkoporov AN, Holohan RK, Daly KM, O’Regan O, Ryan FJ, Draper LA, Plevy SE, Ross RP, et al. Whole-virome analysis sheds light on viral dark matter in inflammatory bowel disease. Cell Host Microbe. 2019; doi: 10.1016/j.chom.2019.10.009. [DOI] [PubMed] [Google Scholar]
- 18.Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD, Lewis JD, Bushman FD. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res. 2011; doi: 10.1101/gr.122705.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, Rohwer F, Gordon JI. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010; doi: 10.1038/nature09199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shkoporov AN, Clooney AG, Sutton TDS, Ryan FJ, Daly KM, Nolan JA, McDonnell SA, Khokhlova EV, Draper LA, Forde A, et al. The human gut virome is highly diverse, stable, and individual specific. Cell Host Microbe. 2019; doi: 10.1016/j.chom.2019.09.009. [DOI] [PubMed] [Google Scholar]
- 21.Manrique P, Bolduc B, Walk ST, Van Oost J, Der, De Vos WM, Young MJ. Healthy human gut phageome. Proc Natl Acad Sci U S A. 2016; doi: 10.1073/pnas.1601060113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, Kambal A, Monaco CL, Zhao G, Fleshner P, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015; doi: 10.1016/j.cell.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shkoporov AN, Ryan FJ, Draper LA, Forde A, Stockdale SR, Daly KM, McDonnell SA, Nolan JA, Sutton TDS, Dalmasso M, et al. Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome 2018; doi: 10.1186/s40168-018-0446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zuo T, Lu XJ, Zhang Y, Cheung CP, Lam S, Zhang F, Tang W, Ching JYL, Zhao R, Chan PKS, et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019; doi: 10.1136/gutjnl-2018-318131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ansari MH, Ebrahimi M, Fattahi MR, Gardner MG, Safarpour AR, Faghihi MA, Lankarani KB. Viral metagenomic analysis of fecal samples reveals an enteric virome signature in irritable bowel syndrome. BMC Microbiol. 2020;20(1):123. doi: 10.1186/s12866-020-01817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gregory AC, Zablocki O, Howell A, Bolduc B, Sullivan MB. The human gut virome database. BioRxiv 2019; doi: 10.1101/655910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCann A, Ryan FJ, Stockdale SR, Dalmasso M, Blake T, Anthony Ryan C, Stanton C, Mills S, Ross PR, Hill C. Viromes of one year old infants reveal the impact of birth mode on microbiome diversity. PeerJ 2018; doi: 10.7717/peerj.4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sutton TDS, Clooney AG, Hill C. Giant oversights in the human gut virome. In Gut. 2019; doi: 10.1136/gutjnl-2019-319067. [DOI] [PubMed] [Google Scholar]
- 29.Colson P, De Lamballerie X, Yutin N, Asgari S, Bigot Y, Bideshi DK, Cheng XW, Federici BA, Van Etten JL, Koonin EV, et al. “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch Virol. 2013; doi: 10.1007/s00705-013-1768-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Legendre M, Bartoli J, Shmakova L, Jeudy S, Labadie K, Adrait A, Lescot M, Poirot O, Bertaux L, Bruley C, et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc Natl Acad Sci U S A. 2014; doi: 10.1073/pnas.1320670111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeffery IB, Quigley EMM, Öhman L, Simrén M, O’Toole PW. The microbiota link to irritable bowel syndrome an emerging story. Gut Microbes. 2012;3(6):572–576. doi: 10.4161/gmic.21772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pittayanon R, Lau JT, Yuan Y, Leontiadis GI, Tse F, Surette M, Moayyedi P. Gut microbiota in patients with irritable bowel syndrome—a systematic review. Gastroenterology. 2019;157(1):97–108. doi: 10.1053/j.gastro.2019.03.049. [DOI] [PubMed] [Google Scholar]
- 33.Colson P, Aherfi S, La Scola B. Evidence of giant viruses of amoebae in the human gut. Human Microbiome J. 2017. doi: 10.1016/j.humic.2017.11.001. [DOI] [Google Scholar]
- 34.Colson P, Fancello L, Gimenez G, Armougom F, Desnues C, Fournous G, Yoosuf N, Million M, La Scola B, Raoult D. Evidence of the megavirome in humans. J Clin Virology. 2013; doi: 10.1016/j.jcv.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 35.Minot S, Grunberg S, Wu GD, Lewis JD, Bushman FD. Hypervariable loci in the human gut virome. Proc Natl Acad Sci U S A. 2012; doi: 10.1073/pnas.1119061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD, Bushman FD. Rapid evolution of the human gut virome. Proc Natl Acad Sci U S A. 2013; doi: 10.1073/pnas.1300833110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roux S, Emerson JB, Eloe-Fadrosh EA, Sullivan MB. Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 2017; doi: 10.7717/peerj.3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sutton TDS, Clooney AG, Ryan FJ, Ross RP, Hill C. Choice of assembly software has a critical impact on virome characterisation. Microbiome 2019; doi: 10.1186/s40168-019-0626-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warwick-Dugdale J, Solonenko N, Moore K, Chittick L, Gregory AC, Allen MJ, Sullivan MB, Temperton B. Long-read viral metagenomics captures abundant and microdiverse viral populations and their niche-defining genomic islands. PeerJ 2019; doi: 10.7717/peerj.6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zuo T, Ng SC. Authors response: giant oversights in the human gut virome. In Gut. 2020; doi: 10.1136/gutjnl-2019-319357. [DOI] [PubMed] [Google Scholar]
- 41.Bin Jang H, Bolduc B, Zablocki O, Kuhn JH, Roux S, Adriaenssens EM, Brister JR, Kropinski AM, Krupovic M, Lavigne R, et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat Biotechnol. 2019; doi: 10.1038/s41587-019-0100-8. [DOI] [PubMed] [Google Scholar]
- 42.Kleiner M, Hooper LV, Duerkop BA. Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. BMC Genomics. 2015; doi: 10.1186/s12864-014-1207-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Draper LA, Ryan FJ, Smith MK, Jalanka J, Mattila E, Arkkila PA, Ross RP, Satokari R, Hill C. Long-term colonisation with donor bacteriophages following successful faecal microbial transplantation. Microbiome 2018; doi: 10.1186/s40168-018-0598-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang T, Breitbart M, Lee WH, Run JQ, Wei CL, Soh SWL, Hibberd ML, Liu ET, Rohwer F, Ruan Y. RNA viral community in human feces: prevalence of plant pathogenic viruses. PLoS Biol. 2006; doi: 10.1371/journal.pbio.0040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Longstreth GF, Thompson WG, Chey WD, Houghton LA, Mearin F, Spiller RC. Functional bowel disorders. Gastroenterology 2006; doi: 10.1053/j.gastro.2005.11.061. [DOI] [PubMed] [Google Scholar]
- 46.Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983; doi: 10.1111/j.1600-0447.1983.tb09716.x. [DOI] [PubMed] [Google Scholar]
- 47.Power SE, Jeffery IB, Ross RP, Stanton C, O’Toole PW, O’Connor EM, Fitzgerald GF. Food and Nutrient Intake of Irish Community-dwelling Elderly Subjects: Who Is at Nutritional Risk? J Nutr Health Aging. 2014; doi: 10.1007/s12603-014-0449-9 [DOI] [PubMed] [Google Scholar]
- 48.Milani C, Casey E, Lugli GA, Moore R, Kaczorowska J, Feehily C, Mangifesta M, Mancabelli L, Duranti S, Turroni F, et al. Tracing mother-infant transmission of bacteriophages by means of a novel analytical tool for shotgun metagenomic datasets: mETAnnotatorX. Microbiome 2018; doi: 10.1186/s40168-018-0527-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016; doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kopylova E, Noé L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012; doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 51.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. MetaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017; doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 2015; doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010; doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guerin E, Shkoporov A, Stockdale SR, Clooney AG, Ryan FJ, Sutton TDS, Draper LA, Gonzalez-Tortuero E, Ross RP, Hill C. Biology and Taxonomy Of Crass-Like Bacteriophages, The Most Abundant Virus In The Human Gut. Cell Host Microbe. 2018; doi: 10.1016/j.chom.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 56.Yutin N, Makarova KS, Gussow AB, Krupovic M, Segall A, Edwards RA, Koonin EV. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat Microbiol. 2018; doi: 10.1038/s41564-017-0053-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Crits-Christoph A, Gelsinger DR, Ma B, Wierzchos J, Ravel J, Davila A, Casero MC, DiRuggiero J. Functional interactions of archaea, bacteria and viruses in a hypersaline endolithic community. Environ Microbiol. 2016; doi: 10.1111/1462-2920.13259. [DOI] [PubMed] [Google Scholar]
- 58.Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015; doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 59.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinform. 2009; doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Von Meijenfeldt FAB, Arkhipova K, Cambuy DD, Coutinho FH, Dutilh BE. Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol. 2019; doi: 10.1186/s13059-019-1817-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 2012;9(8):811–814. Published 2012 Jun 10 doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. In Nat Methods. 2014; doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.