

Abstract
Tumor microenvironment and alternative polyadenylation (APA) have drawn more attention in cancer research. However, their roles in grade II and III gliomas, termed as lower-grade glioma (LGG) in this study, remain to be fully elucidated. Here, we conducted this study and found that stromal and immune scores were elevated in higher grade and isocitrate dehydrogenase (IDH) wild-type glioma. Besides, higher stromal and immune scores indicated a poor prognosis in patients with LGG. APA events in immune-related genes were associated with overall survival, RNA expression, IDH mutation, and disease-free survival. Patients in the high-risk group had poor prognoses, and the risk score could be used to predict the survival rate. The risk score was positively correlated with the expression of immune checkpoints, inflammatory cytokines, and infiltrated immune cells. Moreover, risk stratification could predict the efficacy of radiotherapy and provide a reference for the treatment of grade III glioma. Our study revealed that immune-related genes with APA events in the microenvironment could predict risk stratification and clinical prognosis in patients with LGG.
Keywords: immune-related genes, lower-grade glioma, immune infiltration, alternative polyadenylation
Graphical Abstract
This study revealed that alternative polyadenylation events of immune-related genes in the microenvironment could predict the risk stratification and prognosis of patients with lower-grade glioma. Risk stratification based on the risk signature could predict the efficacy of radiotherapy and provide a reference for the treatment of grade III glioma.
Introduction
Glioma is the most common primary tumor in the central nervous system. According to the World Health Organization (WHO) classification, grade I and grade II gliomas are defined as low-grade glioma, whereas grade III and grade IV gliomas are defined as high-grade glioma.1,2 Notably, with the consideration of the superior malignancy of glioblastoma (grade IV glioma), The Cancer Genome Atlas (TCGA) program defined grade II and grade III gliomas as lower-grade glioma (LGG), which differed from the conventional definition of low-grade glioma by WHO classification. The 10-year survival rate of grade II glioma is 47%, whereas the median overall survival (OS) time of grade III glioma is 3 years.3,4 Patients with LGG enjoy more favorable prognoses compared with those with glioblastoma; however, most LGG will inevitably progress to higher-grade glioma and subsequently lead to death.5 Besides, it was found that glioblastoma with isocitrate dehydrogenase (IDH) mutations indicated a relatively favorable survival.6 Current management of LGG involving maximal surgical resection, radiotherapy, and chemotherapy has achieved great progress. However, tumor recurrence and drug resistance are still inevitable.7 Therefore, it is urgent to identify novel biomarkers to help illustrate the pathological mechanism of LGG and develop therapeutic strategies to treat LGG.
Tumor microenvironment (TME) has attracted more and more attention as a potential target for the treatment of LGG. Various studies have demonstrated the immunosuppressive context of glioma,8 indicating that a better understanding of TME could facilitate the advancement of immunotherapies for the treatment of glioma.9 TME is typically composed of various types of cells, including immune cells, extracellular matrix, and stroma cells. Two algorithms, Estimation of Stromal and Immune Cells in Malignant Tumor Tissues Using Expression Data (ESTIMATE) and Tumor Immune Estimation Resource (TIMER), have been developed to estimate the abundance of infiltrating immune cells and predict tumor purity based on a gene-expression profile.10,11 Multiple studies have investigated the role of immune infiltration in different types of cancer, such as glioblastoma, ovarian cancer, and melanoma.12, 13, 14 The identification of immune-related genes (IRGs) can provide a better understanding of the LGG microenvironment.
Alternative polyadenylation (APA) is an essential step of messenger RNA (mRNA) maturation. APA is highly prevalent in more than 70% of human genes and is involved in various biological processes, such as cell differentiation and proliferation, as well as immune responses.15, 16, 17 A previous study revealed that APA events had a strong power predicting clinical prognosis and molecular variables in seven types of cancer, suggesting their potential as novel prognostic biomarkers.18 Besides, a large-scale analysis showed that APA events are involved in reshaping cellular pathways and regulating specific gene expression in 17 types of cancer, providing new insights into the pathological mechanism of cancer development.19 However, the role of APA events in LGG has not been fully elucidated.
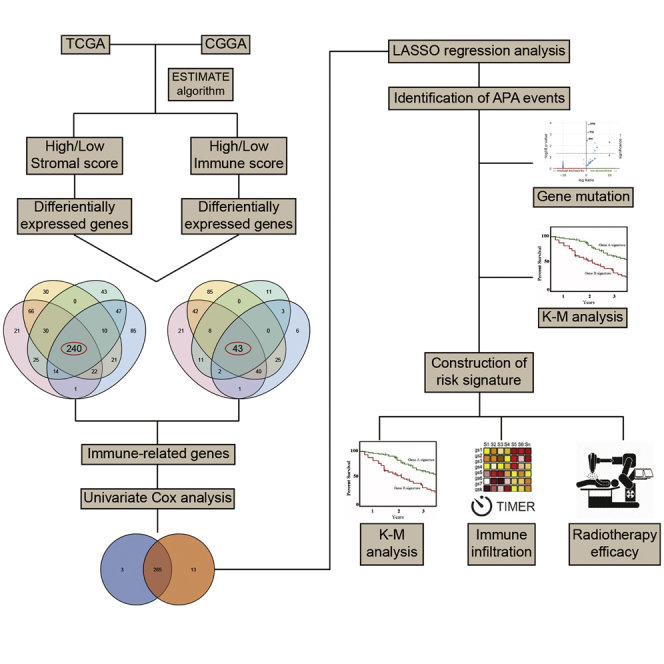
In this study, we applied the ESTIMATE algorithm to calculate stromal and immune scores and identify prognostic IRGs for LGG in TCGA and Chinese Glioma Genome Atlas (CGGA) datasets. Further, we investigated the role of APA events of IRGs in RNA expression and clinical prognosis. The risk signature was constructed based on the expression of IRGs with APA events. The prognostic and predictive values of the risk score were discerned. TIMER and single-sample gene set enrichment analysis (ssGSEA) algorithms were used to assess the immune infiltration landscape of LGG. The association between risk score and immune infiltration was also explored. Moreover, we found that the risk stratification could predict the efficacy of radiotherapy and provide a reference for the treatment of grade III glioma.
Results
TME was associated with LGG features and prognosis
A total of 522 and 623 grade II and grade III glioma patients were extracted from TCGA and CGGA databases, respectively. Characteristics of included patients were summarized in Table 1. The stromal score, immune score, and ESTIMATE score were calculated by ESTIMATE analysis. The stromal score was distributed between −1,860.14 and 1,481.33, and the immune score was −1,596.3 and 2,575.51 in TCGA dataset, whereas the stromal score ranged from −2331.71 to 985.31, and the immune score ranged from −1,720.44 to 1,987.18 in the CGGA dataset (Table S1). Then, patients with LGG were divided into high- and low-score groups according to the stromal and immune scores, with the mean score as the cut-off value. The stromal and immune scores were significantly higher in grade III glioma compared to grade II glioma (p < 0.05) (Figures 1A and 1B). Besides, the stromal and immune scores were significantly higher in IDH wild-type glioma compared to IDH mutant glioma (p < 0.05) (Figures 1C and 1D). Kaplan-Meier analysis revealed that the high stromal and immune scores were associated with poor prognosis in LGG patients (p < 0.05) (Figures 1E−1H). These results suggested that the TME was associated with LGG features and prognosis.
Table 1.
Characteristics of TCGA and CGGA datasets
| TCGA (n = 522) | CGGA (n = 623) | |
|---|---|---|
| Age | ||
| ≤41 | 275 | 353 |
| >41 | 247 | 269 |
| N/A | 0 | 1 |
| Gender | ||
| Male | 289 | 360 |
| Female | 233 | 263 |
| Grade | ||
| II | 256 | 290 |
| III | 265 | 333 |
| N/A | 1 | 0 |
| IDH status | ||
| Mutant | 423 | 438 |
| Wild-type | 96 | 144 |
| N/A | 3 | 41 |
| Radiotherapy | ||
| Yes | 291 | 456 |
| No | 184 | 134 |
| N/A | 47 | 33 |
| Chemotherapy | ||
| Yes | − | 374 |
| No | − | 208 |
| N/A | − | 41 |
IDH, isocitrate dehydrogenase.
Figure 1.

Association of stromal and immune scores with clinical features and prognosis of lower-grade glioma (LGG)
(A and B) The stromal and immune scores in grade II and III gliomas in TCGA (A) and CGGA (B) datasets. (C and D) The stromal and immune scores in isocitrate dehydrogenase (IDH) mutant and wild-type gliomas in TCGA (C) and CGGA (D) datasets. (E−H) Kaplan–Meier analysis stromal and immune scores in patients with LGG. Data are represented as mean ± SD. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001.
Profiling of differentially expressed genes in immune and stromal groups
To profile potential IRGs, differential expression analysis was performed to identify differentially expressed genes between high- and low-score groups. In TCGA dataset, 419 upregulated and 168 downregulated genes were identified in the stromal group, whereas 419 upregulated and 243 downregulated genes were identified in the immune group (Table S2; Figure 2A). In the CGGA dataset, 409 upregulated and 78 downregulated genes were identified in the stromal group, whereas 440 upregulated and 120 downregulated genes were identified in the immune group (Figure 2B). Heatmaps were generated to show expression patterns of differentially expressed genes between high and low immune/stroma groups in TCGA and CGGA datasets (Figures 2C−2F).
Figure 2.

Differentially expressed genes in stromal and immune groups
(A and B) Upregulated and downregulated genes between high- and low-score groups in TCGA and CGGA datasets. (C−F) Heatmaps of identified differentially expressed genes in TCGA (C and D) and CGGA (E and F) datasets.
Identification and enrichment analysis of IRGs
Overlapped genes that were upregulated or downregulated in both immune and stromal groups were identified in TCGA and CGGA datasets (Figures 3A and 3B). A total of 240 upregulated and 43 downregulated genes were identified as IRGs (Table S3). The expression pattern of IRGs in TCGA and CGGA datasets was shown by heatmaps (Figures 3C and 3D). Enrichment analysis revealed that IRGs were enriched in leukocyte activation, major histocompatibility complex (MHC), immunoglobulin binding, and antigen processing and presentation (Figures 3E−3H). These results indicated that the identified IRGs were associated with immune processes.
Figure 3.

Identification and enrichment analysis of immune-related genes
(A and B) Immune-related genes were identified as overlapped genes that were upregulated or downregulated in the stromal and immune groups. (C and D) Expression pattern of immune-related genes shown by heatmaps. (E−G) Enrichment analysis of immune-related genes in Gene Ontology (GO) terms, including biological process (E), cellular component (F), and molecular function (G). (H) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of immune-related genes.
Screening of prognostic IRGs in LGG
The univariate Cox analysis was performed to evaluate the prognostic value of identified IRGs. Among 283 IRGs, 268 genes had a significant impact on the prognosis of LGG patients in TCGA dataset, whereas there were 278 genes in the CGGA dataset. Then, 265 overlapped genes were selected for further investigation (Figure S1). Eight genes (CD74, EMP1, CRTAC1, PCSK2, PGAM2, S100A10, PLEKHA4, and VCAM1) were randomly selected and were significantly associated with the overall survival of LGG patients (Figure 4). The least absolute shrinkage and selection operator (LASSO) analysis was conducted to screen candidate IRGs for further analysis, and 37 IRGs were selected with the minimum lambda value (Figure S2).
Figure 4.

Kaplan-Meier analysis of randomly selected immune-related genes in TCGA and CGGA datasets
APA events of IRGs were associated with LGG prognosis
To explore the role of APA events in the prognosis of LGG, we extracted data of APA events of TCGA-LGG dataset from The Cancer 3′ UTR Atlas (TC3A) database. The frequency of APA events was shown by the percentage of distal polyadenylation site usage index (PDUI), which was based on the DaPars algorithm developed by Xia et al.18 The PDUI value represented the frequency of APA events with a range of 0−1. The greater the PDUI was, the more distal polyadenylation site of a transcript was used and vice versa. Among 37 candidate IRGs, 21 IRGs with APA events were selected, and their PDUI values were shown by the heatmap (Figure 5A). Three clusters were identified based on the distance of PDUI values of each sample. Kaplan-Meier analysis revealed that there was a significant difference in survival rates of patients in three clusters (p < 0.05) (Figure 5B). Moreover, the PDUI value was negatively correlated with gene expression (Figure 5C). Then, multivariate Cox analysis was conducted and identified 14 independent prognostic IRGs of LGG patients (Figure S3). To eliminate IRGs with collinear expression pattern, we calculated the variance inflation factor (Vif) of each IRG, and seven IRGs (PCSK2, CRTAC1, PLEKHA4, VCAM1, EMP1, S100A10, and PGAM2) were selected with Vif < 4.0 for the construction of risk signature (Figure 5D). Enrichment analysis revealed that the seven IRGs were significantly associated with the co-occurrence of ATRX, TP53, and IDH1 mutations (Figure 5E). Cases without alternations in seven IRGs had significantly better disease-free survival compared to those with alternations, whereas the difference was not detected in overall survival (Figures 5F and 5G). Additionally, among seven IRGs, the PDUI value of three IRGs (CRTAC1, EMP1, and PLEKHA4) was significantly associated with overall survival and disease-free survival of LGG patients (p < 0.05), in which the higher PDUI of CRTAC1 and the lower PDUI of EMP1 and PLEKHA4 indicated a relatively favorable prognosis (Figures 6A−6C). These results suggested that APA events were associated with the prognosis of LGG patients.
Figure 5.

Investigation of alternative polyadenylation in LGG
(A) Percentage of distal polyadenylation site usage index (PDUI) value of immune-related genes of each patient in TCGA dataset. Patients were classified into three clusters based on the distance of the PDUI value. (B) Kaplan-Meier analysis of three clusters in overall survival. (C) Correlation between PDUI value and RNA expression of immune-related genes. (D) Multivariate Cox analysis identified seven immune-related genes with poor collinearity. (E) Association between immune-related genes and gene mutations. (F and G) Survival analysis of alternations of immune-related genes in overall survival (F) and disease-free survival (G).
Figure 6.

Survival analysis of PDUI value in LGG
(A−C) Kaplan-Meier analysis of PDUI value of CRTAC1 (A), EMP1 (B), and PLEKHA4 (C) in patients with LGG.
Construction and validation of risk signature based on the expression of IRGs
To further explore the role of IRGs with APA events in LGG patients, we constructed the risk signature using multivariate Cox analysis. As mentioned before, seven IRGs were selected for the construction of risk signature. The risk score of each sample was calculated according to the algorithm, and the proportion of dead patients was higher in the high-risk group compared to the low-risk group (Figures 7A and 7B). Among seven IRGs, PCSK2, PLEKHA4, VCAM1, and EMP1 were termed as risk genes (hazard ratio > 1), whereas CRTAC1, S100A10, and PGAM2 were termed as protective genes (hazard ratio < 1). The expression of risk genes, such as EMP1 and PLEKHA4, was higher in the high-risk group than the low-risk one, whereas that of protective genes, such as CRTAC1 and PGAM2, was markedly higher in the low-risk group than the high-risk one (Figures 7C and 7D). Multivariate Cox analysis revealed that the risk score, glioma grade, and IDH status were independent risk factors of LGG patients (Table 2). Besides, the risk score had a promising value in predicting the survival of LGG patients. In TCGA dataset, the area under curve (AUC) of 1-, 3-, and 5-year survival was 0.881, 0.766, and 0.763, respectively; for the CGGA dataset, the AUC of 1-, 3-, and 5-year survival was 0.726, 0.744, and 0.764, respectively (Figure 7E). Moreover, the risk score was significantly higher in grade III glioma compared with grade II glioma and in IDH wild-type glioma compared with IDH mutant glioma (p < 0.05) (Figures 7F and 7G). Additionally, Kaplan-Meier analysis revealed that the low-risk group had a relatively favorable prognosis compared with the high-risk group in patients with LGG, grade II, and grade III gliomas (p < 0.05) (Figures 7H and 7I). These results indicated that the constructed risk signature could predict the risk stratification and prognosis of LGG patients.
Figure 7.

Construction and validation of risk signature based on the expression of immune-related genes
(A and B) Distribution of risk score and samples in TCGA (A) and CGGA (B) datasets. (C and D) Expression of seven immune-related genes in TCGA (A) and CGGA (B) datasets. (E) Time-dependent receiver operating characteristic (ROC) analysis of risk score in predicting the survival of LGG patients. (F and G) The risk score of different grades (F) and IDH status (G) of gliomas. (H and I) Kaplan-Meier analysis of low-risk and high-risk groups in patients with LGG and grade II and grade III gliomas in TCGA (H) and CGGA (I) datasets. Data are represented as mean ± SD. ∗∗∗∗p < 0.0001.
Table 2.
Multivariate analysis of risk score and clinical features in TCGA and CGGA datasets
| Variables | TCGA (n = 522) |
CGGA (n = 623) |
||||
|---|---|---|---|---|---|---|
| Coefficient | HR (95% CI) | p value | Coefficient | HR (95% CI) | p value | |
| Risk score | 0.880 | 2.411 (1.953−2.975) | <0.001 | 0.590 | 1.804 (1.597−2.038) | <0.001 |
| Age | 0.052 | 1.053 (1.037−1.069) | <0.001 | 0.010 | 1.010 (0.999−1.021) | 0.081 |
| Gender | 0.085 | 1.089 (0.759−1.563) | 0.643 | −0.103 | 0.902 (0.709−1.148) | 0.403 |
| Grade | 0.522 | 1.686 (1.103−2.575) | 0.016 | 0.954 | 2.597 (1.978−3.411) | <0.001 |
| IDH | 0.927 | 2.528 (1.597−4.000) | <0.001 | 0.339 | 1.403 (1.067−1.844) | 0.015 |
HR, hazard ratio; CI, confidence interval.
Risk signature was associated with TME of LGG
To further investigate the relationship between risk signature and TME of LGG, we explored the correlation between risk score and ESTIMATE score, immune checkpoints, inflammatory cytokines, and immune infiltration. Results showed that the risk score was significantly positively associated with the stromal score, immune score, and ESTIMATE score, whereas it was negatively associated with tumor purity, which indicated a high immune infiltration (p < 0.05) (Figures 8A−8D). Besides, the risk score was positively correlated with the expression of immune checkpoints and inflammatory cytokines (Figures 8E and 8F). Moreover, the abundance of infiltrated immune cells was higher in the high-risk group (Figures 8G and 8H). The risk score was especially correlated with the infiltration of dendritic cells (p < 0.05) (Figures 8I and 8J). Multivariate Cox analysis revealed that the risk score and neutrophils were independent prognostic factors of LGG patients (p < 0.05) (Figures S4A and S4B). These results indicated that the risk signature was associated with the TME of LGG.
Figure 8.

Correlation between risks signature and tumor microenvironment of LGG
(A−D) Correlation between risk score and stromal score (A), immune score (B), ESTIMATE score (C), and tumor purity (D). (E and F) Correlation between risk score and immune checkpoints (E) and inflammatory cytokines (F). (G and H) The abundance of infiltrated immune cells revealed by the ssGSEA algorithm in TCGA (G) and CGGA (H) datasets. (I and J) Correlation between risk score and immune infiltration assessed by TIMER algorithm.
Risk stratification correlated with the efficacy of radiotherapy
Then we explored the significance of risk stratification in predicting the efficacy of LGG treatment. Given that the chemotherapy information was not provided in TCGA dataset, we investigated the efficacy of radiotherapy in different risk stratifications. In patients with grade II glioma, those in the low-risk and high-risk groups did not significantly benefit from the radiotherapy (p > 0.05) (Figures 9A−9D). Similarly, radiotherapy did not exhibit significant efficacy for patients in the low-risk group of grade III glioma (p > 0.05) (Figures 9E and 9F). However, radiotherapy could significantly prolong the overall survival in the high-risk group of patients with grade III glioma (p < 0.05) (Figures 9G and 9H). These results suggested that the risk stratification based on the risk signature could predict the efficacy of radiotherapy in the treatment of grade III glioma.
Figure 9.

Association of risk stratification and efficacy of radiotherapy
(A−D) Survival analysis of patients with or without radiotherapy in the low-risk (A and B) and high-risk (C and D) groups of grade II glioma. (E−H) Survival analysis of patients with or without radiotherapy in the low-risk (E and F) and high-risk (G and H) groups of grade III glioma.
Discussion
Although novel therapies, such as immunotherapy and targeted therapy, have achieved great success in cancer treatment, current management of LGG cannot reach a favorable remission. The identification of IDH mutation, O-6-methylguanine-DNA methyltransferase (MGMT) promoter methylation, and 1p/19q co-deletion was found to be closely related to glioma prognosis.20 However, the association between TME and LGG prognosis remains to be fully elucidated. Therefore, we extract data from TCGA and CGGA databases, in combination with ESTIMATE and TIMER algorithms and APA events, to provide novel insights into the role of IRGs in the risk stratification and prognosis of LGG.
Numerous studies have investigated the role of IRG in different types of cancer.14,21, 22, 23 In our study, stromal and immune scores of each patient were calculated and found to be significantly associated with grade, IDH mutation, and prognosis, which was consistent with the result of a previous study.24 Notably, the mutation of IDH was found to be a protective factor for patients with glioblastoma, whereas its prognostic value was not validated in LGG. Based on our extracted data, we found that the IDH mutation indicated a relatively favorable prognosis in LGG patients (Figure S5). Besides, the IDH mutation was identified as an independent risk factor in multivariate Cox analysis. Therefore, the prognostic value of the IDH mutation was preliminarily proven by our results. However, additional prospective studies are needed to verify the prognostic value of the IDH mutation in LGG. Differentially expressed genes were profiled in high/low immune and stroma groups, and IRGs were identified as overlapped up- or downregulated differentially expressed genes. Enrichment analysis showed that selected IRGs were mainly involved in the leukocyte activation, MHC, immunoglobulin binding, and antigen processing and presentation. Therefore, the immune characteristics of IRGs were verified.
APA is a crucial event for gene regulation and is highly involved in cancer progression.25 Previous studies have revealed that APA dynamics were extensively involved in leukemia development at the single-cell level.26 Moreover, APA events could serve as a prognostic marker for early-stage breast cancer.27 Besides, the genome-wide profile revealed that APA events were highly relevant to mitochondrial electron transport and oxidative phosphorylation. Given that energy metabolism, such as adenosine triphosphate (ATP), played a crucial role in glioma,28 and ATP was a substrate of the polyadenylation process,29 we extracted data of APA events from TC3A to explore their roles in LGG. When we classified patients into three clusters based on the distance of PDUI value, Kaplan-Meier analysis revealed that the three clusters had a significant difference in overall survival. Besides, IRGs with APA events were associated with disease-free survival and gene mutations, such as IDH1 and TP53 mutation in LGG patients. Moreover, since the PDUI value was negatively associated with gene expression, the high PDUI value of risk factors (EMP1 and PLEKHA4) and low PDUI value of protective factors (CRTAC1) indicated a favorable prognosis. These results indicated that APA events were associated with prognosis of LGG patients.
The constructed risk signature was shown to be a proper index in stratifying risk groups in LGG, since the low-risk group had a more favorable survival rate compared with the high-risk group. In our study, 37 IRGs were selected by LASSO analysis. In order to demonstrate the superior efficiency of APA-based screening, we constructed the risk signature based on the expression of 37 IRGs without the selection of APA events as a contrast (Figure S6A). Fourteen IRGs were identified as independent prognostic genes, and seven IRGs were selected with Vif < 4.0 (Figure S6B). Kaplan-Meier analysis showed that patients in the high-risk group had poor prognoses, which was consistent with an APA-based signature (p < 0.05) (Figure S6C). However, the predictive accuracy of this signature was worse than an APA-based signature (1-, 3-, and 5-year survival in TCGA dataset: 0.71, 0.608, and 0.627 versus 0.881, 0.766, and 0.763, respectively; for CGGA dataset: 0.572, 0.611, and 0.584 versus 0.726, 0.744, and 0.764, respectively) (Figure S6D). Therefore, genes with APA events might have better efficiencies in predicting the prognosis of LGG patients.
After the construction of the risk signature, we found that the high-risk score indicated low tumor purity, which indicated a high immune infiltration. Therefore, we explored the immune infiltration using TIMER and ssGSEA algorithms. TIMER algorithm was developed in 2017 to comprehensively investigate the molecular characterization of tumor-immune interactions. The abundance of six types of major immune cells precalculated 10,897 tumors from 32 cancer types. Compared with CIBERSORT, another algorithm for evaluating immune infiltration, TIMER was not potentially affected by statistical multicollinearity due to the inclusion of highly correlated immune cell types, leading to high estimations of uncertainty.30 Therefore, we applied the TIMER algorithm to estimate the abundance of immune cells in the LGG microenvironment. The risk score was found to be positively correlated to the expression of immune checkpoints, inflammatory cytokines, and immune infiltrations. Besides, risk score and neutrophils were identified as independent prognostic factors for patients with LGG. These results indicated that the constructed risk signature can serve as an indicator for the infiltration of immune cells and predict clinical prognosis in LGG. Besides, the risk score might be used to indicate the efficacy of immunotherapy, which required further investigation.
Typically, the prognosis of glioma is predicted based on its grade, which is diagnosed by the histological and pathologic features. According to the 2016 WHO classification, diffuse astrocytoma and oligodendroglioma belong to grade II glioma, and anaplastic ones are grade III glioma.31 Although the higher grade of glioma indicates higher malignancy and worse prognosis, few biomarkers can predict the prognosis in the subgroup of grade II and III glioma. Our study revealed that the risk stratification was significantly associated with the prognosis in patients with grade II or grade III glioma. Therefore, the risk score could be applied as a marker to predict the prognosis of LGG patients. Moreover, our study found that the risk stratification was associated with the efficacy of radiotherapy. Current management of grade II glioma is maximal surgical resection, and it remains controversial whether patients with grade II glioma should receive chemotherapy and radiotherapy.32 As a result, the risk stratification did not have a significant association with the efficacy of radiotherapy. However, in grade III glioma, radiotherapy exhibited significant efficacy for patients in the high-risk group but not in the low-risk group. Since radiotherapy was recommended as standard therapy for grade III glioma, the risk stratification could predict the efficacy of radiotherapy. Additionally, a previous meta-analysis suggested that for patients with less aggressive gliomas, radiotherapy might increase the risk of long-term neurocognitive side effects.33 Given that patients in the low-risk group did not significantly benefit from radiotherapy, and radiotherapy might increase the body stress and cause adverse events for those in the low-risk group, the risk stratification could provide a reference for the treatment of grade III glioma.
To sum up, our study revealed that IRGs with APA events in the microenvironment could be used to construct a risk signature and subsequently predict the prognosis of LGG with high accuracy. Besides, risk stratification based on the risk signature could predict the efficacy of radiotherapy and provide a reference for the treatment of grade III glioma.
Materials and methods
Data extraction
In total, 522 and 623 grade II and grade III glioma samples from TCGA (https://portal.gdc.cancer.gov/) and CGGA databases (http://www.cgga.org.cn/) were included in our study, respectively. RNA sequencing (RNA-seq) data and clinical features of these samples were downloaded from databases for further investigation.
Differentially expressed gene profile
The infiltration of stromal and immune cells was measured by ESTIMATE analysis, which is performed by the “estimate” R package.10 Tumor purity was calculated according to the algorithm. All patients were classified into high/low immune groups and high/low stroma groups based on their immune score and stroma score, respectively. Then, the differential expression gene profile was conducted between high and low immune/stroma groups by the “limma” R package. Genes with log2 |fold change| >1.0 and adjusted p <0.05 were selected. The volcano plot was generated by TBtools to visualize the selected genes.34
Functional enrichment analysis
Functional enrichment analysis was performed using the “clusterProfiler” R package.35 The enrichment analysis contains Gene Ontology (GO) terms, including biological process, cellular component, and molecular function categories and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The false discovery rate (FDR) < 0.05 was used as the cut-off.
Identification of APA events
APA events in RNA-seq data were evaluated by the PDUI value. The PDUI value represented the frequency of APA events with the range of 0−1, in which the greater the PDUI was, the more distal polyadenylation site of a transcript was used and vice versa. The PDUI value for all genes in each patient of TCGA-LGG dataset was downloaded from TC3A (http://tc3a.org/).36 The enrichment analysis of selected genes with APA events was performed on TC3A website.
Construction of risk signature
LASSO analysis was employed to identify candidate risk genes. The role of risk genes in prognosis was determined by multivariate Cox regression analysis. Vif and hypothesis testing were used to filter out genes with high collinearity. The risk score for each patient was calculated based on the gene expression according to the following algorithm:
Immune cell infiltration analysis
TIMER and ssGSEA algorithms were applied to estimate immune infiltration. The TIMER algorithm contained six types of immune cells, including B cells, CD4+ T cells, CD8+ T cells, dendritic cells, neutrophils, and macrophages.30 The ssGSEA algorithm contained 28 types of immune cells.
Statistical analysis
Statistical analyses and visualization were conducted using R software v4.0.2, GraphPad Prism v8.0, and TBtools. Measurement data were represented as mean ± standard deviation. Student’s t test was used to estimate the differences between the two groups. Kaplan-Meier and log-rank analyses were used to evaluate the survival differences between grouped patients. Univariate and multivariate Cox regression analyses were conducted to profile independent prognostic genes. Receiver operating characteristic (ROC) analysis was used to predict risk signature and overall survival time using the “survivalROC” R package. p <0.05 was regarded as statistically significant.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81873635 and 81902553), Natural Science Foundation of Hunan Province (2019JJ50963 and 2019JJ50942), and Key Research and Development Program of Hunan Province (2018SK2101).
Author contributions
Z.L. and C.L. conceived, designed, and supervised the study. S.X. drafted the manuscript and performed data analysis. S.X., L.T., and G.D. collected the data.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2021.01.033.
Contributor Information
Chengke Luo, Email: ck_luo@csu.edu.cn.
Zhixiong Liu, Email: zhixiongliu@csu.edu.cn.
Supplemental information
References
- 1.Ostrom Q.T., Cioffi G., Gittleman H., Patil N., Waite K., Kruchko C., Barnholtz-Sloan J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro-oncol. 2019;21(Suppl 5):v1–v100. doi: 10.1093/neuonc/noz150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brat D.J., Verhaak R.G., Aldape K.D., Yung W.K., Salama S.R., Cooper L.A., Rheinbay E., Miller C.R., Vitucci M., Morozova O., Cancer Genome Atlas Research Network Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015;372:2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohgaki H., Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J. Neuropathol. Exp. Neurol. 2005;64:479–489. doi: 10.1093/jnen/64.6.479. [DOI] [PubMed] [Google Scholar]
- 4.Bleeker F.E., Molenaar R.J., Leenstra S. Recent advances in the molecular understanding of glioblastoma. J. Neurooncol. 2012;108:11–27. doi: 10.1007/s11060-011-0793-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Claus E.B., Walsh K.M., Wiencke J.K., Molinaro A.M., Wiemels J.L., Schildkraut J.M., Bondy M.L., Berger M., Jenkins R., Wrensch M. Survival and low-grade glioma: the emergence of genetic information. Neurosurg. Focus. 2015;38:E6. doi: 10.3171/2014.10.FOCUS12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z., Bao Z., Yan W., You G., Wang Y., Li X., Zhang W. Isocitrate dehydrogenase 1 (IDH1) mutation-specific microRNA signature predicts favorable prognosis in glioblastoma patients with IDH1 wild type. J. Exp. Clin. Cancer Res. 2013;32:59. doi: 10.1186/1756-9966-32-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osuka S., Van Meir E.G. Overcoming therapeutic resistance in glioblastoma: the way forward. J. Clin. Invest. 2017;127:415–426. doi: 10.1172/JCI89587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma Q., Long W., Xing C., Chu J., Luo M., Wang H.Y., Liu Q., Wang R.F. Cancer Stem Cells and Immunosuppressive Microenvironment in Glioma. Front. Immunol. 2018;9:2924. doi: 10.3389/fimmu.2018.02924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu S., Tang L., Li X., Fan F., Liu Z. Immunotherapy for glioma: Current management and future application. Cancer Lett. 2020;476:1–12. doi: 10.1016/j.canlet.2020.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Yoshihara K., Shahmoradgoli M., Martínez E., Vegesna R., Kim H., Torres-Garcia W., Treviño V., Shen H., Laird P.W., Levine D.A. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li B., Severson E., Pignon J.C., Zhao H., Li T., Novak J., Jiang P., Shen H., Aster J.C., Rodig S. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17:174. doi: 10.1186/s13059-016-1028-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen S., Wang G., Zhang R., Zhao Y., Yu H., Wei Y., Chen F. Development and validation of an immune gene-set based Prognostic signature in ovarian cancer. EBioMedicine. 2019;40:318–326. doi: 10.1016/j.ebiom.2018.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia D., Li S., Li D., Xue H., Yang D., Liu Y. Mining TCGA database for genes of prognostic value in glioblastoma microenvironment. Aging (Albany NY) 2018;10:592–605. doi: 10.18632/aging.101415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang R., Mao M., Lu Y., Yu Q., Liao L. A novel immune-related genes prognosis biomarker for melanoma: associated with tumor microenvironment. Aging (Albany NY) 2020;12:6966–6980. doi: 10.18632/aging.103054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoque M., Ji Z., Zheng D., Luo W., Li W., You B., Park J.Y., Yehia G., Tian B. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods. 2013;10:133–139. doi: 10.1038/nmeth.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elkon R., Ugalde A.P., Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat. Rev. Genet. 2013;14:496–506. doi: 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- 17.Huang G., Huang S., Wang R., Yan X., Li Y., Feng Y., Wang S., Yang X., Chen L., Li J. Dynamic Regulation of Tandem 3′ Untranslated Regions in Zebrafish Spleen Cells during Immune Response. J. Immunol. 2016;196:715–725. doi: 10.4049/jimmunol.1500847. [DOI] [PubMed] [Google Scholar]
- 18.Xia Z., Donehower L.A., Cooper T.A., Neilson J.R., Wheeler D.A., Wagner E.J., Li W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat. Commun. 2014;5:5274. doi: 10.1038/ncomms6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiang Y., Ye Y., Lou Y., Yang Y., Cai C., Zhang Z., Mills T., Chen N.Y., Kim Y., Muge Ozguc F. Comprehensive Characterization of Alternative Polyadenylation in Human Cancer. J. Natl. Cancer Inst. 2018;110:379–389. doi: 10.1093/jnci/djx223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen X., Yan Y., Zhou J., Huo L., Qian L., Zeng S., Li Z., Wei J., Xu Z., Gong Z. Clinical prognostic value of isocitrate dehydrogenase mutation, O-6-methylguanine-DNA methyltransferase promoter methylation, and 1p19q co-deletion in glioma patients. Ann. Transl. Med. 2019;7:541. doi: 10.21037/atm.2019.09.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z., Zhu J., Liu Y., Liu C., Wang W., Chen F., Ma L. Development and validation of a novel immune-related prognostic model in hepatocellular carcinoma. J. Transl. Med. 2020;18:67. doi: 10.1186/s12967-020-02255-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.She Y., Kong X., Ge Y., Yin P., Liu Z., Chen J., Gao F., Fang S. Immune-related gene signature for predicting the prognosis of head and neck squamous cell carcinoma. Cancer Cell Int. 2020;20:22. doi: 10.1186/s12935-020-1104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wan B., Liu B., Huang Y., Yu G., Lv C. Prognostic value of immune-related genes in clear cell renal cell carcinoma. Aging (Albany NY) 2019;11:11474–11489. doi: 10.18632/aging.102548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng X., Lin D., Zhang X., Shen X., Yang Z., Yang L., Lu X., Yu L., Zhang N., Lin J. Profiles of immune-related genes and immune cell infiltration in the tumor microenvironment of diffuse lower-grade gliomas. J. Cell. Physiol. 2020;235:7321–7331. doi: 10.1002/jcp.29633. [DOI] [PubMed] [Google Scholar]
- 25.Sun M., Ding J., Li D., Yang G., Cheng Z., Zhu Q. NUDT21 regulates 3′-UTR length and microRNA-mediated gene silencing in hepatocellular carcinoma. Cancer Lett. 2017;410:158–168. doi: 10.1016/j.canlet.2017.09.026. [DOI] [PubMed] [Google Scholar]
- 26.Ye C., Zhou Q., Hong Y., Li Q.Q. Role of alternative polyadenylation dynamics in acute myeloid leukaemia at single-cell resolution. RNA Biol. 2019;16:785–797. doi: 10.1080/15476286.2019.1586139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim N., Chung W., Eum H.H., Lee H.O., Park W.Y. Alternative polyadenylation of single cells delineates cell types and serves as a prognostic marker in early stage breast cancer. PLoS ONE. 2019;14:e0217196. doi: 10.1371/journal.pone.0217196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang R., Li G., Wang Z., Hu H., Zeng F., Zhang K., Wang K., Wu F. Identification of an ATP metabolism-related signature associated with prognosis and immune microenvironment in gliomas. Cancer Sci. 2020;111:2325–2335. doi: 10.1111/cas.14484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Millevoi S., Vagner S. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 2010;38:2757–2774. doi: 10.1093/nar/gkp1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li T., Fan J., Wang B., Traugh N., Chen Q., Liu J.S., Li B., Liu X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017;77:e108–e110. doi: 10.1158/0008-5472.CAN-17-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Louis D.N., Perry A., Reifenberger G., von Deimling A., Figarella-Branger D., Cavenee W.K., Ohgaki H., Wiestler O.D., Kleihues P., Ellison D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 32.Jiang T., Nam D.H., Ram Z., Poon W.S., Wang J., Boldbaatar D., Mao Y., Ma W., Mao Q., You Y., Chinese Glioma Cooperative Group (CGCG) Society for Neuro-Oncology of China (SNO-China) Chinese Brain Cancer Association (CBCA) Chinese Glioma Genome Atlas (CGGA) Asian Glioma Genome Atlas (AGGA) network Clinical practice guidelines for the management of adult diffuse gliomas. Cancer Lett. 2021;499:60–72. doi: 10.1016/j.canlet.2020.10.050. [DOI] [PubMed] [Google Scholar]
- 33.Lawrie T.A., Gillespie D., Dowswell T., Evans J., Erridge S., Vale L., Kernohan A., Grant R. Long-term neurocognitive and other side effects of radiotherapy, with or without chemotherapy, for glioma. Cochrane Database Syst. Rev. 2019;8:CD013047. doi: 10.1002/14651858.CD013047.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen C., Chen H., Zhang Y., Thomas H.R., Frank M.H., He Y., Xia R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020;13:1194–1202. doi: 10.1016/j.molp.2020.06.009. [DOI] [PubMed] [Google Scholar]
- 35.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng X., Li L., Wagner E.J., Li W. TC3A: The Cancer 3′ UTR Atlas. Nucleic Acids Res. 2018;46(D1):D1027–D1030. doi: 10.1093/nar/gkx892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.