

Abstract
Introduction:
Sporadic Creutzfeldt Jakob Disease, the most common reported prion disease, is a fatal neurodegenerative disease caused by the misfolding of protein PrPC to PrPSC. EEG is the first in vivo test to support the clinical diagnosis of sporadic CJD (sCJD). This study is carried out to describe the comprehensive electroencephalography changes in a cohort of patients with probable sCJD from south India.
Methodology:
This retrospective observational study is conducted by reviewing case records from 2013–2020. Demographic, clinical, imaging details were noted. Electroencephalogram (EEG) recordings were retrieved and reviewed independently by two qualified epileptologists and the findings were visually analyzed and correlated with clinical and imaging abnormalities.
Results:
Fifty patients with probable sCJD with the availability of artefact-free EEG were included in the study. The mean age was 59.76 ± 8.17 years and M: F was 31:19. Magnetic resonance imaging (MRI) brain showed abnormality defining CJD in 48/50, i.e., 96%patients. EEG showed specific and or nonspecific abnormalities in 88%. The specific abnormalities, i.e., periodic sharp wave complexes (PSWC), were noted 66%. PSWC were mostly asymmetric (90%) and more frequently seen in the right hemisphere (57.6%). Amplitude maximum in the occipital region was noted in 42.4% and in frontal region in 36.4%. The sensitivity of EEG specific abnormalities to diagnose probable sCJD according to CDC 2018 criteria with positive MRI findings was 68.75%.
Conclusion:
This study showed that EEG is a relatively inexpensive and sensitive tool and assists in the diagnosis of sCJD. However, it can be normal or show nonspecific abnormalities in the early stages of the disease.
Keywords: CJD, EEG, electroencephalography, prion disease, sporadic CJD
INTRODUCTION
Creutzfeldt–Jakob disease (CJD), the most common reported prion disease, is a fatal neurodegenerative disease caused by misfolding of protein PrPC to PrPSC.[1] The exact etiopathogenesis is not known. It has a wide phenotypic spectrum caused by genetic and acquired causes. Broadly, it can be classified in to sporadic, familial, iatrogenic, and variant types depending on the mode of transmission. Sporadic CJD (sCJD) is the most common form with characteristic electroencephalogram (EEG) and imaging abnormalities. sCJD is classified into six major variants with distinctive clinic-pathological features correlating with the genotype at the polymorphic codon 129 (methionine, M or valine, V) in the prion protein gene and two PrPSc profiles (type 1 and type 2), i.e., MM1/MV1, MM2 cortical, MM2 thalamic, MV2, VV1, and VV2.[2]
EEG is the first in vivo test to support the clinical diagnosis of sCJD. The typical EEG appearances in sCJD are periodic, triphasic sharp wave complexes. However, the EEG has been shown to be positive in only a subset of sCJD patients, usually MM1 or MV1 cases and these changes may not be seen in the early stages of the disease. Typically, periodic sharp wave complexes are detected late in the disease and the median time to positive EEG is around 12 weeks. The overall sensitivity of EEG ranged from 40%–70% in various studies.[3] Nonetheless, it remains an important, noninvasive surrogate marker for sCJD. There is a wide range of literature from all over the world about electrophysiological characteristics of CJD; however, the studies from India include few case reports and case series.[4] This study is carried out to describe the comprehensive electroencephalography changes in a cohort of patients with probable sCJD as per the CDC 2018 criteria.[3]
METHODOLOGY
This retrospective observational study is conducted in the Department of Neurology, NIMHANS from 2013–2020. The study is approved by Institute ethics committee. Patients coded with CJD were reviewed from Medical record section from 2013–2020 and those fulfilling the criteria for probable sCJD by CDC2018 criteria were recruited in the study.[3]
Study cohort
Fifty patients with probable sCJD with the availability of artefact-free EEG were included in the study. Following neurological illnesses known to CJD mimickers were excluded: hepatic encephalopathy in 50/50, autoimmune encephalitis in 37/50, paraneoplastic encephalitis 34/50, and thyroid disorders in 45/50 by the available panel of investigations. None of the patients had 14.3.3 protein level test results in this cohort. The demographic, clinical, and imaging data were collected from the case files. The clinical data included age, duration of symptoms, interval between EEG and symptom onset, as well as death, clinical symptoms, i.e., dementia, psychiatric disturbance, myoclonus, ataxia, extrapyramidal symptoms, pyramidal, visual disturbance, etc.
Scalp EEGs were recorded on 40-channel “Galileo NT (EBN)” machine, employing the International 10-20 system of electrode placement using standard parameters and procedures, e.g., high filter: 70 Hz; low filter: 0.1 Hz; recording time: 30 min; sensitivity: 7 μV/mm; sweep speed: 10 s/page; impedance <10 kΩ, Notch OFF, and sampling rate: 256 Hz. Forty-seven patients had an awake record, with additional sleep recording in five patients. One patient had only sleep record and two patients were in altered sensorium at the time of recording. Activation procedures included hyperventilation in 20 and photic stimulation (1–30 Hz for 5 s at a stretch with eyes open and closed) done in all records. Sound or nociceptive stimuli were also given in 39/50 of the patients. EEG recordings were retrieved and reviewed independently by two qualified epileptologists and the findings were tabulated in an excel sheet. The EEGs were assessed for specific and nonspecific abnormalities reported in the literature for CJD. The specific abnormalities were presence or absence of periodic sharp wave complexes (PSWC). The characteristics of PSWC, i.e., abundance, spatial distribution, early peak, maximum voltage distribution, influence of environmental or physiological stimuli, were assessed by visual analysis. EEG recordings were recorded at the time of admission and stored in an electronic database.
Descriptive statistics like mean, standard deviation, and frequency were used to express the data. The relationship between different nominal variables was assessed using the Chi-square test. An independent sample t-test was used to test the significance between nominal and continuous variables. The sensitivity and specificity of EEG and magnetic resonance imaging (MRI) for diagnosing sCJD by CDC criteria were calculated.
RESULTS
Study cohort
After screening the records, a total of 50 patients were included in the study fulfilling the criteria of sCJD. None of the patients had 14.3.3 protein level test results in this cohort. Hence, we included patients with progressive dementia with either EEG showing period complexes or MRI showing changes characteristic of CJD or both.
Clinical profile [Table 1]
Table 1.
Clinical, imaging, and EEG characteristics of the cohort
| Clinical variable (n=50) | Value (%) |
|---|---|
| Age (mean±SD) | 59.76±8.17 (37-78) |
| Gender M: F | 31:19 |
| Interval between EEG and clinical onset in days (mean±SD) | 141.94±220.29 (7-1440) |
| Interval between EEG and death in days n=19 (mean±SD) | 261.68±343.70 |
| Median Survival duration in days n=19 | 270 (60-1980) |
| Myoclonus n (%) | 35 (70) |
| Dementia n (%) | 44 (88) |
| Psychiatric disturbances n (%) | 38 (76) |
| Extrapyramidal symptoms/signs n (%) | 28 (56) |
| Ataxia n (%) | 15 (30) |
| Pyramidal symptoms/signs n (%) | 18 (36) |
| Akinetic mutism n (%) | 16 (32) |
| Visual symptoms n (%) | 9 (18) |
| Seizures n (%) | 2 (4) |
| Previous surgery | 2 (4) |
| Abnormal MRI supporting CJD | 48 (96) |
| Bilateral MRI changes n=44 | 44 (100) |
| Symmetric n=45 | 8 (17.8) |
| Right > left n=45 | 22 (48.9) |
| Left > right n=45 | 15 (33.3) |
| Cortical ribbon | 46 (92) |
| Cortical ribbon distribution anteriorly prominent n=41 | 4 (9.8) |
| Cortical ribbon distribution posteriorly prominent n=41 | 28 (70.7) |
| Cortical ribbon distribution uniform n=41 | 8 (19.5) |
| Basal ganglia signal changes | 38 (76) |
| Caudate and lentiform uniform involvement n=37 | 8 (21.6) |
| Caudate more than lentiform involvement n=37 | 25 (67.6) |
| Caudate less than lentiform involvement n=37 | 4 (10.8) |
| Bilateral thalamic signal changes | 17 (34) |
| Overall MRI is suggestive of cortical more than BG involvement n=46 | 25 (54.3) |
| Overall MRI is suggestive of BG more than cortical involvement n=46 | 10 (21.7) |
| Overall MRI is suggestive of BG and cortical uniform involvement n=46 | 11 (23.9) |
| Signal changes only in DWI n=44 | 14 (30.2) |
| Signal changes DWI and T2 FLAIR n=44 | 30/43 (69.8) |
| Abnormal EEG | 44 (88) |
| Background abnormal | 42 (84) |
| PDR absent | 31 (62) |
| Predominant BG frequency | |
| Beta | 1 (2) |
| Alpha | 13 (26) |
| Theta | 27 (54) |
| Delta | 9 (18) |
| Background reactivity | |
| Nonreactive | 5 (10) |
| Poorly reactive | 34 (68) |
| Well reactive | 11 (22) |
| FIRDA | 8 (16) |
| Periodic complexes | 33 (66) |
| Morphology (n=33) | |
| Biphasic | 23 (69.7) |
| Triphasic | 10 (30.3) |
| Abundance of PSWC (n=33) | |
| Infrequent | 11 (33.3) |
| Frequent but not continuous | 9 (27.2) |
| Continuous | 13 (39.4) |
| PSWC peak gradient (n=33) | |
| No gradient | 15 (45.5) |
| Anterior to posterior | 8 (24.2) |
| Posterior to anterior | 2 (6.1) |
| Mixed | 5 (15.2) |
| Central to periphery | 3 (9.1) |
| PSWC amplitude (n=33) | |
| Frontal maximum | 12 (36.4) |
| Occipital maximum | 14 (42.4) |
| Temporal maximum | 3 (9.1) |
| Mixed | 4 (12.1) |
| PSWC symmetry (n=33) | |
| Symmetric | 3 (9.1) |
| Right >left | 19 (57.6) |
| Left >right | 11 (33.3) |
| PSWC synchrony (n=33) | |
| Unilateral/Bilateral independent | 2 (6.1) |
| Bilateral synchronous | 13 (39.4) |
| Independent as well as synchronous | 18 (54.5) |
The mean age was 59.76 ± 8.17 years and M: F was 31:19. Only 19 of the 50 patients had follow-up data about survival duration. The mean duration of clinical onset and EEG recording was 141.94 ± 220.29. The median survival duration was 270 days with a range of 60–1980 days. The common clinical symptoms were dementia in 88%, psychiatric disturbances in 76%, myoclonus in 70%, akinetic mutism in 32%, and ataxia in 30% [Table 1].
Imaging abnormalities [Table 1]
In this cohort, 5/50 MRI brain images could not be retrieved from PACS and the available findings from the case records were analyzed for these patients. MRI brain showed abnormality defining CJD in 48/50, i.e., 96%patients. MRI abnormalities were found to be 100% bilateral, and asymmetric in 88.2%, predominantly involved right hemisphere (48.9%) when it was asymmetric. Cortical ribbon was present in 92%, basal ganglia signal changes in 76%, and thalamic signal changes were noted in 34%. Cortical ribbon was prominent in the posterior head region in 70.7% [Figure 1 d and e]. In the basal ganglia, caudate was more hyperintense than in lentiform in 70.7%. Overall, the cortex was more involved than basal ganglia in 54.3%. The MRI signal changes were seen in FLAIR and DWI imaging in 68.2%, and 31.8% showed signal changes exclusively in DWI. The classical MRI brain abnormalities noted are shown in Figure 1 (a-f). When the UCSF 2011 revised criteria are applied to the cohort of 45 patients with available images in PACS for review, it showed MRI definitely CJD in 41/45 (91.1%), MRI probably CJD in 2/45 (4.4%), MRI probably not CJD in none, and MRI definitely not CJD in 2/45.[5]
Figure 1.

Classical MRI abnormalities a) FLAIR, b) DWI, and c) ADC images: showing signal changes in the bilateral asymmetric left > right caudate, putamen, and medial thalami. d) FLAIR and e) ADC images: showing classical cortical ribboning with diffusion restriction, asymmetric right > left, posterior > anterior involvement. f) DWI image showing the hyperintense signal changes in the bilateral medial thalami, basal ganglia and precuneus, and temporooccipital cortex
EEG findings [Table 1]
EEG showed specific and or nonspecific abnormalities in 88%. The mean duration of illness during EEG evaluation was 142 days. The specific abnormalities, i.e., PSWC, were noted 66%. The characteristics of PSWC were analyzed visually. The morphology was predominantly biphasic in around 70% and triphasic in 30.3%. Continuous PSWC, i.e., greater than 80% of the record, were noted in 39.4% [Figure 2a]. PSWC were mostly asymmetric (90%) and more frequently seen in the right hemisphere (57.6%). In 54.5%, the PSWC were bilateral independent and as well as synchronous. Periodic lateralized complexes were noted in 6.1% [Figure 1c]. Early peak gradient from anterior to posterior was noted in 24.2% [Figure 4a]. Amplitude maximum in the occipital region was noted in 42.4% [Figure 4b] and frontal region in 36.4% [Figure 4c]. The amplitude ranged from 100–400 μV, and the interval between periodic complexes ranged from 0.5 to 4 s. In most of the patients 25/33 (75.8%), the periodic complexes were reactive to the environmental stimuli or body movement. In one of the patients, PSWC appeared only during hyperventilation.
Figure 2.

(a-d): a) EEG showing classical periodic sharp wave complexes (PSWC) with an interval of 1 s. b) EEG showing less frequent PSWC with variable interval ranging from 1–4 s, c) EEG showing PSWC confined to right posterior head region with resemblance to lateralized periodic complexes LPDs. d) Periodic complexes confined to bilateral posterior head region leads
Figure 4.

(a-c): a) EEG showing PSWC with early peak in the frontocentral leads, b) PSWC with maximum amplitude in the occipital leads (O1), c) PSWC with early peak in the occipital leads but maximum amplitude in the frontocentral leads
The nonspecific abnormalities noted in the cohort were abnormal background in 84% and FIRDA 16%. The background abnormalities were in the form of absent posterior dominant rhythm in 62%, decreased reactivity in 78%, and abnormal focal or diffuse delta-theta slowing in 72%. None of the patients had electrographic seizures. One patient showed focal spikes mixed with PSWC [Figure 3a]. The representative EEG images describing the characteristics of PSWC are provided in Figures 2 and 3.
Figure 3.
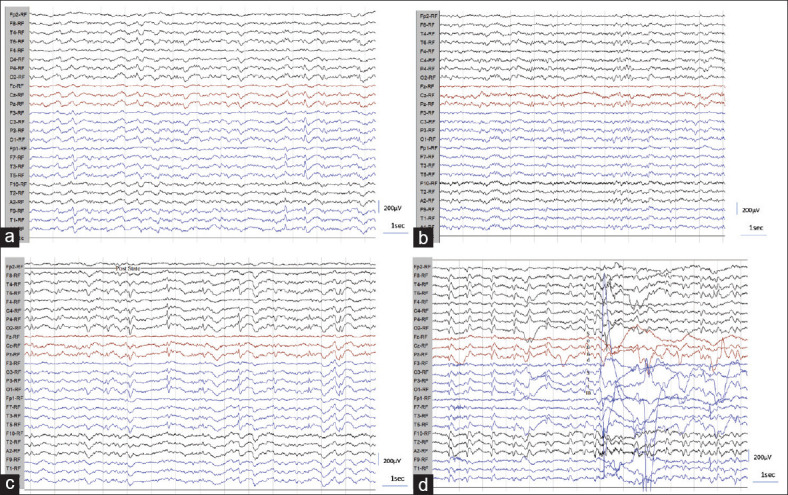
(a-g): a) EEG showing left temporal focal spike appearance along with infrequent bilateral independent PSWC. b and c) PSWC appearing only during and post hyperventilation in graph “c” and absent before hyperventilation as noted in graph “b.” d) EEG showing decreased frequency of PSWC following sound stimuli and body movements
Relationship of PSWC with clinical and imaging data [Tables 2 and 3]
Table 2.
Relationship of PSWC with duration of illness, interval between EEG and survival
| PSWC | n | Mean | SD | MD | P | 95%CI limits | ||
|---|---|---|---|---|---|---|---|---|
| Duration of presentation | Absent | 17 | 115.00 | 92.837 | −40.82 | .540 | −173.90 | 92.26 |
| Present | 33 | 155.82 | 263.475 | |||||
| Time interval between EEG and death | Absent | 6 | 415.00 | 185.338 | 224.08 | .194 | −125.891 | 574.1 |
| Present | 13 | 190.92 | 381.718 | |||||
| Survival duration | Absent | 6 | 500.00 | 202.287 | 140.23 | .541 | −334.230 | 614.7 |
| Present | 13 | 359.77 | 526.372 | |||||
*Independent sample t-test, PSWC: Periodic complexes, SD: Standard Deviation, MD: Mean Difference, CI: Confidence Interval
Table 3.
Relationship of periodic complexes with clinical and imaging variables
| Variable | PSWC present | PSWC absent | Chi-square test value | df | P | |
|---|---|---|---|---|---|---|
| Gender | Male | 20 | 11 | .08 | 1 | 0.78 |
| Female | 13 | 6 | ||||
| Duration of illness | < 3 months | 19 | 11 | 0.24 | 1 | 0.63 |
| > 3 months | 14 | 6 | ||||
| Myoclonus | Present | 23 | 10 | 0.004 | 1 | 0.95 |
| Absent | 12 | 5 | ||||
| Dementia | Present | 30 | 14 | |||
| Absent | 3 | 3 | ||||
| Psychiatric disturbances | Present | 26 | 12 | 0.41 | 1 | 0.52 |
| Absent | 7 | 5 | ||||
| Extrapyramidal Symptoms/signs | Present | 20 | 8 | 0.84 | 1 | 0.36 |
| Absent | 13 | 9 | ||||
| Ataxia | Present | 6 | 9 | 6.45 | 1 | 0.01 |
| Absent | 27 | 8 | ||||
| Pyramidal signs | Present | 16 | 2 | |||
| Absent | 17 | 15 | ||||
| Akinetic mutism | Present | 12 | 4 | 0.85 | 1 | 0.36 |
| Absent | 21 | 13 | ||||
| Visual symptoms | Present | 4 | 5 | 2.27 | 1 | 0.13 |
| Absent | 29 | 12 | ||||
| Seizures | Present | 2 | 0 | |||
| Absent | 31 | 17 | ||||
| Cortical ribbon in MRI brain | Present | 31 | 15 | |||
| Absent | 2 | 2 | ||||
| Basal ganglia signal changes | Present | 23 | 15 | |||
| Absent | 10 | 2 | ||||
| Thalamic signal changes | Present | 9 | 8 | 1.96 | 1 | 0.16 |
| Absent | 24 | 9 |
There is no significant relation for mean duration of disease, interval of death from EEG, or survival time with the occurrence of periodic complexes. However, the mean duration of illness was more, i.e., 155 vs. 115 days, survival duration was shorter, i.e., 359 vs. 500 days, in patients with PSWC compared with those without. In addition, there is no significant association of PSWC with any of the clinical variables except ataxia (P = 0.01). However, the sample size is not adequate to draw any specific conclusions.
The asymmetry of PSWC distribution is corresponding with MRI signal asymmetric changes. In 19 patients with right hemispheric predominant PSWCs, 14 (73.7%) had MRI brain signal asymmetry, with right hemispheric predominance.
Sensitivity of EEG and MRI in this cohort
The sensitivity of EEG specific abnormalities to diagnose probable sCJD according to CDC 2018 criteria with positive MRI findings was 68.75%. Similarly, the sensitivity of MRI was 93.93% in this cohort for diagnosing probable sCJD with PSWC in EEG.
DISCUSSION
This study described the EEG findings in 50 patients of probable from a single center. This is the largest cohort describing the EEG findings from India. The study included patients evaluated over 7 years from 2013 to 2020 and diagnosed as probable CJD fulfilling the CDC criteria.
The key findings noted in the study were abnormal EEG in around 88% patients, with CJD specific abnormalities, i.e., periodic complexes in 66% of probable sCJD. Steinhoff et al. reported a sensitivity of 67% and 64% for periodic complexes in a series of 29 patients and 150 patients, respectively, with confirmed and suspected cases of CJD.[6,7] The presence of normal EEG in few patients (12%) in our study could be due to the lack of serial follow-up EEGs.
The morphology of periodic complexes was predominantly biphasic (70%). Typical early peak in the frontal leads with anterior to posterior gradient was noted in only one-fourth of the patients. Maximum amplitude in the occipital leads was noted in 42%. A regional voltage maximum over occipital areas was shown in the Heidenhain variant of sCJD.[8] In most of the patients (54.5%), the PSWC were asymmetric and independent as well as synchronous. Only independent PSWC in either or one hemisphere, appearing as periodic lateralized complexes were noted in 6.1%. Chiofalo et al. reported asymmetric EEG abnormalities in 23 of 27 patients with sCJD.[9] It has been suggested that the occurrence of lateralized PSWC in sCJD may reflect an early state of disease, when the pathology is not yet progressed to diffuse cortical disease.
In three-fourths of the patients, PSWC showed disappearance during any kind of environmental stimuli or body movement. Earlier studies showed that PSWC in sCJD is usually decreased by external stimuli as well as sedative medication like benzodiazepines.[10,11] In addition, PSWC are prominent during wakefulness and tend to disappear during sleep.[12] These observations will help some times to differentiate PSWC of other etiology from CJD.[13]
The exact pathophysiology and source of PSWC are not known. Traub and Pedley (1981) suggested that fusion of dendritic membranes of affected neurons could result in increased electrotonic coupling and pathologically synchronized bursting activity.[14] Tschampa et al. 2002 hypothesized that the predominant reduction of PV+ cells in the reticular nucleus with preserved functional integrity of the subcortico-cortical or thalamo-cortical network is a prerequisite for generation of PSWC (associated with myoclonus) in patients with sCJD.[15] Source localization of periodic complexes using independent component analysis by Jung et al. showed dipole source in the dorsolateral and medial frontal cortices, basal ganglia, and thalami.[16] The predominant occurrence of PSWC ipsilateral to the cortical signal changes noted in this study supports the hypothesis of the role of the cortex in the generation of PSWC.
The spectrum of EEG findings, sensitivity and specificity of periodic discharges depend on the type of CJD as well as clinical staging of the disease. In ataxic variant of sCJD and variant CJD, periodic complexes may not be seen during the entire course of illness. In patients with Heidenhain variant of CJD, which involves predominantly occipital cortex, EEG may show slowing of the background activity and PSWCs over the posterior head region. In the study cohort, two-thirds of the patients had a prominent cortical ribbon in the posterior head region and amplitude of the periodic complexes was maximum in the posterior head region, which might suggest more prevalence of Heidenhain variant of CJD in this geographical region. However, further studies with histopathological and genotyping are needed to confirm these observations.
With respect to clinical staging, first stage (mean duration of 9 weeks) is characterized by dementia and variety of focal symptoms, in which EEG shows nonspecific changes like background abnormalities in the form of generalized or focal delta theta slowing and poor reactivity. During the second stage (mean duration of 10 weeks), the focal features tend to disappear, and progressive dementia, widespread myoclonus, bilateral rigidity, and pyramidal signs evolve; EEG shows characteristic generalized PSWC with 0.5–1 s interval. During the third stage (mean duration of 14 weeks), the patient becomes bedbound with deepening stupor, increasing rigidity and weakness of the limbs; EEG shows a simplification of PSWC in the form of broadening of the sharp wave complexes and increase of the interval duration.[13,17]
The sensitivity of EEG specific abnormality in this cohort was around 69%. Earlier studies by Steinhoff et al. reported a similar pattern in a cohort of confirmed and suspected sCJD.[7] The MRI signal changes appear to be more sensitive than EEG for diagnosing sCJD (94%vs. 69%), but this cannot be generalized as the study cohort included subjects with either specific EEG abnormality or with MRI abnormality. Diagnostic sensitivity of MRI in clinically probable or autopsy-proven sCJD was reported as 59.7%, 58.3%, and 70.8% by three independent observers.[18]
The strengths of the study include one of the largest single center studies from South Asia. The study described the EEG visual analysis finding systematically and correlated with clinical and imaging abnormalities. The limitations of the study were lack of autopsy/tissue diagnosis, retrospective nature of the study, lack of autoimmune work up in few patients, and lack of survival data in more than half of the cohort. Serial EEGs were not carried out. In addition, none of the patients had 14.3.3 level estimation levels or prion protein genetic analysis.
CONCLUSION
This study showed that EEG is a relatively inexpensive and sensitive tool and assists in the diagnosis of sCJD. However, it can be normal or show nonspecific abnormalities in the early stages of the disease. Prospective studies in cohorts with comprehensive evaluation including genetics, 14.3.3 protein levels, and autopsy along with quantitative EEG methods may add to our understanding and to the literature in future.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
We acknowledge the Electrophysiology lab technicians of NIMHANS for the support.
REFERENCES
- 1.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;0(95):13363–83. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: Classification and characterisation. Br Med Bull. 2003;66:213–39. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- 3.Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132:2659–68. doi: 10.1093/brain/awp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chandra SR, Issac TG, Philip M, Gadad V. Creutzfeldt–Jakob disease phenotype and course: Our experience from a tertiary center. Indian J Psychol Med. 2016;38:438–42. doi: 10.4103/0253-7176.191376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vitali P, Maccagnano E, Caverzasi E, Henry RG, Haman A, Torres-Chae C, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011;76:1711–9. doi: 10.1212/WNL.0b013e31821a4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinhoff BJ, Räcker S, Herrendorf G, Poser S, Grosche S, Zerr I, et al. Accuracy and reliability of periodic sharp wave complexes in Creutzfeldt-Jakob disease. Arch Neurol. 1996;53:162–6. doi: 10.1001/archneur.1996.00550020074017. [DOI] [PubMed] [Google Scholar]
- 7.Steinhoff BJ, Zerr I, Glatting M, Schulz-Schaeffer W, Poser S, Kretzschmar HA. Diagnostic value of periodic complexes in Creutzfeldt-Jakob disease. Ann Neurol. 2004;56:702–8. doi: 10.1002/ana.20261. [DOI] [PubMed] [Google Scholar]
- 8.Furlan AJ, Henry CE, Sweeney PJ, Mitsumoto H. Focal EEG abnormalities in Heidenhain's variant of Jakob-Creutzfeldt disease. Arch Neurol. 1981;38:312–4. doi: 10.1001/archneur.1981.00510050078015. [DOI] [PubMed] [Google Scholar]
- 9.Chiofalo N, Fuentes A, Gálvez S. Serial EEG findings in 27 cases of Creutzfeldt-Jakob disease. Arch Neurol. 1980;37:143–5. doi: 10.1001/archneur.1980.00500520041005. [DOI] [PubMed] [Google Scholar]
- 10.Rossini PM, Caltagirone C, David P, Macchi G. Jakob-Creutzfeldt disease: Analysis of EEG and evoked potentials under basal conditions and neuroactive drugs. Eur Neurol. 1979;18:269–79. doi: 10.1159/000115089. [DOI] [PubMed] [Google Scholar]
- 11.Elliott F, Gardner-Thorpe C, Barwick DD, Foster JB. Jakob-Creutzfeldt disease.Modification of clinical and electroencephalographic activity with methylphenidate and diazepam. J Neurol Neurosurg Psychiatry. 1974;37:879–87. doi: 10.1136/jnnp.37.8.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terzano MG, Parrino L, Pietrini V, Mancia D, Spaggiari MC, Rossi G, et al. Precocious loss of physiological sleep in a case of Creutzfeldt Jakob disease: A serial polygraphic study. Sleep. 1995;18:849–58. [PubMed] [Google Scholar]
- 13.Wieser HG, Schindler K, Zumsteg D. EEG in Creutzfeldt–Jakob disease. Clin Neurophysiol. 2006;117:935–51. doi: 10.1016/j.clinph.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Traub RD, Pedley TA. Virus-induced electrotonic coupling: Hypothesis on the mechanism of periodic EEG discharges in Creutzfeldt-Jakob disease. Ann Neurol. 1981;10:405–10. doi: 10.1002/ana.410100502. [DOI] [PubMed] [Google Scholar]
- 15.Tschampa HJ, Herms JW, Schulz-Schaeffer WJ, Maruschak B, Windl O, Jastrow U, et al. Clinical findings in sporadic Creutzfeldt-Jakob disease correlate with thalamic pathology. Brain. 2002;125:2558–66. doi: 10.1093/brain/awf253. [DOI] [PubMed] [Google Scholar]
- 16.Jung K-Y, Seo D-W, Na DL, Chung C-S, Lee IK, Oh K, et al. Source localization of periodic sharp wave complexes using independent component analysis in sporadic Creutzfeldt-Jakob disease. Brain Res. 2007;1143:228–37. doi: 10.1016/j.brainres.2007.01.127. [DOI] [PubMed] [Google Scholar]
- 17.Roos R, Gajdusek DC, Gibbs CJ. The clinical characteristics of transmissible Creutzfeldt-Jakob disease. Brain. 1973;96:1–20. doi: 10.1093/brain/96.1.1. [DOI] [PubMed] [Google Scholar]
- 18.Tschampa HJ, Kallenberg K, Urbach H, Meissner B, Nicolay C, Kretzschmar HA, et al. MRI in the diagnosis of sporadic Creutzfeldt–Jakob disease: A study on inter-observer agreement. Brain. 2005;128:2026–33. doi: 10.1093/brain/awh575. [DOI] [PubMed] [Google Scholar]