

A striking feature of severe forms of coronavirus disease 2019 (COVID-19), the current pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is severe endothelial injury with micro- and macrothrombotic disease in the lung and other organs, including the heart. This has led to speculation that viral infection may damage the endothelium through 2 mechanisms: indirectly, via neighborhood effects, circulating mediators, and immune mechanisms or directly by viral infection of endothelial cells (ECs).
To support the hypothesis of direct viral damage of ECs via virus-induced infection, the cells should express the main receptor for SARS-CoV-2, ACE2 (angiotensin-converting enzyme 2), a metalloprotease component of the renin–angiotensin hormone system and a critical regulator of cardiovascular homeostasis.1 Indeed, several recent review articles propose that SARS-CoV-2 binding to ACE2 on ECs is the mechanism through which the virus may cause direct endothelial damage and endothelialitis.1 However, expression of ACE2 in ECs has not been convincingly demonstrated to support this assumption, nor has there been sufficient evidence to support a direct infection of ECs by SARS-CoV-2.
To address the questions of ACE2 expression in human ECs and of the ability of SARS-CoV-2 to infect the endothelium, we interrogated transcriptomic and epigenomic data on human ECs and studied the interaction and replication of SARS-Cov-2 and its viral proteins with ECs in vitro. The data, analytic methods, and study materials will be maintained by the corresponding author and made available to other researchers on reasonable request.
Analysis of RNA sequencing was carried out on ENCODE data from ECs from arterial, venous, and microvascular beds, in comparison with epithelial cells from respiratory, gastrointestinal, and skin sources. Very low or no basal ACE2 expression was found in ECs compared with epithelial cells (Figure A and B). Moreover, in vitro exposure of ECs to inflammatory cytokines reported as elevated in the plasma of patients with severe COVID-19 failed to upregulate ACE2 expression (Figure C).
Figure.
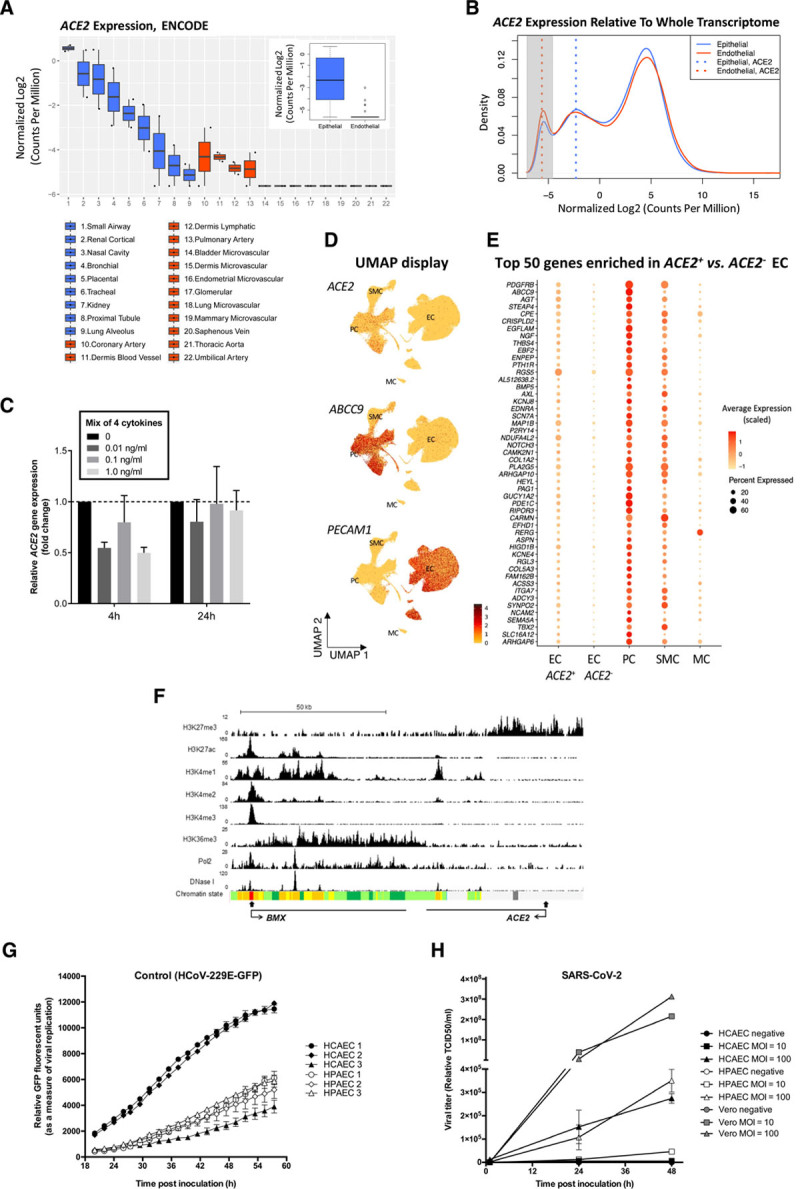
Analysis of ACE2 expression in human ECs and of coronavirus replication in primary human ECs. A and B, Comparison of ACE2 expression in human primary epithelial cells and ECs using total RNA sequencing data from the ENCODE database shows low or absent expression in ECs. A, The difference of ACE2 expression in epithelial cells and ECs is shown in boxplots with individual, as well as grouped, samples (inner boxplot). Each dot represents a single sample (n=2 per cell type). B, Transcriptome profiles of epithelial cells and ECs are shown in a density plot, using the median of all samples per group (n=19 360 genes). ACE2 expression in each group is marked with a dotted line: ACE2 expression in ECs (red) overlaps with the peak for nonexpressed transcripts (highlighted in grey), while ACE2 expression in epithelial cells (blue) is to the right, indicating detectable expression. Median ACE2 expression in endothelial cells equals -5.6 in log2 CPM, which corresponds with 0 raw read counts, signifying undetectable ACE2 expression in the majority of ECs. Expression values in all plots are represented as log2-transformed CPM, normalized by trimmed mean of M-value (blue: epithelial, red: endothelial). C, ACE2 expression is not regulated by inflammatory cytokines in HUVECs. qPCR analysis of ACE2 mRNA expression in HUVECs treated with a mix of 4 cytokines/chemokines (TNF-α, IL1-β, IL8, and IL6/IL6R chimeric protein) for 4 hours or 24 hours at 0, 0.01, 0.1, or 1.0 ng/mL. Data are normalized to GAPDH and presented as mean±SEM of 3 independent experiments. D and E, Very low-level, rare, and likely contaminating ACE2 transcripts are seen in ECs. D, ACE2 transcript reads are detected preferentially in PCs. UMAP landscapes of publicly available human heart data sets2 include 100 579 ECs, 77 856 PCs, 16 242 SMCs, and 718 MCs (https://www.heartcellatlas.org/). ACE2 transcript reads are detected preferentially in the PC cluster (enriching for ABCC9) and are rare in the EC cluster (enriching for PECAM1). E, PC transcripts are enriched together with ACE2 in 0.47% of ECs. Dot plot displaying the abundance of top-50 transcripts enriched ACE2+ versus ACE2- ECs, across cell types indicated in D. (The Wilcoxon rank-sum tests with Bonferroni-corrected P values are < 1E-60 for each). F, Epigenetic profiling indicates that the ACE2 gene is inactive in ECs. ChIP-seq binding profiles in HUVECs for histone modifications, RNA Pol2 enrichment, and deoxyribonuclease I hypersensitivity. The x axis represents the genomic position, the transcription start sites are indicated by closed arrow, and the direction of transcription is indicated by open arrows; the y axis shows ChIP-seq signal in reads per million per base pair (rpm/bp). The bottom row represents the chromatin state segmentation. Color key: active promoter, red; enhancers, yellow; transcriptional elongation, green; repressed, grey. G and H, Coronavirus replication in primary human cardiac and pulmonary ECs shows limited replication of SARS-CoV-2. G, Viral replication curves in human pulmonary (HPAEC) and cardiac (HCAEC) endothelial cells following infection with control HCoV-229E GFP (green fluorescent protein) reporter virus (MOI=0.6). Virus replication was measured via GFP fluorescence every 2 hours from 20 to 58 hours postinoculation. Mean±SEM of 3 technical replicates are shown at each time point for each biological replicate. H, Viral growth curves in HPAEC (n=3), HCAEC (n=3), and nonendothelial Vero cells (n=1) after infection with SARS-CoV-2 at MOI=10 or 100. Supernatant were collected at 1, 24, and 48 hours postinfection and virus copy number quantified by RT-qPCR detection of the SARS-CoV-2 N3 gene. ChIP-seq indicates chromatin immunoprecipitation sequencing; CPM, counts per million; EC, endothelial cell; HUVEC, human umbilical vein endothelial cell; IL, interleukin; MC, mesothelial cell; MOI, multiplicity of infection; PC, pericyte; qPCR, quantitative polymerase chain reaction; RT, real time; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; SMC, smooth muscle cell; TCID, tissue culture infectivity dose; TNF, tumor necrosis factor; and UMAP, Uniform Manifold Approximation and Projection.
Publicly available single-cell RNA sequencing of human organ donor hearts2 showed that, while ACE2 sequence reads are abundant in pericytes, they are rare in ECs (Figure D). Of 100,579 ECs, only 468 (0.47%) were ACE2+, and in the majority (424), only a single ACE2 transcript was detected. This could reflect true low and rare endothelial ACE2 expression but also contamination from adherent pericyte fragments, a common confounder in vascular single-cell RNA sequencing data.3 If such fragments contributed to the ACE2 transcripts observed in certain ECs, we would expect to detect other pericyte transcripts in the same cells. Among the top-50 gene transcripts enriched in ACE2+ versus ACE2− ECs, we noticed several known pericyte markers, including PDGFRB, ABCC9, KCNJ8, and RGS5 (Figure E). Comparison of transcript abundance across the 3 major vascular and mesothelial cells showed that the top-50 gene transcripts were expressed at the highest levels in pericytes (Figure E). This suggests that the rare occurrence of ACE2 transcripts in human heart ECs is likely caused by pericyte contamination. Similar conclusions have been reached previously in mouse tissues.3
Analysis of the chromatin landscape at the ACE2 gene locus in human umbilical vein ECs using data from ENCODE further supports this concept. The histone modification mark H3K27me3, which indicates repressed chromatin, was enriched at the ACE2 transcription start site; conversely, promoter, enhancer, and gene body activation marks (H3K27ac, H3K4me1, H3K4me2, H3K4me3, and H3K36me3), RNA polymerase-II and deoxyribonuclease I hypersensitivity were absent or low, suggesting that ACE2 is inactive in ECs. In marked contrast, the adjacent gene BMX, an endothelial-restricted nonreceptor tyrosine kinase, displays an epigenetic profile consistent with active endothelial expression (Figure F). Thus, transcriptomic and epigenomic data indicate that ACE2 is not expressed in human ECs.
Other cell surface molecules have been suggested as possible receptors for the virus, but their role in supporting SARS-CoV-2 cell infection remains to be demonstrated. We therefore tested directly whether ECs could be capable of supporting coronavirus replication in vitro. Productive levels of replication in primary human cardiac and pulmonary ECs were observed for the human coronavirus 229E GFP (green fluorescent protein) reporter virus,4 which uses CD13 as its receptor, demonstrating directly that human ECs can support coronavirus replication in principle (Figure G). However, when cells were exposed to SARS-CoV-2, replication levels were extremely low for ECs, even after exposure to very high concentrations of virus compared with more permissive VeroE6 cells (Figure H). The observed low levels of SARS-CoV-2 replication in ECs are likely explained by viral entry via a non-ACE2–dependent route, attributable to exposure to extremely high concentrations of virus in these experiments (multiplicity of infection, 10 and 100).
These data indicate that direct endothelial infection by SARS-CoV-2 is not likely to occur. The endothelial damage reported in severely ill patients with COVID-19 is more likely secondary to infection of neighboring cells and/or other mechanisms, including immune cells, platelets and complement activation, and circulating proinflammatory cytokines. Our hypothesis is corroborated by recent evidence that plasma from critically ill and convalescent patients with COVID-19 causes EC cytotoxicity.5 These findings have implications for therapeutic approaches to tackle vascular damage in severe COVID-19 disease.
Acknowledgments
For the ENCODE database, please see www.encodeproject.org.
Sources of Funding
This research was supported by grants from the Imperial College COVID response fund (to Dr Randi), the Imperial College British Heart Foundation (BHF) Research Excellence Award (RE/18/4/34215) (to Drs Randi, Birdsey, and Noseda); BHF Program grant funding (RG/17/4/32662) (to Drs Randi and Birdsey); the National Institute for Health Research Imperial Biomedical Research Center (to Drs Randi and Birdsey); BHF/German Center for Cardiovascular Research grant (SP/19/1/34461) and Chan Zuckerberg Initiative (grant 2019–202666) (to Dr Noseda); the Swedish Science Council, The Swedish Cancer Society, the Knut and Alice Wallenberg, and Erling-Persson Family Foundation (to Drs Betsholtz and Lendahl); the University of Edinburgh BHF Research Excellence Award; BHF Chair of Translational Cardiovascular Sciences and H2020 European Union grant COVIRNA (agreement DLV-101016072); IMI-2 CARE (to Drs Tait-Burkard and Haas); Biotechnology and Biological Sciences Research Council Institute Strategic Program grant funding to The Roslin Institute (BBS/E/D/20241866, BBS/E/D/20002172, and BBS/E/D/20002174) (to Dr Tait-Burkard); and BHF Center for Vascular Regeneration (RM/17/3/33381) (to Drs Baker and Randi).
Disclosures
None.
Footnotes
I.R. McCracken and Drs Saginc, He, and Huseynov share first authorship.
A. Daniels and Dr Fletcher contributed equally.
Drs Birdsey, Betsholtz, Noseda, Baker, and Randi share senior authorship.
Contributor Information
Ian R. McCracken, Email: ian.mccracken@ed.ac.uk.
Gaye Saginc, Email: g.saginc-giannoustas@imperial.ac.uk.
Liqun He, Email: liqun.he@ki.se.
Alik Huseynov, Email: a.huseynov@imperial.ac.uk.
Alison Daniels, Email: A.Daniels@sms.ed.ac.uk.
Sarah Fletcher, Email: Sarah.Fletcher@roslin.ed.ac.uk.
Claire Peghaire, Email: c.peghaire@imperial.ac.uk.
Viktoria Kalna, Email: v.kalna15@imperial.ac.uk.
Maarja Andaloussi-Mäe, Email: maarja.andaloussi_mae@igp.uu.se.
Lars Muhl, Email: Lars.Muhl@ki.se.
Nicky M. Craig, Email: Nicky.craig@roslin.ed.ac.uk.
Samantha J. Griffiths, Email: samantha.griffiths@ed.ac.uk.
Jürgen G. Haas, Email: juergen.haas@ed.ac.uk.
Christine Tait-Burkard, Email: Christine.Burkard@roslin.ed.ac.uk.
Urban Lendahl, Email: urban.lendahl@ki.se.
Graeme M. Birdsey, Email: g.birdsey@imperial.ac.uk.
Christer Betsholtz, Email: christer.betsholtz@igp.uu.se.
Michela Noseda, Email: m.noseda@imperial.ac.uk.
References
- 1.Evans PC, Rainger GE, Mason JC, Guzik TJ, Osto E, Stamataki Z, Neil D, Hoefer IE, Fragiadaki M, Waltenberger J, et al. Endothelial dysfunction in COVID-19: a position paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc Res. 2020;116:2177–2184. doi: 10.1093/cvr/cvaa230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Litviňuková M, Talavera-López C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, et al. Cells of the adult human heart. Nature. 2020;588:466–472. doi: 10.1038/s41586-020-2797-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He L, Mäe MA, Muhl L, Sun Y, Pietilä R, Nahar K, Liébanas EV, Fagerlund MJ, Oldner A, Liu J, et al. Pericyte-specific vascular expression of SARS-CoV-2 receptor ACE2: implications for microvascular inflammation and hypercoagulopathy in COVID-19. bioRxiv. 20202020. doi: 10.1101/2020.05.11.088500 [Google Scholar]
- 4.Cervantes-Barragan L, Zust R, Maier R, Sierro S, Janda J, Levy F, Speiser D, Romero P, Rohrlich PS, Ludewig B, et al. Dendritic cell-specific antigen delivery by coronavirus vaccine vectors induces long-lasting protective antiviral and antitumor immunity. mBio. 2010;14e00171–10. doi: 10.1128/mBio.00171-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rauch A, Dupont A, Goutay J, Caplan M, Staessens S, Moussa M, Jeanpierre E, Corseaux D, Lefevre G, Lassalle F, et al. ; Lille COVID Research Network (LICORNE); Members of the LICORNE Scientific Committee. Endotheliopathy is induced by plasma from critically Ill patients and associated with organ failure in severe COVID-19. Circulation. 2020;142:1881–1884. doi: 10.1161/CIRCULATIONAHA.120.050907 [DOI] [PMC free article] [PubMed] [Google Scholar]