

Summary
This study focused on characterizing the potential mechanism of valvular toxicity caused by TGFβ receptor inhibitors (TGFβRis) using rat valvular interstitial cells (VICs) to evaluate early biological responses to TGFβR inhibition. Three TGFβRis that achieved similar exposures in the rat were assessed. Two dual TGFβRI/-RII inhibitors caused valvulopathy, whereas a selective TGFβRI inhibitor did not, leading to a hypothesis that TGFβ receptor selectivity may influence the potency of valvular toxicity. The dual valvular toxic inhibitors had the most profound effect on altering VIC phenotype including altered morphology, migration, and extracellular matrix production. Reduction of TGFβ expression demonstrated that combined TGFβ2/β3 inhibition by small interfering RNA or neutralizing antibodies caused similar alterations as TGFβRis. Inhibition of TGFβ3 transcription was only associated with the dual TGFβRis, suggesting that TGFβRII inhibition impacts TGFβ3 transcriptional regulation, and that the potency of valvular toxicity may relate to alteration of TGFβ2/β3-mediated processes involved in maintaining proper balance of VIC phenotypes in the heart valve.
Subject areas: molecular physiology, molecular biology, cell biology
Graphical Abstract
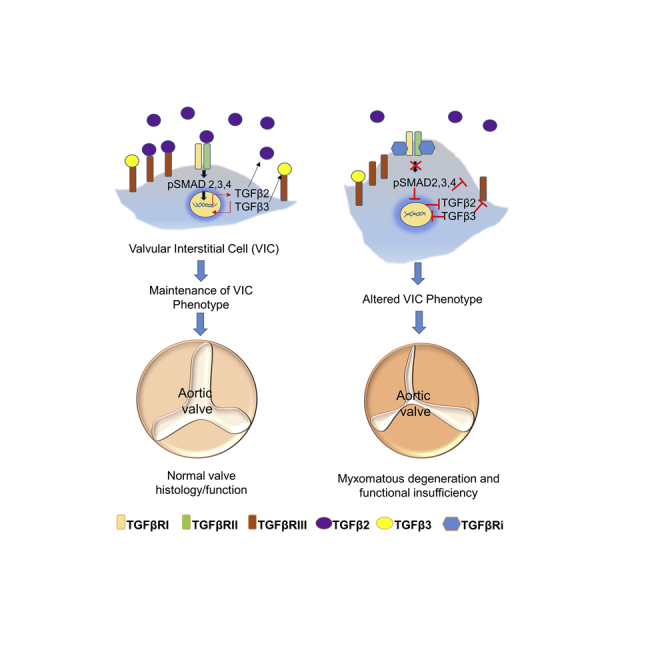
Highlights
-
•
TGFβ signaling blockade causes valvulopathy; VICs may be the cellular target
-
•
VICs express TGFβ receptors, ligands, and pSMAD2/3, indicating autocrine regulation
-
•
TGFβ2 and TGFβ3 maintain VIC phenotype; TGFβRis altered shape, migration, and ECM
-
•
Maintaining TGFβ3 transcription may reduce the potency of toxicity
Molecular physiology; molecular biology; cell biology
Introduction
The TGFβ superfamily consists of at least 40 structurally and functionally related cytokines that are involved in various biological processes including embryonic development, extracellular matrix (ECM) formation, immune regulation, inflammation, and cancer invasion and metastasis (Massague and Chen, 2000). TGFβ is produced by leukocytes and other cell types, and its receptor-mediated signaling involves both autocrine and paracrine regulation (Letterio and Roberts, 1998). TGFβ signals through TGFβ receptors as a heterodimer, TGFβRI, also called ALK5, and TGFβRII, to activate downstream signaling pathways (Hinck, 2012). Canonical TGFβ signaling involves binding of the ligand to TGFβRII and subsequent dimerization of the TGFβRI and TGFβRII at the cell surface, and then phosphorylation of SMADs (Shi and Massague, 2003) with translocation to the nucleus to induce transcription of downstream targets. TGFβRIII, which is not directly part of canonical TGFβR signaling, can impact this pathway as it can bind TGFβ ligands to the cell surface and serve as a reservoir of ligand for signaling. TGFβRIII also has high affinity to the glycoprotein, endoglin, which can also bind TGFβRI and modulate signaling (Gordon and Blobe, 2008). TGFβs can activate non-SMAD-dependent signaling pathways (non-canonical pathways), including the mitogen-activated protein kinase (MAPK) pathway mediated by p38, c-Jun amino terminal kinase (JNK), extracellular signal-regulated kinases (ERK), nuclear factor-κB (NF-κB), Rho, phosphatidylinositol 3-kinase (PI3K) (Letterio and Roberts, 1998), and protein kinase B (Akt) (Zhang, 2009), with respective downstream signaling leading to various cellular events. Both canonical and non-canonical pathways are highly species conserved (Massague, 2008).
The therapeutic potential of inhibiting TGFβ signaling has been well characterized with encouraging results in animal models of fibrosis (Gellibert et al., 2006) and cancer (Yoshioka et al., 2011). However, maintaining safety when targeting this pathway can be challenging given the role of TGFβ signaling in homeostatic maintenance of tissues. Earlier work with various TGFβ receptor inhibitors (TGFβRis) evaluated efficacy in tumor regression/metastasis and fibrosis as well as potential toxicities (Frazier et al., 2007; Herbertz et al., 2015; Yingling et al., 2004; Rak et al., 2020). Pre-clinical toxicology studies with TGFβ inhibitors that block TGFβRI have reported cardiovascular toxicities in rats and dogs including aortic pathologies, aneurysms, and valvulopathy (Anderton et al., 2011; Herbertz et al., 2015; Stauber et al., 2014; Rak et al., 2020). Such findings are considered directly related to the target biology due to similar presentation of cardiovascular changes in both mouse and human inactivation mutations and roles of certain members of the pathway in cardiovascular development. For instance, conditional TGFβRII knockout mice present thoracic aortic dissection and aneurysm (Chen et al., 2013; Lin and Yang, 2010). TGFβ2 knockout mice exhibited a range of cardiovascular malformations resulting from failure of normal completion of looping and separation of the outflow tract and the atrioventricular canal, as well as abnormalities of valve differentiation and arterial growth (Bartram et al., 2001). TGFβ2 expression correlated with leaflet thickness in human myxomatous mitral valves suggesting the role of TGFβ in cardiac and valvular morphogenesis in humans (Hulin et al., 2012, 2013). Genetic mutations associated with de-regulation of the TGFβ signaling pathway cause aortic dilatation and aneurysms and mitral valve disease such as observed in patients with Marfan and Loeys-Dietz syndromes (Leger et al., 2014; Loeys et al., 2005; Kovacs et al., 2015; Dietz et al., 1991), the latter of which has five sub-classes of mutations with each class associated with mutations in TGFβRI,TGFβRII, TGFβ2, TGFβ3, or SMAD3 genes, respectively.
Valvular interstitial cells (VICs) are the most prevalent cell type in the heart valve and have context-dependent phenotypes including stem cell-like VICs, osteogenic VICs, myofibroblast-like VICs (also known as activated VICs [aVICs]), and quiescent VICs (qVICs). Together, the multiple phenotypes are responsible for maintaining elasticity, structure, and repair/renewal processes essential in maintaining functional integrity of the valve (Han et al., 2013; Liu et al., 2007). The exquisite interplay among the VIC phenotypes achieves a structure that is flexible enough to change shape easily and strong enough to withstand elevated hemodynamic stress (Schoen, 1997). VIC activation is tightly regulated by cytokines that control their differentiation, proliferation, and migration to the site of tissue remodeling and repair (Powell et al., 1999). In injured or diseased valves, qVICs become aVICs with myofibroblast characteristics including increased α-SMA expression, increased secretion of ECM, and expression of matrix metalloproteinases (MMPs) (Gotlieb et al., 2002). TGFβ is a central mediator that drives fibroblasts to differentiate into myofibroblasts in wound healing, scarring, and fibrosis. Additionally, epithelial-to-mesenchymal transition (EMT) (Walker et al., 2004), which is essential in embryogenesis, tumorigenesis, and fibrosis, is also primarily mediated by TGFβ signaling (Turini et al., 2019; Massague and Chen, 2000). Cells undergoing EMT lose E-cadherin expression and undergo cytoskeletal and morphological changes to become mesenchymal to enable invasion (Thiery et al., 2009).
In this study we evaluated three TGFβR inhibitors that possessed different potencies in causing valvulopathy in the rat. LY2109761 and BMT-A are dual TGFβRI/RII inhibitors that caused valvulopathy, whereas BMT-B is a selective TGFβR1 inhibitor, which did not cause valvulopathy at similar exposures that caused valvular toxicity with the other inhibitors. With these inhibitors, the biological effects of inhibiting TGFβ signaling in VICs were studied to better understand this mechanism of toxicity. We propose that TGFβRII inhibition may impact transcriptional regulation of TGFβ3 and that an interplay between TGFβ2 and TGFβ3 may maintain the proper context-dependent microenvironment, EMT-MET (mesenchymal-to-epithelial transition) homeostasis, and VIC phenotype in the rat, where alteration of this balance could lead to valvulopathy.
Results
Pharmacology and toxicology profiles of the respective TGFβRis
Three TGFβRis were evaluated in this study. LY2109761 is a potent dual TGFβRI/-RII inhibitor previously evaluated in rat toxicology evaluations and reported to cause valvulopathy (Herbertz et al., 2015; Tan et al., 2009; Maratera, 2009). LY2109761 was used as a benchmark compound to explore optimum dose and necropsy times that would be sufficient to induce and histologically detect valvulopathy, respectively. BMT-A and BMT-B are azaindoles, a distinct chemotype from LY2109761. BMT-A is a potent dual TGFβRI/-RII inhibitor, whereas BMT-B is a potent, selective TGFβRI inhibitor (Table 1). All three compounds presented no substantial off-target activity but presented similar potency to respective TGFβ receptors (Table 1). BMT-A and BMT-B presented acceptable pharmacokinetic profiles in the rat and were selected for toxicological evaluation.
Table 1.
Relative binding and in vitro cellular potencies of three TGFβRis compared with rat in vivo exposures and valvular toxicity profiles
| TGFβR inhibitor | Potency TGFβRI kinase filter IC50 μM |
Potency TGFβRII kinase filter IC50 μM |
Potency NIH3T3 proliferation IC50 μM | Dose (mg/kg/day) | Toxicokinetics (Day 1) |
Incidence of rats presenting valvular histopathology | |
|---|---|---|---|---|---|---|---|
| Cmax (μM) | AUClast (μM∗h) | ||||||
| BMT-A | 0.01 | 0.03 | 0.79 | 10 QD | 5.6 | 36.3 | 1/5 |
| 50 QD | 21.9 | 180 | 5/5 | ||||
| BMT-B | 0.012 | >50 | 0.12 | 10 QD | 10.7 | 84.3 | 0/5 |
| 50 QD | 16 | 137 | 0/5 | ||||
| LY2109761 | 0.007 | 0.073 | 0.33 | 50 BID | 7 | 16 | 5/5 |
| 150 QD | 11 | 63 | 7/8 | ||||
Sprague-Dawley rats were dosed orally with the respective TGFβRi for 4 days, and hearts were collected on Days 4 and 15 (LY2109761) or Day 14 (BMT-A and BMT-B) and evaluated histologically (Table 1, Figure 1). Early studies with LY2109761 dosed daily for 4 days indicated that although no valvular histopathology was observed at 4 days, by Day 15 LY2109761 caused valvular changes including thickening of the aortic and atrioventricular valves as reported for this compound and other TGFβRis (Herbertz et al., 2015; Maratera, 2009). Subsequently, the BMT compounds were dosed for 4 consecutive days followed by a 10-day dose holiday before necropsy and heart collection. To this end, treatment with BMT-A also caused similar valvulopathy. Most of the rats treated with 50 mg/kg twice a day (BID) or 150 mg/kg once a day (QD) LY2109761 presented valvulopathy, whereas 50 mg/kg BMT-A caused valvulopathy in all five treated animals (Table 1, Figure 1). In contrast, there was no evidence of valvulopathy in rats treated with up to 50 mg/kg BMT-B, which achieved similar exposures as 50 mg/kg BMT-A (BMT-A: AUClast 180 μM∗h versus BMT-B: AUClast 137 μM∗h) and were substantially higher than the 150 mg/kg QD LY2109761 treatment, which caused valvulopathy in seven of eight rats (AUClast 63 μM ∗h) (Table 1). A representative set of atrioventricular valves from a rat treated with LY2109761 and BMT-A are presented in Figure 1 and shows the characteristic myxomatous change (thickening) of the valve leaflets, whereas no myxomatous change was observed in rats treated with BMT-B (Figure 1). Following treatment with TGFβRis, the affected valvular histology included the characteristic thickening of the valve.
Figure 1.

Representative histology of heart valves from rats treated with TGFβR inhibitors
(A and E) (A) Left atrioventricular (AV) valves and (E) close-up of a vehicle-treated rat presenting normal histology. Valves are predominately populated with VICs surrounded by ECM.
(B and F) (B) Left AV valves from a rat treated with 50 mg/kg BID LY2109761 with close-up (F) presenting myxomatous changes, including thickening of the valve.
(C and G) (C) Left AV valves from a rat treated with BMT-A and (G) close-up presenting similar myxomatous changes as LY2109761.
(D and H) (D) Left AV valves from a rat treated with BMT-B and (H) close-up presenting normal valvular histology.
Scale bar, 120 μm.
Rat valvular interstitial cells: characterization of cultured cells and morphologies
Rat VICs were harvested and cultured according to previously described methods (Liu et al., 2015) (Figure S1). VICs are stem cell-like and grow through clonal expansion (Figure S1A) and have the potential to differentiate into cells with distinct morphology and functions reminiscent of mesenchymal stem cells (Chen et al., 2009). We have found that sub-culture by scraping maintains mesenchymal-like morphology and by maintaining cells in conditioned media, enables VICs to be cultured up to 11 passages without an obvious decrease in proliferation. There are at least three morphologies of VICs observed in primary cultures: spindle-shaped (Figure S1C); flattened, elongated-shaped (Figure S1D); and round-shaped, of which the last is typical of endothelial cells, which may also be present in the primary cultures (Liu et al., 2015) (Figure S1E). However, the cells did not label positive for the endothelial cell marker, CD-31, suggesting the cultures represented predominantly VICs and not endothelial cells (Figures S1I and S1 J). When each morphology of cell was sub-cloned, the cells returned to their original morphologies, suggesting that the observed morphologies do not represent cells in various stages of differentiation (data not shown). When the cells were labeled with a marker that distinguishes the activated phenotype (myofibroblast marker, α-SMA) from the other VIC phenotypes (using mesenchymal marker, vimentin), all cells were positive for vimentin with some additionally positive for α-SMA (Figures S1F–S1H). This observation aligns with an earlier study that characterized the phenotype of cultured porcine VICs, where 64% of the VICs express α-SMA and 98% expressed vimentin (Witt et al., 2012).
Based on several studies (Liu et al., 2007; Taylor et al., 2003), maintaining VICs in a quiescent state is important for maintaining normal structure and function of the valve, and qVICs are considered to have the closest biology to endogenous VICs in the valve in vivo (Liu et al., 2007). In this study, all the experiments were performed with cultures that promoted mostly qVICs (Figure S1B). This was achieved by serum-starving the cells in culture media containing 0.5% FBS for at least 24 h before treatment, which slowed proliferation. The cells used in the experiments were from passages 3–8. Working with early passages of VICs are optimal in maintaining quiescence, because over several passages, the cultured cells have been reported to become more activated in phenotype (Latif et al., 2006).
TGFβ-mediated signaling is intact in cultured VICs
TGFβ and TGFβR expression in VICs: Transcriptional profiles of TGFβ and TGFβR expression were evaluated in cultured VICs, compared with expression in rat thoracic aorta, another target of TGFβR-induced toxicity, and both assessed relative to rat heart muscle expression (not considered a direct target of TGFβR inhibitor toxicity). As in aorta, VICs transcriptionally expressed all three TGFβ ligands and TGFβ receptors. All TGFβ ligands were robustly expressed in VICs at a range of ∼10- to 50-fold higher expression relative to heart. Additionally, TGFβ1 and TGFβ2 were 2-fold and 10-fold higher in VICs than thoracic aorta, respectively, whereas TGFβ3 expression was similar in VICs and thoracic aorta (Figure 2A). In contrast TGFβRI, TGFβRII, and TGFβRIII expression was the highest in thoracic aorta (∼5- to 7-fold relative to heart) and expression of TGFβRI and TGFβRII was about 2-fold higher relative to VICs. However, VICs presented low expression of TGFβRIII, which was considerably less relative to heart and aorta (Figure 2B).
Figure 2.

TGFβ-mediated signaling is intact in cultured VICs
(A) Relative expression of TGFβ ligands in rat VICs and thoracic aorta compared with heart muscle RNA.
(B) Relative expression of TGFβ receptors in rat VICs and thoracic aorta compared with heart muscle RNA.
(C) Exogenous hTGFβ stimulation in VICs causes concentration-dependent increases in SMAD3 phosphorylation with peak response reached at 1 h and sustained for at least 48 h.
(D) After 48-h treatment with 10 ng/mL hTGFβ, myofibroblast targets, ACTA2 and Tagln, were upregulated, indicative of enhanced activation of VICs.
(E) Following 1-h treatment with TGFβR inhibitors that included 10 ng/mL hTGFβ stimulation during the last 30 min of compound treatment, the inhibitors caused concentration-dependent decreases in SMAD3 phosphorylation.
(F) Decreases in SMAD3 phosphorylation without hTGFβ stimulation.
All transcriptional data in (A and B) were normalized with housekeeping gene, Hrpt1, and calculated relative to rat heart muscle RNA. Represented data presented in (A–F) are the average of three experiments ± SEM. Statistical comparisons employed one-way or two-way ANOVA followed by Dunnett's test. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.005; ∗∗∗∗p ≤ 0.0001.
TGFβ induced p-SMAD3 activation in VICs: Phosphorylation of SMAD2 and SMAD3 is well characterized in TGFβ-induced EMT. Activation of SMAD3 was demonstrated in VICs with dose-responsive increases in p-SMAD3 protein following hTGFβ stimulation, with peak response in p-SMAD activity within 1 h post stimulation (Figure 2C). The response was sustained for at least 48 h.
TGFβ drives activation of VICs: TGFβ is a central activator of the myofibroblast phenotype and EMT during wound healing processes (Martin, 1997). aVICs are similar to myofibroblasts and express α-SMA and Tagln, maintaining stretch, elasticity, and matrix in the valve using similar processes as those in repair mechanisms (Hosper et al., 2013; Sappino et al., 1990). When VICs were stimulated with 10 ng/mL hTGFβ for 48 h to mimic cell activation, ACTA2 (which encodes α-SMA) and Tagln increased ∼2- and ∼4-fold, respectively, indicating enhanced activation of VICs (Figure 2D).
TGFβRis pharmacologically mediate both canonical and non-canonical signaling: TGFβRis effectively decreased p-SMAD3 in a concentration-dependent fashion, which was achieved with (Figure 2E) or without (Figure 2F) pre-stimulation with hTGFβ. Considering the strong transcriptional expression of TGFβ ligands in these cells, these results suggested that TGFβ signaling is intact and possesses constitutive, autocrine activity. Based on these results, we subsequently performed biological assessments in VICs without pre-stimulation with hTGFβ as a means to better reflect endogenous response to TGFβR inhibition.
To assess whether non-canonical TGFβ signaling may be present in VICs, VICs were treated with a concentration range of TGFβRis and profiled for p-SMAD2, p-AKT, and p-ERK1/2 (Table 2) and mTOR phosphoproteins (Table S3). All the phosphoproteins were detected in VICs, and all compounds were potent inhibitors of p-SMAD2, with varying potency in inhibiting p-AKT and mTOR phosphoproteins. Only LY2109761 presented inhibitory, albeit relatively weak, activity on p-ERK1/2. Altogether the studies indicate both intact canonical and non-canonical signaling are present in the VICs, but the respective inhibitors may have differential potency in inhibiting phosphoproteins associated with non-canonical signaling.
Table 2.
IC50 (μM) of phosphoprotein expression at 48 h post TGFβRi treatment
| TGFβR inhibitor | p-Akt | p-ERK1/2 | p-Smad2 |
|---|---|---|---|
| LY2109761 | 0.42 ± 0.08 | 2.08 | 0.04 |
| BMT-A | 0.13 ± 0.05 | >10 | 0.02 ± 0.01 |
| BMT-B | 0.85 ± 0.31 | >10 | 0.14 ± 0.03 |
SEM represents the average of four experiments with the exception of p-ERK1/2 (all compounds) and p-SMAD2 (for LY2109761), which were only run once.
Early alterations in biological responses to TGFβRi treatment in VICs
TGFβRis may induce metabolic remodeling: TGFβRis cause myxomatous changes in valves typically within 1–2 weeks in rats. These changes include increased VIC proliferation and glycosaminoglycan production in the valve leaflets (Walker et al., 2004). We evaluated the early effects of TGFβRis on cellular proliferation and mitochondrial function of VICs. VICs were treated with TGFβRis for 48 h and evaluated for cytotoxicity, ATP activity, oxidative stress, mitochondrial respiration, and proliferation. The cells were treated at a concentration range of 0.01–10 μM, evaluated for viability by microscopy, then labeled with markers for apoptosis and necrosis, and evaluated by flow cytometry. Following TGFβRi treatment, cells appeared to be growing normally with no obvious impact on viability, which was confirmed by flow cytometry analysis, where there were no changes on percentages of apoptotic or necrotic populations (Table 3 and Table S4). Despite the lack of cytotoxicity, all inhibitors caused concentration-responsive decreases in ATP levels, decreased reactive oxygen species (ROS) production, and decreased mitochondrial respiration (Figures 3A and 3B). Proliferation measured by CyQUANT analysis indicated that TGFβRi treatment caused concentration-dependent decreases in cellular proliferation (Figure 3A). Together these results suggest that early responses of TGFβ signaling inhibition may not involve increased proliferation but repress cellular metabolism, perhaps initiating metabolic remodeling associated with other biological changes (Trefely and Wellen, 2018).
Table 3.
Flow cytometric analysis of apoptosis and necrosis of VICs at 48 h following LY2109761 treatment
| μM | % of Dead Cells | % of Apoptosis |
|---|---|---|
| 0 | 4.4 | 1.4 |
| 0.01 | 3.0 | 0.6 |
| 0.1 | 3.9 | 1.1 |
| 1 | 3.7 | 1.3 |
| 10 | 2.6 | 0.7 |
Figure 3.

TGFβRi treatment alters mitochondrial function and cell proliferation in VICs
(A) Assessment of mitochondrial function and cell proliferation following 48-h treatment with TGFβR inhibitors. Mitochondrial function was measured by ATP production, oxidative stress (ROS generation), and cellular mitochondrial respiration. Proliferation was evaluated by CyQUANT. Concentration-responsive decreases in production of ATP and ROS and proliferation were observed following TGFβRi treatment.
(B) Cellular mitochondrial respiration evaluation by Seahorse. Additional evaluation of mitochondrial respiration with LY2109761 following 48-h treatment demonstrated concentration-related decreases, which was also observed with other TGFβR inhibitors (data not shown).
Represented data are the average of three experiments ±SEM. Statistical comparisons employed one-way or two-way ANOVA followed by Dunnett's test. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.005; ∗∗∗∗p ≤ 0.0001.
TGFβRis alter autocrine TGFβ signaling: Following 48-h treatment with TGFβRis, VICs were evaluated for impact on TGFβ receptor/ligand gene expression. TGFβRis decreased TGFβ signaling including concentration-related inhibition of p-SMAD and decreased transcription of all TGFβ ligands and TGFβRI expression (Figure 4A). In contrast, TGFβRis upregulated TGFβRII and TGFβRIII expression, the latter with a remarkable ∼8- to ∼30-fold upregulation in expression, which may have been a compensatory response to the depletion of ligand expression. Although all three inhibitors presented similar responses, the only exception was that TGFβ3 transcriptional expression remained unaltered following BMT-B treatment.
Figure 4.

TGFβRi treatment alters TGFβ receptor, ligand, and ECM expression in VICs
(A) Comparison of transcriptional changes of TGFβ ligands and receptors in VICs following 48-h treatment with TGFβRis. All inhibitors caused similar transcriptional alterations of ligand and receptor expression with the exception of BMT-B, which caused less increase in TGFβRII and TGFβRIII transcription and had no inhibitory response on TGFβ3 transcription.
(B) Effect on collagen production in VICs following 48-h treatment (represented with LY2109761).
(C) Elastin protein production increased following LY2109761 and BMT-A treatment but decreased following treatment with BMT-B.
For each transcriptional target, at least three TGFβRi-treated VIC samples were analyzed with two technical replicates (≥6 PCR tests) in calculating ± SEM. For the protein measurements, represented data are the average of three experiments ± SEM.
TGFβRis alter ECM production from VICs: Elastin fiber disruption in the heart valves and aorta has been associated with TGFβ inhibitor treatment (Anderton et al., 2011; Rak et al., 2020). TGFβ has also been found to stimulate transcriptional expression of elastin (ELN) (Kahari et al., 1992). All inhibitors effectively decreased ELN transcription in a concentration-related manner (Table 4 and Figure S2). Additionally, the transcriptional expression of the collagens that comprise ∼90% of ECM protein in the heart valve, COL 1A1 and COL 11A1, were decreased (Table 4 and Figure S2) (Wiltz et al., 2013; Hosper et al., 2013; Vazquez-Villa et al., 2015). On a protein level, collagens were likewise decreased following TGFβRi treatment (represented with LY2109761 in Figure 4B). In contrast, elastin protein levels presented concentration-related increases with LY2109761 and BMT-A treatment but mild decrease with BMT-B treatment (Figure 4B).
Table 4.
Average fold change in transcriptional expression relative to DMSO control at 48 h following treatment with 10 μM TGFβRis
| Gene | Category | Ly2109761 | BMT-A | BMT-B |
|---|---|---|---|---|
| TGFβ1 | TGFβ ligands | −3.86 ± 0.47 | −1.80 ± 0.30 | −1.64 ± 0.42 |
| TGFβ2 | TGFβ ligands | −2.18 ± 0.24 | −1.94 ± 0.26 | −1.54 ± 0.3 |
| TGFβ3 | TGFβ ligands | −5.33 ± 0.28 | −9.34 ± 0.34 | 0.98 ± 0.13 |
| TGFβRI | TGFβ receptors | −1.53 ± 0.14 | −1.34 ± 0.22 | −1.46 ± 0.22 |
| TGFβRII | TGFβ receptors | 5.16 ± 0.18 | 7.57 ± 0.53 | 2.74 ± 0.16 |
| TGFβRIII | TGFβ receptors | 18.24 ± 10.73 | 23.75 ± 2.39 | 8.93 ± 0.52 |
| ACTA2 | Myofibroblast marker | −2.75 ± 0.20 | −7.85 ± 0.64 | −1.85 ± 0.16 |
| Tagln | Myofibroblast marker | −4.3 ± 0.46 | −10.75 ± 1.02 | −3.32 ± 0.38 |
| Cdh1 | Epithelial marker | 34.29 ± 4.99 | 13.38 ± 2.02 | 18.43 ± 1.48 |
| VIM | Mesenchymal marker | 1.14 ± 0.05 | 2.18 ± 0.06 | 1.64 ± 0.11 |
| MMP2 | MMP2 EMT marker | 1.98 ± 0.13 | 2.53 ± 0.19 | 2.03 ± 0.22 |
| COL1A1 | Collagens | −2.61 ± 0.11 | −4.05 ± 0.42 | −2.24 ± 0.17 |
| COL11A1 | Collagens | −4.88 ± 0.26 | −7.99 ± 0.99 | −2.94 ± 0.20 |
| ELN | Elastin | −87.63 ± 7.50 | −58.11 ± 4.51 | −12.25 ± 1.73 |
Bold text denotes notable differences (≥1.75-fold) between BMT-B compared with BMT-A and LY2109761 responses (average from three experiments ±SEM).
TGFβRis alter VIC phenotypes: All TGFβRis caused concentration-dependent decreases in ACTA2 and Tagln and increased E-cadherin transcription, a marker of epithelial cells (Frixen et al., 1991) (Figure S2), suggesting that the VIC phenotype may be changing toward an epithelial phenotype, such as observed in MET. The mesenchymal marker, vimentin, along with MMP2, an EMT inducer, presented mild transcriptional up-regulation (ranging up to 2.5× at 10 μM), but this was not as pronounced as the down regulation of the myofibroblast markers (Table 4, Figure S2, Table S5) (Cheng and Lovett, 2003). These responses may reflect a potential imbalance of EMT/MET because EMT/MET are multi-step reciprocal processes with cells in different phases of differentiation, where some cells may be more advanced in transitioning toward epithelial transition, whereas others are more mesenchymal (Willis and Borok, 2007).
VICs were labeled with antibodies against protein markers of aVICs (α-SMAhigh/vimentin+) versus other VIC phenotypes (α-SMAlow/vimentin+) and evaluated by confocal microscopy to better characterize the effects of the transcriptional changes on VIC phenotypes, cytoskeletal architecture, and morphology. LY2109761 and BMT-A caused concentration-dependent changes in cellular morphology and labeling pattern, where α-SMAhigh-positive cells presented progressively disorganized actin filaments, whereas vimentin patterns remained unchanged or were slightly more pronounced (Figure 5A). In contrast, BMT-B appeared to have less effect on cellular morphology and α-SMA/vimentin staining. Following TGFβRi treatment, cell impedance was additionally assessed over a time course. Impedance will change as cells proliferate, become more adherent, or undergo morphological changes. Cellular impedance increased in DMSO-treated control VICs but presented concentration-related decrease with LY2109761 and BMT-A treatments. In contrast, there was minimal change in impedance with BMT-B (Figure 5B), suggesting that BMT-B had less of inhibition of adhesion and morphological change.
Figure 5.

TGFβRi treatment induces altered morphology and migration of VICs
(A) Morphological changes of VICs labeled with α-SMAgreen and vimentinred following 48-h treatment with 10 μM TGFβRis. Nuclei were labeled with DAPIblue. Middle panels show 2.5× enlargement of boxed area of α-SMA, vimentin, and DAPI merged. Bottom panels show boxed area with only vimentin and DAPI signal.
(B) VICs were assessed over a time course for changes in cell membrane impedance. Impedance will increase as cells become more adherent. Cellular impedance increased in DMSO-treated controls but decreased following treatment with LY2109761 and BMT-A. In contrast, there was minimal change in impedance following BMT-B treatment.
(C) VIC migration was quantitated over a 48 h time course using the scratch test. The percent cell density corrected from T0 baseline is presented. LY2109761 and BMT-A presented less cell migration compared with DMSO and BMT-B.
Represented data in (B and C) are the average of three experiments ± SEM. Statistical comparisons employed one-way or two-way ANOVA followed by Dunnett's test. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.005; ∗∗∗∗p ≤ 0.0001. Scale bars, 100 μm.
VICs treated with 10 μM of each TGFβRi were evaluated by flow cytometry with antibodies against α-SMA and vimentin. The relative brightness of α-SMA and vimentin staining would indicate whether VICs may be becoming more activated in phenotype. BMT-A treatment caused a slight increase in intensity of α-SMA staining, whereas there was no notable change in staining intensity following treatment with LY2109761 and BMT-B. All compounds caused an increase in vimentin staining intensity, agreeing with the observed increased transcriptional expression of vimentin that suggested an alteration of EMT-MET balance. VICs were additionally stained with antibodies against epithelial markers, EpCAM, E-cadherin, and the endothelial marker, CD-31. VICs normally do not express these markers, and following TGFβRi treatment they remained negative for staining by both flow cytometry and immunohistochemical evaluation (data not shown). This indicated that the VICs were not definitively changed into an epithelial or endothelial phenotype (Figure S3).
Considering the morphological changes and alteration of EMT/MET markers, a scratch test was undertaken to determine whether TGFβRi treatment altered cell migration (Liang et al., 2007). Following preparation of VIC cultures, a scratch was made in the middle of the well followed by administration of 10 μM TGFβRi. Three wells per treatment were photographed at 0, 6, 24, and 48 h post scratch, where photographs were taken at the same marked location. The annotation of the scratched open area was performed for the 0 h image, and then the annotation was copied and pasted for its sequential images at 6, 24, and 48 h. Thus, the area of the cells was measured and the percentage of cell density was calculated and compared to the Time 0 (T0) density. DMSO and BMT-B treatments showed similar and significant increases in cell density in the scratch region over the entire time course, suggesting cell migration was intact. In contrast, both LY2109761 and BMT-A did not present significant migration at the 6 h time point and reduced migration at 24 and 48 h, suggesting that migration was inhibited with these treatments (Figure 5C).
TGFβ2 and TGFβ3 may mediate signaling involved in maintaining VIC phenotype
In context of the finding that TGFβ ligands were robustly transcribed in VICs and VICs present autocrine signaling, small interfering RNA (siRNA) experiments were undertaken to determine whether loss of function of specific ligands may produce similar biological changes as observed with the inhibitors. Multiple siRNA constructs for each respective ligand were evaluated in rat VICs to identify constructs with low toxicity and high specificity to the respective ligands (Figure S4A). During the course of these evaluations, it was found that multiple constructs directed against rat TGFβ2 or TGFβ3 caused decreased transcription of both ligands in VICs despite lack of homology of the respective constructs to other TGFβ sequences (Figures 6A and S4A). Together this suggested that transcriptional expression inhibition of either ligand may influence transcriptional regulation or mRNA stability of TGFβ2 or TGFβ3.
Figure 6.

Small interfering RNA knockdown of respective TGFβs cause differential alterations of myofibroblast and collagen target expression
(A) Small interfering RNA (siRNA) knockdown of TGFβ mRNAs at 48 h post transfection with respective TGFβ siRNA construct.
(B) Effect of TGFβ siRNA knockdown on myofibroblast and ECM target transcription at 48 h post transfection in VICs.
Represented data are the average of three experiments ±SEM. Statistical comparisons employed one-way or two-way ANOVA followed by Dunnett's test. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.005; ∗∗∗∗p ≤ 0.0001.
Silent RNA knockdown of TGFβ1 had no significant effect on transcription of myofibroblast targets or Col1 A1, whereas knockdown of TGFβ2 or TGFβ3 caused decreases in expression of ACTA2, Tagln and Col 1A1, and ELN that were similar to responses observed with TGFβRis (Figures 6B and S4B).
Owing to the co-down regulation of TGFβ2/β3 transcriptional expression observed with the TGFβ2 or β3 siRNA constructs, it was unclear from the siRNA experiments whether either or both ligands mediated these responses. However, the finding that TGFβ3 transcriptional expression was unaltered following treatment with the non-valvular toxic TGFβRi (BMT-B), suggested the need for further evaluation of potential roles of these ligands in maintaining VIC phenotype.
To this end, VICs were treated with a pan-TGFβ neutralizing antibody, 1D11, previously demonstrated to cause valvulopathy in the rat (Martin et al., 2020). Additionally, VICs were also treated with TGFβ ligand-specific neutralizing antibodies to determine impact on secreted TGFβs, p-SMAD signaling, and transcription of phenotypic markers.
Initially, VICs were treated with a concentration range of respective neutralizing antibody and evaluated for p-SMAD3 inhibition at 1 and 48 h post treatment. At 10 μg/mL, all antibodies inhibited p-SMAD3, which was detectible by 1 h and retained effective inhibition at 48 h (Table 5). Therefore an antibody concentration of 10 μg/mL was selected to the evaluate effects of ligand neutralization on VIC phenotype.
Table 5.
Percentage of p-SMAD3 activity relative to PBS controls at 1 and 48h post TGFβ neutralizing antibody treatment
| 1D11ˆ | Anti-TGFβ1 | Anti-TGFβ2 | Anti-TGFβ3 | Anti-TGFβ2+3 | |
|---|---|---|---|---|---|
| 1 h | 37 ± 6.85∗∗∗∗ | 66.9 ± 6.45∗ | 63.89 ± 7.55∗ | 77.48 ± 11.31 | 102.29 ± 10.72 |
| 48 h | 32.58 ± 6.38∗∗∗∗ | 66.45 ± 13.57∗ | 42.93 ± 7.65∗∗∗∗ | 46.54 ± 3.13∗∗∗∗ | 39 ± 8.98∗∗∗∗ |
Average of three experiments ±SEM; 10 μg/mL neutralizing antibody used in respective treatments. Statistical comparisons employed one-way or two-way ANOVA followed by Dunnett's test. ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.005;∗∗∗∗p ≤ 0.0001
TGFβ1, 2, and 3 was measured in cell culture media (starving media containing 0.5% FBS in M199) as well as conditioned media following 5 days of VIC culture. The conditioned media represents the 3 days VICs are cultured in serum-starved conditions plus the 2 days duration for running TGFβRi experiments. Rat serum was used as a positive control for confirming measurement of each respective ligand (Table 6). Because the cell culture media contained 0.5% FBS, the media was measured for respective ligand to determine background levels. The cell culture media contained low levels of TGFβ1 and TGFβ2 representing approximately 18% and 1% of the levels measured in conditioned media, respectively. There was no detectible TGFβ3 measured in the cell culture media or conditioned media (Table 6).
Table 6.
Amount of TGFβ ligands present in rat serum and VIC media and effect of 48-h TGFβ neutralizing antibody or TGFβRi treatment on secreted ligands into VIC media
| TGFβ1 | TGFβ2 | TGFβ3 | |
|---|---|---|---|
| Control (pg/mL) | |||
| Rat serum | 5302.8 ± 191 | 3336.7 ± 93 | 187.5 ± 11.3 |
| Media alone (0.5% FBS/M199)a | 624.5 ± 20.3 | 22.21 ± 2.3 | <BDL |
| Conditioned mediab | 3558 ± 61.3 | 1676.6 ± 56.8 | <BDL |
| Neutralization antibodies (percentage of controld) | |||
| PBS control | 100 | 100 | <BDL |
| 1D11 (10 μg/mL) | ≤0 | 45 | NA |
| TGFβ1 (10 μg/mL) | 57.4 | 72.5 | NA |
| TGFβ2 (10 μg/mL) | 77.4 | 26.4 | NA |
| TGFβ3 (10 μg/m)L | 65.5 | 51.5 | NA |
| TGFβ2+TGFβ3 (10 μg/mL each) | 100.1 | 29.9 | NA |
| TGFβRis (percentage of controld) | |||
| DMSOc | 100 | 100 | <BDL |
| LY2109761 (10 μM) | 55 | 27.5 | NA |
| BMT-B (10 μM) | 59 | 31.9 | NA |
<BDL, below detectible level; NA, not applicable due to lack of detectable ligand in cell media.
Average of three experiments ± SEM.
Baseline TGFβ concentration was measured with pre-test culture media containing 0.5% FBS in M199.
PBS in 0.5% FBS/M199 was used as an experimental control for VICs treated with TGFβ neutralizing antibodies. The amount of TGFβ in these samples reflects 4–5 days' accumulation (seeding + starving).
0.05% DMSO in 0.5% FBS/M199 is used as an experimental control for VICs treated with TGFβRis.
Percent change calculation: (actual concentration – media alone/control concentration (PBS or DMSO)) × 100.
Following 48-h treatment with TGFβ neutralizing antibodies, the pan-neutralizing antibody-depleted TGFβ1 and TGFβ2 levels were 45% of vehicle control. The TGFβ1- and TGFβ3-specific antibodies attenuated (<50% reduction) TGFβ1 and TGFβ2 protein levels. However, the TGFβ2 neutralizing antibody reduced TGFβ2 levels to 26% of control but only attenuated TGFβ1 levels. Combination of TGFβ2 + TGFβ3 neutralizing antibodies had no effect on TGFβ1 levels and caused a similar reduction of TGFβ2 as observed with the TGFβ2 neutralizing antibody, suggesting TGFβ3 protein neutralization had no additive or synergistic impact on reducing ligand levels (Table 6).
The effects of 48-h 10-μM treatments with LY2109761 and BMT-B on TGFβ ligands were additionally evaluated, and both inhibitors had similar impact on reducing TGFβ1 and TGFβ2 as the TGFβ2 neutralizing antibody (Table 6).
At 48 h following TGFβ neutralizing antibody treatment, VICs were collected and evaluated for transcriptional changes of phenotypic markers. Interestingly, 1D11 significantly reduced the transcription of myofibroblast and ECM targets and increased TGFβ3 transcription. In contrast, TGFβ3 neutralizing antibody treatment only significantly increased TGFβ3 transcriptional expression, whereas TGFβ2 neutralizing antibody treatment caused significant but slight decreases in TagIn and ELN and a slight increase in TGFβ3 expression. However, co-treatment with TGFβ2 and TGFβ3 neutralizing antibodies resulted in similar changes as 1D11 (Table 7).
Table 7.
Average fold change of transcriptional expression relative to DMSO control at 48-h following TGFβ neutralizing antibody treatment
| Genes | 1D11a | Anti-TGFβ2+3b | Anti-TGFβ1b | Anti-TGFβ2b | Anti-TGFβ3b |
|---|---|---|---|---|---|
| ACTA2 | 0.79 ± 0.03∗∗ | 0.79 ± 0.05∗ | 1.12 ± 0.03 | 0.92 ± 0.03 | 1.00 ± 0.04 |
| Tagln | 0.88 ± 0.01∗∗∗∗ | 0.92 ± 0.04∗∗ | 0.97 ± 0.02 | 0.95 ± 0.02∗ | 0.97 ± 0.02 |
| Col1A1 | 0.90 ± 0.05∗ | 0.87 ± 0.08 | 1.11 ± 0.03 | 1.04 ± 0.03 | 1.07 ± 0.07 |
| ELN | 0.37 ± 0.01∗∗∗∗ | 0.71 ± 0.01∗∗∗ | 1.14 ± 0.05 | 0.86 ± 0.02∗ | 0.91 ± 0.03 |
| TGFβ1 | 0.88 ± 0.03∗ | 0.92 ± 0.08 | 0.97 ± 0.03 | 0.95 ± 0.04 | 0.97 ± 0.04 |
| TGFβ2 | 0.87 ± 0.03∗∗ | 0.91 ± 0.06 | 1.02 ± 0.04 | 1.05 ± 0.04 | 1.11 ± 0.04 |
| TGFβ3 | 1.50 ± 0.04∗∗∗∗ | 1.10 ± 0.04 | 1.00 ± 0.03 | 1.18 ± 0.04∗ | 1.23 ± 0.04∗ |
Average of three experiments ± SEM. Statistical comparisons employed one-way or two-way ANOVA followed by Dunnett's test. The values in bold indicate statisticially significant transcriptional changes compared to vehicle control.
∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.005; ∗∗∗∗p ≤ 0.0001.
1D11 previously demonstrated to cause valvulopathy in rats.
VICs were treated with 10 μg/mL of respective TGFβ neutralizing antibody.
Discussion
Potential mechanisms of toxicity associated with TGFβR inhibition in VICs
VICs exhibit a constitutive level of p-SMAD activity and respond robustly to TGFβ stimulation, and they do not need pre-stimulation with ligand to assess inhibition of canonical signaling with the TGFβR inhibitors due to TGFβ synthesis and secretion in VICs. These findings, together with confirmation that the VICs express all three TGFβ ligands and receptors, suggest that the TGFβ signaling is constitutively active and the cells have autocrine regulation of this pathway. Very few cell types have shown constitutive p-SMAD expression. Instead, most cells/tissues require pre-stimulation with TGFβ ligand before measurement of inhibitory activity. Interestingly, the exceptions encountered in our hands have been tumor cells/tissue, vascular aortic smooth muscle cells/aortic tissue, and heart valves/VICs (data not shown). It is possible that the dependency of these cell types/tissues on constitutive TGFβ signaling may be the basis for why tumor efficacy and the cardiovascular toxicity tend to occur at similar exposures, making safety windows very narrow or non-existent.
The biological response of TGFβ signaling depends on the cell type and microenvironment. TGFβ could either promote or suppress cancer progression and metastasis depending upon the context of the tumor type and associated microenvironment (David and Massague, 2018; Stover et al., 2007). In the heart valve, TGFβ will promote VIC proliferation in response to injury, but when VICs are cultured in low-density monolayers without TGFβ stimulation and other growth factors, there is decreased proliferation, independent of injury (Li and Gotlieb, 2011). In vivo, treatment of rats with TGFβRis produced valvulopathy that included VIC proliferation (Frazier et al., 2007; Anderton et al., 2011). However treatment of cultured VICs with TGFβRis, reduced proliferation compared with vehicle controls. Proliferative response was profiled in vitro over a 48-h period, whereas the proliferation response observed in vivo was typically not histologically apparent until ∼1–2 weeks post initial dose. Similar observations have been reported with cultured porcine VICs treated with the TGFβRI inhibitors, SB431542 and SD208, which resulted in concentration-dependent decreases in VIC proliferation and had no effect on apoptosis (Li and Gotlieb, 2011).
TGFβ can induce ROS production and suppress antioxidant response inducing oxidative stress or redox imbalance (Liu and Desai, 2015; Weidinger and Kozlov, 2015). Although the VICs presented relatively unaffected proliferation and viability following TGFβRi treatment, there was a concentration-dependent decrease in mitochondrial respiration, ATP production, and ROS generation within 48 h, suggesting lack of oxidative stress but possible alteration in mitochondrial function that included decreased energy production without cytotoxicity. This response may be possibly related to a change in VIC phenotype and function, which may require cellular metabolism shifts and other biological changes similar to those encountered in differentiation response (Trefely and Wellen, 2018).
During injury or abnormal hemodynamic/mechanical stress, local accumulation of cytokines and growth factors, including TGFβ, will stimulate qVICs to change to an activated, myofibroblast-type cell, expressing α-SMA and producing increased ECM, which engages in repair mechanisms (Elangbam, 2010). Increased numbers of VICs were observed in valves of rats treated with TGFβR inhibitors and have altered morphology that have changed from fibroblast-like to more rounded morphology as well as increased presence of glycosaminoglycans in the surrounding matrix (Anderton et al., 2011) (Figure 1). Following TGFβRi treatment, cultured VICs presented similar changes in shape as well as decreased collagen and elastin transcription. Although collagens were also decreased on a protein level, interestingly, elastin production increased with the valvular toxic TGFβRis but not with BMT-B. Elastin is a direct transcriptional target of TGFβ signaling, where TGFβ affects both elastin transcription and mRNA stability (Davidson, 2002). Elastin is translated into a precursor protein, tropoelastin, which was the form of elastin that was measured in our assessment. Tropoelastin together with fibulin-5 deposits onto microfibrils in elastin fiber assembly (Noda et al., 2013), and disruption of elastin filament organization in heart valves and aorta is commonly identified with the histopathology of TGFβR inhibitors and animals/humans possessing inactivation mutations. The increased tropoelastin production associated with LY2109761 and BMT-A may reflect a compensatory response to alteration in elastin mRNA levels but will likely decrease with sustained TGFβR inhibition. It is possible that the less potent phenotypic changes observed following BMT-B treatment may also reflect less impact on elastin transcription and tropoelastin production leading to less effect on downstream elastin fiber assembly.
The interaction between elastin and collagens is critical to valvular function. Elastin is mechanically coupled to collagen and important in maintaining specific collagen fiber configuration for imposing tensile forces on collagen fibers during valve unloading. Furthermore, it is important in restoring collagen fiber geometry back to resting confirmation between loading cycles (Vesely, 1998). Additionally, glycosaminoglycans bind to the collagens, altogether making up a delicately balanced ECM that must maintain appropriate levels of stiffness and elasticity to withstand constant sheer stress, expansion, and contraction associated with the load cycle.
Additional changes occurred suggesting phenotypic change, particularly with the valvular toxic TGFβRis, LY2109761 and BMT-A. These included alterations of the active VIC phenotype including decreased myofibroblast marker transcription along with increases in MMP2, vimentin, and E-cadherin expression. Phenotypic changes were further supported by assessment of α-SMA protein organization, where following TGFβRi treatment, there was disruption of the striated α-SMA filament pattern with associated rounded or tile-like morphological changes along with decreased cellular impedance, indicative of reduction in cell-cell adhesion. Furthermore, the valvular toxic inhibitors presenting reduced cell migration in the scratch test indicates that the cellular responses were associated with altered EMT-MET homeostasis. This suggests that TGFβRis have a direct effect on the balance of aVICs and other VIC phenotypes without influence from exogenous cytokines or inflammatory processes. Inhibition of TGFβ signaling may drive the VICs toward MET resulting in phenotypic and functional alterations that could have a damaging effect on the integrity of the valve in vivo. Altogether, the TGFβRi-induced valvular toxicities observed in the rat may be due to deregulation and possible switching of the aVIC phenotype to other phenotypes that lose stretch and elasticity and homeostatic repair/renewal mechanisms associated with response to hemodynamic shear and mechanical stress.
Maintenance of TGFβ3-mediated signaling may be protective against TGFβRi-induced valvulopathy
Small-molecule TGFβRis have been reported to cause valvulopathy in nonclinical toxicology species (Anderton et al., 2011; Frazier et al., 2007; Stauber et al., 2014), and we have additionally encountered similar toxicities in the rat with BMT-A and LY2109760. Given this precedence and the similarity of these toxicities to rodent and human inactivation mutations affecting the TGFβ signaling pathway, it is likely that these toxicities are pharmacologically mediated. A difference between small molecules is that the class-based toxicities can occur at different exposures, and sometimes the toxicity can be mitigated by inclusion of dose holidays in the administration schedule (Stauber et al., 2014). In our experience, we found that BMT-B did not cause cardiovascular toxicity within exposure ranges that effectively produced valvular toxicity with LY2109761 and BMT-A, even though all three TGFβRis were similar in TGFβRI binding and biological potencies. As such, we compared the effects of the inhibitors on biological response in cultured VICs to determine if there were any differences that may relate to the lack of valvular toxicity associated with BMT-B.
Although the three inhibitors induced some similar biological responses, a striking difference was identified with TGFβ3 expression profiles following TGFβRi treatment. All three inhibitors caused mild reduction of TGFβ1 and TGFβ2 transcriptional expression. However, only the valvular toxicants, LY2109761 and BMT-A, caused potent and steep concentration-dependent reduction of TGFβ3 transcription, whereas BMT-B treatment had no effect on TGFβ3 expression. Importantly, BMT-B presented less effects on phenotypic change of VICs compared with the valvular toxic inhibitors, suggesting that sparing TGFβ3 may protect the VICs from these alterations.
Furthermore, both the siRNA and TGFβ neutralizing antibody studies demonstrated that combined loss of function of TGFβ2 and TGFβ3 on an RNA or protein level caused similar transcriptional changes indicative of EMT-MET imbalance and altered ECM, implying them as the primary ligands driving TGFβ signaling for maintaining appropriate context-dependent phenotype of VICs. In addition, the observed co-reduction in TGFβ2/β3 transcription associated with TGFβ2 or -β3 RNA silencing suggests potential existence of transcriptional co-regulation or regulation of mRNA stability of these ligands.
Only TGFβ1 and TGFβ2 were secreted at detectable levels from VICs, where the valvular toxic pan-neutralizing antibody, 1D11, effectively depleted TGFβ1 and reduced TGFβ2 levels by ∼75%. The TGFβ2 neutralizing antibody had a similar effect on reducing TGFβ2, whereas the TGFβ3 antibody administered alone or in combination with the TGFb2 antibody had considerably less inhibitory effect on TGFβ2 levels. However, combination treatment of TGFβ2 and TGFβ3 neutralizing antibodies synergized transcriptional deregulation of phenotypic markers and increased TGFβ3 transcription, indicating that both ligands have important involvement in this mechanism. In VICs, TGFβ2 appears to be the prominent secreted ligand responsible for driving signaling involved in maintaining the VIC phenotype. However, our studies also suggest that TGFβ3 may also have a role in this biology. It is possible that TGFβ3 function is more relevant on the cellular level where its transcriptional regulation can impact transcription of TGFβ2 and associated feedback mechanisms. It is also possible that its protein may be expressed at low levels and kept in reserve on the cell membrane via TGFβIII for signaling purposes.
It is unclear why BMT-B presented the distinct profile where it had no effect on inhibiting TGFβ3 transcription and possessed lack of valvular toxicity at relatively high exposures. All three inhibitors presented similar potencies on TGFβRI, yet BMT-B generally was the least potent in altering the profiled biological responses. BMT-B and BMT-A are both azaindoles with only slight differences in chemical structure (Fink et al., 2017; Zhang et al., 2018). However, unlike LY2109761 and BMT-A, BMT-B is a selective TGFβRI inhibitor with no significant activity on the TGFβRII receptor. In contrast, both LY2109761 and BMT-A were potent dual receptor inhibitors with ≤10 fold selectivity on TGFβRI over TGFβRII, respectively. Given overall preclinical experience evaluating both dual and TGFβR1-selective (ALK5) inhibitors, both pharmacological classes will likely cause valvulopathy as dose and/or duration of treatment increases. Interestingly, the only TGFβR inhibitors currently in clinical development are selective TGFβRI inhibitors, which require dose holidays to achieve an acceptable but narrow safety window for avoiding cardiovascular toxicity (Herbertz et al., 2015; Keedy et al., 2018; Rak et al., 2020).
TGFβ1 and TGFβ3 bind to TGFβRII to initiate RI/RII dimerization and canonical signal transduction and may include feedback mechanisms as indicated in this study. It is possible that transcriptional regulation of TGFβ3 is dependent upon TGFβRII signaling and that the lack of inhibitory activity of BMT-B on TGFβRII allowed appropriate feedback signaling for maintaining TGFβ3 transcription. Alternately, the selective TGFβRI inhibitory activity of BMT-B might have allowed for non-canonical signaling mediated by TGFβ1 and TGFβ3 that was not dependent upon TGFβRI activation. All three of the TGFβRis presented inhibitory activity on p-AKT; however BMT-B had the least potent inhibitory activity. Activation of AKT is associated with regulation of TGFβRII, where it causes phosphorylation of p-FAF1, which leads to decreased cell surface FAF1 and an increase of TGFβRII (Xie et al., 2017). The more potent inhibitory activity of BMT-A and LY2109761 on p-AKT, together with direct inhibitory activity on TGFβRII, may have further repressed feedback regulation of TGFβ3 expression.
Additionally, TGFβRII expression levels have been found to impact both SMAD and non-SMAD signaling pathways and alter biological effects of TGFβ signaling (Rojas et al., 2009). For instance, activation of the MAP/ERK pathway is dependent upon TGFβRII (Rojas et al., 2009). The MAP/ERK pathway has been implicated in calcification processes, and ERK1/2 is upregulated in VICs cultured in conditions promoting calcification processes (Gu and Masters, 2010). Activation of ERKs are also implicated in valvulopathy caused by 5-hydroxytryptamine receptor 2B (5-HT2B) receptor agonists, where ERK activation occurs as part of the 5-HT2B receptor signaling cascade and leads to mitogenic responses in the heart valve (Elangbam, 2010). Serotonin can enhance the activity of TGFβ receptors, suggesting an intersection between the two pathways (Elangbam, 2010). However, inhibition of TGFβRII would be thought to down regulate ERK signaling with less detrimental effects on the heart valve. Only LY2109761 presented weak inhibitory activity on p-ERK1/2 in VICs (Table 2) and did not show any protective activity on altered biological responses in the VICs or valvulopathy in the rat in vivo. Further work will be required to better understand the underlying basis for how BMT-B maintained TGFβ3 transcriptional expression and how TGFβ3-mediated signaling protects the VICs from responses that lead to valvulopathy.
In summary, we have characterized TGFβ signaling in cultured rat VICs and have used this model to better understand the impact of inhibition of this pathway on biological function of these cells. We have found that blockade of TGFβR signaling results in EMT-MET imbalance, impacting morphology and function, and that TGFβ2 and TGFβ3 may mediate TGFβ signaling in VICs necessary to maintain appropriate context-dependent phenotypes. The TGFβRi, which did not produce valvulopathy at the evaluated dose/exposures, was more resistant to the EMT-MET imbalance and ECM-related changes and did not alter TGFβ3 expression, suggesting that the degree of these responses may potentially predict potency in inducing valvulopathy in vivo. Taken together, these results suggest that the pathogenesis of TGFβRi-induced valvular toxicities may involve redirection of the context-dependent VIC phenotype and impede homeostatic processes essential for maintaining integrity of the valve, where potency of an inhibitor in driving phenotypic change may reflect its potential to cause valvulopathy in vivo.
Significance of the study
TGFβR inhibitors (TGFβRis) have promise for treating cancer and fibrotic diseases, but can cause cardiovascular toxicity including valvulopathy in preclinical species, leading to capping of clinical doses or impede progression to the clinic. Although dose-holiday schedules sometimes mitigate toxicity, safety margins remain narrow. VICs can change phenotype pliable based upon the context of their anatomical location and microenvironment in the valve; maintain appropriate elasticity and ECM production; and engage in repair mechanisms essential in maintaining integrity of the valve. VICs express TGFβRs and are suspected to be the cellular target of toxicity of TGFβRis. Using VICs harvested from rats, a sensitive species to TGFβRi -induced valvular toxicity, this study provided additional insight into the mechanism of toxicity. Canonical and non-canonical signaling are intact in VICs. TGFβ ligands are expressed with SMAD2/3 presenting phosphorylation without stimulation, suggesting that TGFβ signaling is constitutive. TGFβRi treatment inhibited transcription of myofibroblast and ECM targets, induced E-cadherin transcription, and altered smooth muscle actin organization, resulting in changes in morphology, cellular impedance, and migration. Together these changes suggest that TGFβR inhibition alters EMT-MET homeostasis in VICs. Additionally, using siRNA and TGFβ neutralizing antibodies, the results suggest that TGFβ2 and-β3 may mediate signaling essential for maintaining appropriate balance of context-dependent VIC phenotype within the valve. Altogether these findings provide additional understanding regarding which ligands are essential in mediating TGFβ signaling in VICs and the mechanism of toxicity of TGFβRis and how the pathway could be probed to potentially reduce the potential for valvulopathy.
Limitations of the study
VICs present context-dependent phenotypes based upon their anatomical location, microenvironment, and mechanical stresses in the valve. Multiple VIC phenotypes are important to maintain appropriate elasticity and ECM production and engage in repair mechanisms essential in maintaining integrity of the valve. The in vitro approaches used in this study do not fully model the diverse microenvironments and physiological stresses encountered in vivo; however the model demonstrates how the biology of VICs maintained in a static condition can be altered by interference of TGFβ signaling. Also, we have demonstrated that TGFβ2 and TGFβ3 have roles in driving VIC biology; however, we could only demonstrate detectable levels of secreted TGFβ2 protein. This may be related to limitations in sensitivity of detection of the secreted protein with Luminex technology, that TGFβ3 protein may have its functional relevance on the cellular membrane in association with TGFβ receptors (including TGFβRIII), or in a different microenvironment or under different physiological stresses, TGFβ3 may have more robust expression. Further study will be required to better characterize this function.
Resource availability
Lead contact
Karen Augustine-Rauch, Department of Discovery Toxicology, Bristol-Myers Squibb, Route 206 & Province Line Road, F1.4107B, Princeton, NJ 08543; E-mail: karen.augustine@bms.com; Ph: 609-252-3089.
Materials availability
This study did not generate unique reagents. However, all protocols associated with the work are available upon request. In addition, BMT-A and BMT-B are not available due to limited supply of synthesized compound. However, these azaindole series inhibitors can be synthesized according to information provided in Zhang et al. (2018). The structure of BMT-A and BMT-B can be found as Example 14B and 47, respectively, in Fink et al., (2017), Patent US 9,708,316 B2.
Data and code availability
The raw datasets supporting the current study have not been deposited in a public repository due to our company's information protection policies but are available from the corresponding author upon requests.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Inclusion and diversity
We worked to include sex balance in the selection of non-human subjects. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Acknowledgments
We thank Miguel Sanjuan, Myrtle Davis, and Greg Rak for their critical review of the manuscript. We are grateful to Steven Stryker, Yimer Callejas, Karen Granaldi, Christopher Hasson, and colleagues at Biocon BMS R&D Center for their technical assistance in participation of the in vivo studies and histological preparation of tissues. We thank Jia Zhu for his initial establishment of the rat valve surgical dissection method. All reported research was funded by Bristol-Myers Squibb Company.
Author contributions
F.W., C.Z., J.K., B.S., J.L., M.H., Y.S., and B.L. performed the experiments. F.W., J.K., C.Z., B.S., R.W., K.P., V.K.H., R.B., and K.A-R. contributed to experimental design and data analysis. K.A-R. supervised the study. F.W. and K.A-R. wrote the manuscript, which was edited by all authors.
Declaration of interests
All authors are employees of BMS and may own shares of company stocks, but there are no conflicts of interest in regard to the disclosure of data associated with this study.
Published: March 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102133.
Supplemental information
References
- Anderton M.J., Mellor H.R., Bell A., Sadler C., Pass M., Powell S., Steele S.J., Roberts R.R., Heier A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol. 2011;39:916–924. doi: 10.1177/0192623311416259. [DOI] [PubMed] [Google Scholar]
- Bartram U., Molin D.G., Wisse L.J., Mohamad A., Sanford L.P., Doetschman T., Speer C.P., Poelmann R.E., Gittenberger-De Groot A.C. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation. 2001;103:2745–2752. doi: 10.1161/01.cir.103.22.2745. [DOI] [PubMed] [Google Scholar]
- Chen J.H., Yip C.Y., Sone E.D., Simmons C.A. Identification and characterization of aortic valve mesenchymal progenitor cells with robust osteogenic calcification potential. Am. J. Pathol. 2009;174:1109–1119. doi: 10.2353/ajpath.2009.080750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Lu H., Rateri D.L., Cassis L.A., Daugherty A. Conundrum of angiotensin II and TGF-beta interactions in aortic aneurysms. Curr. Opin. Pharmacol. 2013;13:180–185. doi: 10.1016/j.coph.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S., Lovett D.H. Gelatinase A (MMP-2) is necessary and sufficient for renal tubular cell epithelial-mesenchymal transformation. Am. J. Pathol. 2003;162:1937–1949. doi: 10.1016/S0002-9440(10)64327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David C.J., Massague J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018;19:419–435. doi: 10.1038/s41580-018-0007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson J.M. Smad about elastin regulation. Am. J. Respir. Cell Mol. Biol. 2002;26:164–166. doi: 10.1165/ajrcmb.26.2.f228. [DOI] [PubMed] [Google Scholar]
- Dietz H.C., Cutting G.R., Pyeritz R.E., Maslen C.L., Sakai L.Y., Corson G.M., Puffenberger E.G., Hamosh A., Nanthakumar E.J., Curristin S.M. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- Elangbam C.S. Drug-induced valvulopathy: an update. Toxicol. Pathol. 2010;38:837–848. doi: 10.1177/0192623310378027. [DOI] [PubMed] [Google Scholar]
- Fink B.E., Zhao Y., Borzilleri R.M., Zhang L., Kim K.S., Kamau M.G., Tebben A.J., Zhang Y., Donnell A.F., (2017) Bristol-Myers Squibb Company. TGFbR antagonists. US Patent 09708316.
- Frazier K., Thomas R., Scicchitano M., Mirabile R., Boyce R., Zimmerman D., Grygielko E., Nold J., Degouville A.C., Huet S. Inhibition of ALK5 signaling induces physeal dysplasia in rats. Toxicol. Pathol. 2007;35:284–295. doi: 10.1080/01926230701198469. [DOI] [PubMed] [Google Scholar]
- Frixen U.H., Behrens J., Sachs M., Eberle G., Voss B., Warda A., Lochner D., Birchmeier W. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J. Cell Biol. 1991;113:173–185. doi: 10.1083/jcb.113.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellibert F., De Gouville A.C., Woolven J., Mathews N., Nguyen V.L., Bertho-Ruault C., Patikis A., Grygielko E.T., Laping N.J., Huet S. Discovery of 4-{4-[3-(pyridin-2-yl)-1H-pyrazol-4-yl]pyridin-2-yl}-N-(tetrahydro-2H- pyran-4-yl)benzamide (GW788388): a potent, selective, and orally active transforming growth factor-beta type I receptor inhibitor. J. Med. Chem. 2006;49:2210–2221. doi: 10.1021/jm0509905. [DOI] [PubMed] [Google Scholar]
- Gordon K.J., Blobe G.C. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim. Biophys. Acta. 2008;1782:197–228. doi: 10.1016/j.bbadis.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Gotlieb A.I., Rosenthal A., Kazemian P. Fibroblast growth factor 2 regulation of mitral valve interstitial cell repair in vitro. J. Thorac. Cardiovasc. Surg. 2002;124:591–597. doi: 10.1067/mtc.2002.123812. [DOI] [PubMed] [Google Scholar]
- Gu X., Masters K.S. Regulation of valvular interstitial cell calcification by adhesive peptide sequences. J. Biomed. Mater. Res. A. 2010;93:1620–1630. doi: 10.1002/jbm.a.32660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han R.I., Clark C.H., Black A., French A., Culshaw G.J., Kempson S.A., Corcoran B.M. Morphological changes to endothelial and interstitial cells and to the extra-cellular matrix in canine myxomatous mitral valve disease (endocardiosis) Vet. J. 2013;197:388–394. doi: 10.1016/j.tvjl.2013.01.027. [DOI] [PubMed] [Google Scholar]
- Herbertz S., Sawyer J.S., Stauber A.J., Gueorguieva I., Driscoll K.E., Estrem S.T., Cleverly A.L., Desaiah D., Guba S.C., Benhadji K.A. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 2015;9:4479–4499. doi: 10.2147/DDDT.S86621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck A.P. Structural studies of the TGF-betas and their receptors - insights into evolution of the TGF-beta superfamily. FEBS Lett. 2012;586:1860–1870. doi: 10.1016/j.febslet.2012.05.028. [DOI] [PubMed] [Google Scholar]
- Hosper N.A., Van Den Berg P.P., De Rond S., Popa E.R., Wilmer M.J., Masereeuw R., Bank R.A. Epithelial-to-mesenchymal transition in fibrosis: collagen type I expression is highly upregulated after EMT, but does not contribute to collagen deposition. Exp. Cell Res. 2013;319:3000–3009. doi: 10.1016/j.yexcr.2013.07.014. [DOI] [PubMed] [Google Scholar]
- Hulin A., Deroanne C.F., Lambert C.A., Dumont B., Castronovo V., Defraigne J.O., Nusgens B.V., Radermecker M.A., Colige A.C. Metallothionein-dependent up-regulation of TGF-beta2 participates in the remodelling of the myxomatous mitral valve. Cardiovasc. Res. 2012;93:480–489. doi: 10.1093/cvr/cvr337. [DOI] [PubMed] [Google Scholar]
- Hulin A., Deroanne C., Lambert C., Defraigne J.O., Nusgens B., Radermecker M., Colige A. Emerging pathogenic mechanisms in human myxomatous mitral valve: lessons from past and novel data. Cardiovasc. Pathol. 2013;22:245–250. doi: 10.1016/j.carpath.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Kahari V.M., Olsen D.R., Rhudy R.W., Carrillo P., Chen Y.Q., Uitto J. Transforming growth factor-beta up-regulates elastin gene expression in human skin fibroblasts. Evidence for post-transcriptional modulation. Lab. Invest. 1992;66:580–588. [PubMed] [Google Scholar]
- Keedy V.L., Bauer T.M., Clarke J.M., Hurwitz H., Baek I., Ha I., Ock C.Y., Nam S.Y., Kim M., Park N. Association og TGFb responsive signature with anti-tumor effect of vactosertib, a potent oral TGFb receptor type 1 (TGFbR1) inhibitor in patients with advance solid tumors. J. Clin. Oncol. 2018;36:3031. [Google Scholar]
- Kovacs R.J., Maldonado G., Azaro A., Fernandez M.S., Romero F.L., Sepulveda-Sanchez J.M., Corretti M., Carducci M., Dolan M., Gueorguieva I. Cardiac safety of TGF-beta receptor I kinase inhibitor LY2157299 monohydrate in cancer patients in a first-in-human dose study. Cardiovasc. Toxicol. 2015;15:309–323. doi: 10.1007/s12012-014-9297-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latif N., Sarathchandra P., Taylor P.M., Antoniw J., Brand N., Yacoub M.H. Characterization of molecules mediating cell-cell communication in human cardiac valve interstitial cells. Cell Biochem. Biophys. 2006;45:255–264. doi: 10.1385/CBB:45:3:255. [DOI] [PubMed] [Google Scholar]
- Leger J., Olivieri A., Donaldson M., Torresani T., Krude H., Van Vliet G., Polak M., Butler G., ESPE-PES-SLEP-JSPE-APEG-APPES-ISPAE. Congenital Hypothyroidism Consensus Conference Group European society for paediatric endocrinology consensus guidelines on screening, diagnosis, and management of congenital hypothyroidism. J. Clin. Endocrinol. Metab. 2014;99:363–384. doi: 10.1210/jc.2013-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letterio J.J., Roberts A.B. Regulation of immune responses by TGF-beta. Annu. Rev. Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Li C., Gotlieb A.I. Transforming growth factor-beta regulates the growth of valve interstitial cells in vitro. Am. J. Pathol. 2011;179:1746–1755. doi: 10.1016/j.ajpath.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C.C., Park A.Y., Guan J.L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- Lin F., Yang X. TGF-beta signaling in aortic aneurysm: another round of controversy. J. Genet. Genomics. 2010;37:583–591. doi: 10.1016/S1673-8527(09)60078-3. [DOI] [PubMed] [Google Scholar]
- Liu R.M., Desai L.P. Reciprocal regulation of TGF-beta and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol. 2015;6:565–577. doi: 10.1016/j.redox.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A.C., Joag V.R., Gotlieb A.I. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am. J. Pathol. 2007;171:1407–1418. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.M., Flanagan T.C., Lu C.C., French A.T., Argyle D.J., Corcoran B.M. Culture and characterisation of canine mitral valve interstitial and endothelial cells. Vet. J. 2015;204:32–39. doi: 10.1016/j.tvjl.2015.01.011. [DOI] [PubMed] [Google Scholar]
- Loeys B.L., Chen J., Neptune E.R., Judge D.P., Podowski M., Holm T., Meyers J., Leitch C.C., Katsanis N., Sharifi N. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- Maratera K. Perdue University Graduate School, 2009 UMI; 2009. The Effect of Transforming Growth Factor Beta Type 1 Receptor Kinase Inhibition on the Heart Valves of Sprague-Dawley Rats; p. 3403130. PhD Thesis. [Google Scholar]
- Martin P. Wound healing--aiming for perfect skin regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- Martin C.J., Datta A., Littlefield C., Kalra A., Chapron C., Wawersik S., Dagbay K.B., Brueckner C.T., Nikiforov A., Danehy F.T., Jr. Selective inhibition of TGFbeta1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci. Transl Med. 2020;12:eaay8456. doi: 10.1126/scitranslmed.aay8456. [DOI] [PubMed] [Google Scholar]
- Massague J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J., Chen Y.G. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- Noda K., Dabovic B., Takagi K., Inoue T., Horiguchi M., Hirai M., Fujikawa Y., Akama T.O., Kusumoto K., Zilberberg L. Latent TGF-beta binding protein 4 promotes elastic fiber assembly by interacting with fibulin-5. Proc. Natl. Acad. Sci. U S A. 2013;110:2852–2857. doi: 10.1073/pnas.1215779110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell D.W., Mifflin R.C., Valentich J.D., Crowe S.E., Saada J.I., West A.B. Myofibroblasts. I. Paracrine cells important in health and disease. Am. J. Physiol. 1999;277:C1–C9. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- Rak G.D., White M.R., Augustine-Rauch K., Newsome C., Graziano M.J., Schulze G.E. Intermittent dosing of the transforming growth factor beta receptor 1 inhibitor, BMS-986260, mitigates class-based cardiovascular toxicity in dogs but not rats. J. Appl. Toxicol. 2020;40:931–946. doi: 10.1002/jat.3954. [DOI] [PubMed] [Google Scholar]
- Rojas A., Padidam M., Cress D., Grady W.M. TGF-beta receptor levels regulate the specificity of signaling pathway activation and biological effects of TGF-beta. Biochim. Biophys. Acta. 2009;1793:1165–1173. doi: 10.1016/j.bbamcr.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappino A.P., Schurch W., Gabbiani G. Differentiation repertoire of fibroblastic cells: expression of cytoskeletal proteins as marker of phenotypic modulations. Lab. Invest. 1990;63:144–161. [PubMed] [Google Scholar]
- Schoen F.J. Aortic valve structure-function correlations: role of elastic fibers no longer a stretch of the imagination. J. Heart Valve Dis. 1997;6:1–6. [PubMed] [Google Scholar]
- Shi Y., Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Stauber A.J., Credille K., Truex L.L., Ehlhardt W.J., Young J.K. Non¬clinical safety evaluation of a transforming growth factor β receptor I kinase inhibitor in fischer 344 rats and beagle dogs. J. J. Clin. Pract. 2014;4:1000196. doi: 10.4172/2161-0495.196. [DOI] [Google Scholar]
- Stover D.G., Bierie B., Moses H.L. A delicate balance: TGF-beta and the tumor microenvironment. J. Cell. Biochem. 2007;101:851–861. doi: 10.1002/jcb.21149. [DOI] [PubMed] [Google Scholar]
- Tan A.R., Alexe G., Reiss M. Transforming growth factor-beta signaling: emerging stem cell target in metastatic breast cancer? Breast Cancer Res. Treat. 2009;115:453–495. doi: 10.1007/s10549-008-0184-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor P.M., Batten P., Brand N.J., Thomas P.S., Yacoub M.H. The cardiac valve interstitial cell. Int. J. Biochem. Cell Biol. 2003;35:113–118. doi: 10.1016/s1357-2725(02)00100-0. [DOI] [PubMed] [Google Scholar]
- Thiery J.P., Acloque H., Huang R.Y., Nieto M.A. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Trefely S., Wellen K.E. Metabolite regulates differentiation. Science. 2018;360:603–604. doi: 10.1126/science.aat6663. [DOI] [PubMed] [Google Scholar]
- Turini S., Bergandi L., Gazzano E., Prato M., Aldieri E. Epithelial to mesenchymal transition in human mesothelial cells exposed to asbestos fibers: role of TGF-beta as mediator of malignant mesothelioma development or metastasis via EMT event. Int. J. Mol. Sci. 2019;20:150. doi: 10.3390/ijms20010150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Villa F., Garcia-Ocana M., Galvan J.A., Garcia-Martinez J., Garcia-Pravia C., Menendez-Rodriguez P., Gonzalez-Del Rey C., Barneo-Serra L., De Los Toyos J.R. COL11A1/(pro)collagen 11A1 expression is a remarkable biomarker of human invasive carcinoma-associated stromal cells and carcinoma progression. Tumour Biol. 2015;36:2213–2222. doi: 10.1007/s13277-015-3295-4. [DOI] [PubMed] [Google Scholar]
- Vesely I. The role of elastin in aortic valve mechanics. J. Biomech. 1998;31:115–123. doi: 10.1016/s0021-9290(97)00122-x. [DOI] [PubMed] [Google Scholar]
- Walker G.A., Masters K.S., Shah D.N., Anseth K.S., Leinwand L.A. Valvular myofibroblast activation by transforming growth factor-beta: implications for pathological extracellular matrix remodeling in heart valve disease. Circ. Res. 2004;95:253–260. doi: 10.1161/01.RES.0000136520.07995.aa. [DOI] [PubMed] [Google Scholar]
- Weidinger A., Kozlov A.V. Biological activities of reactive oxygen and nitrogen species: oxidative stress versus signal transduction. Biomolecules. 2015;5:472–484. doi: 10.3390/biom5020472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis B.C., Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;293:L525–L534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- Wiltz D., Alexander Arevalos C., Balaoing L.R., Blancas A.A., Sapp M.C., Zhang X., Jane Grande-Allen K. Extracellular Matrix Organization, Structure, and Function. In: Aikawa E., editor. Calcific Aortic Valve Disease. 2013. [Google Scholar]
- Witt W., Jannasch A., Burkhard D., Christ T., Ravens U., Brunssen C., Leuner A., Morawietz H., Matschke K., Waldow T. Sphingosine-1-phosphate induces contraction of valvular interstitial cells from porcine aortic valves. Cardiovasc. Res. 2012;93:490–497. doi: 10.1093/cvr/cvs002. [DOI] [PubMed] [Google Scholar]
- Xie F., Jin K., Shao L., Fan Y., Tu Y., Li Y., Yang B., Van Dam H., Ten Dijke P., Weng H. FAF1 phosphorylation by AKT accumulates TGF-beta type II receptor and drives breast cancer metastasis. Nat. Commun. 2017;8:15021. doi: 10.1038/ncomms15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingling J.M., Blanchard K.L., Sawyer J.S. Development of TGF-β signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 2004;3:1011. doi: 10.1038/nrd1580. [DOI] [PubMed] [Google Scholar]
- Yoshioka N., Kimura-Kuroda J., Saito T., Kawamura K., Hisanaga S., Kawano H. Small molecule inhibitor of type I transforming growth factor-beta receptor kinase ameliorates the inhibitory milieu in injured brain and promotes regeneration of nigrostriatal dopaminergic axons. J. Neurosci. Res. 2011;89:381–393. doi: 10.1002/jnr.22552. [DOI] [PubMed] [Google Scholar]
- Zhang Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Zhao Y., Tebben A.J., Sheriff S., Ruzanov M., Fereshteh M.P., Fan Y., Lippy J., Swanson J., Ho C.P. Discovery of 4-azaindole inhibitors of TGFbetaRI as immuno-oncology agents. ACS Med. Chem. Lett. 2018;9:1117–1122. doi: 10.1021/acsmedchemlett.8b00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw datasets supporting the current study have not been deposited in a public repository due to our company's information protection policies but are available from the corresponding author upon requests.