

Abstract
Critical limb ischemia is a condition in which tissue necrosis occurs due to arterial occlusion, resulting in limb amputation in severe cases. Both endothelial cells (ECs) and vascular smooth muscle cells (SMCs) are needed for the regeneration of peripheral arteries in ischemic tissues. However, it is difficult to isolate and cultivate primary EC and SMC from patients for therapeutic angiogenesis. Induced pluripotent stem cells (iPSCs) are regarded as useful stem cells due to their pluripotent differentiation potential. In this study, we explored the therapeutic efficacy of human iPSC‐derived EC and iPSC‐derived SMC in peripheral artery disease model. After the induction of mesodermal differentiation of iPSC, CD34+ progenitor cells were isolated by magnetic‐activated cell sorting. Cultivation of the CD34+ progenitor cells in endothelial culture medium induced the expression of endothelial markers and phenotypes. Moreover, the CD34+ cells could be differentiated into SMC by cultivation in SMC culture medium. In a murine hindlimb ischemia model, cotransplantation of EC with SMC improved blood perfusion and increased the limb salvage rate in ischemic limbs compared to transplantation of either EC or SMC alone. Moreover, cotransplantation of EC and SMC stimulated angiogenesis and led to the formation of capillaries and arteries/arterioles in vivo. Conditioned medium derived from SMC stimulated the migration, proliferation, and tubulation of EC in vitro, and these effects were recapitulated by exosomes isolated from the SMC‐conditioned medium. Together, these results suggest that iPSC‐derived SMC enhance the therapeutic efficacy of iPSC‐derived EC in peripheral artery disease via an exosome‐mediated paracrine mechanism.
Keywords: differentiation, endothelial cells, induced pluripotent stem cells, peripheral artery diseases, smooth muscle cells
Co‐transplantation of the induced pluripotent stem cell (iPSC)‐derived endothelial cells and smooth muscle cells enhanced the therapeutic efficacies of endothelial cells and smooth muscle cells in a murine hindlimb ischemia model. Smooth muscle cells promoted angiogenesis and therapy of peripheral artery disease through exosome‐mediated paracrin mechanism.
Significance statement.
Ischemic disease is a peripheral artery disease, which indicates pain during rest with necrosis and ulcers leading to amputation. Because of the ability of endothelial cells and vascular smooth muscle cells to regenerate peripheral artery in ischemic tissues, cell‐based therapies provide a promising means to treat patients with ischemic disease. This study demonstrates that cotransplantation of induced pluripotent stem cell‐derived endothelial cells and smooth muscle cells improve blood perfusion and limb salvage rate by promoting neovascularization in the ischemic limb through smooth muscle cells derived exosome‐mediated paracrine mechanism. This study presents a new cell therapy technique for treating peripheral vascular diseases.
1. INTRODUCTION
Peripheral artery disease (PAD) is a vascular disease in which narrowed arteries reduce blood flow to peripheral tissues, including the head, organs, and limbs. In these patients, normal arteries become hardened, and certain risk factors, such as obesity, smoking, diabetes, and hypertension, can increase a person's risk of developing PAD. 1 It is estimated that >200 million people have PAD worldwide with a spectrum of symptoms from none to severe. 2 PAD can progress into critical limb ischemia (CLI), its more severe disease stage, which is associated with higher risk of limb amputation and cardiovascular death. 3 Impaired regeneration contributes to poor outcomes in patients with diabetic PAD. 4
Blood vessels are composed of two interacting cell types. Endothelial cells (EC) form the inner lining of the vessel wall, and smooth muscle cells (SMC) envelop the surface of the vascular tube. EC have been shown to promote tissue regeneration and recovery of vascular function through angiogenesis, neovascularization, and vascular repair, leading to the formation of new blood vessels and increased branching of existing blood vessels after vascular disease. 5 , 6 In addition, like EC, cell therapy with SMC has been shown to be useful for tissue regeneration. 7 , 8 SMC stabilize newly formed blood vessels and improve the therapeutic potential of EC in ischemic diseases. 9 , 10 Accumulating evidence suggests that both EC and SMC are highly useful for the treatment of PAD. 9 Both EC and SMC have many advantages for vascular therapy; however, obtaining human tissues for isolation and cultivation of EC and SMC is difficult.
Induced pluripotent stem cells (iPSC) are highly useful for cellular therapy as they have unlimited self‐renewal potential and the ability to differentiate into almost all cell types in human body. 11 Induced PSC can be prepared from somatic cells isolated from patients and are crucial for medical researches, such as those involving disease modeling and drug screening. 12 Recent studies have reported that iPSC‐derived EC (iPSC‐EC) exhibit better therapeutic efficacy than cord blood‐derived EC in the treatment of vascular disease. 13 Therapeutic effects of SMC derived from embryonic stem cells in neovascularization and tissue regeneration have also been observed. 14 Cooperation of EC and SMC derived from human iPSC enhances neovascularization in dermal wounds. 15 It has been reported that human PSCs can be differentiated into CD34+ vascular progenitors, which can be further differentiated into EC and SMC. 16 , 17 , 18 However, there is no study on combination therapy using EC and SMC, simultaneously differentiated from the same iPSC, for the treatment of PAD. Moreover, it is still unclear how iPSC‐derived EC and SMC interact during angiogenesis.
Exosomes are extracellular vesicles that emerge from a variety of cells and tissues, and exosome‐mediated signaling and cross‐talk have been reported in several pathophysiological states. 19 Exosomes may contain DNA, proteins, and other bioactive molecules that are involved in signal transduction. Exosomes released from stem cells have been reported to stimulate regenerative processes in injured cells through the modulation of relevant cellular processes, including proliferation, angiogenesis, inflammation, and differentiation. 20 , 21 Accumulating evidence suggests that exosomes are involved in therapeutic angiogenesis in ischemic diseases. 22 , 23 , 24 Therefore, it is necessary to elucidate the role of exosomes in the therapeutic angiogenesis mediated by EC and SMC in PAD.
In this study, both EC and SMC were differentiated from an iPSC line derived from human adipose tissue‐derived mesenchymal stem cells (MSC). After cotransplantation of the differentiated iPSC‐EC and iPSC‐SMC in a murine hindlimb ischemia model, the therapeutic efficacies of and paracrine interaction between iPSC‐EC and iPSC‐SMC were explored.
2. MATERIALS AND METHODS
2.1. Materials
Dulbecco's modified Eagle's medium: Nutrient Mixture F‐12 (DMEM/F12, #11330032), fetal bovine serum (FBS), penicillin‐streptomycin, 1× L‐glutaMAX, penicillin‐streptomycin, β‐mercaptoethanol, 1× nonessential amino acids (#11140), knockout serum replacement (#10828), collagenase type IV (#17104019), Dispase (#17105041), Accutase (#A1110501), Live/dead viability assay kit (L3224) were purchased from Thermo Fisher Scientific (Waltham, Massachusetts). EGM‐2 medium (#CC‐3202) and SmGM‐2 medium (#CC‐3182) were purchased from Lonza (Basel, Switzerland). Basic fibroblast growth factor (100‐18B) was purchase from PeproTech (Rocky Hill, New Jersey). CHIR99021 (#CT‐99021) was purchased from Selleckchem (Houston, Texas), and CD34 microbeads (#130‐046‐702) was purchased from Miltenyi Biotec (Bergisch Gladbach, Germany). Mitomycin C (#M4287), Y‐27632 (#Y0503), ascorbic acid (#A8960), and other unlisted reagents were purchased from Sigma‐Aldrich (St. Louis, Missouri).
2.2. Generation and cultivation of human iPSC
Mouse embryonic fibroblasts (MEFs) were purchased from the CEFO Corporation (www.cefobio.com, Seoul, Republic of Korea). For preparation of feeder layers, MEFs at passage 4 were treated with 5 μg/mL Mitomycin C for 80 minutes and seeded at 4 × 105 cells/60 mm dish (0.1% gelatin‐coated) in DMEM/F12 containing 10% FBS and penicillin‐streptomycin (125 U/mL). Human iPSC was generated from human adipose tissue‐derived MSC by retroviral overexpression of four stemness factors including Octamer‐binding transcription factor 4 (OCT4), SRY‐Box transcription factor 2 (SOX2), Krüppel‐like factor 4 (KLF4), and c‐MYC. The MSC was isolated from subcutaneous adipose tissues of a healthy donor after receiving approval from the Institution Review Board of Pusan National University Hospital (H‐2008‐116; Pusan, Korea) as previously described. 25 Undifferentiated iPSC colonies were cultured on mitomycin C‐treated un‐mitogenic MEF feeder layers. Induced PSC colonies were maintained in DMEM/F12 medium containing 1× L‐glutaMAX, penicillin‐streptomycin (125 U/mL), 60 μM β‐mercaptoethanol, 1× nonessential amino acids, 20% knockout serum replacement, and 5 ng/mL basic fibroblast growth factor with daily change of medium for 4 days. On day 5, iPSC colonies were passaged by treatment with dissociation medium (DMEM/F12 containing 200 μg/mL collagenase type IV and 1 mg/mL Dispase) for 10 minutes. After dissociation, the colonies were harvested by sedimentation and washed with the maintenance medium twice. The collected human iPSC colonies were fragmented by pipetting and plated onto MEF feeders at a 1:3 or 1:4 split ratio; then, they were incubated at 37°C under 5% CO2. The study protocol was approved by the Public Institutional Bioethics Committee designated by the MOHW. All experiments were performed in accordance with relevant guidelines and regulations.
2.3. Differentiation of iPSC into EC and SMC
Differentiation of iPSC into EC and SMC was conducted based on a previously reported protocol. 16 The iPSC was maintained on a feeder layer in DMEM/F12 medium, dissociated into single cells with Accutase at 37°C for 5 minutes, and seeded onto a Matrigel‐coated cell culture dish at 50 000 cells/cm2 in mTeSR1 supplemented with 5 μM ROCK inhibitor Y‐27632. Cells were then cultured in mTeSR1 for 3 days before induction of cell differentiation. At day 0 defined as the first day of cell differentiation, the iPSC was treated with 6 μM CHIR99021 for 2 days in LaSR basal medium, which consists of Advanced DMEM/F12, 2.5 mM GlutaMAX, and 60 mg/mL ascorbic acid. After 2 days, CHIR99021‐containing medium was aspirated, and cells were maintained in LaSR basal medium without CHIR99021 for 3 additional days. At day 5, the differentiated cells were dissociated by treatment with Accutase for 10 minutes, and the CD34+ cell population was isolated by magnetic‐activated cell sorting using CD34 microbeads, according to the manufacturer's instructions. The purified CD34+ cells were seeded on fibronectin‐coated dishes and maintained in EGM‐2 medium or SmGM‐2 medium for differentiation into EC or SMC, respectively.
2.4. Flow cytometry analysis
For flow cytometry analysis, the iPSC‐derived CD34+ progenitor cells and the iPSC‐EC were dissociated with 0.05% trypsin‐EDTA, followed by filtration through a 40 μm cell strainer. The dissociated cells were fixed with 4% paraformaldehyde for 20 minutes, washed with PBS, and then incubated with APC‐conjugated antibodies (anti‐CD34 and anti‐VE‐cadherin antibodies) or PE‐conjugated antibodies (anti‐KDR and anti‐CD31 antibodies) (BD Biosciences, Bedford, Massachusetts) at 1:200 dilution for 20 minutes. The fluorescence intensity of the stained cells was measured using CANTO II (BD Biosciences) and analyzed using FlowJo (ver 10, Tree Star Inc).
2.5. Reverse transcription‐polymerase chain reaction (RT‐PCR)
Cells were harvested at the indicated times, and total RNA was extracted using TRIzol (Sigma‐Aldrich). For cDNA synthesis, 1 μg total RNA from each sample was reverse transcribed into cDNA using an Easy cDNA Synthesis Kit (NanoHelix Co, Ltd, Daejeon, Republic of Korea), according to the manufacturer's instructions. The cDNA (1 μL each) was added into a PCR tube containing HelixAmp Ready‐2 × GO (NanoHelix, Co, Ltd) and 10 pmol of sense and antisense primers. PCR was performed using the following thermal cycle: denaturation at 95°C for 30 seconds, annealing at i°C for 30 seconds depending on the primers used, and extension at 72°C for 30 seconds. Each PCR reaction involved 25 to 30 cycles. Primer sequences are listed in Supplementary Table 1. PCR products were analyzed by electrophoresis on a 1% agarose gel.
Quantitative RT‐PCR was performed using an ABI7500 (Applied Biosystems, Waltham, Massachusetts) sequence‐detection system, using SYBR Green PCR Master Mix (Applied Biosystems), according to the manufacturer's instructions. Experiments were performed in triplicate, and the data were normalized to the expression of the GAPDH mRNA. Data were analyzed using the Δ(ΔCT) method.
2.6. Murine hindlimb ischemia model
BALB/CA‐nu/nu mice, athymic immunodeficient mice which are unable to produce T cells, were used for xenograft transplantation of iPSC‐derived cells into hindlimb ischemia animal model. The BALB/CA‐nu/nu mice (an average weight of 20‐24 g) were purchased from Orient Bio (Seongnam‐city, Republic of Korea). All animals were housed in an air‐conditioned animal room with constant relative humidity and were provided a standard laboratory diet and water. Animal treatment and maintenance were performed in accordance with the Principles of Laboratory Animal Care, and animal experiments were performed using protocols approved by the Pusan National University Institutional Animal Use and Care Committee. All mice were anesthetized using an intraperitoneal injection of 400 mg/kg 2,2,2 tribromoethanol (Avertin; Sigma‐Aldrich) for operative resection of the femoral artery and laser Doppler perfusion imaging. The femoral artery was excised from its proximal origin at the branch of the external iliac artery to the distal point where it bifurcates into the saphenous and popliteal arteries. Right after arterial ligation, ischemic hindlimbs were injected with either iPSC‐EC or iPSC‐SMC (1.2 × 106 cells each). For coadministration of iPSC‐EC and iPSC‐SMC, equal numbers (0.6 × 106 cells each) of iPSC‐EC and iPSC‐SMC were mixed to 1:1 ratio and co‐injected into ischemic hindlimbs. In each animal, 80 μL HBSS containing the iPSC‐derived cells or 80 μL HBSS only were injected into four sites (20 μL/each site) of the gracilis muscle in the medial thigh. Before transplantation of cells, the percentage of viable cells was confirmed to be more than 95% using a Live/dead viability assay kit (Invitrogen).
2.7. Measurement of blood flow and tissue necrosis
Blood flow of the ischemic and normal limbs was measured using a laser Doppler perfusion imaging (LDPI) analyzer (Moor Instruments Ltd, Devon, UK) on days 0, 7, 14, 21, and 28 after induction of hindlimb ischemia. The perfusion of the ischemic and nonischemic limbs was calculated based on colored histogram pixels. Red and blue colors indicate high and low perfusion, respectively. Blood perfusion is expressed as the LDPI index, representing the ratio of ischemic vs nonischemic limb blood flow. A ratio of 1 before operation indicates equal blood perfusion of both legs. The extent of necrosis in the ischemic hindlimb was recorded on day 28 after surgery. The necrosis was scored as follows: 0, limb salvaged; 1, toe amputation; 2, foot amputation; and 3, limb amputation.
2.8. Histological and immunofluorescence analyses
For immunostaining of the tissues, hindlimb muscles were removed, formalin‐fixed, paraffin‐embedded, and sectioned at 5 μm. The blood vessels were stained with biotinylated‐ILB4 (Vector Laboratories) or rabbit anti‐α‐SMA (Abcam plc., Cambridge, UK) antibodies. The specimens were incubated with Alexa 488 streptavidin, Alexa 488, or Alexa 568 goat anti‐rabbit secondary antibodies (Thermo Fischer Scientific), followed by washing and mounting in Vectashield medium containing DAPI for visualization of nuclei. The stained sections were visualized using a laser scanning confocal microscope. The injected iPSC‐EC and iPSC‐SMC were identified in the ischemic limbs by staining with anti‐human nuclear antigen (HNA) antibody (Millipore, Burlington, Massachusetts) and Alexa 488 goat anti‐mouse secondary antibody. The injected cells in tissue sections were quantified by counting the number of cells positive for both HNA and ILB4 expression or HNA and α‐SMA expression. Blood vessel densities were counted by identifying ILB4‐ or α‐SMA‐positive vascular structures per high‐power field. Four randomly selected microscopic fields from three serial sections in each tissue block were examined by two independent observers blinded to the experimental conditions. in vitro differentiation of iPSC to iPSC‐EC and iPSC‐SMC was confirmed by immunostaining and confocal microscopy analysis. The iPSC‐EC or iPSC‐SMC were fixed with 4% paraformaldehyde for 15 minutes, washed with PBS, and permeabilized with 1% Triton‐X‐100 in PBS for 20 minutes. The cells were blocked with 5% BSA for 1 hour and incubated with primary antibodies overnight and secondary antibodies conjugated with fluorescent dyes for 1 hour. Cells were mounted using Vectashield medium with DAPI for nuclear staining. Images were obtained using a laser scanning confocal microscope (Olympus FluoView FV1000).
2.9. Preparation of SMC‐CM and isolation of exosomes
To obtain SMC‐CM, iPSC‐SMCs were cultured in 100 mm culture dishes until subconfluent and were then washed twice with HBSS to remove serum components. The cells were incubated in 10 mL serum‐free SMC basal medium for 48 hours. The collected SMC‐CM was filtered through a 0.45 μm syringe filter and immediately centrifuged at 500g for 10 minutes and 10 000g for 30 minutes to remove cells and debris. The collected culture supernatant was centrifuged at 100 000g for 70 minutes at 4°C to precipitate exosomal fractions, and the exosome‐depleted supernatant (exosome‐depleted SMC‐CM) was collected for experiments. The resulting exosomal pellets were washed with HBSS, precipitated by ultracentrifugation at 100 000g for 70 minutes at 4°C to obtain exosomes, and resuspended in HBSS. The protein concentration of exosomal fractions was measured using a Bradford protein assay kit (Bio‐Rad, Hercules, California).
2.10. Statistical analysis
The results of multiple observations are presented as mean ± SD. For multivariate data analysis, group differences were assessed using one‐way or two‐way ANOVA, followed by Scheffé's post hoc test.
3. RESULTS
3.1. Differentiation of human iPSC into CD34 + cells
To induce the differentiation of human iPSC to EC and SMC, iPSC were differentiated into a mesodermal lineage by treatment with the GSK3 inhibitor CHIR99021 for 2 days, followed by incubation in a defined media without CHIR99021 for an additional 3 days. On day 5, CD34+ multipotent mesodermal progenitor or vascular progenitor cells (VPCs) were purified through magnetic associated cell sorting using CD34 magnetic beads and further differentiated to either EC or SMC (Figure 1A). Upon mesodermal differentiation of iPSC, the mRNA levels of the pluripotency markers OCT4, SOX2, and NANOG were reduced (Figure 1B). The mRNA levels of mesodermal markers, including Bruchury, MIXL1, and TBX6, were increased 24 hours after CHIR99021 treatment (Figure 1C). Expression of the vascular progenitor markers, CD34 and KDR, was detected on day 4 and increased till day 5 (Figure 1D). To determine the differentiation efficiency of the cells, the percentage of the cells expressing CD34 and KDR was measured by flow cytometry. As shown in Figure 1E, approximately 30% of cells expressed both CD34 and KDR at day 5. The CD34+ cells were isolated using magnetic beads conjugated with an anti‐CD34 antibody. The CD34+ cells were cultured for 4 days, following which approximately 96% were found to be CD34‐ and KDR‐double positive which indicates their differentiation into VPC.
FIGURE 1.
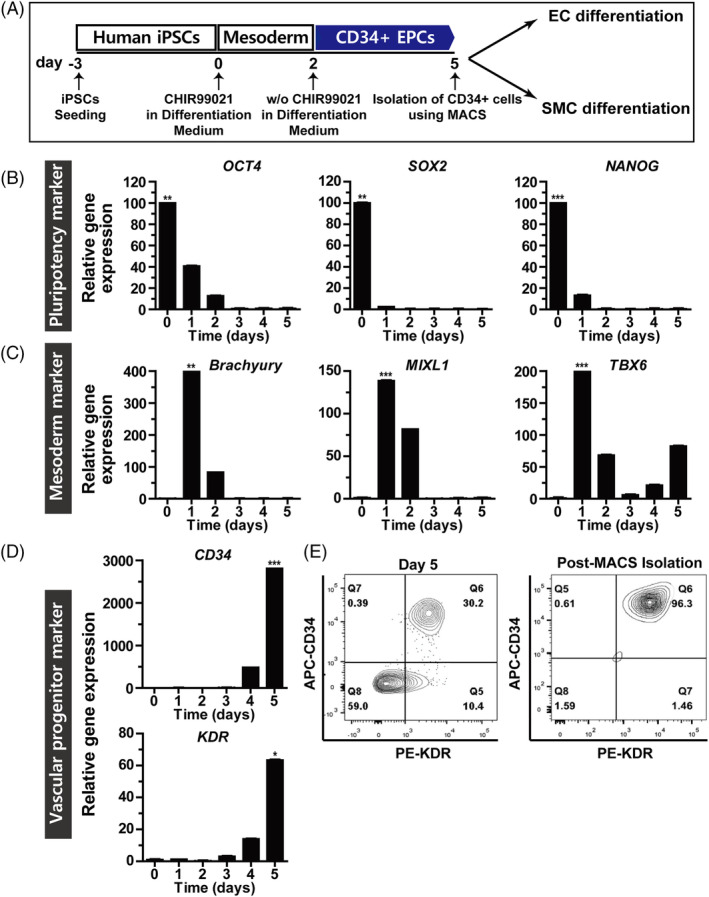
Differentiation of induced pluripotent stem cells to vascular progenitor cells. A, Overview of the differentiation protocol for induced pluripotent stem cells (iPSCs) to vascular progenitor cells (VPC). At day 0, iPSC was treated with CHIR99021 for 2 days in a differentiation medium, and followed by incubation in the differentiation medium without CHIR99021 for 3 days. The CD34+ cells were purified using CD34 magnetic beads at day 5. For differentiation into EC and SMC, CD34+ cells were cultured in the endothelial cell (EC) and smooth muscle cell (SMC) culture medium, respectively. B, Gene expression at different time points during the differentiation of CD34+ cells was assessed by quantitative RT‐PCR of pluripotency markers (OCT4, SOX2, and NANOG); C, mesodermal markers (Bruchury, MIXL1, and TBX6); and D, vascular progenitor markers (CD34 and KDR). Results are presented as mean ± SD (n = 3). *P < .05; **P < .01; ***P < .001. E, Flow cytometric analysis of CD34 and KDR in iPSC‐VPC on day 5 after induction of differentiation. Flow cytometric analysis of CD34 and KDR in iPSC‐VPC cultured for 4 days
3.2. Characterization of human iPSC‐derived ECs and iPSC‐derived SMCs
To explore whether the CD34+ VPC can differentiate into EC and SMC by cultivation in EC and SMC culture medium, the isolated CD34+ VPCs were cultured in EC and SMC culture medium, respectively, on fibronectin‐coated plates. The CD34 and KDR expression was drastically reduced in iPSC‐EC and iPSC‐SMC compared to that in iPSC‐VPC (Figure 2A). The mRNA levels of the EC markers, including CD31 and VEGFR1, in iPSC‐EC were higher than those in iPSC‐VPC and iPSC‐SMC (Figure 2B). The expression of SMC markers, such as SM22α and Desmin, was found to be the highest in iPSC‐SMC compared to those iPSC‐VPC and iPSC‐EC (Figure 2C). These results suggest that iPSC‐VPC can be further differentiated to EC and SMC by cultivation in EC‐ and SMC‐selective media.
FIGURE 2.
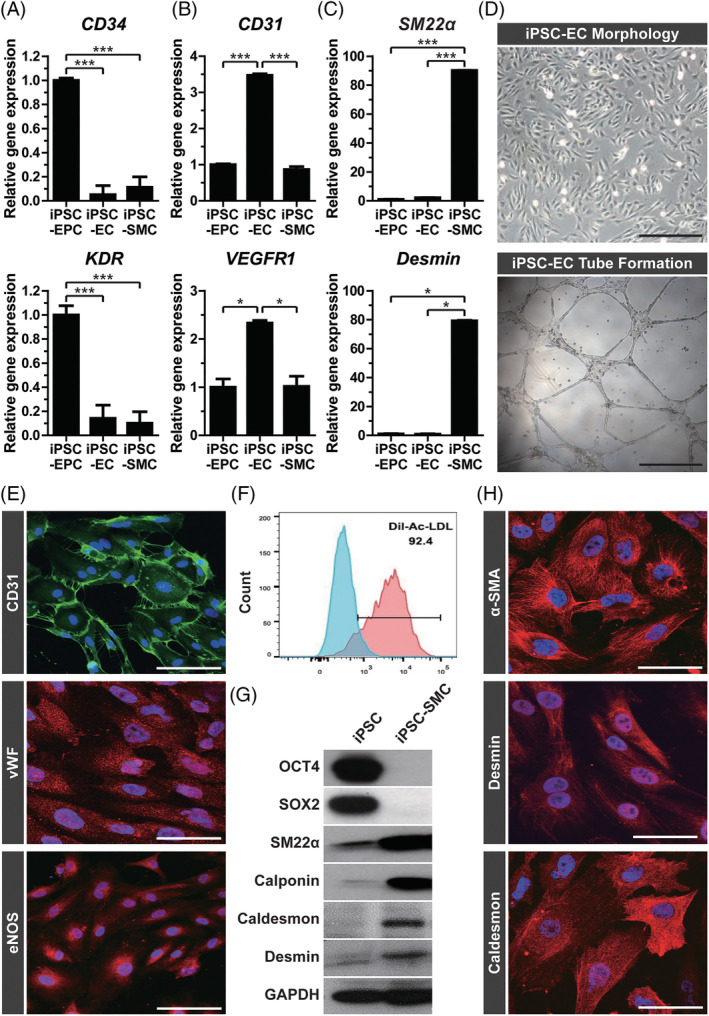
Characterization of induced pluripotent stem cell (iPSC)‐derived endothelial cells and iPSC‐derived smooth muscle cells. Gene expression of iPSC‐vascular progenitor cell (VPC), ‐endothelial cell (EC), and ‐smooth muscle cell (SMC) was assessed by quantitative reverse transcription‐polymerase chain reaction (RT‐PCR) of (A) vascular progenitor cell markers (CD34 and KDR), (B) endothelial markers (CD31 and VEGFR1), and (C) smooth muscle markers (SM22α and Desmin). Results are presented as mean ± SD (n = 3). *P < .05; **P < .01; ***P < .001. D, Representative images of adherent cells and tube formation by iPSC‐EC are shown. Scale bar = 200 μm. E, The iPSC‐EC, on day 3 after sorting, were subjected to immunofluorescence staining using antibodies against CD31 (green), vWF (red), and eNOS (red). Nuclei were counterstained with DAPI (blue), and the merged images are shown. Scale bar = 50 μm. F, Flow cytometric analysis of iPSC‐EC after sorting are shown. Ac‐LDL uptake was measured after incubating the cells with Ac‐LDL. No Ac‐LDL treatment is a negative control. G, Western blot analysis of iPSC and iPSC‐SMC using pluripotency markers (OCT4 and SOX2), smooth muscle markers (SM22α, Calponin, Caldesmon, Desmin), and GAPDH is shown. H, Representative images for immunocytochemistry of iPSC‐SMC at day 4 after sorting are shown. Confocal images of iPSC‐SMC after immunolabeling with antibodies against α‐SMA, Desmin, and Caldesmon are shown (red). Nuclei were counterstained with DAPI (blue), and the merged images are shown. Scale bar = 50 μm
To further describe the differentiation of iPSC to EC, the levels of differentiated EC markers were measured by RT‐PCR. After differentiation of iPSC to EC, pluripotency markers, such as OCT4 and SOX2, were not expressed in differentiated iPSC‐EC (Supplementary Figure 1A). However, iPSC‐EC exhibited increased expression of endothelial markers, such as vWF, VE‐Cadherin, Prominin, and E‐selectin (Supplementary Figure 1B). Using FACS analysis, we found that the iPSC‐EC was positive for not only CD31 but also VE‐cadherin (Supplementary Figure 2A). Consistently, the immunostaining and western blot analyses confirmed the expression of both VE‐cadherin and CD31 in iPSC‐EC (Supplementary Figure 2B,C). To characterize the functional properties of the iPSC‐EC, we evaluated their endothelial tube‐forming ability. As shown in Figure 2D, the iPSC‐EC exhibited endothelial morphology and tube forming ability. Further endothelial phenotypes of the iPSC‐EC were confirmed by immunostaining with antibodies against CD31 and vWF, as both CD31 and vWF were expressed in iPSC‐EC. Additionally, the expression of endothelial nitric oxide synthase (eNOS), an EC‐specific marker, could be detected in the iPSC‐EC by immunofluorescence (Figure 2E). Finally, we measured acetylated low‐density lipoprotein (Ac‐LDL) uptake, which is a predominantly endothelial phenotype. We found that more than 92% of the cells were Ac‐LDL‐positive (Figure 2F). These results suggest that the differentiated iPSC‐ECs exhibit phenotypes and functional properties of EC.
To demonstrate differentiation of the iPSC to SMC, the sorted CD34+ cells were seeded on fibronectin‐coated plates and cultured in SMC culture medium. After 4 days, the levels of smooth muscle markers were determined by western blotting. The protein levels of smooth muscle markers, such as SM22α, Calponin, h‐caldesmon, and Desmin, were higher in the iPSC‐SMC compared to those in the iPSC, whereas the expression of pluripotency markers, including OCT4 and SOX2, was not detected in the iPSC‐SMC (Figure 2G). To further verify the SMC phenotypes of the iPSC‐SMC, we measured the expression of the smooth muscle‐specific markers α‐SMA, Desmin, and Caldesmon by immunostaining. The iPSC‐SMC showed the expression of α‐SMA, Desmin, and Caldesmon, (Figure 2H). These results suggest that the iPSC‐SMCs differentiated from iPSC exhibit phenotypes of SMC.
3.3. Synergistic effects of coadministration of iPSC‐EC and iPSC‐SMC on the ischemic hindlimb
To explore the therapeutic effects of iPSC‐EC and iPSC‐SMC on ischemic tissue injury, we confirmed their functional properties, such as angiogenesis and tissue repair, in the murine hindlimb ischemia model. The iPSC‐EC and iPSC‐SMC were administered into the ischemic hindlimb by intramuscular injection, and blood flow was measured for 4 weeks using an LDPI analyzer. As shown in Figure 3A,B, intramuscular injection of iPSC‐EC, but not that of iPSC‐SMC, resulted in improved blood perfusion in ischemic limbs. However, the cotransplantation of iPSC‐EC and iPSC‐SMC augmented blood flow compared to the transplantation of iPSC‐EC alone. Furthermore, a combined injection of iPSC‐EC and iPSC‐SMC into the ischemic limb significantly inhibited tissue amputation, as demonstrated by reduced necrosis score 4 weeks after induction of ischemia compared to those in the other groups (Figure 3C). These results suggest that cotransplantation of iPSC‐EC with iPSC‐SMC synergistically increased blood perfusion and reduced tissue necrosis.
FIGURE 3.
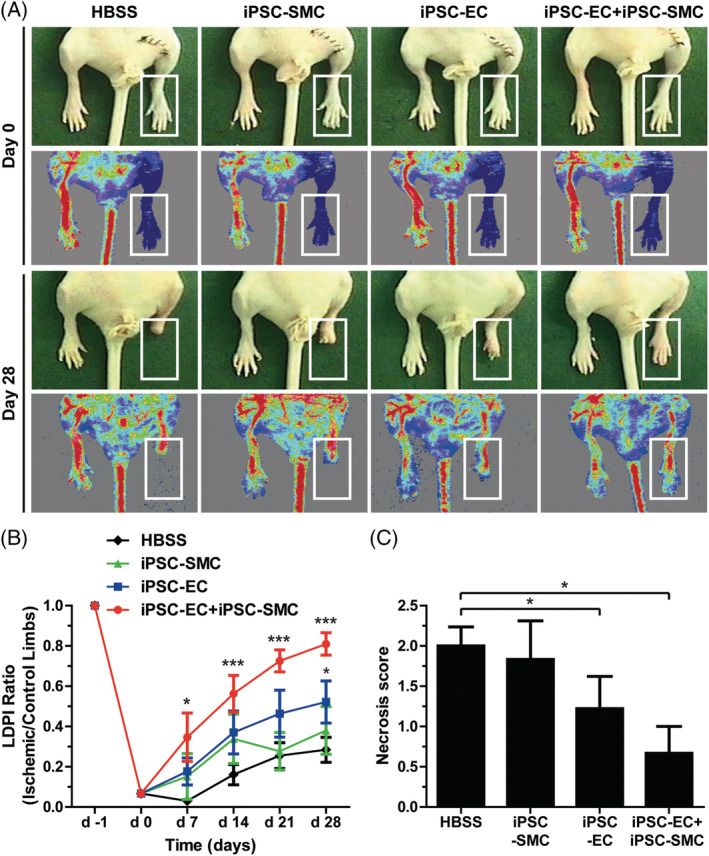
Effect of combined induced pluripotent stem cell‐endothelial cell (iPSC‐EC) and iPSC‐smooth muscle cell (SMC) treatment on blood perfusion and tissue necrosis in hindlimb ischemia. A, Representative photographs and laser Doppler perfusion imaging (LDPI) of mouse hindlimbs on day 0 and 28 after injection of HBSS, iPSC‐EC, iPSC‐SMC, or iPSC‐EC with iPSC‐SMC. B, Quantitative analysis of the blood perfusion recovery measured using an LDPI analyzer. The LDPI ratio was calculated as the ratio of ischemic to nonischemic hindlimb blood perfusion. (n = 6‐9 per experimental group). C, Statistical analysis of the necrosis score on day 28. Results are presented as mean ± SD (n = 6‐9). *P < .05; ***P < .001
3.4. Coadministration of iPSC‐EC and iPSC‐SMC promotes neovascularization in the ischemic limb
Angiogenesis is crucial for ischemic tissue repair and the recovery of blood perfusion. 26 To evaluate whether cotransplantation of iPSC‐EC and iPSC‐SMC can stimulate angiogenesis in vivo, we determined blood vessel density in the ischemic limbs by immunostaining. Intramuscular injection of iPSC‐EC increased the number of ILB4+ capillaries in the ischemic limbs (Figure 4A,B). Furthermore, the number of α‐SMA+ arteries/arterioles was significantly increased in iPSC‐EC‐injected limbs compared with that in the control group limbs (Figure 4A,C). However, administration of iPSC‐SMC had no significant effect on the number of ILB4+ and α‐SMA+ blood vessels. Interestingly, the cotransplantation of iPSC‐EC and iPSC‐SMC increased the numbers of ILB4+ capillaries and α‐SMA+ arteries/arterioles more than those in the control groups, which were injected with either iPSC‐EC or iPSC‐SMC. These results suggest that the combination of iPSC‐EC and iPSC‐SMC synergistically enhances angiogenesis in ischemic limbs.
FIGURE 4.
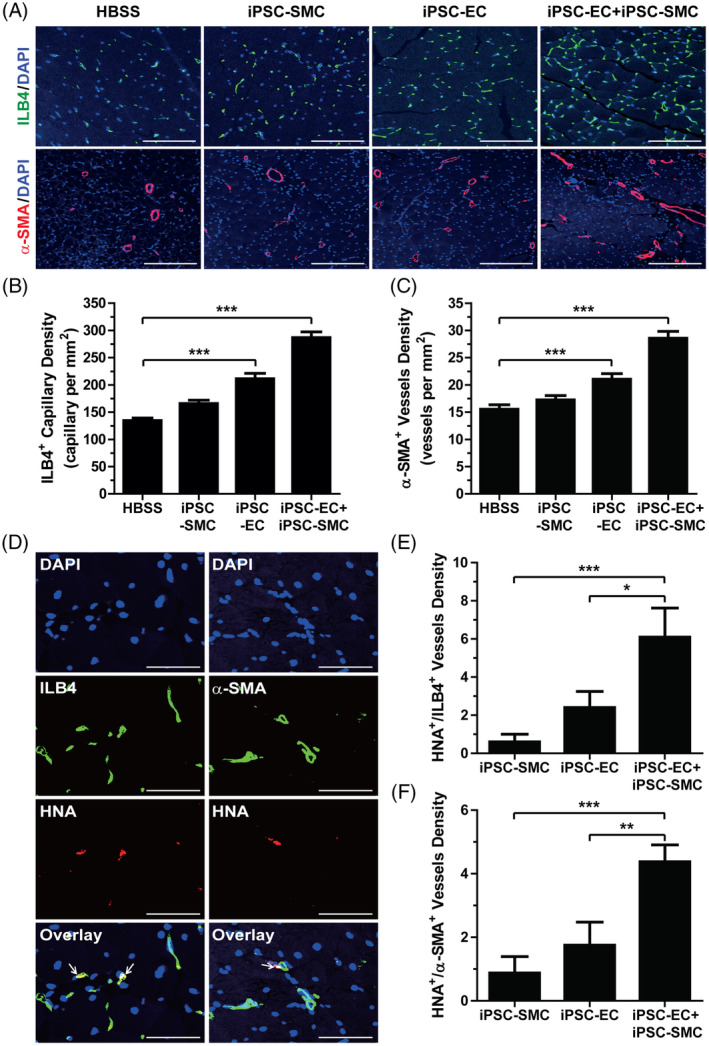
Effect of combined induced pluripotent stem cell‐endothelial cell (iPSC‐EC) and iPSC‐(smooth muscle cell) SMC treatment on angiogenesis in hindlimb ischemia. A, Immunostaining of ILB4+ capillaries (green) and α‐SMA+ blood vessels (red) in ischemic limbs injected with HBSS, iPSC‐EC, iPSC‐SMC, or iPSC‐EC with iPSC‐SMC at 28 days after surgery. Nuclei (blue) were counterstained with DAPI. Scale bar = 100 μm (ILB4/DAPI) or 200 μm (α‐SMA/DAPI). B, Quantification of ILB4+ capillaries in the ischemic limb by immunohistochemistry. C, Quantification of α‐SMA+ arteries in the ischemic limb by immunohistochemistry. Results are presented as mean ± SD (n = 6‐9 per each group). D, Representative images of hindlimb tissues double stained with anti‐HNA (red) and anti‐ILB4 (green) antibodies or anti‐α‐SMA (green) antibody 28 days after surgery. Nuclei were counterstained with DAPI (blue). Scale bar = 50 μm. E, Number of HNA+/ILB4+ capillaries in the ischemic limbs. F, Number of HNA+/α‐SMA+ vessels in the ischemic limbs. Results are presented as mean ± SD (n = 9). *P < .05; **P < .01; ***P < .001
To further determine whether transplanted iPSC‐EC and iPSC‐SMC are incorporated into the blood vessels of ischemic limbs, we investigated iPSC‐EC‐ and iPSC‐SMC‐mediated neovascularization by quantifying the number of capillaries and vessels double positive for human specific nuclear antigen (HNA) and either ILB4 or α‐SMA. As shown in Figure 4D, some ILB4+ capillaries and α‐SMA+ vessels were also HNA+. Compared to the ischemic limbs injected with either iPSC‐EC or iPSC‐SMC, cotransplantation of iPSC‐EC and iPSC‐SMC synergistically increased the number of HNA+/ILB4+ and HNA+/α‐SMA+ blood vessels in the ischemic limbs (Figure 4E,F). These results suggested that the cotransplantation of EC and SMC synergistically accelerates neovascularization in ischemic limbs.
3.5. The iPSC‐SMC stimulates the angiogenic activity of iPSC‐EC through a paracrine mechanism
To explore how iPSC‐SMCs stimulate iPSC‐EC‐mediated angiogenesis in vivo, we examined the effect of SMC‐CM on the migration of iPSC‐EC using a chemotaxis assay. As shown in Figure 5A, SMC‐CM stimulated the chemotactic migration of iPSC‐EC in a dose‐dependent manner. Consistently, SMC‐CM significantly enhanced tube formation of iPSC‐EC at 20% with a slight inhibition of EC tube formation at 50% concentration (Figure 5B), suggesting the existence of inhibitory factors of EC tube formation in SMC‐CM. We next measured the effect of SMC‐CM on the proliferation of iPSC‐EC by evaluating PCNA expression. Treatment of iPSC‐EC with SMC‐CM dose‐dependently promoted cell proliferation (Figure 5C). These results suggest that iPSC‐SMC promotes the angiogenic activity of iPSC‐EC through a paracrine mechanism.
FIGURE 5.
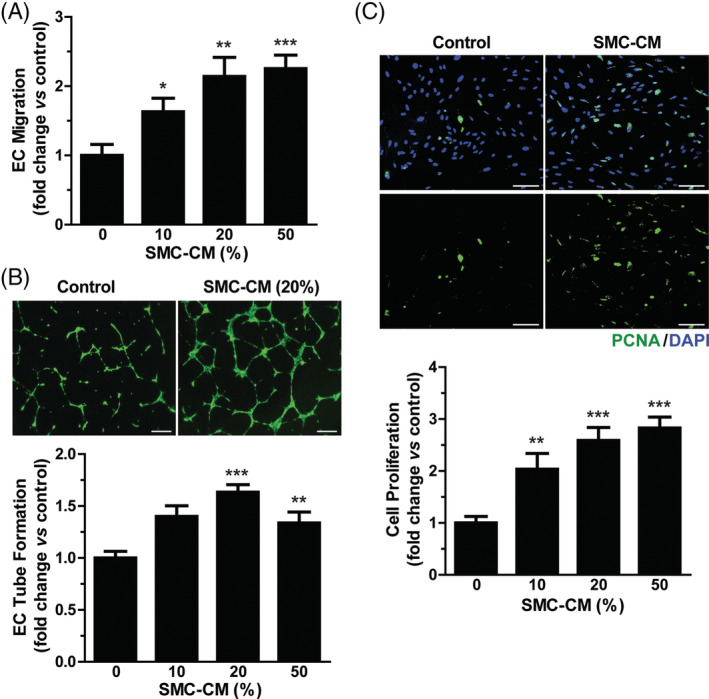
Effects of smooth muscle cell (SMC)‐CM on migration, tube formation, and proliferation of induced pluripotent stem cell‐endothelial cell (iPSC‐EC). A, Representative images of chemotactic migration in response to SMC‐CM are shown. Migration of iPSC‐EC was measured using a chemotaxis chamber in response to various concentrations of SMC‐CM after a 12‐hours incubation. B, Representative images of tube formation in response to SMC‐CM are shown (upper panels). Tube formation was quantified by measuring the length of the tubes formed (lower panel). Scale bar = 500 μm. C, The proliferative effect of SMC‐CM on iPSC‐EC was measured by staining with anti‐PCNA antibody (green). Nuclei were counterstained with DAPI (blue) (upper panel). Scale bar = 100 μm. Number of PCNA+ nuclei per field were counted and expressed as a relative percentage of the total cells (lower panel). Data indicate mean ± SD (n = 4). *P < .05; **P < .01; ***P < .001
To verify the specificity of SMC‐CM‐stimulated pro‐angiogenesis, we compared the effects of SMC‐CM and human dermal fibroblast‐conditioned medium (hDF‐CM) on angiogenic activities of iPSC‐EC. In contrast to the pro‐angiogenic activities of SMC‐CM, hDF‐CM did not stimulate migrating and tube forming abilities of iPSC‐EC (Supplementary Figure 3). Furthermore, cotransplantation of iPSC‐EC together with hDF exhibits less potent blood perfusion than transplantation of iPSC‐EC alone (Supplementary Figure 4). These results support the specificity of SMC‐CM for stimulation of iPSC‐EC‐mediated angiogenesis.
3.6. SMC‐CM stimulates the migration and proliferation of iPSC‐EC through an exosome‐dependent mechanism
Exosomes are composed of various components, including proteins, DNA, miRNA, and lipids. To explore whether protein components are involved in the SMC‐CM‐mediated pro‐angiogenic activity, SMC‐CM was heated at 95°C for 10 minutes to denature proteins. As shown in Figure 6A, the SMC‐CM‐stimulated chemotaxis of iPSC‐EC was abrogated by heat denaturation of SMC‐CM proteins, suggesting that protein factors are involved in the SMC‐CM‐stimulated migration of iPSC‐EC.
FIGURE 6.
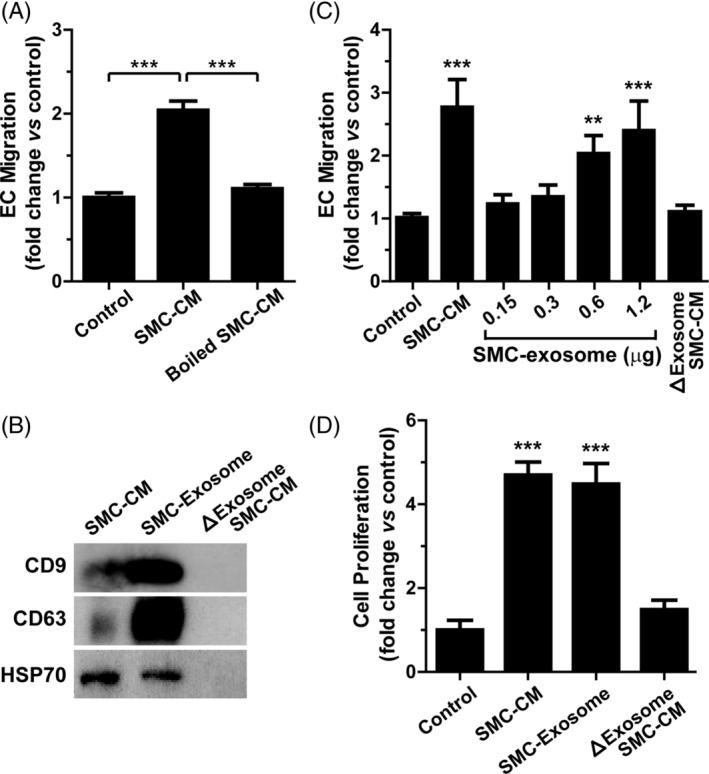
Role of exosomes in the smooth muscle cell (SMC)‐CM‐stimulated migration and proliferation of induced pluripotent stem cell‐endothelial cell (iPSC‐EC). A, Role of protein factors in SMC‐CM‐mediated angiogenesis. SMC‐CM was heated at 95°C for 10 minutes to denature protein components. The effects of SMC‐CM and heated SMC‐CM on the chemotactic migration of iPSC‐EC were determined. B, Isolation of exosomes from SMC‐CM. Exosomes were isolated from SMC‐CM by centrifugation. The levels of exosomal markers (CD9, CD63, and HSP70) in the SMC‐CM, SMC‐exosomes, and exosome‐depleted SMC‐CM (ΔExosome SMC‐CM) were determined by western blotting. C, Dose‐dependent effects of SMC‐exosomes on the chemotactic migration of iPSC‐EC. The effects of SMC‐CM and ΔExosome SMC‐CM on EC migration were examined. D, Role of exosomes in SMC‐CM‐stimulated proliferation of iPSC‐EC. The effects of SMC‐CM, SMC‐exosome, and ΔExosome SMC‐CM on the proliferation of iPSC‐EC were examined. Data indicate mean ± SD (n = 4). **P < .01; ***P < .001
Exosomes derived from various cells, such as MSC, 27 CD34+ stem cells, 28 and cancer cells, 29 have been reported to stimulate angiogenesis. To investigate whether iPSC‐SMC‐derived exosomes (SMC‐exosomes) mediate SMC‐CM‐induced angiogenesis, we isolated exosomes from SMC‐CM and examined their effects on the chemotactic migration and proliferation of iPSC‐EC. SMC‐exosomes were isolated by ultracentrifugation, and exosomal markers, such as CD9, CD63, and HSP70, could be detected in the SMC‐exosomes, but not in exosome‐depleted SMC‐CM (Figure 6B). The CD9 and CD63 proteins were highly enriched in the SMC‐exosomes compared to those in the SMC‐CM. The SMC‐CM‐stimulated migration of iPSC‐EC was markedly attenuated by the depletion of exosomes from the SMC‐CM, whereas the SMC‐exosomes dose‐dependently stimulated the chemotactic migration of iPSC‐EC (Figure 6C). In addition, SMC‐exosomes were as potent in promoting the proliferation of iPSC‐EC as the SMC‐CM (Figure 6D). However, exosome‐depleted SMC‐CM had no significant effect on the proliferation of iPSC‐EC. These results suggest that SMC‐exosomes may mediate the SMC‐CM‐stimulated migration and proliferation of iPSC‐EC.
To explore whether SMC‐exosomes mediate the iPSC‐SMC‐stimulated therapeutic activities of iPSC‐EC, we examined the effects of coadministration of iPSC‐EC and SMC‐exosome on blood perfusion and tissue necrosis in hindlimb ischemia. Coinjection of iPSC‐EC along with SMC‐exosomes significantly augmented blood perfusion and alleviated tissue necrosis (Supplementary Figure 5). Moreover, coinjection of exosomes derived from both iPSC‐EC and iPSC‐SMC exhibited less potent blood perfusion than the co‐injection of iPSC‐EC and SMC‐exosomes. These results suggest that SMC‐exosomes promote iPSC‐EC‐stimulated blood perfusion in ischemic limbs.
4. DISCUSSION
Cell therapy is gaining a lot of attention as a new treatment for PAD mediated by an increase in angiogenesis. 8 In this study, we demonstrated that EC and SMC could be produced from the same human iPSC clone by serial differentiation steps. The iPSC was differentiated to the mesodermal progenitor cell lineage by treatment with the GSK3‐β inhibitor CHIR99021, followed by differentiation to the CD34+ VPC by culture in the absence of CHIR99021. The CD34+ cells were further differentiated into either EC or SMC by culture in EC or SMC culture medium, respectively. The differentiation protocol used in the present study was adopted from a previous report demonstrating that human iPSC can be differentiated into CD34+ VPC via modulation of Wnt signaling and further differentiated to either EC or SMC, depending on culture conditions. 16 Similarly, it has been reported that GSL3 inhibition and BMP4 treatment committed PCSs to a mesodermal lineage and that the subsequent exposure of these cells to VEGF or PDGF‐BB resulted in the differentiation to EC or SMC, respectively. 30 Moreover, human PCS‐derived CD34+ VPCs have been reported to give rise to EC and SMC depending on culture conditions. 17 , 18 These results suggest that both EC and SMC could be produced from the same pool of iPSC‐derived CD34+ progenitor cells by modulating culture conditions.
For treatment of patients with CLI, various clinical trials using progenitor cells isolated from bone marrow or peripheral blood have been made. 31 , 32 Although the cell therapy of CLI significantly improved clinical parameters in majority of the studies, the therapeutic efficacy of primary progenitor cells is highly variable from patient to patient due to its heterogeneity. Therefore, differentiation of iPSC to specific cell types is highly useful approach for treatment of ischemic diseases. EC and SMC are promising cell therapeutics for the regeneration of ischemic tissues. 9 It has been shown that SMC stabilized newly formed blood vessels and improved the therapeutic potential of EC in ischemic diseases. 9 , 10 In the present study, transplantation of iPSC‐EC, but not that of iPSC‐SMC, promoted blood perfusion, tissue protection, and neovascularization in the ischemic hindlimb. Moreover, cotransplantation of iPSC‐EC with iPSC‐SMC further enhanced therapeutic efficacy in ischemic limb repair by increasing blood vessel density. Consistently, it has been reported that cotransplantation of human iPSC‐derived EC and SMC synergistically promoted angiogenesis in dermal wounds. 15 Coinjection of iPSC‐EC with normal human lung fibroblasts and a fibrin matrix into the dorsal flank of severe combined immunodeficiency mice led to the formation of functional microvasculature in vivo. 33 The potential of EC and SMC derived from human iPSC for clinical application in PAD needs to be explored further.
In this study, we showed that conditioned medium derived from iPSC‐SMC promoted migration, tube formation, and proliferation of iPSC‐EC in vitro, suggesting a paracrine activation of iPSC‐EC by iPSC‐SMC. In agreement, the conditioned medium from SMC promoted proliferation and migration of EC in vitro. 10 Growth factors and cytokines secreted from SMC may have a key role in the regulation of the proliferation and migration of EC. Many studies have demonstrated that MSC stimulates neovascularization and blood perfusion in ischemic tissues by secreting pro‐angiogenic cytokines and growth factors, including VEGF, FGF‐2, HGF, and IGF‐1. 34 Accumulating evidence suggests exosomes secreted from different cell types carry vast different varieties of bioactive molecular cargoes and subsequently provide different biological responses. 35 For instance, exosomes secreted from human CD34+ stem cells, which were isolated from peripheral blood mononuclear cells, promoted the repair of the ischemic hindlimb. 28 The exosomes derived from CD34+ cells contain proangiogenic miRNAs, such as miR‐126‐3p, which enhance angiogenesis by regulating the expression of pro‐angiogenic genes, including VEGF, angiopoietins, and matrix metallopeptidase 9. MicroRNA exchange via exosomes has been shown to modulate the cross‐talk between EC and SMC. 36 Exosomes contain not only miRNAs but also various protein factors, including Wnt4, Stromal cell derived factor 1, Notch‐Dll4, Tetraspanin 8, heat shock protein 20, and hypoxia‐inducible factor‐1α, which have been implicated in EC regeneration. 37 In this study, we demonstrated that heat denaturation of protein factors abrogated SMC‐exosome‐stimulated angiogenesis in vitro. These results suggest that protein factors present in exosomes as cargo may mediate exosome‐stimulated angiogenesis. The molecular identity of these protein factors that are involved in the SMC‐exosome‐stimulated angiogenic effects is still unknown and it needs to be clarified in future studies.
In summary, our findings suggest that the combined use of iPSC‐EC and iPSC‐SMC is useful for neovascularization and regeneration in PAD. In addition, we demonstrate, for the first time, that iPSC‐SMC promotes the angiogenic and regenerative activities of iPSC‐EC via exosome‐dependent mechanisms. Identification of the factors implicated in the SMC‐exosome‐mediated angiogenesis is crucial for the therapeutic application of SMC‐exosomes. In addition, development of mass production of iPSC‐derived EC and SMC and efficient isolation methods of SMC‐exosomes will be needed for clinical application of the present study.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
J.J.P., Y.W.K.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing; J.W.K., G.T.P., J.W.Y., Y.S.K., D.S.K.: collection of data, data analysis and interpretation, manuscript writing; S.M.K., S.S.B., K.K., C.‐S.K.: conception and design, data analysis and interpretation; J.H.K.: conception and design, financial support, administrative support, manuscript writing, final approval of manuscript.
Supporting information
Appendix S1: Supplementary Information
ACKNOWLEDGMENTS
Support for this research was provided by the MRC programs (NRF‐2015R1A5A2009656) of the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (NRF‐2015M3A9C6030280; NRF‐2017R1A6A3A11034390), and a fund (2018ER610300) by the Research of Korea Centers for Disease Control and Prevention.
Park JJ, Kwon YW, Kim JW, et al. Coadministration of endothelial and smooth muscle cells derived from human induced pluripotent stem cells as a therapy for critical limb ischemia. STEM CELLS Transl Med. 2021;10:414–426. 10.1002/sctm.20-0132
Jin Ju Park, Yang Woo Kwon, and Jeong Won Kim contributed equally to the study.
Funding information Research of Korea Centers for Disease Control and Prevention, Grant/Award Number: 2018ER610300; Ministry of Education, Science and Technology, Grant/Award Numbers: NRF‐2017R1A6A3A11034390, 2015M3A9C6030280; National Research Foundation of Korea, Grant/Award Number: NRF‐2015R1A5A2009656
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Campia U, Gerhard‐Herman M, Piazza G, Goldhaber SZ. Peripheral artery disease: past, present, and future. Am J Med. 2019;132(10):1133‐1141. [DOI] [PubMed] [Google Scholar]
- 2. Shu J, Santulli G. Update on peripheral artery disease: epidemiology and evidence‐based facts. Atherosclerosis. 2018;275:379‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frangogiannis NG. Cell therapy for peripheral artery disease. Curr Opin Pharmacol. 2018;39:27‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fadini GP, Spinetti G, Santopaolo M, Madeddu P. Impaired regeneration contributes to poor outcomes in diabetic peripheral artery disease. Arterioscler Thromb Vasc Biol. 2020;40(1):34‐44. [DOI] [PubMed] [Google Scholar]
- 5. Kwon YW, Heo SC, Lee TW, et al. N‐acetylated Proline‐glycine‐Proline accelerates cutaneous wound healing and neovascularization by human endothelial progenitor cells. Sci Rep. 2017;7:43057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kwon YW, Heo SC, Jeong GO, et al. Tumor necrosis factor‐alpha‐activated mesenchymal stem cells promote endothelial progenitor cell homing and angiogenesis. Biochim Biophys Acta. 2013;1832(12):2136‐2144. [DOI] [PubMed] [Google Scholar]
- 7. Park HS, Choi GH, Hahn S, Yoo YS, Lee JY, Lee T. Potential role of vascular smooth muscle cell‐like progenitor cell therapy in the suppression of experimental abdominal aortic aneurysms. Biochem Biophys Res Commun. 2013;431(2):326‐331. [DOI] [PubMed] [Google Scholar]
- 8. Merkulova‐Rainon T, Broqueres‐You D, Kubis N, Silvestre JS, Levy BI. Towards the therapeutic use of vascular smooth muscle progenitor cells. Cardiovasc Res. 2012;95(2):205‐214. [DOI] [PubMed] [Google Scholar]
- 9. Foubert P, Matrone G, Souttou B, et al. Coadministration of endothelial and smooth muscle progenitor cells enhances the efficiency of proangiogenic cell‐based therapy. Circ Res. 2008;103(7):751‐760. [DOI] [PubMed] [Google Scholar]
- 10. Joo HJ, Seo HR, Jeong HE, et al. Smooth muscle progenitor cells from peripheral blood promote the neovascularization of endothelial colony‐forming cells. Biochem Biophys Res Commun. 2014;449(4):405‐411. [DOI] [PubMed] [Google Scholar]
- 11. Yu J, Vodyanik MA, Smuga‐Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917‐1920. [DOI] [PubMed] [Google Scholar]
- 12. Choi KD, Yu J, Smuga‐Otto K, et al. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells. 2009;27(3):559‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Park SJ, Moon SH, Lee HJ, et al. A comparison of human cord blood‐ and embryonic stem cell‐derived endothelial progenitor cells in the treatment of chronic wounds. Biomaterials. 2013;34(4):995‐1003. [DOI] [PubMed] [Google Scholar]
- 14. Cheung C, Sinha S. Human embryonic stem cell‐derived vascular smooth muscle cells in therapeutic neovascularisation. J Mol Cell Cardiol. 2011;51(5):651‐664. [DOI] [PubMed] [Google Scholar]
- 15. Kim KL, Song SH, Choi KS, Suh W. Cooperation of endothelial and smooth muscle cells derived from human induced pluripotent stem cells enhances neovascularization in dermal wounds. Tissue Eng Part A. 2013;19(21–22):2478‐2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lian X, Bao X, Al‐Ahmad A, et al. Efficient differentiation of human pluripotent stem cells to endothelial progenitors via small‐molecule activation of WNT signaling. Stem Cell Rep. 2014;3(5):804‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marchand M, Anderson EK, Phadnis SM, et al. Concurrent generation of functional smooth muscle and endothelial cells via a vascular progenitor. Stem Cells Translational Medicine. 2014;3(1):91‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hill KL, Obrtlikova P, Alvarez DF, et al. Human embryonic stem cell‐derived vascular progenitor cells capable of endothelial and smooth muscle cell function. Exp Hematol. 2010;38(3):246‐257.e241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. S ELA, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12(5):347‐357. [DOI] [PubMed] [Google Scholar]
- 21. Bruno S, Kholia S, Deregibus MC, Camussi G. The role of extracellular vesicles as paracrine effectors in stem cell‐based therapies. Adv Exp Med Biol. 2019;1201:175‐193. [DOI] [PubMed] [Google Scholar]
- 22. Manuel GE, Johnson T, Liu D. Therapeutic angiogenesis of exosomes for ischemic stroke. Int J Physiol Pathophysiol Pharmacol. 2017;9(6):188‐191. [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao L, Johnson T, Liu D. Therapeutic angiogenesis of adipose‐derived stem cells for ischemic diseases. Stem Cell Res Ther. 2017;8(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson TK, Zhao L, Zhu D, et al. Exosomes derived from induced vascular progenitor cells promote angiogenesis in vitro and in an in vivo rat hindlimb ischemia model. Am J Physiol Heart Circ Physiol. 2019;317(4):H765‐H776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jeon ES, Park WS, Lee MJ, Kim YM, Han J, Kim JH. A rho kinase/myocardin‐related transcription factor‐A‐dependent mechanism underlies the sphingosylphosphorylcholine‐induced differentiation of mesenchymal stem cells into contractile smooth muscle cells. Circ Res. 2008;103(6):635‐642. [DOI] [PubMed] [Google Scholar]
- 26. Annex BH. Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol. 2013;10(7):387‐396. [DOI] [PubMed] [Google Scholar]
- 27. Zhang K, Zhao X, Chen X, et al. Enhanced therapeutic effects of Mesenchymal stem cell‐derived exosomes with an injectable hydrogel for hindlimb ischemia treatment. ACS Appl Mater Interfaces. 2018;10(36):30081‐30091. [DOI] [PubMed] [Google Scholar]
- 28. Mathiyalagan P, Liang Y, Kim D, et al. Angiogenic mechanisms of human CD34(+) stem cell exosomes in the repair of ischemic hindlimb. Circ Res. 2017;120(9):1466‐1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kalluri R. The biology and function of exosomes in cancer. J Clin Invest. 2016;126(4):1208‐1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Patsch C, Challet‐Meylan L, Thoma EC, et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat Cell Biol. 2015;17(8):994‐1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dubsky M, Jirkovska A, Bem R, Nemcova A, Fejfarova V, Jude EB. Cell therapy of critical limb ischemia in diabetic patients ‐ state of art. Diabetes Res Clin Pract. 2017;126:263‐271. [DOI] [PubMed] [Google Scholar]
- 32. Persiani F, Paolini A, Camilli D, et al. Peripheral blood mononuclear cells therapy for treatment of lower limb ischemia in diabetic patients: a single‐center experience. Ann Vasc Surg. 2018;53:190‐196. [DOI] [PubMed] [Google Scholar]
- 33. Bezenah JR, Rioja AY, Juliar B, Friend N, Putnam AJ. Assessing the ability of human endothelial cells derived from induced‐pluripotent stem cells to form functional microvasculature in vivo. Biotechnol Bioeng. 2019;116(2):415‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pankajakshan D, Agrawal DK. Mesenchymal stem cell paracrine factors in vascular repair and regeneration. J Biomed Technol Res. 2014;1(1). 10.19104/jbtr.2014.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fan C, Zhang E, Joshi J, Yang J, Zhang J, Zhu W. Utilization of human induced pluripotent stem cells for cardiac repair. Front Cell Dev Biol. 2020;8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Caputo M, Saif J, Rajakaruna C, Brooks M, Angelini GD, Emanueli C. MicroRNAs in vascular tissue engineering and post‐ischemic neovascularization. Adv Drug Deliv Rev. 2015;88:78‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baruah J, Wary KK. Exosomes in the regulation of vascular endothelial cell regeneration. Front Cell Dev Biol. 2019;7:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.