

Summary
HIF1-alpha expression defines metabolic compartments in the developing heart, promoting glycolytic program in the compact myocardium and mitochondrial enrichment in the trabeculae. Nonetheless, its role in cardiogenesis is debated. To assess the importance of HIF1-alpha during heart development and the influence of glycolysis in ventricular chamber formation, herein we generated conditional knockout models of Hif1a in Nkx2.5 cardiac progenitors and cardiomyocytes. Deletion of Hif1a impairs embryonic glycolysis without influencing cardiomyocyte proliferation and results in increased mitochondrial number and transient activation of amino acid catabolism together with HIF2α and ATF4 upregulation by E12.5. Hif1a mutants display normal fatty acid oxidation program and do not show cardiac dysfunction in the adulthood. Our results demonstrate that cardiac HIF1 signaling and glycolysis are dispensable for mouse heart development and reveal the metabolic flexibility of the embryonic myocardium to consume amino acids, raising the potential use of alternative metabolic substrates as therapeutic interventions during ischemic events.
Subject areas: Animal Physiology, Biological Sciences, Cellular Physiology, Developmental Biology
Graphical abstract
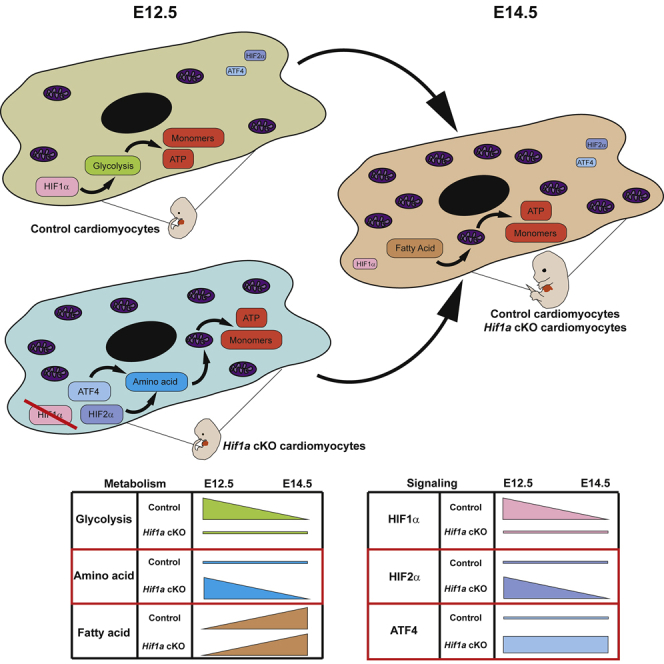
Highlights
-
•
Loss of cardiac Hif1a does not preclude heart development or cardiac function
-
•
Embryonic Hif1a-deficient hearts transiently upregulate amino acid catabolism
-
•
Amino acid catabolism activation sustains heart growth in the absence of glycolysis
-
•
HIF2α and ATF4 are transiently upregulated in the developing heart upon Hif1a loss
Animal Physiology; Biological Sciences; Cellular Physiology; Developmental Biology
Introduction
The heart is the first organ to form in utero, and it is essential to deliver oxygen and nutrients to embryonic tissues from early stages of development. Different subsets of cardiac progenitors proliferate, migrate, and differentiate into the diverse cell types that form the mature heart (Martin-Puig et al., 2008; Watanabe and Buckingham, 2010). Nkx2.5 cardiovascular progenitors give rise to the majority of cardiac cells, contributing to epicardium, myocardium, and endocardium (Moses et al., 2001). Cardiogenesis is a complex process that can result in malformations, and congenital heart defects occur in 1% of live births. Several factors have been involved in developmental cardiac failure, and among them hypoxia has been previously described as an environmental factor associated with cardiac defects during pregnancy (Cerychova and Pavlinkova, 2018; Nanka et al., 2008). Hypoxia-inducible factors (HIFs) are known to mediate a well-characterized transcriptional response to low oxygen tensions. HIF heterodimers are formed by a constitutively expressed β subunit (HIFβ or ARNT) and an oxygen-regulated α subunit, with three different isoforms (1α, 2α, and 3α) (Kaelin and Ratcliffe, 2008). Under normoxic conditions, the oxygen sensors prolyl hydroxylases (PHDs) hydroxylate HIFα in specific proline residues (Jiang et al., 1997). These modifications are recognized by the von Hippel-Lindau/E3 ubiquitin ligase complex, which polyubiquitinates and drives α subunits to proteasomal degradation. In hypoxic conditions, α subunits evade degradation due to the inhibited PHD activity, dimerize with β subunits, and mediate the adaptive response to hypoxia by activating the transcription of their target genes (Pouyssegur et al., 2006).
Poorly oxygenated areas appear during heart development (Lee et al., 2001) and physiological hypoxia is involved in outflow tract remodeling (Sugishita et al., 2004). However, chronic exposure of pregnant females to low oxygen causes embryonic myocardial thinning and epicardial detachment (Menendez-Montes et al., 2016; Ream et al., 2008) and excessive embryonic hypoxia also increases the vulnerability to suffer common septation and conotruncal heart defects (Kenchegowda et al., 2014). Genetic-based overactivation of HIF signaling by inactivation of Vhl in cardiac progenitors using different drivers (Mlc2vCre, Nkx2.5Cre) causes morphological, metabolic, and functional cardiac alterations that result in embryonic lethality (Lei et al., 2008; Menendez-Montes et al., 2016). On the other hand, Hif1a conditional loss-of-function models in cardiac progenitors and cardiomyocytes also causes several embryonic cardiac alterations (Guimarães-Camboa et al., 2015; Huang et al., 2004; Krishnan et al., 2008), suggesting that a controlled balance in oxygen levels and hypoxia signaling is required for proper cardiac development. However, important phenotypic discrepancies between the different published loss-of-function models exist. On one hand, the use of cardiac-specific Mlc2vCre driver in combination with a Hif1a floxed mice does not affect embryonic survival but causes cardiac hypertrophy with reduced cardiac function in the adulthood, together with decreased glycolysis and ATP and lactate levels (Huang et al., 2004). On the other hand, when a null Hif1a allele in germline is used in combination with a Hif1a floxed allele and the Mlc2vCre driver, mutant embryos show several cardiac alterations and increased cardiomyocyte proliferation, with associated embryonic lethality by E12.0 (Krishnan et al., 2008). The combination of null and floxed Hif1a alleles under the control of Nkx2.5Cre driver provides a more homogeneous recombination than Mlc2vCre and results in the activation of cell stress pathways that inhibit cardiomyocyte proliferation and lead to myocardial thinning, resulting in embryonic lethality by E15.5 (Guimarães-Camboa et al., 2015). Considering the importance of hypoxia pathway in early hematopoiesis, placentation, and vascular development (Llurba Olive et al., 2018), the potential secondary effect of using Hif1a-null alleles on cardiac development cannot be ruled out when interpreting data from some of these mutants. Therefore, although it is clear that sustained HIF1 signaling in the embryonic heart is detrimental for proper cardiac development, there is still disagreement and open debate about the impact of Hif1a loss during cardiogenesis and subsequent effects on cardiac function.
HIF1 signaling is an important regulator of cellular metabolism in physiological and pathological contexts. In addition to glycolytic activation (Majmundar et al., 2010), HIF1 reduces mitochondrial metabolism by repressing pyruvate entry into the mitochondria (Kim et al., 2006) and by promoting COX4 isoform switch from COX4-1 to COX4-2 (Fukuda et al., 2007). Moreover, HIF1 can also limit oxidative metabolism through an inhibitory role on mitochondrial biogenesis (Zhang et al., 2007). Several in vitro studies have demonstrated that the embryonic heart relies on glycolysis for energy supply (Chung et al, 2010, 2011), in contrast with the adult heart that sustains most of ATP production through mitochondrial oxidation of fatty acids (FA) (Lopaschuk and Jaswal, 2010). This metabolic switch is coincident with the change in oxygen levels after birth (Puente et al., 2014). However, a recent report from our group has shown that an earlier metabolic shift toward FA oxidation occurs during development at around E14.5, through a mechanism dependent on a decrease in HIF1 signaling in the embryonic myocardium (Menendez-Montes et al., 2016). Despite the importance of glucose and FAs as cardiac energy sources, amino acids can also be used as bioenergetics fuel. Hence, amino acids have the capacity to enter the Krebs Cycle at different levels, a phenomenon known as anaplerosis, and to replenish metabolic intermediates that warrant both NADH/FADH2 and building blocks production that enable the cells to continue growing under amino acid metabolism. The importance of amino acids as catabolic substrates has been described in tumor growth (Yue et al., 2017), pulmonary hypertension (Piao et al., 2013), or limited oxygen supply conditions (Bing et al., 1954; Julia et al., 1990). However, the ability of the embryonic heart to catabolize amino acids remains unexplored.
Herein, we describe that Hif1a loss in Nkx2.5 cardiovascular progenitors or cardiomyocytes during heart development blunts glycolysis and drives a compensatory metabolic adaptation based on transient activation of amino acid transport and catabolism associated with increased ATF4 and HIF2α to maintain energy production and growth. Our results demonstrate that HIF1 signaling in Nkx2.5 progenitors and cardiomyocytes is dispensable for cardiogenesis and show the relevance of amino acid metabolism during cardiac development in the absence of glycolysis, opening future research horizons toward studying the ability of the heart to use amino acids as an alternative energy fuel and biosynthetic precursor source in different pathophysiological contexts like cardiac ischemia and regeneration.
Results
HIF1 signaling in Nkx2.5 progenitors is dispensable for cardiac development
HIF1α is expressed in the developing myocardium, with a temporal dynamics along midgestation (Guimarães-Camboa et al., 2015; Krishnan et al., 2008; Menendez-Montes et al., 2016). We and others have described the heterogeneous regional distribution of HIF1 signaling with high HIF1α levels in the compact myocardium in contrast with low expression in the trabeculae (Guimarães-Camboa et al., 2015; Menendez-Montes et al., 2016). To investigate the role of HIF1 in heart development we generated a cardiac-specific loss-of-function model using two floxed alleles of Hif1a gene in combination with the cardiac progenitor-specific Cre recombinase driver Nkx2.5Cre (Hif1aflox/flox/Nkx2.5Cre/+, from here on Hif1a/Nkx2.5). Cre-mediated recombination was analyzed by agarose gel electrophoresis of cardiac and non-cardiac tissue at E12.5 (Figure S1A). A 400-bp product, corresponding to the processed Hif1a allele was only obtained in cardiac tissue in the presence of Cre recombinase activity, indicating that the deletion is specific of cardiac tissue and there is no ectopic recombination. To confirm deletion efficiency, we analyzed the expression of the floxed Hif1a exon by qPCR at E14.5, confirming its correct elimination despite a signal increase outside of the floxed region, probably caused by compensatory mechanisms (Figure S1B). Furthermore, Phd3, whose expression is dependent on HIF1, showed decreased expression in the Hif1a/Nkx2.5 mutants (Figure S1B). We also determined HIF1α protein distribution and abundance within cardiac tissue by immunostaining in mutant and control littermates by E12.5 (Figure S1C). Hif1a deletion in the mutant embryos resulted in reduced HIF1α staining, with a displacement of the HIF1α channel intensity curve to lower fluorescence intensity (Figure S1D). It is noteworthy that deletion efficiency, as measured by qPCR, was higher than that estimated by immunostaining. This is probably because the mutant Hif1a mRNA produced after Cre recombination is sufficiently stable to be translated, although the resulting protein is not functional as it lacks the DNA-binding domain located in the N-terminal region of HIF1α. Efficient deletion was further confirmed by western blot of Hif1a-deficient heart lysates at E12.5 (Figure 1A). The lower molecular weight of the HIF1α protein band confirms the presence of a truncated protein in mutant embryos, which is recognized by HIF1 antibodies raised against the C-terminal domain.
Figure 1.

Embryonic phenotype of Hif1a-deficient embryos at E12.5
(A) Representative immunoblot against HIF1α (upper panel) and smooth muscle actin (SMA, lower panel) in heart lysates of control (Hif1af/f/Nkx2.5+/+) and Hif1a/Nkx2.5 (Hif1af/f/Nkx2.5Cre/+) mutant embryos at E12.5.
(B) E12.5 control (upper panels) and mutant (lower panels) heart sections stained with hematoxylin and eosin (H&E). Scale bars, 100 μm (overview) and 20 μm (insets).
(C) H&E quantification of ventricular walls and interventricular septum width in E12.5 control (black bars, n = 4) and mutant (white bars, n = 3) embryos.
(D) Quantification of BrdU immunostaining, represented as percentage of BrdU+ cells in the compact myocardium and trabeculae of E12.5 control (black) and Hif1a/Nkx2.5 mutant (white) embryos (n = 3). In all graphs, bars represent mean ± SEM, Student's t test, n.s: non-significant. LV: left ventricle; RV: right ventricle; IVS: interventricular septum. Similar amount of male and female embryos has been used in these analyses.
Hif1a/Nkx2.5 mutants were viable and recovered in the expected Mendelian proportions from E14.5 to weaning (Table 1). Histological analysis of control and Hif1a/Nkx2.5 mutant hearts at E12.5 (Figure 1B) did not reveal differences between genotypes in terms of ventricular wall thickness (Figure 1C) or chamber sphericity (data not shown). Proliferation analysis by bromodeoxyuridine (BrdU) staining at E12.5 proved comparable proliferation index between control and Hif1a-deficient hearts (Figure 1D). Similar results were obtained at E14.5, when Hif1a-deficient embryos showed neither morphological alterations nor differences between genotypes in terms of cell size and proliferation by means of BrdU staining (data not shown). These results indicate that the lack of Hif1a in Nkx2.5 cardiac progenitors does not influence cardiomyocyte proliferation and suggest that HIF1 signaling in this cell population is dispensable for proper cardiac development.
Table 1.
Analysis of Hif1a/Nkx2.5 mutant embryo recovery
| Stage | Hif1af/f/Nkx2.5Cre/+ | Total | Litters | Observed % | Expected % | p value |
|---|---|---|---|---|---|---|
| E14.5 | 20 | 90 | 13 | 24.4 ± 4.9 | 25 | 0.905 |
| Weaning | 10 | 38 | 6 | 28.6 ± 5.8 | 25 | 0.787 |
For each stage, table shows the number of mutant embryos recovered, the total number of embryos/pups collected, and the number of litters analyzed. The percentage of recovered mutants and the expected recovery percentage (25% in all cases) were compared by the Wilcoxon signed rank test. Equivalent proportion of male and female embryos and mice has been considered in the analysis.
Embryonic loss of Hif1a in Nkx2.5 progenitors does not influence adult cardiac function or morphology
Albeit deletion of Hif1a in Nkx2.5 cardiovascular progenitors did not hamper embryonic ventricular chamber formation, we wondered whether mutant mice might develop heart alterations in the adulthood. Histology analysis by hematoxylin-eosin staining at five months of age did not indicate evident changes in cardiac morphology of Hif1a-deficient hearts relative to control animals (Figure 2A). Moreover, Masson's trichrome staining analysis excluded the presence of fibrotic areas in any of the genotypes (Figure 2B). Nevertheless, the lack of macroscopic malformations does not rule out that cardiac performance could be affected. To determine if embryonic deletion of Hif1a influences cardiac function during adulthood, we performed echocardiography in 5-month-old control and mutant mice. Both 2D and M mode analysis (Figure 2C) and the quantification of several morphological parameters confirmed the absence of anatomical alterations (Figures 2D and 2E). Furthermore, conserved cardiac function in Hif1a/Nkx2.5 mutant versus control mice was demonstrated by means of ejection fraction and fractional shortening (Figure 2F). On the other hand, electrocardiographic analysis showed normal PR and QRS segment length in Hif1a/Nkx2.5 mice (data not shown), ruling out the existence of conduction or coupling defects.
Figure 2.

Cardiac morphology and function in adult Hif1a/Nkx2.5 mutants
(A and B) H&E (A) and Masson's trichrome (B) staining of the left ventricle from a representative 5-month-old control (Hif1af/f/Nkx2r+/+) and Hif1a/Nkx2.5 mutant (Hif1af/f/Nkx2.5Cre/+) heart. Even distribution of male and female mice was used in each experimental group (n = 6 females and 6 males). No differences associated with sex were observed in cardiac structure or fibrosis.
(C) Representative echocardiography imaging of 5-month-old control and Hif1a-deficient mutant mice in 2D mode (upper panels) and M mode (lower panels).
(D and E) Echocardiography-based quantification of interventricular septum (IVS) thickness (D) and left ventricle (LV) posterior wall thickness (E) in controls (black bars, n = 9) and Hif1a/Nkx2.5 mutants (white bars, n = 11) by 5 months of age.
(F) Quantification of ejection fraction (EF) and fractional shortening (FS) in controls (black bars, n = 9) and Hif1a/Nkx2.5 mutants (white bars, n = 11) by 5 months of age. Uniform distribution of male and female mice was used in each experimental group (n = 10 females and 10 males). No differences associated with sex were observed for cardiac structural or functional parameters. In all graphs bars represent mean ± SEM, Student's t test, n.s: non-significant. For all images, scale bars, 50 μm.
These results indicate that active HIF1 signaling in cardiovascular Nkx2.5 progenitors during heart development is not required for proper cardiac morphogenesis or normal heart function in the adulthood.
Cardiac deletion of Hif1a prevents the expression of glycolytic enzymes in the compact myocardium
To investigate the adaptive mechanisms operating upon Hif1a loss in Nkx2.5 cardiovascular progenitors to allow normal cardiac development, we performed massive expression analysis by RNA sequencing (RNA-seq) of E12.5 ventricular tissue from control and Hif1a/Nkx2.5 mutant embryos. Subsequent bioinformatics analysis identified 14,406 genes being expressed. Among them, 201 genes showed differential expression: 118 were downregulated in mutant hearts relative to control hearts and 83 were upregulated, representing positively and negatively regulated targets respectively, dependent on functional HIF1 signaling in direct or indirect fashion (Table S1). To obtain a summarized view of their function, the list of 201 genes was subjected to enrichment analyses, which found significant associations with several metabolic processes, such as nucleotide/nucleoside and monocarboxylic acid metabolism, amino acid metabolism, and, especially, carbohydrate metabolism. Complete results are presented in Table S2, and a selection of significantly enriched processes is shown in Figure 3A, connected with the subset of differentially expressed genes that are involved in those processes. We have previously described the existence of metabolic territories in the embryonic myocardium with an enhanced glycolytic signature in the compact myocardium by E12.5 (Menendez-Montes et al., 2016). Here we found that glucose transporter 1 (GLUT1) protein levels were significantly reduced in the compact myocardium of the Hif1a/Nkx2.5 mutant embryos by E12.5 (Figure 3B). Because Nkx2.5 cardiac progenitors contribute to different cardiac layers including myocardium, epicardium, and endocardium, to determine if the glycolytic inhibition observed in the Hif1a/Nkx2.5 mutant embryos was associated with Hif1a loss in the myocardial layer, we specifically deleted Hif1a in cardiomyocytes using cTnT-Cre (Jiao et al., 2003) (Hif1aflox/flox/cTnTCre/+, hereon Hif1a/cTnT). Hif1a/cTnT mutants also showed reduced HIF1α levels by E14.5 without cardiac developmental defects compared with control littermates (data not shown). The inhibition of the glycolytic program in both Hif1a/Nkx2.5 and Hif1a/cTnT mutants was further confirmed by mRNA expression analysis of the critical enzymes Glut1, Pdk1, and Ldha by in situ hybridization at E12.5 (Figure 3C) and E14.5 (data not shown) in controls and Hif1a mutants. Results showed strong inhibition of glycolytic gene expression in the compact myocardium of Hif1a-deficient hearts at both stages. This sustained glycolytic inhibition at E14.5 was further validated by qPCR in both genetic models (Figures 3D and 3E), including the downregulation of Slc16a3, responsible for the transport of monocarboxylic acids, such as lactate, across mitochondrial membrane.
Figure 3.

Glycolytic metabolism alterations in cardiac Hif1a-deficient embryos
(A) Circular plot representing logFC value for genes detected as differentially expressed in mutant (Hif1af/f/Nkx2.5Cre/+) embryos, relative to controls (Hif1af/f/Nkx2.5+/+), at E12.5 (left side), associated to Gene Ontology (GO) terms related to carbohydrate metabolism, selected among those detected as enriched with p value < 0.001 with GOrilla (right side). logFC values for genes are color coded: blue color denotes lower expression in mutant samples. Ribbons connecting genes and biological processes are colored by process.
(B) Representative GLUT1 immunofluorescence on E12.5 heart sections of controls (left panels) and Hif1a/Nkx2.5 mutants (right panels). Nuclei shown in blue, Troponin T in green, and GLUT1 in red. Insets show left ventricle. Scale bars, 100 μm and 20 μm in insets.
(C) E12.5 in situ hybridization of Glut1 (top panels), Pdk1 (middle panels), and Ldha (bottom panels) in control and Hif1a/Nkx2.5 mutant right ventricles (left) and in control and Hif1a/cTnT mutant right ventricles (right). Scale bar, 20μm
(D and E) RT-qPCR analysis of glycolytic genes from E14.5 Hif1a/Nkx2.5 (D) and Hif1a/cTnT (E) mutant ventricles. Bars (mean ± SEM, n = 3) represent fold induction relative to baseline expression in littermate controls (red line). Student's t test. ∗p value<0.05; ∗∗0.005<p value<0.01, ∗∗∗p value<0.005. Equivalent proportion of male and female embryos have been included in all experiments.
Taken together, these results confirm our previous findings that HIF1 signaling controls the expression of glycolytic genes in the embryonic heart and indicate that an active glycolytic program in the compact myocardium is not essential for proper cardiogenesis.
Lipid metabolism is preserved in cardiac Hif1a mutant mice
We have previously reported that sustained HIF1 signaling in the embryonic myocardium results in severe alterations of mitochondrial amount and function (Menendez-Montes et al., 2016). To evaluate the bioenergetics adaptations in response to the lack of cardiac HIF1 activation we investigated mitochondrial network and activity in Hif1a/NKx2.5 mutants. Analysis and quantification of ventricular ultrastructure by transmission electron microscopy at E12.5 indicated a moderate increase in mitochondrial content in Hif1a-deficient embryos compared with control littermates. Images also confirmed our previously reported observation that mitochondrial number is higher in the trabecular layer than in the compact myocardial layer (Menendez-Montes et al., 2016), both in control and Hif1a/Nkx2.5 mutant hearts (Figure 4A). Enriched mitochondrial content in Hif1a/NKx2.5 mutants correlated with reduced expression levels of HIF1 target genes involved in mitophagy like Nix and Bnip3 (Zhang et al., 2008) or genes that negatively regulate mitochondrial biogenesis through Myc transcriptional repression like Mxi1 (max interacting protein 1) (Figure 4B). Furthermore, we observed a significant enriched expression of genes related to oxidative phosphorylation determined by gene set enrichment analysis (Table S3, Figure 4C).
Figure 4.

Mitochondrial content and lipid metabolism in Hif1a/Nkx2.5 mutants at E12.5
(A) Transmission electron micrographs of ventricular tissue from a representative E12.5 control embryo (Hif1af/f/Nkx2.5+/+, left) and a mutant littermate (Hif1af/f/Nkx2.5Cre/+, right), showing compact myocardium (top panels) and trabeculae (bottom panels), and quantification of total mitochondria in electron micrographs from E12.5 controls (black bars) and mutants (white bars). Results are expressed as number of mitochondria per tissue area (px2). Scale bars, 5 μm. Bars represent mean ± SEM (n = 4). Student's t test, ∗p value<0.05
(B) RT-qPCR analysis of mitophagy-related genes in E12.5 Hif1a/Nkx2.5 mutant ventricles. Bars (mean ± SEM, n = 3 for Bnip3 and Mxi1 and n = 4 for Nix) represent fold induction relative to baseline expression in littermate controls (red line). Student's t test, ∗p value<0.05.
(C) GSEA enrichment plot for the Hallmark database Oxidative Phosphorylation gene set. The red to blue stripe represents 14,406 genes detected as expressed after differential expression analysis, ranked by logFC. Genes at the left side (colored in red) are more expressed in Hif1a/Nkx2.5 mutants, and those located at the right side (colored in blue) are more expressed in control littermates. Vertical black lines represent the position of members of the Oxidative Phosphorylation gene set along the ranked collection of genes. The green curve represents cumulative enrichment score.
(D) Fold change gene expression determined by RNA-seq of genes involved in fatty acid uptake and catabolism in Hif1a/Nkx2.5 mutants. Red line represents baseline expression in control littermates. Bars represent mean ± SEM (n = 2). All experiments were performed using a comparable number of male and female embryos at each stage.
As mature cardiomyocytes rely on FA oxidation (FAO) for ATP production and cardiac performance, one possible metabolic adaptation of Hif1a-deficient hearts associated with increased mitochondrial content could be an early utilization of FA to provide sufficient ATP levels in the absence of effective glycolysis. However, RNA-seq (Figure 4D) and proteomic results (data not shown) demonstrated that the expression of genes involved in lipid catabolism was not different in control and Hif1a/Nkx2.5 genotypes, at E12.5. These results indicate that inhibited glycolysis due to Hif1a loss is not associated with a compensatory increase in FAO.
In summary, our observations demonstrate that reduced HIF1 signaling promotes an increment of cardiac mitochondrial network and suggest the activation of metabolic compensatory mechanisms other than FAO activation upon glycolytic inhibition in Hif1a/Nkx2.5 mutant embryos.
Amino acid metabolic program is transiently enhanced in cardiac Hif1a-deficient embryos
As indicated earlier, enrichment analysis identified several metabolic processes that could be altered upon deletion of Hif1a in cardiovascular progenitors, some of them related with amino acid metabolism and, specifically, to the “cellular response to amino acid starvation” (Table S2). Complementary functional enrichment analyses allowed to pinpoint more precise functional terms, which suggested alterations in Ala, Leu, Val, Ile, Asn, Asp, Ser, and Gly biosynthesis (Figure 5A, Table S4). These results lead us to hypothesize about the activation of a metabolic reprogramming toward amino acid oxidative catabolism in embryonic cardiomyocytes in the absence of effective glycolysis associated with Hif1a deletion.
Figure 5.

Metabolic adaptations in Hif1a-deficient hearts
(A) Circular plot representing logFC values for genes detected as differentially expressed in mutant (Hif1af/f/Nkx2.5Cre/+) embryos, relative to controls (Hif1af/f/Nkx2.5+/+) at E12.5 (left side), associated to a selection of functional terms related to amino acid metabolism (right side). Functions were detected with Panther by comparison against the Biological Process component of the Gene Ontology database, as well as against the Panther Pathway and Reactome databases. All functional terms were enriched with p value < 0.05. logFC values are color coded: red color denotes higher expression in mutant samples. Ribbons connecting genes and functional terms are colored by process.
(B) Representation of protein statistical weights (wq’) grouped by functional categories (FDR<1%, n = 6) versus protein abundance in Hif1a-deficient hearts relative to control embryos (zq’) at E12.5, as determined by MS/MS proteomics. A displacement right from the experimental curve indicates increased pathway in mutant embryos, whereas a left displacement represents a reduction.
(C) Heatmap representation of mRNA (quantified by RNA-seq) and protein (quantified by MS/MS) of components of glucose (left) and amino acid (right) metabolic pathways. Color code indicated in the legend is calculated as the value found in Hif1a/Nkx2.5 mutants relative to control littermates.
(D) Representative 1H-NMR spectra in ventricular samples from E12.5 control (bottom) and Hif1a/Nkx2.5 mutant embryos (top). The inset highlights the differences in glutamine and total GLU [glutamine (Gln) + glutamate (Glu)] NMR signals.
(E and F) (E) 1H-NMR spectroscopy quantification of glutamine and total GLU abundance in control (black) and Hif1a/Nkx2.5 mutant embryos (white). Bars represent mean ± SEM (n = 3). (F) RT-qPCR analysis of amino acid transporter gene expression in Hif1a mutant ventricular tissue at E14.5 (black bars) and E17.5 (white bars). Bars (mean ± SEM, n = 2–4 for E14.5 and n = 3 for E17.5) represent fold induction relative to baseline expression in littermate controls (red line). For all graphs, Student's t test, ∗p value<0.05, ∗∗∗p value<0.005, n.s. non-significant. Even proportion of male and female embryos has been included to carry out these experiments at each gestational stage.
To validate this hypothesis, we performed global proteomic analysis in Hif1a/Nkx2.5 embryos and control littermates by E12.5. Quantitative analyses revealed a significant increase in the abundance of proteins related to amino acid metabolism and, specifically, glutamine family metabolism (Figure 5B and Table S5). In addition, the decrease observed for proteins related to glycolysis confirmed the inhibited glycolytic gene expression program upon Hif1a deletion (Figure 3). Therefore, quantitative results for both gene expression, by RNA-seq, and protein abundance, by tandem mass spectrometry (MS/MS) proteomics, correlate to a certain extent, showing inhibited glycolysis (Figure 5C, left) and increased amino acid catabolism (Figure 5C, right). Specifically, Hif1a/Nkx2.5 mutants showed increased expression and protein levels of genes contributing to anaplerosis of amino acids into Krebs cycle. These contributions (Figure S2) included aromatic amino acids (Phe, Tyr), polar amino acids (Asn, Asp, Gln, Glu, Ser, and Cys), Pro, Gly, and branched-chain amino acids (Val, Leu, and Ile). Urea cycle was also upregulated, resulting in increased contribution of Arg to Krebs cycle by the generation of fumarate, which can also act as a Krebs cycle intermediate. To determine differences between the amino acid profiles of Hif1a/Nkx2.5 mutant and control hearts, we performed proton nuclear magnetic resonance (1H-NMR) spectroscopy. The ventricular chambers of E12.5 Hif1a/Nkx2.5 mutants showed higher levels of glutamine and total GLU (glutamine + glutamate) (Figures 5D and 5E). This metabolic signature is consistent with the Hif1a/Nkx2.5 transcriptional and proteomic profiles. Finally, to investigate whether this amino acid signature was maintained over time in Hif1a-deficient hearts, we analyzed the expression levels of amino acid transporters by RT-qPCR at E14.5 and E17.5 (Figure 5F). The results showed that gene expression levels of several transporters, such as Slc7a5 (Lat1) (transporter of Trp, Phe, Tyr, and His, and also Met, Val, Leu, and Ile [Yanagida et al., 2001]), Slc7a11 (transporter of Cys [Lim and Donaldson, 2011]), and Slc3a2 (transporter of Val, Leu and Ile by association with Slc7a5 [Kanai et al., 1998]), as well as the leucyl-tRNA synthetase Lars, were still upregulated by E14.5 in Hif1a-deficient hearts, but returned to control-like expression levels by E17.5.
These data indicate that upregulation of amino acid transport is transient and suggest that temporary increase of amino acid catabolism and anaplerosis could act as a compensatory mechanism to overcome the loss of glycolytic metabolism upon Hif1a loss until the FAO is established later in gestation, reflecting the metabolic flexibility of the embryonic heart to adapt to different substrates for energy supply.
ATF4 signaling is upregulated in Hif1a/Nkx2.5 mutant embryos
The fact that there is a transient upregulation of general amino acid catabolism upon Hif1a loss in cardiovascular progenitors suggests the existence of upstream regulators that are temporally induced in the Hif1a/Nkx2.5 mutants. Amino acid metabolism and its transport is tightly regulated through several pathways, including mTOR (mammalian target of rapamycin), GCN2 (general control non-derepresable 29), and ATF4 (Activating Transcription Factor 4), among others (Bröer and Bröer, 2017). Upstream regulator analysis of our RNA-seq data using Ingenuity Pathway Analysis (IPA) in fact shows that ATF4 and CHOP could function as main regulators of a gene set implicated in amino acid metabolism in Hif1a-deficient hearts (Figure 6A and Table S6). ATF4 is a transcriptional regulator that activates the expression of genes involved in amino acid transport and metabolism (Harding et al., 2003) and also responds to nutrient and metabolic stress in hypoxia (Weidemann and Johnson, 2007). Atf4 gene expression, and mRNA levels of its target genes, such as Slc7a11, Slc7a3, Asns, Lars, or Trib3, among others, are upregulated in our Hif1a/Nkx2.5 deletion model by E12.5 (Figure 6B). Furthermore, ATF4 protein levels were increased at E12.5 and E14.5 in cardiac lysates from Hif1a/Nkx2.5 mutant hearts compared with control littermates (Figure 6C). Interestingly, transcriptional upregulation of ATF4 is sustained at E14.5, but its expression returned to control levels by E17.5 (Figure 6D), following an expression pattern similar to that of amino acid transporters (Figure 5F). Because glucose deprivation upon glycolytic inhibition conditions is known to promote the activation of unfolded protein response (UPR) (Badiola et al., 2011; Ikesugi et al., 2006; Vavilis et al., 2016), we decided to examine the expression levels of genes involved in UPR in the Hif1a/Nkx2.5 mutant versus control hearts. The results show a significant upregulation in the expression of genes involved in UPR such as ATF3, ATF4, or CHAC1 among others (Figure 6E).
Figure 6.

Upstream regulators of amino acid catabolism activation in Hif1a-deficient hearts
(A) Regulatory network summarizing the interactions between ATF4 and CHOP with a collection of genes related to amino acid metabolism, detected as differentially expressed at E12.5 in Hif1a/Nkx2.5-deficient hearts relative to controls. The graph is a simplified version of a mechanistic network predicted after IPA's upstream regulator analysis on the complete set of 201 differentially expressed genes. Intensity of red color in target genes is proportional to logFC. Intensity of orange color in regulator genes (ATF4 and CHOP) is proportional to the predicted activation Z score. Arrow-pointed and flat-headed lines represent positive and negative regulation interactions, respectively. Orange and yellow lines represent congruent and non-congruent connections, respectively, relative to the predicted activation state of regulators. The inset below summarizes Z score value and enrichment p value for ATF4 and CHOP, as well as the number of differentially expressed genes that are regulated by each of them.
(B) Relative expression of genes related to amino acid metabolism downstream of ATF4 determined by RNA-seq at E12.5 in Hif1a/Nkx2.5 mutant versus control ventricles (n = 2). Student's t test. ∗p value<0.05, ∗∗∗ p value<0.005.
(C) Representative immunoblot out of 5 against ATF4 (upper panel) and Vinculin (lower panel) from ventricular heart lysates of control (Hif1af/f/Nkx2.5+/+) and Hif1a/Nkx2.5 mutant (Hif1af/f/Nkx2.5Cre/+) embryos at E12.5 and E14.5.
(D) RT-qPCR analysis of Atf4 gene expression at E14.5 (black bar) and E17.5 (white bar) in Hif1a/Nkx2.5 mutant ventricular tissue. Bars (mean ± SEM, n = 5 for E14.5 and n = 3 for E17.5) represent fold induction relative to baseline expression in littermate controls (red line). Student's t test, ∗ p value<0.05, n.s. non-significant.
(E) Heatmap representing RNA-seq based, normalized expression levels for genes involved in the unfolded protein response (UPR). The UPR gene set, as defined in the Hallmark database, was detected as enriched in mutant embryos after GSEA, although enrichment was not statistically significant (nominal p value = 0.31). Genes presented in the heatmap correspond to the leading-edge subset, this is, those mostly contributing to the calculated enrichment score. All experiments and analyses were performed using equivalent amount of male and female embryos at each stage.
All together these results led us to suggest that Hif1a loss in cardiovascular progenitors induces the temporal activation of ATF4 expression and ATF4-mediated amino acid response probably trough UPR activation in response to glycolytic impairment.
Loss of Hif1a in Nkx2.5 progenitors leads to transient induction of HIF2 by midgestation
In the absence of active HIF1 cascade, HIF2α, an alternative HIFα isoform able to form functional heterodimers with ARNT, could play a compensatory role. To explore this possibility, we analyzed HIF2α abundance by western blot and found protein expression induction by E12.5 in the Hif1a/Nkx2.5-deficient hearts and comparable levels by E14.5 relative to controls (Figures 7A and 7B). A similar induced expression by E12.5 was observed for the HIF2 target gene, PAI-1 (plasminogen activator inhibitor 1) (Figures 7C and 7D). Interestingly, glucose deprivation has been shown to activate HIF2 signaling in an acetylation-dependent manner (Chen et al., 2015). We hypothesize that this adaptive activation of HIF2 signaling could partially contribute together with ATF4 upregulation to the transcriptional induction of amino acid transporters observed in the cardiac Hif1a/Nkx2.5-deficient model. Indeed, HIF2α has been involved in the direct control of Slc7a5 (Lat1) expression by binding to the proximal promoter of the gene in renal clear cell carcinoma, as well as in lung, liver, and glioblastoma cells (GBCs) (Corbet et al., 2014; Elorza et al., 2012; Zhang et al., 2020). Additionally, HIF2 signaling has been related to the expression of other amino acid metabolism-related genes that are increased in Hif1a/Nkx2.5 mutants compared with controls, such as Mthfd2 or Atf3 (Green et al., 2019; Turchi et al., 2008). Hence, these results suggest that HIF2 could further participate in the metabolic reprogramming toward amino acid catabolic pathways in the absence of effective HIF1 signaling.
Figure 7.

HIF2 signaling induction upon Hif1a deletion
(A) Representative immunoblot against HIF2α (upper panel) and Tubulin (lower panel) in heart lysates of control (Hif1af/f/Nkx2.5+/+) and Hif1a/Nkx2.5 mutant (Hif1af/f/Nkx2.5Cre/+) embryos at E12.5 and E14.5.
(B) Quantification of HIF2α band intensity normalized by Tubulin as loading control (n = 3).
(C) Representative immunoblot against PAI-1 (upper panel) and Vinculin (lower panel) in heart lysates of control (Hif1af/f/Nkx2.5+/+) and Hif1a/Nkx2.5 mutant (Hif1af/f/Nkx2.5Cre/+) embryos at E12.5 and E14.5.
(D) Quantification of PAI-1 band intensity normalized by Vinculin as loading control (n = 3). For all graphs, bars (mean ± SEM, n = 3) represent fold induction relative to baseline expression in littermate controls at E12.5 or E14.5. Student's t test. ∗p value<0.05, n.s: non-significant. Comparable proportion of male and female embryos has been included to perform these experiments at each gestational stage.
(E) Model representing the embryonic myocardium by E12.5. Compact myocardium is mainly glycolytic (yellowish) by the action of HIF1 signaling, whereas trabeculae rely more on mitochondrial metabolism (orange) in control embryos (left). In Hif1a mutants (right), glycolysis is compromised and the whole myocardium relies on mitochondrial metabolism, displaying higher mitochondrial content, and favoring the use of amino acids as energy source parallel to the activation of ATF4 and HIF2 signaling.
In summary, our data demonstrate that HIF1 signaling in Nkx2.5 cardiac progenitors and cardiomyocytes is dispensable for proper heart formation and that the absence of Hif1a triggers a cardiac metabolic reprogramming, enhancing temporal amino acid catabolism to ensure sufficient ATP and biosynthetic precursors to sustain cardiac growth and function even in the absence of glycolysis (Figure 7E). Importantly, these adaptations might be relevant in the adulthood under pathological scenarios associated with oxygen signaling like pulmonary hypertension or cardiomyopathy toward the development of novel drugs against new metabolic targets.
Discussion
Here, we describe that Hif1a loss in Nkx2.5 cardiovascular progenitors or cardiomyocytes causes glycolytic program inhibition in the compact myocardium (CM) by E12.5, without compromising normal cardiac development and embryonic viability. Our results show that upon Hif1a deletion, the embryonic myocardium conserves FAO capacity but exhibits the ability to activate metabolic programs oriented to amino acid catabolism, together with an increase in the mitochondrial content by E12.5. Taken together, our findings point out the metabolic versatility of the embryonic heart and conciliate the discrepancies from previous deletion models of Hif1a in cardiovascular progenitors.
Integration with previous Hif1a deletion model in the embryonic heart
As outlined in the introduction, there is a lack of consensus between previous reports on cardiac embryonic mouse models of Hif1a loss (Guimarães-Camboa et al., 2015; Huang et al., 2004; Krishnan et al., 2008). The Hif1a haploinsufficiency of some of these models outside the heart Nkx2.5 territories might cause extracardiac affections, such as vascular or placental, that could significantly influence the described phenotype. Indeed, the use of a Hif1a-null allele has been reported to cause cardiovascular malformations associated with maternal diabetes (Bohuslavova et al., 2013). In this regard, the exhaustive characterization by Guimaraes-Camboa et al. at gene expression level extensively overlaps with our RNA-seq data, except in terms of stress and apoptotic pathways, upregulated only in the null-allele context of their mutant.
Moreover, we have also analyzed a Hif1a null/floxed model in Nkx2.5 progenitors in parallel with the homozygous floxed model described here. We found that although glycolytic inhibition by E14.5 is comparable in both models (data not shown), only the null/floxed mice exhibited embryonic lethality (5% retrieved versus 25% expected, p value 0.0029, n = 7 litters), in contrast with the observed viability for the homozygous floxed mice. This observation, together with other results, supports the notion that cardiac HIF1 is dispensable for heart development, although an extensive comparison, in terms of gene expression in placental and vascular embryonic tissue, between the homozygous floxed and the null/floxed models would be necessary to exclude extracardiac influences of Hif1a deficiency affecting heart development as reported by Guimaraes-Camboa and colleagues, and also observed by the lab of Steven Fisher using a global conditional Hif1a knockout mice (Kenchegowda et al., 2014).
Amino acid catabolism and metabolic versatility of the embryonic heart
A key finding of our investigation is the fact that the embryonic myocardium is able to upregulate alternative metabolic pathways (amino acids transportation and catabolism), and to promote mitochondrial enrichment that could support the ATP demand upon glycolytic inhibition subsequent to Hif1a loss. The use of amino acids as cardiac metabolic fuel has been proposed mainly in oxygen-deprived scenarios (Bing et al., 1954; Julia et al., 1990). Amino acids provide, by deamination, carbon skeletons that can be converted into pyruvate, alpha-ketoglutarate, succinyl-CoA, fumarate, oxalacetate, acetyl-CoA, and acetoacetyl-CoA, all of them metabolites that can be incorporated into the Krebs cycle (Evans and Heather, 2016; Neubauer, 2007). As detailed earlier, our cardiac Hif1a-deficient model upregulates, both at the transcriptional and protein levels, a variety of amino acid transporters and biosynthetic and catabolic enzymes, which can replenish the Krebs cycle upon glucose deprivation. Moreover, the fact that this upregulation is accompanied by an increase in mitochondrial content indicates that the embryonic heart, in the absence of Hif1a, readapts its metabolism to maintain enough ATP levels and building blocks, without compromising the normal protein synthesis required for myocardium development and embryo viability.
Interestingly, this adaptation is transient and reversible, as revealed by the control-like levels of amino acid transporters and Atf4 transcripts found at later stages by E17.5, without precluding the embryonic metabolic switch toward FAO previously described (Menendez-Montes et al., 2016). These observations indicate that the embryonic myocardium has the plasticity to modulate its metabolism to adapt to the energetic demand and nutrient availability. Moreover, in addition to this interesting role of amino acid catabolism activation in the embryonic context, the use of amino acids as an alternative energy source could be an attracting option to achieve cardioprotection and recovery after cardiac injury. In this regard, some of the enzymes upregulated in our massive screenings in Hif1a-deficient hearts are involved in Ser biosynthesis and one-carbon cycle, including Phgdh, Psph, and Shmt1. These pathways have been previously described to increase glutathione levels and protect the heart against oxidative stress (Zhou et al., 2017), also in a context of myocardial hypertrophy (Padrón-Barthe et al., 2018).
Origin of catabolized amino acids in Hif1a-deficient hearts
An interesting open aspect of the metabolic adaptation exhibited by the Hif1a-deficient hearts is the source of amino acid supply during glycolytic inhibition upon Hif1a loss. In this regard, two potential sources could be considered. First, protein-forming amino acids could be recycled through autophagy. This hypothesis is reasonable considering the context of the embryonic heart, where protein turnover, especially transcription factors, occurs fast and at a high rate (Merz et al., 1981). Moreover, a positive nitrogen balance has been reported in both adult rat and human hearts, indicative of rapid turnover of tissue proteins (Sprinson and Rittenberg, 1949). Interestingly, our Hif1a mutant embryos showed increased transcription of p62, a cargo-recognizing protein involved in autophagic degradation of cellular proteins (Lim et al., 2015). Although an extensive characterization analyzing autophagy, pro- and anti-autophagic signaling pathways, and protein labeling and turnover would be needed to further investigate this hypothesis, the fact that autophagy could be involved in this metabolic adaptation suggests an exciting link between cardiac metabolism, hypoxia, and autophagy.
Another possible source of amino acids in Hif1a-deficient hearts is fetal circulation. Even though an extensive characterization of fetal blood nutrient content over gestation has not been reported, the transcriptional increase in several membrane amino acid transporters observed in Hif1a/Nkx2.5 mutants suggests that Hif1a-deficient cardiomyocytes could be obtaining amino acids directly from embryonic circulation. Interestingly, cardiac amino acids uptake in human subjects infused intravenously with protein hydrolysate increases by 245% (Bing et al., 1954), showing that the heart can respond to blood amino acids levels. Moreover, the regulation of amino acid transporters' expression in the placenta is essential for maintaining high levels of amino acids in the fetal blood to sustain embryo growth (Díaz et al., 2014). In this regard, an increased cardiac uptake of amino acids in the Hif1a-deficient embryo could result in increased amino acids supply through the placenta that might respond to some secreted cues in the absence of cardiac HIF1 signaling.
Molecular determinants of amino acid catabolism activation
Amino acid metabolism and transport is tightly regulated through several pathways, including mTOR, GCN2, and G-protein-coupled receptors, among others (Bröer and Bröer, 2017). ATF4 is a transcriptional regulator that activates the expression of genes involved in amino acid transport and metabolism (Harding et al., 2003) and also responds to nutrient and metabolic stress in hypoxia (Weidemann and Johnson, 2007). Atf4 gene expression is positively regulated in the Hif1a/Nkx2.5 deletion model at both transcriptional and protein levels. In addition, upstream regulators' analysis identified ATF4 and CHOP (Ddit3) as putative regulators of amino acid metabolism in Hif1a-deficient hearts. ATF4 is an essential factor for amino acid starvation, by activating the gene expression of genes containing amino acid response elements (AARE) (Zhang et al., 2010) and regulates CHOP expression (Averous et al., 2004).
Loss of HIF1α signaling results in downregulated expression of glycolytic enzymes. Glucose deprivation, in turn, has been shown to cause activation of UPR (Badiola et al., 2011; Ikesugi et al., 2006; Vavilis et al., 2016). In our Hif1a deletion model, reduced glycolysis was accompanied by increased expression of UPR genes, suggesting that Hif1a deletion could contribute to ATF4 activation through glycolytic inhibition and UPR activation in the embryonic myocardium.
Interestingly, the Hif1a deletion model by Guimarães-Camboa et al. (2015) also shows increased ATF4 signaling in Hif1a-deficient hearts, supporting ATF4 as one of the main regulators of the described metabolic adaptation upon loss of effective glycolysis in Hif1a-deficient hearts. Moreover, as ATF4 is upregulated in both animal models, and considering the lack of lethality of our floxed/floxed mice despite an efficient Cre-mediated recombination of Hif1a floxed exon 2, the upregulation of ATF4 does not seem to be responsible for the embryonic lethality reported by Zambon's/Evan's groups that, in contrast, reported the activation of p53 stress pathway. Further analysis of p53-associated effects in the Hif1a null/floxed mice, as well as extracardiac impact of elimination of one Hif1a copy in the germ line might contribute to understand the phenotype of null/floxed mice.
Previous work in renal clear cell carcinoma, liver, and lung has identified a novel axis connecting HIF2 with mTOR through the action of the L-type amino acid transporter LAT1 (Elorza et al., 2012). Furthermore, mTORC1 has been reported to activate purine synthesis through the activation of ATF4 via a cellular stress-independent mechanism (Ben-Sahra et al., 2016). Hence, we hypothesized that upregulation of HIF2α in the Hif1a/Nkx2.5 mutant hearts might lead to the activation of mTOR pathway that could further connect with ATF4 activation. However, analysis of the phosphorylation state of mTOR targets Factor 4E Binding Protein (4EBP1) and S6 Kinase (S6K) by western blot and immunohistochemistry revealed inhibition of these downstream effectors of mTOR in the Hif1a/Nkx2.5 mutant hearts (data not shown), demonstrating that HIF2/LAT1/mTOR axis is not conserved in the embryonic myocardium and suggesting that ATF4 upregulation occurs in a HIF1 and mTORC1-independent manner, probably associated to enhanced UPR activation due to impaired glycolysis.
We also show that Hif1a/Nkx2.5 mutants display higher levels of glutamine and glutamate compared with controls. Both amino acids are important carbon and nitrogen sources that can be used for energy production as well as for nucleotide and protein synthesis in cancer cells. In some tissues like the brain, part of glutamate can arise from branched-chain amino acids like leucine, isoleucine, or valine, which are transported into the cytosol via L-type amino acid transporters (LAT1-4) and converted into branched-chain alpha ketoacids through the action of cytosolic (BCAT1) or mitochondrial (BCAT2) branched-chain aminotransferases (Yudkoff, 1997). A recent article on GBCs describes the regulation of branched-chain amino acid reprogramming by HIFs and shows that the main LAT isoform expressed in GBC is LAT1. The authors show that Lat1 promoter can be transactivated by both HIF1 and HIF2, although BCAT1, associated to GBC proliferation, is only regulated by HIF1 in this cell type (Zhang et al., 2020). In contrast, in the Hif1a/Nkx2.5 mutant hearts we have observed upregulation of LAT1 and BACT1 independently of HIF1 abrogation, suggesting that in the embryonic heart HIF2 might play an important role in controlling the expression of these key elements of BCAA metabolic reprogramming. Further functional and molecular characterization of HIF1/HIF2 double mutant mice would contribute to confirm this hypothesis. Another important aspect for future research would be to determine the relative contribution of HIF2 versus ATF4 in the amino acid metabolic reprogramming observed in the Hif1a/Nkx2.5 mutant mice as both transcription factors share several common target genes related to amino acid transport and catabolism.
In summary, our work demonstrates that HIF1 elimination in cardiac progenitors and cardiomyocytes is dispensable for heart development and function, and that impaired glycolysis during cardiogenesis in Hif1a-deficient mice induces a transient reprogramming of amino acid metabolism concomitant with HIF2 and ATF4 activation. These observations uncover the metabolic flexibility of the embryonic heart that might share common adaptive bioenergetics responses to cancer cells under compromised glucose and FAO availability.
Limitations of the study
Performing metabolic flow studies during embryonic development is challenging as metabolites can be utilized by the maternal metabolism before reaching the embryo. Hence, future functional assays on amino acids utilization will contribute to reinforce our transcriptomic and proteomic data on amino acids import and catabolism activation.
The definitive role of ATF4 and HIF2 in metabolic reprogramming to amino acid catabolism in the absence of glycolytic program in the Hif1a cKO will require future genetic analysis of single and double knockout models.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Silvia Martín-Puig (silvia.martin@cnic.es; silvia.martin@ufv.es).
Materials availability
Mouse lines generated in this study are available upon request from the Lead Contact with a completed Materials Transfer Agreement and might require additional payment.
Data and code availability
RNA-seq data have been deposited in NCBI's Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo) and are accessible through GEO Series accession number GSE164453.
Proteomic datasets (raw files, protein databases, search parameters, and results) have been deposited in the Peptide Atlas repository (http://www.peptideatlas.org) and are available through the accession number PASS01236.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors thank Lorena Flores for echocardiography technical assistance, Raquel Baeza and Mercedes de la Cueva for animal housing and handling, and CNIC Microscopy, Genomics and Histology Core Facilities for technical assistance. We thank Miguel Torres Sanchez, Jose Antonio Enriquez, Luke Szweda, and Hesham Sadek for critical discussion of the manuscript. This project has been supported by Fundación Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC), Spain and by grants to S.M.-P. from the European Research Council, European Union, FP7-PEOPLE-2010-RG_276891; Fundación TV3 La Marató, Spain, 201507.30.31; Comunidad de Madrid (CAM); Spain and European Union (EU), B2017/BMD-3875; Instituto de Salud Carlos III, Spain, PI17/01817 cofunded by European Regional Development Fund (ERDF); Universidad Francisco de Vitoria (UFV), Spain and LeDucq Foundation, France, 17CVD04. I.M.-M. was supported by La Caixa-CNIC and Fundacion Alfonso Martín Escudero fellowships, Spain. T.A.-G. was supported by a predoctoral award granted by CAM/EU and UFV, Spain, PEJD-2018-PRE/SAL-9529 and SM-P by a Contrato de Investigadores Miguel Servet (CPII16/00050) and UFV, Spain.
Author contributions
S.M.-P. conceived and supervised the project. I.M.-M. performed most of the in vitro and in vivo experiments with assistance from B.E., B.P., and T.A.-G., prepared the figures, and together with S.M.-P. analyzed the data and co-wrote the manuscript. M.J.G. performed all bioinformatic analysis and contributed to manuscript editing and discussion. E.B. and J.V. performed proteomic analysis. A.V.A. and L.J.J.-B. carried out echocardiographic studies. J.L.I.-G., A.F., and J.R.-C. performed metabolic analysis. Funding acquisition by S.M.-P.
Declaration of interests
The authors declare no competing interests.
Published: February 19, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2021.102124.
Supplemental information
Total detected transcript quantification and differentially expressed genes (DEG) in control (Hif1af/f/Nkx2.5+/+) and Hif1a/Nkx2.5 mutant (Hif1af/f/Nkx2.5Cre/+) hearts at E12.5. The table is composed of two spreadsheets that describe raw and normalized expression values for each gene in each sample, averaged values per condition and for all samples, as well as fold change, logFC, p value, and Benjamini-Hochberg-adjusted p value, in a mutant versus control comparison. The first spreadsheet describes expression values for 201 genes detected as differentially expressed with Benjamini-Hochberg adjusted p values < 0.055. 83 genes were upregulated (red background) in Hif1a-deficient hearts versus control littermates, and 118 genes were downregulated (blue background). The second spreadsheet describes expression values for the whole collection of 14,406 genes detected in the RNA-seq.
Over-represented Biological Process GO terms associated to the collection of 201 differentially expressed genes (adjusted p value < 0.055), as detected with GOrilla, with p value < 0.001 (default threshold). The table describes, for each GO term, the number of mapped annotated genes in the reference dataset, the number of mapped annotated genes in the target set, its fold enrichment, and p value. Dispensable terms were eliminated after processing the list with REVIGO.
Enriched gene sets from the Hallmark, Biocarta, and Reactome databases associated to the collection of 14,406 expressed genes, as detected with GSEA, with p value < 0.05. The table describes, for each gene set, the calculated normalized enrichment score and associated p value.
Over-represented Reactome pathways, PANTHER pathways, and Biological Process Gene Ontology terms associated to the collection of 201 differentially expressed genes (adjusted p value < 0.055), as detected with PANTHER, with p value < 0.05. The table describes, for each pathway or GO term, the number of mapped annotated genes in the reference dataset, the number of mapped annotated genes in the target set, its fold enrichment, and associated p value.
The table shows the list of proteins quantified by mass spectrometry analysis. ZQ (KO/WT) values are standardized log2-ratio averages of proteins from mutant (Hif1af/f/Nkx2.5Cre/+) samples relative to embryos (Hif1af/f/Nkx2.5+/+), merged from the 3 biological replicates. 1% FDR is used as criterion for protein statistically significant abundance change.
The table presents, in three spreadsheets, functional enrichment results generated with IPA for the set of 201 genes detected as differentially expressed (adjusted p value < 0.055), for the following analysis types: Canonical Pathways, Upstream Regulators and Diseases, and Biofunctions (Downstream Effect analysis). The key parameters provided for the first three analysis types describe enrichment significance (p value and Benjamini-Hochberg-adjusted p value) and activation Z score (a prediction of the activity of the pathway, regulator or function; in this case, positive and negative values denote higher activity in mutant or control samples, respectively). Absolute Z score values higher than 2 are considered relevant.
References
- Averous J., Bruhat A., Jousse C., Carraro V., Thiel G., Fafournoux P. Induction of CHOP expression by amino acid limitation requires both ATF4 expression and ATF2 phosphorylation. J. Biol. Chem. 2004;279:5288–5297. doi: 10.1074/jbc.M311862200. [DOI] [PubMed] [Google Scholar]
- Badiola N., Penas C., Miñano-Molina A., Barneda-Zahonero B., Fadó R., Sánchez-Opazo G., Comella J.X., Sabriá J., Zhu C., Blomgren K., et al. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011;2:e149. doi: 10.1038/cddis.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I., Hoxhaj G., Ricoult S.J.H., Asara J.M., Manning B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–733. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing R.J., Siegel A., Ungar I., Gilbert M. Metabolism of the human heart: II. Studies on fat, ketone and amino acid metabolism. Am. J. Med. 1954;16:504–515. doi: 10.1016/0002-9343(54)90365-4. [DOI] [PubMed] [Google Scholar]
- Bohuslavova R., Skvorova L., Sedmera D., Semenza G.L., Pavlinkova G. Increased susceptibility of HIF-1α heterozygous-null mice to cardiovascular malformations associated with maternal diabetes. J. Mol. Cell. Cardiol. 2013;60:129–141. doi: 10.1016/j.yjmcc.2013.04.015. [DOI] [PubMed] [Google Scholar]
- Bröer S., Bröer A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem. J. 2017;474:1935–1963. doi: 10.1042/BCJ20160822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerychova R., Pavlinkova G. HIF-1, metabolism, and diabetes in the embryonic and adult heart. Front. Endocrinol. (Lausanne) 2018;9:460. doi: 10.3389/fendo.2018.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., Xu M., Nagati J.S., Hogg R.T., Das A., Gerard R.D., Garcia J.A. The acetate/ACSS2 switch regulates HIF-2 stress signaling in the tumor cell microenvironment. PLoS One. 2015;10:e0116515. doi: 10.1371/journal.pone.0116515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S., Arrell D.K., Faustino R.S., Terzic A., Dzeja P.P. Glycolytic network restructuring integral to the energetics of embryonic stem cell cardiac differentiation. J. Mol. Cell. Cardiol. 2010;48:725–734. doi: 10.1016/j.yjmcc.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S., Dzeja P.P., Faustino R.S., Perez-terzic C., Behfar A., Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Natl. Institutes Heal. 2011;4:1–12. doi: 10.1038/ncpcardio0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbet C., Draoui N., Polet F., Pinto A., Drozak X., Riant O., Feron O. The SIRT1/HIF2α axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy. Cancer Res. 2014;74:5507–5519. doi: 10.1158/0008-5472.CAN-14-0705. [DOI] [PubMed] [Google Scholar]
- Díaz P., Powell T.L., Jansson T. The role of placental nutrient sensing in maternal-fetal resource Allocation1. Biol. Reprod. 2014;91:82. doi: 10.1095/biolreprod.114.121798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elorza A., Soro-Arnáiz I., Meléndez-Rodríguez F., Rodríguez-Vaello V., Marsboom G., de Cárcer G., Acosta-Iborra B., Albacete-Albacete L., Ordóñez A., Serrano-Oviedo L., et al. HIF2α acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol. Cell. 2012;48:681–691. doi: 10.1016/j.molcel.2012.09.017. [DOI] [PubMed] [Google Scholar]
- Evans R.D., Heather L.C. Surg. (United Kingdom); 2016. Metabolic Pathways and Abnormalities. [Google Scholar]
- Fukuda R., Zhang H., Kim J.W., Shimoda L., Dang C.V., Semenza G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- Green N.H., Galvan D.L., Badal S.S., Chang B.H., LeBleu V.S., Long J., Jonasch E., Danesh F.R. MTHFD2 links RNA methylation to metabolic reprogramming in renal cell carcinoma. Oncogene. 2019;38:6211–6225. doi: 10.1038/s41388-019-0869-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimarães-Camboa N., Stowe J., Aneas I., Sakabe N., Cattaneo P., Henderson L., Kilberg M.S., Johnson R.S., Chen J., McCulloch A.D., et al. HIF1α represses cell stress pathways to allow proliferation of hypoxic fetal cardiomyocytes. Dev. Cell. 2015;33:507–521. doi: 10.1016/j.devcel.2015.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H.P., Zhang Y., Zeng H., Novoa I., Lu P.D., Calfon M., Sadri N., Yun C., Popko B., Paules R., et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Huang Y., Hickey R.P., Yeh J.L., Liu D., Dadak A., Young L.H., Johnson R.S., Giordano F.J. Cardiac myocyte-specific HIF-1alpha deletion alters vascularization, energy availability, calcium flux, and contractility in the normoxic heart. FASEB J. 2004;18:1138–1140. doi: 10.1096/fj.04-1510fje. [DOI] [PubMed] [Google Scholar]
- Ikesugi K., Mulhern M.L., Madson C.J., Hosoya K.I., Terasaki T., Kador P.F., Shinohara T. Induction of endoplasmic reticulum stress in retinal pericytes by glucose deprivation. Curr. Eye Res. 2006;31:947–953. doi: 10.1080/02713680600966785. [DOI] [PubMed] [Google Scholar]
- Jiang B.H., Zheng J.Z., Leung S.W., Roe R., Semenza G.L. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J. Biol. Chem. 1997;272:19253–19260. doi: 10.1074/jbc.272.31.19253. [DOI] [PubMed] [Google Scholar]
- Jiao K., Kulessa H., Tompkins K., Zhou Y., Batts L., Baldwin H.S., Hogan B.L.M. An essential role of Bmp4 in the atrioventricular septation of the mouse heart service. Genes Dev. 2003;17:2362–2367. doi: 10.1101/gad.1124803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julia P., Young H., Buckberg G., Kofsky E., Bugyi H. Studies of myocardial protection in the immature heart. II. Evidence for importance of amino acid metabolism in tolerance to ischemia. J. Thorac. Cardiovasc. Surg. 1990;100:888–895. [PubMed] [Google Scholar]
- Kaelin W.G., Ratcliffe P.J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kanai Y., Segawa H., Miyamoto K.I., Uchino H., Takeda E., Endou H. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98) J. Biol. Chem. 1998;273:23629–23632. doi: 10.1074/jbc.273.37.23629. [DOI] [PubMed] [Google Scholar]
- Kenchegowda D., Liu H., Thompson K., Luo L., Martin S.S., Fisher S.A. Vulnerability of the developing heart to oxygen deprivation as a cause of congenital heart defects. J. Am. Heart Assoc. 2014;3:e000841. doi: 10.1161/JAHA.114.000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.W., Tchernyshyov I., Semenza G.L., Dang C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Krishnan J., Ahuja P., Bodenmann S., Knapik D., Perriard E., Krek W., Perriard J.-C. Essential role of developmentally activated hypoxia-inducible factor 1 for cardiac morphogenesis and function. Circ. Res. 2008;103:1139–1146. doi: 10.1161/01.RES.0000338613.89841.c1. [DOI] [PubMed] [Google Scholar]
- Lee Y.M., Jeong C.-H., Koo S.-Y., Son M.J., Song H.S., Bae S.-K., Raleigh J.A., Chung H.-Y., Yoo M., Kim K.-W. Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: a possible signal for vessel development. Dev. Dyn. 2001;220:175–186. doi: 10.1002/1097-0177(20010201)220:2<175::AID-DVDY1101>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Lei L., Mason S., Liu D., Huang Y., Marks C., Hickey R., Jovin I.S., Pypaert M., Johnson R.S., Giordano F.J. Hypoxia-inducible factor-dependent degeneration, failure, and malignant transformation of the heart in the absence of the von Hippel-Lindau protein. Mol. Cell Biol. 2008;28:3790–3803. doi: 10.1128/MCB.01580-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J., Lachenmayer M.L., Wu S., Liu W., Kundu M., Wang R., Komatsu M., Oh Y.J., Zhao Y., Yue Z. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015;11:e1004987. doi: 10.1371/journal.pgen.1004987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J.C., Donaldson P.J. Focus on Molecules: the cystine/glutamate exchanger (System xc -) Exp. Eye Res. 2011;92:162–163. doi: 10.1016/j.exer.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Llurba Olive E., Xiao E., Natale D.R., Fisher S.A. Oxygen and lack of oxygen in fetal and placental development, feto–placental coupling, and congenital heart defects. Birth Defects Res. 2018;110:1517–1530. doi: 10.1002/bdr2.1430. [DOI] [PubMed] [Google Scholar]
- Lopaschuk G.D., Jaswal J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010;56:130–140. doi: 10.1097/FJC.0b013e3181e74a14. [DOI] [PubMed] [Google Scholar]
- Majmundar A.J., Wong W.J., Simon M.C. Hypoxia-Inducible factors and the response to hypoxic stress. Mol. Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Puig S., Wang Z., Chien K.R. Lives of a heart cell: tracing the origins of cardiac progenitors. Cell Stem Cell. 2008;2:320–331. doi: 10.1016/j.stem.2008.03.010. [DOI] [PubMed] [Google Scholar]
- Menendez-Montes I., Escobar B., Palacios B., Gómez M.J., Izquierdo-Garcia J.L., Flores L., Jiménez-Borreguero L.J., Aragones J., Ruiz-Cabello J., Torres M., Martin-Puig S. Myocardial VHL-HIF signaling controls an embryonic metabolic switch essential for cardiac maturation. Dev. Cell. 2016;39:724–739. doi: 10.1016/j.devcel.2016.11.012. [DOI] [PubMed] [Google Scholar]
- Merz E.A., Brinster R.L., Brunner S., Chen H.Y. Protein degradation during preimplantation development of the mouse. J. Reprod. Fertil. 1981;61:415–418. doi: 10.1530/jrf.0.0610415. [DOI] [PubMed] [Google Scholar]
- Moses K.A., DeMayo F., Braun R.M., Reecy J.L., Schwartz R.J. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis. 2001;31:176–180. doi: 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- Nanka O., Krizova P., Fikrle M., Tuma M., Blaha M., Grim M., Sedmera D. Abnormal myocardial and coronary vasculature development in experimental hypoxia. Anat. Rec. 2008;291:1187–1199. doi: 10.1002/ar.20738. [DOI] [PubMed] [Google Scholar]
- Neubauer S. The failing heart--an engine out of fuel. N. Engl. J. Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- Padrón-Barthe L., Villalba-Orero M., Gómez-Salinero J.M., Acín-Pérez R., Cogliati S., López-Olañeta M., Ortiz-Sánchez P., Bonzón-Kulichenko E., Vázquez J., García-Pavía P., et al. Activation of serine one-carbon metabolism by calcineurin Aβ1 reduces myocardial hypertrophy and improves ventricular function. J. Am. Coll. Cardiol. 2018;71:654–667. doi: 10.1016/j.jacc.2017.11.067. [DOI] [PubMed] [Google Scholar]
- Piao L., Fang Y.H., Parikh K., Ryan J.J., Toth P.T., Archer S.L. Cardiac glutaminolysis: a maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J. Mol. Med. 2013;91:1185–1197. doi: 10.1007/s00109-013-1064-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouyssegur J., Dayan F., Mazure N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- Puente B.N., Kimura W., Muralidhar S.A., Moon J., Amatruda J.F., Phelps K.L., Grinsfelder D., Rothermel B.A., Chen R., Garcia J.A., et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ream M., Ray A.M., Chandra R., Chikaraishi D.M. Early fetal hypoxia leads to growth restriction and myocardial thinning. AJP Regul. Integr. Comp. Physiol. 2008;295:R583–R595. doi: 10.1152/ajpregu.00771.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinson D.B., Rittenberg D. The rate of utilization of ammonia for protein synthesis. J. Biol. Chem. 1949;180:707–714. [PubMed] [Google Scholar]
- Sugishita Y., Watanabe M., Fisher S.A. Role of myocardial hypoxia in the remodeling of the embryonic avian cardiac outflow tract. Dev. Biol. 2004;267:294–308. doi: 10.1016/j.ydbio.2003.11.017. [DOI] [PubMed] [Google Scholar]
- Turchi L., Aberdam E., Mazure N., Pouysségur J., Deckert M., Kitajima S., Aberdam D., Virolle T. Hif-2alpha mediates UV-induced apoptosis through a novel ATF3-dependent death pathway. Cell Death Differ. 2008;15:1472–1480. doi: 10.1038/cdd.2008.74. [DOI] [PubMed] [Google Scholar]
- Vavilis T., Delivanoglou N., Aggelidou E., Stamoula E., Mellidis K., Kaidoglou A., Cheva A., Pourzitaki C., Chatzimeletiou K., Lazou A., et al. Oxygen–glucose deprivation (OGD) modulates the unfolded protein response (UPR) and inflicts autophagy in a PC12 hypoxia cell line model. Cell. Mol. Neurobiol. 2016;36:701–712. doi: 10.1007/s10571-015-0250-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Buckingham M. The formation of the embryonic mouse heart: heart fields and myocardial cell lineages. Ann. N. Y. Acad. Sci. 2010;1188:15–24. doi: 10.1111/j.1749-6632.2009.05078.x. [DOI] [PubMed] [Google Scholar]
- Weidemann A., Johnson R.S. A wrinkle in the unfolding of hypoxic response: HIF and ATF4. Blood. 2007;110:3492–3493. [Google Scholar]
- Yanagida O., Kanai Y., Chairoungdua A., Kim D.K., Segawa H., Nii T., Cha S.H., Matsuo H., Fukushima J., Fukasawa Y., et al. Human L-type amino acid transporter 1 (LAT1): characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta - Biomembr. 2001;1514:291–302. doi: 10.1016/s0005-2736(01)00384-4. [DOI] [PubMed] [Google Scholar]
- Yudkoff M. Brain metabolism of branched-chain amino acids. Glia. 1997;21:92–98. doi: 10.1002/(sici)1098-1136(199709)21:1<92::aid-glia10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Yue M., Jiang J., Gao P., Liu H., Qing G. Oncogenic MYC activates a feedforward regulatory loop promoting essential amino acid metabolism and tumorigenesis. Cell Rep. 2017;21:3819–3832. doi: 10.1016/j.celrep.2017.12.002. [DOI] [PubMed] [Google Scholar]
- Zhang B., Chen Y., Shi X., Zhou M., Bao L., Hatanpaa K.J., Patel T., DeBerardinis R.J., Wang Y., Luo W. Regulation of branched-chain amino acid metabolism by hypoxia-inducible factor in glioblastoma. Cell. Mol. Life Sci. 2020 doi: 10.1007/s00018-020-03483-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Bosch-Marce M., Shimoda L.A., Tan Y.S., Baek J.H., Wesley J.B., Gonzalez F.J., Semenza G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang H., Gao P., Fukuda R., Kumar G., Krishnamachary B., Zeller K.I., Dang C.V., Semenza G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–420. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Jin Y., Williams T.A., Burtenshaw S.M., Martyn A.C., Lu R. Amino acid deprivation induces CREBZF/Zhangfei expression via an AARE-like element in the promoter. Biochem. Biophys. Res. Commun. 2010;391:1352–1357. doi: 10.1016/j.bbrc.2009.12.059. [DOI] [PubMed] [Google Scholar]
- Zhou X., He L., Wu C., Zhang Y., Wu X., Yin Y. Serine alleviates oxidative stress via supporting glutathione synthesis and methionine cycle in mice. Mol. Nutr. Food Res. 2017;61 doi: 10.1002/mnfr.201700262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Total detected transcript quantification and differentially expressed genes (DEG) in control (Hif1af/f/Nkx2.5+/+) and Hif1a/Nkx2.5 mutant (Hif1af/f/Nkx2.5Cre/+) hearts at E12.5. The table is composed of two spreadsheets that describe raw and normalized expression values for each gene in each sample, averaged values per condition and for all samples, as well as fold change, logFC, p value, and Benjamini-Hochberg-adjusted p value, in a mutant versus control comparison. The first spreadsheet describes expression values for 201 genes detected as differentially expressed with Benjamini-Hochberg adjusted p values < 0.055. 83 genes were upregulated (red background) in Hif1a-deficient hearts versus control littermates, and 118 genes were downregulated (blue background). The second spreadsheet describes expression values for the whole collection of 14,406 genes detected in the RNA-seq.
Over-represented Biological Process GO terms associated to the collection of 201 differentially expressed genes (adjusted p value < 0.055), as detected with GOrilla, with p value < 0.001 (default threshold). The table describes, for each GO term, the number of mapped annotated genes in the reference dataset, the number of mapped annotated genes in the target set, its fold enrichment, and p value. Dispensable terms were eliminated after processing the list with REVIGO.
Enriched gene sets from the Hallmark, Biocarta, and Reactome databases associated to the collection of 14,406 expressed genes, as detected with GSEA, with p value < 0.05. The table describes, for each gene set, the calculated normalized enrichment score and associated p value.
Over-represented Reactome pathways, PANTHER pathways, and Biological Process Gene Ontology terms associated to the collection of 201 differentially expressed genes (adjusted p value < 0.055), as detected with PANTHER, with p value < 0.05. The table describes, for each pathway or GO term, the number of mapped annotated genes in the reference dataset, the number of mapped annotated genes in the target set, its fold enrichment, and associated p value.
The table shows the list of proteins quantified by mass spectrometry analysis. ZQ (KO/WT) values are standardized log2-ratio averages of proteins from mutant (Hif1af/f/Nkx2.5Cre/+) samples relative to embryos (Hif1af/f/Nkx2.5+/+), merged from the 3 biological replicates. 1% FDR is used as criterion for protein statistically significant abundance change.
The table presents, in three spreadsheets, functional enrichment results generated with IPA for the set of 201 genes detected as differentially expressed (adjusted p value < 0.055), for the following analysis types: Canonical Pathways, Upstream Regulators and Diseases, and Biofunctions (Downstream Effect analysis). The key parameters provided for the first three analysis types describe enrichment significance (p value and Benjamini-Hochberg-adjusted p value) and activation Z score (a prediction of the activity of the pathway, regulator or function; in this case, positive and negative values denote higher activity in mutant or control samples, respectively). Absolute Z score values higher than 2 are considered relevant.
Data Availability Statement
RNA-seq data have been deposited in NCBI's Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo) and are accessible through GEO Series accession number GSE164453.
Proteomic datasets (raw files, protein databases, search parameters, and results) have been deposited in the Peptide Atlas repository (http://www.peptideatlas.org) and are available through the accession number PASS01236.