

Abstract
Cross-links formed within and between proteins are a major cause of protein dysfunction, and are postulated to drive the accumulation of protein aggregates in some human pathologies. Cross-links can be formed from multiple residues and can be reversible (usually sulfur-sulfur bonds) or irreversible (typically carbon-carbon or carbon-heteroatom bonds). Disulfides formed from oxidation of two Cys residues are widespread, with these formed both deliberately, via enzymatic reactions, or as a result of unintended oxidation reactions. We have recently demonstrated that new protein-glutathione mixed disulfides can be formed through oxidation of a protein disulfide to a thiosulfinate, and subsequent reaction of this species with glutathione. Here we investigate whether similar reactions occur between an oxidized protein disulfide, and a Cys residues on a second protein, to give novel protein cross-links. Singlet oxygen (1O2)-mediated oxidation of multiple proteins (α-lactalbumin, lysozyme, beta-2-microglobulin, C-reactive protein), and subsequent incubation with the Cys-containing protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH), generates inter-protein cross-links as detected by SDS-PAGE, immunoblotting and mass spectrometry (MS). The cross-link yield is dependent on the 1O2 concentration, the presence of the original protein disulfide bond, and the free Cys on GAPDH. MS with 18O-labeling has allowed identification of the residues involved in some cases (e.g. Cys25 from the Cys25-Cys80 disulfide in beta-2-microglobulin, with Cys149 or Cys244 of GAPDH). The formation of these cross-links results in a loss of GAPDH enzymatic activity. These data provide ‘proof-of-concept’ for a novel mechanism of protein cross-link formation which may help rationalize the accumulation of cross-linked proteins in multiple human pathologies.
Keywords: Disulfide, Singlet oxygen, Protein cross-links, Protein aggregation, Thiol-disulfide exchange, Post-translational modification, Photo-oxidation
Abbreviations: ACN, acetonitrile; aLA, α-Lactalbumin; B2M, β-2 microglobulin; CRP, recombinant human C-reactive protein; DTT, DL-Dithiothreitol; FA, formic acid; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IAM, iodoacetamide; LYSO, lysozyme; NEM, N-ethylmaleimide; PBST, phosphate-buffered saline containing Tween 20; PDB, protein data bank; RB, Rose Bengal; TCEP, tris(2-carboxyethyl)phosphine hydrochloride; TFA, trifluoroacetic acid
Graphical abstract

Highlights
-
•
Disulfide bonds (DSBs) are critical to protein structure and function.
-
•
DSBs are rapidly oxidized by singlet oxygen and other oxidants to reactive intermediates.
-
•
These intermediates react with Cys-containing proteins to give new protein-protein cross-links.
-
•
This novel disulfide cross-linking pathway affects the functional activity of the proteins.
-
•
These cross-links can be diminished by reductants, but this does not repair the DSB damage.
1. Introduction
Disulfide bonds formed between two cysteine (Cys) residues, are essential for the stability of many secreted and transmembrane proteins [1], and are often categorized into two types: redox-active (with these being either catalytic or allosteric) and structural. The former are often involved in the regulation of protein function, while the latter play a critical role in protein folding and stabilization [2,3]. The formation of these (intentional) disulfide bonds occurs primarily during and immediately after protein synthesis in the (relatively oxidizing) environment of the endoplasmic reticulum (ER) in animal cells, with the correct formation of disulfides driven by a complex mixture of oxido-reductase proteins and chaperones [4,5]. Dysregulation of this synthetic system, can result in aberrant disulfide formation and misfolded proteins [4]. Similarly, alterations in redox status can increase the level of non-native disulfide bonds within and between proteins, with this being particularly prevalent under conditions of oxidative stress due to the rapid reaction of most oxidants with the thiol group (-SH) of Cys residues [6,7]. Both one-electron (radical) and two-electron reactions can give rise to new disulfides, via the intermediacy of thiyl radicals (RS., and subsequent dimerization of these species [6,7] (reaction 1) or oxidation products such as sulfenic acids (RS-OH) or nitroso adducts (RS-NO) [6,[8], [9], [10]]. Subsequent reactions of RS-OH (reaction 2) or RS-NO species with another thiol, on another protein or a low-molecular-mass species such as glutathione, yields a new disulfide. Although many of these native and non-intended disulfide bonds can be reduced enzymatically [1,11,12], and also slowly via uncatalyzed thiol-disulfide reactions [13], the levels of these species appear to accumulate in some diseases, with this linked with protein and tissue dysfunction [14,15].

Singlet oxygen (1O2) is a highly reactive oxidant that reacts with a wide range of biomolecules including proteins, lipids and DNA/RNA [[16], [17], [18]]. 1O2 is generated both by exposure of O2 to short wavelength UV, visible light in the presence of a sensitizer, and also via a range of other biologically-relevant processes, including termination reactions of peroxyl radicals, via reaction of H2O2 with HOCl, and via peroxidase enzyme catalyzed reactions [[16], [17], [18]]. Proteins are a major target for 1O2 due to their high abundance in nearly all biological matrices, with this species reacting most rapidly with the side chains of Cys, Met, His, Tyr, Trp [16,17,19]; reaction with the disulfide bond of cystine is also a facile process, though this process is less well characterized [20,21].
Studies on the reaction of 1O2 with low-molecular-mass disulfides have provided evidence for the formation of zwitterionic peroxides [RS+(OO−)SR’], thiosulfinates (disulfide-S-monoxides, RS(=O)SR′) and thiosulfonates (disulfide-S-dioxides, RS(=O)2SR′) [20,21] (reaction 3). Both the zwitterionic peroxides [RS+(OO−)SR’] and thiosulfinates are unstable and can undergo further reaction with another thiol to give a new disulfide bond with cleavage of the original disulfide [22,23] (reaction 4). In the absence of added thiol, cleavage of the disulfide bond can also occur with formation of sulfinic (RSO2H) and sulfonic acids (RSO3H) (reaction 5). Similar reactions of disulfides to give thiosulfinates and subsequently new thiolated (e.g. glutathionylated) species have been detected with a range of other oxidants (e.g. hypochlorous acid, peroxynitrous acid, H2O2) [[22], [23], [24], [25]], indicating that these ‘oxidant-mediated thiol-disulfide exchange reactions’ are a common pathway after initial oxidation of a disulfide bond (reaction 6).

In the present study, we hypothesized that analogous thiol adduction reactions to initial zwitterionic peroxides and/or thiosulfinates might occur with Cys residues present on proteins and therefore that these reactions would give rise to new protein-protein disulfide cross-links. These reactions provide a novel pathway to protein aggregation in addition to the well-established formation of intra- and inter-molecular protein-protein disulfide bonds arising from Cys oxidation. This mechanism allows for the cross-linking of proteins that do not contain an initial free Cys residue. The studies reported here provide ‘proof-of-concept’ of this new pathway for a range of disulfide-containing proteins, which are shown to form 1O2-mediated cross-links with an archetypal Cys-containing protein, glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
2. Materials and methods
2.1. Materials
α-Lactalbumin (aLA) from bovine milk (Type I, ≥85%), lysozyme from chicken egg white (LYSO, ≥90%), Rose Bengal (RB), Coomassie brilliant blue G, DL-Dithiothreitol (DTT), tris (2-carboxyethyl) phosphine hydrochloride (TCEP), iodoacetamide (IAM), N-ethylmaleimide (NEM), glyceraldehyde-3-phosphate dehydrogenase from rabbit muscle (GAPDH), catalase from bovine liver and GAPDH activity assay kit were purchased from Sigma Aldrich (St Louis, MO). Beta-2 microglobulin (B2M, > 98%) and recombinant human C-reactive protein (CRP) were obtained from Lee Biosolutions (USA, MO). 18O water (95.7% pure) was purchased from Sercon. Trifluoroacetic acid (TFA), ammonium bicarbonate, acetonitrile and formic acid for mass spectrometry were obtained from VWR. Beta-2 Microglobulin antibody (4H5L5) and GAPDH monoclonal antibody (GA1R) were obtained from Thermo Fisher. Anti-C Reactive Protein antibody (ab32412) was obtained from Abcam. Sequencing grade trypsin and rLys-C (mass spec grade) were purchased from Promega (Denmark). NuPAGE MES SDS running buffer (20×), NuPAGE LDS sample buffer (4×), NuPAGE 4-12% Bis-tris gel were obtained from Thermo Fisher. Sheep anti-mouse IgG HRP-linked whole Ab (NXA931-1 ML) was obtained from VWR. Anti-rabbit IgG HRP-linked antibody was obtained from Cell Signaling Technology. All solvents employed were HPLC grade.
2.2. Sensitized photo-oxidation of disulfide-containing proteins
Photo-oxidation experiments were performed as described previously [26] with minor modifications. In brief, solutions containing disulfide-containing proteins (aLA, LYSO, CRP and B2M, in 10 mM phosphate buffer, pH 7.4) and Rose Bengal (RB, final concentration 10 μM) were exposed to the light from a Leica P 150 slide projector through a 345-nm cut-off filter. After photolysis, samples were treated with catalase (1 mg mL−1) to remove H2O2 generated during the photo-oxidation which might also react with the added thiol-containing proteins. Catalase was not added before the photolysis as this enzyme has a significant optical absorption, and therefore interferes with the photosensitization reaction, as well as acting as a competitive target for 1O2. In experiments where the role of the disulfide bond in B2M was examined, a combination of DTT and NEM (both 100-fold molar excess over the protein; in 10 mM phosphate buffer, pH 7.4, reaction for 1 h at 21 oC) were used to reduce and alkylate the disulfide bonds of B2M (20 μM), with excess DTT and NEM subsequently removed using mini dialysis devices (3.5 kDa molecular mass cut-off; ThermoFisher) and 10 mM phosphate buffer, pH 7.4, prior to photo-oxidation.
2.3. SDS-polyacrylamide gel electrophoresis and immunoblotting analysis
SDS–PAGE was performed using NuPAGE MES SDS running buffer and NuPAGE 4–12% Bis-tris gels under non-reducing or reducing conditions. Briefly, protein samples were mixed with non-reducing (10 mM phosphate buffer) or reducing sample buffer (NuPAGE reducing agent, Thermo; containing a final concentration of 50 mM dithiothreitol, pH 8.5) and heated at 60 °C for 10 min with NuPAGE LDS sample buffer, prior to electrophoresis at 200 V for 35 min. After electrophoresis, gels were either stained with colloidal Coomassie Blue, or blotted to a polyvinylidene fluoride (PVDF) membrane using an iBlot 2 system (Thermo Fisher, 20 V, 7 min). For immunoblotting, the membrane was blocked with 5% skim milk in 0.1% PBST containing 2.5 mM NEM for 1 h with agitation at 21 °C. The membranes were incubated with a primary mouse monoclonal anti-GAPDH antibody (1:5000 diluted in 1% PBST, overnight, 4 °C), rabbit monoclonal anti-B2M antibody (1:2000 diluted in 1% PBST, overnight, 4 °C) or rabbit monoclonal anti-CRP antibody (1:5000 diluted in 1% PBST, overnight, 4 °C), and subsequent incubation with anti-mouse secondary antibody or anti-rabbit secondary antibody (1:5000 diluted in 1% PBST). Chemiluminescence from the enhanced chemiluminescence (ECL plus) solution was recorded using either a GeneSys system (Frederick, USA) or an Azure Biosystems (USA) imager. Quantification of the protein bands was performed using Image J software.
2.4. In-gel digestion for mass spectrometric characterization of protein cross-links after SDS-PAGE
In-gel digestion for mass spectrometric (MS) characterization of protein peptides was performed according to a method described previously [27]. Control protein samples (100 μM aLA, 40 μM B2M and 40 μM CRP) and photo-oxidized cross-linked samples (LYSO-GAPDH, B2M-GAPDH and CRP-GAPDH) were separated by SDS-PAGE as described above and stained with Coomassie blue. After 4 h, the gels were washed in Milli-Q (MQ) H2O until the cross-linked protein bands were clearly detected. These bands were then excised from the gel, placed in Eppendorf tubes, and incubated with acetonitrile (ACN) for 10 min. Afterwards the samples were treated with 10 mM dithiothreitol (DTT, in 100 mM ammonium bicarbonate, 30 min at 56 °C), followed by incubation with 55 mM IAM (in 100 mM ammonium bicarbonate) for 20 min at 21 °C in the dark. The gel pieces were then dehydrated in 500 μL ACN before enzymatic digestion with 13 ng μL−1 trypsin (in 10 mM ammonium bicarbonate containing 10% ACN v/v) at 4 °C for 2 h, and then incubated at 37 °C overnight. For the CRP-GAPDH cross-links, Lys-C (13 ng μL−1 Lys-C in 10 mM ammonium bicarbonate, 24 h at 37 °C) was also added after the trypsin digestion. At the end of the digestion, 100 μL extraction buffer (5% formic acid in ACN) was added to each tube and mixed for 15 min to the extract peptides. The supernatant was then transferred to new tubes and dried down in a vacuum centrifuge at 30 °C. The peptides were then subjected to solid-phase extraction using StageTip C18 reverse-phase discs packed into pipette tips before analysis using nanoLC-MS/MS.
2.5. 18O isotopic labeling with trypsin digestion
Mass spectrometric analysis for the identification of cross-linked peptides between B2M and GAPDH was performed as described previously with minor modifications [28,29]. Briefly, dried proteins (20 μg for pure B2M sample, 40 μg for B2M-GAPDH sample) were dissolved in 20 μL of 8 M Urea and 50 mM Tris-HCl (pH = 8.0), and incubated for 30 min at 21 °C. The protein samples were then divided into two aliquots (10 μL each) and dried using a SpeedVac at 30 °C. The two aliquots were then reconstituted separately into 80 μL of either 18O-water or 16O-water containing 100 mM ammonium bicarbonate at pH 7, then 0.1 μg μL−1 trypsin in 18O-water or 16O-water was added to achieve a 1:50 (w/w) enzyme/substrate ratio, respectively. The reaction mixtures were then incubated at 37 °C overnight. Where applicable, these samples were mixed in 1:1 ratio immediately before analysis. For the analysis of intra-protein disulfides, 18O labeling was used for both MS1 level spectra and the MS/MS spectra, whereas for the inter-protein disulfide analyses, 18O labeling was used for MS1 level spectra and 16O labeling for the MS2 level spectra.
2.6. Mass spectrometric analysis
Peptide samples were analyzed on an Impact II Q-TOF mass spectrometer (Bruker, Billerica, MA) coupled to a Dionex Ultimate 3000RSLCnano chromatography system (Thermo Fisher Scientific, Waltham, MA). Peptides were loaded onto a Nanoelute C18 column (75 μm × 15 cm, 1.9 μm particle size, Bruker) and eluted over 65 min using a gradient elution system consisting of mobile phase A (0.1% FA in H2O) and B (80% ACN in H2O containing 0.1% FA) at a flow rate of 0.3 μL min−1. A linear gradient was used consisting of: 5% B at 4 min, 61% B at 35 min, 99% B at 40 min, 99% B at 45 min, and then re-equilibration at 4% B from 55 min over 10 min. The operation conditions for the CaptiveSpray source were as follows: capillary voltage (4.5 kV) and temperature (180 °C); dry gas 4 L min−1. A high-resolution TOF-MS scan over a mass range of 150–1750 m/z was followed by MS/MS scans of the top 10 most intense precursor ions per cycle. The acquisition rate was set to 2 Hz (MS) and 2.03 Hz (MS/MS). MaxQuant (version 1.6.1.0; www.maxquant.org) was used as a database search engine for identification of peptides with the following parameters: carbamidomethylation of Cys (fixed modification); Met oxidation (variable modification); allowed number of missed cleavages, 3; first search mass tolerance, 20 ppm; main search peptide tolerance, 4.5 ppm. MassAI software (version 1.0; www.massai.dk) was used for identification of cross-linked peptides with the following settings: fixed (carbamidomethylation of Cys when IAM was used) and variable (Met oxidation) modifications; maximum number of missed tryptic cleavages, 3; parent mass tolerance, 20 ppm; MS2 peak tolerance, 0.2 m/z, respectively. Cys-Cys (−2.016 Da) was selected as a potential cross-link. Peptides were searched against the following UniProt protein accession numbers: CRP: P02741; Lyso: P00698; B2M: P61769; GAPDH: P46406. GPMAW software version 9.5 (Lighthouse Data) was used as a reference for identification of fragmentation patterns of potential cross-linked peptides.
2.7. Determination of GAPDH activity
GAPDH activity was determined using a commercial kit using the instructions provided by the manufacturer (Sigma, St. Louis, MO). In brief, the conversion of glyceraldehyde-3-phosphate (GAP) to 1,3-bisphosphateglycerate was quantified by measuring absorbance at 450 nm (A450) using a RF-20AS detector, with the rate calculated from the initial, linear part of the curve. The (low) background signals detected in the absence of each of the reaction components was subtracted from the data presented.
2.8. Errors and statistics
Statistical analyses were carried out using statistical package GraphPad Prism version 6 for Windows (GraphPad Software, La Jolla, USA). Quantitative data are presented as mean ± SD, from three independent experiments. Differences between groups were determined using one-way analysis of variance (ANOVA) with Dunnett's multiple comparison test. Statistical significance was set at P < 0.05.
3. Results
3.1. Detection of 1O2-induced B2M-GAPDH cross-links
Beta-2 microglobulin (B2M) contains a single disulfide bond (Cys 25-Cys 80) and has no free Cys residues in its structure [30], allowing this to be readily used as a model to investigate 1O2-mediated disulfide oxidation in the absence of reactions at free Cys. Samples of B2M (20 μM) were exposed to O2 and visible light in the presence of Rose Bengal (10 μM) for defined periods of time and then treated with catalase (1 mg mL−1) to remove any photo-generated peroxides. The samples were then examined by both SDS-PAGE and immunoblotting using an anti-B2M antibody. These experiments resulted in the detection of dimers and higher oligomers of B2M, and loss of the parent B2M bands, using either Coomassie staining (Fig. 1A) or an anti-B2M antibody (Fig. 1B). These data indicate that B2M undergoes facile aggregation when subject to photo-oxidation.
Fig. 1.

Detection of 1O2-induced B2M aggregation and formation of B2M-GAPDH cross-links. Panel A: representative image of B2M (20 μM in 10 mM phosphate buffer, pH 7.4) subjected to increasing periods of illumination in the absence or presence of Rose Bengal/visible light and O2 (as indicated), with subsequent separation by SDS-PAGE and Coomassie staining. Panel B: As panel A, but with subsequent transfer of the proteins separated by SDS-PAGE to a PVDF membrane and probing with an anti-B2M antibody. Panel C: representative immunoblot image of B2M-GAPDH cross-link detection upon exposure of B2M to 1O2 for different time points, subsequent reaction with GAPDH, separation by SDS-PAGE and immunoblotting using anti-B2M antibody. Panel D: As panel C, but with detection using an anti-GAPDH antibody. Panel E: Optical density ratio (OD n min/OD 0 min) of B2M-GAPDH cross-linking bands from immunoblotting data in panels C, D (n = 0 min, 5 min, 15 min, 30 min, 60 min and 90 min). Each gel image or blot is representative of one of three, obtained from three independent experiments. Quantitative data are presented as mean ± SD from three independent experiments. Statistical differences were examined using one-way analysis of variance (ANOVA) with Dunnett's multiple comparison test and are indicated as follows: *P < 0.05 treatment vs. lane 4 in panels C, D.
To examine whether the photo-oxidized samples of B2M undergo subsequent reaction with a thiol-containing protein, to give protein-protein dimers, GAPDH (20 μM, in 10 mM phosphate buffer, pH 7.4) was added to B2M (20 μM) after the cessation of photolysis and incubated for 60 min, before analysis by either immunoblotting using antibodies against both B2M (Fig. 1C) and GAPDH (Fig. 1D) or Coomassie staining (Supplementary Fig. 1). Comparison of the data obtained with photo-oxidized B2M alone (and corresponding controls) with the samples incubated with GAPDH (Fig. 1B versus 1C, Supplementary Fig. 1) showed the presence of additional bands that are consistent with the presence of B2M-GAPDH cross-linked species. Thus, analysis of the Coomassie-stained gel, or probing the membranes generated from the samples with both proteins present with appropriate antibodies showed the presence of new bands at ~48 kDa and at ~96 kDa (band at ~48 kDa in Supplementary Fig. 1, lanes 6,8,10; bands at ~48 kDa and at ~96 kDa in Fig. 1C and D; lanes 5–9 in both panels). As the intensity of the bands was much stronger from the membranes probed with the antibodies than the Coomassie staining (cf. images in Fig. 1 versus Supplementary Fig. 1) most subsequent studies used only the latter approach to detect the new cross-links, though some direct Coomassie staining was carried out in some cases (see below). As the bands on the membranes were recognized by both antibodies, this is consistent with the presence of crossed-dimers, with these assigned to the formation of cross-linked B2M-GAPDH species containing one, and two, molecules of each protein respectively. These bands are distinct from the formation of the B2M dimers and oligomers (cf. Fig. 1A and B), and also the monomer (~36k), dimer and tetramer bands of GAPDH (Fig. 1D). The formation of these new bands increased with longer extents of the initial light exposure of the B2M (and hence 1O2 concentration), with the species that gives rise to the band ~48 kDa appearing at earlier time points than the ~96 kDa band, consistent with the latter being a downstream (dimeric) form of the former. Densitometric analysis of both the anti-B2M and anti-GAPDH immunoblots showed a significant increase in the intensity of B2M-GAPDH cross-linking band when compared to the 0 min RB-illuminated samples for both antibodies, and also a significant increase with longer illumination times of the B2M (Fig. 1E).
Additional bands were also detected, especially with the anti-B2M antibody (Fig. 1C), at ~60 kDa, assigned to a possible B2M dimer-GAPDH (trimeric) species, and ~84 kDa assigned to a B2M-GAPDH dimer. The former band is believed to arise from reaction of an initial B2M dimer formed during photolysis (~23 kDa, see Fig. 1A), and subsequent reaction with GAPDH, and the B2M-GAPDH dimer is ascribed to reaction of an oxidized B2M species with the dimeric form of GAPDH present in the commercial GAPDH samples (as detected in the control lanes in Fig. 1D, lane 2). No cross-links were observed in control samples where B2M was illuminated in the absence of RB, or where B2M was incubated with RB in the absence of light, without or with subsequent reaction with GAPDH. Each antibody showed high specificity towards its target (Fig. 1C and D; lanes 1 and 2), with no cross-reactivity observed with the other protein. These data indicate that the formation of these B2M-GAPDH cross-linked species requires the formation of photo-generated species on B2M, with these intermediates undergoing subsequent reaction with GAPDH. The absence of cross-links in the control samples containing the native proteins (Fig. 1C and D; lane 3) establishes that direct thiol-disulfide exchange reactions are insignificant under the conditions employed and that RB alone, in the absence of light, does not act as a cross-linking agent (Fig. 1C and D; lane 4).
3.2. Effect of 1O2-induced B2M-GAPDH cross-links on GAPDH activity
In order to assess the effect of B2M-GAPDH cross-link formation on GAPDH activity, this was assessed without and with incubation with photo-oxidized B2M. The GAPDH alone was not subject to photo-oxidation. As shown in Fig. 2, a decrease in GAPDH activity was observed, relative to incubation with non-treated B2M, with B2M samples subjected to increasing periods of prior photo-oxidation, with this being significant after 30 min photolysis of B2M, and decreasing to ~6% of control levels in the 90 min photo-oxidized samples (Fig. 2). No statistically-significant difference was detected for the control samples, where GAPDH was incubated with RB alone (10 μM, in 10 mM phosphate buffer, pH 7.4) or with incomplete oxidation systems.
Fig. 2.
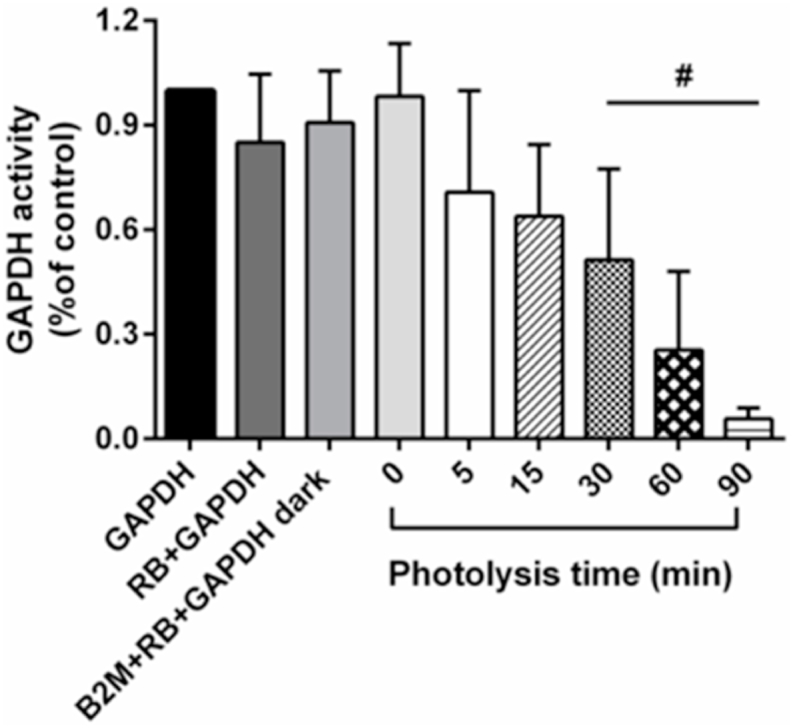
GAPDH activity is reduced after reaction with pre-oxidized B2M. GAPDH (0.2 μM in 10 mM phosphate buffer, pH 7.4) was incubated with photo-oxidized B2M (0.2 μM in 10 mM phosphate buffer, pH 7.4, 5 min, 15 min, 30 min, 60 min and 90 min photolysis) for 60 min and then residual GAPDH activity was determined. The control samples were untreated GAPDH (0.2 μM) alone. Bar labelled ‘RB + GAPDH’ indicates GAPDH samples incubated with RB alone. Bar labelled B2M + RB + GAPDH dark’ indicates B2M samples incubated with RB and GAPDH in the absence of light. Data are presented as mean ± SD from three independent experiments. Statistical differences were examined using one-way analysis of variance (ANOVA) with Dunnett's multiple comparison test and are indicated as follows: #P < 0.05 vs. GAPDH alone samples.
3.3. Role of the B2M disulfide bond and GAPDH thiols in the formation of B2M-GAPDH cross-links
In order to examine the role of disulfide in B2M in the formation of the observed cross-linking, DTT and NEM (both 100-fold molar excess over the protein) were used in combination to reduce (DTT) and alkylate (NEM) the disulfide bonds of B2M (20 μM), with the excess DTT and NEM subsequently removed by dialysis prior to photo-oxidation. The oxidized B2M was then incubated with GAPDH, and subjected to immunoblotting with detection using either the anti-B2M or anti-GAPDH antibodies. A significant decrease in the extent of B2M-GAPDH cross-link formation was detected for the reduced and alkylated B2M (~50% for the B2M antibody and ~78% for the GAPDH antibody), when compared to the non-reduced/non-alkylated systems (Fig. 3A–C, lanes 7 vs. 3 for each blot).
Fig. 3.

Role of disulfide in B2M and thiol residues on GAPDH in the formation of B2M-GAPDH cross-links. Panel A: representative immunoblot image of B2M-GAPDH cross-link detected by the anti-B2M antibody for samples without and with prior DTT/NEM treatment (both 100-fold molar excess over the protein) before exposure to 1O2 for 60 min, followed by reaction with GAPDH (20 μM in 10 mM phosphate buffer, pH 7.4). Panel B: as panel A, but with detection of cross-links (and parent GAPDH) by use of an anti-GAPDH antibody. Panel C: Optical density ratio (OD lane 7/OD lane 3) of B2M-GAPDH cross-link bands (~48k Da) from the immunoblotting data presented in panels A and B. Panel D: representative immunoblot image of B2M-GAPDH cross-link detection upon exposure of B2M (20 μM in 10 mM phosphate buffer, pH 7.4) to 1O2 for 60 min, and then reaction with GAPDH (20 μM in 10 mM phosphate buffer, pH 7.4), or GAPDH pretreated with NEM (200 μM in 10 mM phosphate buffer, pH 7.4). The formation of cross-links (and parent B2M) was analyzed by use of an anti-B2M antibody. Panel E: as panel D, but with detection of cross-links (and parent GAPDH) by use of an anti-GAPDH antibody. Panel F: Optical density ratio (OD lane 11/OD lane 10) of B2M-GAPDH cross-link bands (~48k Da) from the immunoblotting data presented in panels D and E. Each blot is representative of one of three, obtained from three independent experiments. Quantitative data are presented as mean ± SD from three independent experiments. Statistical differences were examined using a Student's t-test and are indicated as follows: *P < 0.05 for lane 7 vs. lane 3 in panels 3A and 3B. #P < 0.05 for lane 11 vs. lane 10 in panels D and E.
The role of the GAPDH Cys residues was examined in experiments where the GAPDH was pretreated with the thiol-blocking reagent NEM (10-fold excess over GAPDH concentration) before addition to the photo-oxidized B2M samples; control samples of untreated GAPDH were also examined. Preincubation of the GAPDH with NEM for 1 h significantly decreased the formation of the bands assigned to the B2M-GAPDH cross-links (at ~48k Da) after 60 min illumination as detected by both immunoblotting and Coomassie staining of SDS-PAGE gels (Fig. 3D and E; lanes 10 vs. 11 in each panel; Supplementary Fig. 1). Quantification of the immunoblots showed a reduction of ~45% (for the B2M antibody) and ~80% (for the GAPDH antibody) for the B2M-GAPDH cross-link yield, when compared to control samples without pre-treatment of GAPDH with NEM (Fig. 3F). A similar decrease was observed for the band assigned to the cross-linked B2M dimer-GAPDH species detected at ~60 kDa. For incomplete oxidation systems, where photolysis of B2M was carried out in the absence of RB for 60 min, or where B2M was incubated with RB for 60 min in the dark, before addition of GAPDH or NEM-treated GAPDH, these changes were not observed (Fig. 3D and E; lanes 3–9).
3.4. Reversibility of cross-links formed between B2M and GAPDH
Information on the role of (reversible) disulfide bonds in the B2M-GAPDH cross-links was examined using non-reducing and reducing SDS-PAGE, with subsequent immunoblot analysis of the samples using either the anti-B2M or anti-GAPDH antibodies. A decrease in the intensity of B2M-GAPDH cross-links was detected by both anti-B2M (~39%, Fig. 4A) and anti-GAPDH antibodies (~34%, Fig. 4B) under reducing conditions when compared to non-reducing gel condition (lanes 7 vs. 3 for each blot, Fig. 4C). The lower % loss of cross-links observed under these conditions, when compared to the DTT/NEM treatment reported above (section 3.3), is likely to be due to the shorter incubation period employed (10 vs. 60 min), the absence of any reagent (e.g. NEM) to prevent disulfide re-oxidation in the reducing gel protocol, and also potential differences in accessibility between the native B2M disulfide bond and the postulated B2M-GAPDH disulfide.
Fig. 4.

Reversibility of pre-formed B2M-GAPDH cross-links. B2M (20 μM in 10 mM phosphate buffer, pH 7.4) was subjected to photo-oxidation for 60 min and then incubated with GAPDH (20 μM in 10 mM phosphate buffer, pH 7.4) for 1 h. Subsequently the cross-linked samples were subjected to SDS-PAGE under non-reducing (panel A and B, lanes 1–3) or reducing condition (panel A and B, lanes 5–7), or incubated with DTT (2 mM in 10 mM phosphate buffer, pH 7.4; panels D and E, lanes 6–8) or TCEP (2 mM in 10 mM phosphate buffer, pH 7.4; panels D and E, lanes 9–11) for a further 1 h before analysis by immunoblotting. Panels A and D: detection with the anti-B2M antibody. Panels B and E: as panels A and D, respectively, but with detection using an anti-GAPDH antibody. Panel C: Optical density ratio (ODlane 7/ODlane 3) of cross-linked bands (~48k Da) from immunoblotting data in panels A and B. Panel F: Optical density ratio (ODlane 8 or lane 11/ODlane 5) of cross-linked bands (~48k Da) from immunoblotting data in panels D and E. Each blot is representative of one of three, obtained from three independent experiments. Quantitative data are presented as mean ± SD from three independent experiments. Statistical differences were examined using a Student's t-test and are indicated as follows: *P < 0.05 for lane 7 vs. lane 3 in panels A and B. #P < 0.05 vs lane 5 in panels D, E.
The potential reversibility of pre-formed GAPDH adducts to B2M was examined further using (separately) DTT or TCEP (both at a 100-fold molar excess over the B2M concentration). B2M-GAPDH cross-linked species were generated as described above (section 3.3) with native GAPDH, and then left either untreated or incubated with either DTT (Fig. 4D and E; lanes 6–8) or TCEP (Fig. 4D and E; lanes 9–11) for 1 h, before separation by SDS-PAGE and probing of the membranes with either the anti-B2M antibody (Fig. 4D) or the anti-GAPDH antibody (Fig. 4E). Quantification of the pixel densities of the B2M-GAPDH dimer band are presented in (Fig. 4F). Treatment with either DTT or TCEP after dimer generation resulted in a significant decrease in the level of cross-links detected by both two antibodies. The densitometric analysis showed a ~38% (anti-B2M antibody) and ~26% (anti-GAPDH antibody) reduction in yield of B2M-GAPDH cross-links with post-generation DTT treatment when compared to control samples without the DTT reduction step (Fig. 4F). Similarly, treatment with TCEP resulted in a ~63% (anti-B2M antibody) and ~42% (anti-GAPDH antibody) decrease in yield of B2M-GAPDH cross-links (Fig. 4F). The incomplete loss of the protein cross-links with these treatments may reflect the relatively modest excesses of reagents and mild reaction conditions employed (37 oC, pH 7.4), compared to those used in other protocols.
3.5. Characterization of disulfide bond oxidation in B2M by mass spectrometry and 18O labelling
Mass spectrometry with 18O-labeling was used to characterize both the initial presence and subsequent photo-oxidation of the intra-chain disulfide present in B2M, and the newly-formed inter-protein bonds between B2M and GAPDH. Identification of the peptides corresponding to the site of disulfide bond was achieved by tryptic digestion to peptides with the use of 18O-labelling and MassAI software. Tryptic digestion in 18O-water results in the incorporation of two 18O atoms into tryptic peptides containing a new (single) C-terminus (i.e. at both oxygen atoms of the carboxyl group), whereas with a cross-linked peptide with two new C-termini, four 18O atoms are incorporated (for further information on this method see Refs. [29,31]). The former peptides therefore show a +4 Da shift relative to the peptides generated by tryptic digestion in 16O-water, and the latter (cross-linked) species show a +8 Da shift; this difference allows facile detection of the cross-linked peptides. The MS/MS fragment ions arising from these peptides were identified through manual validation, according to their theoretical m/z calculated by the GPMAW software. The experimental and theoretical masses of the cross-linked peptides, together with their charge states and mass errors are collected in Supplementary Table 1.
For B2M, one Cys-Cys (disulfide) cross-link was identified, as expected on the basis of the known protein structure, with two different charge states (quintuply- and sextuply-charged). As shown in Fig. 5A, the isotopic distribution of the peptide at m/z 906.5876 (sextuply-charged, experimental mass 5433.5256 Da, mass error 7.65 ppm) shows a mass shift of +8 Da (due to the incorporation of four 18O atoms) after digestion of B2M in 18O-water compared to that in 16O-water, indicating it is a cross-linked peptide. Sequence analysis of this m/z 906.5876 ion by MS/MS confirmed the detection and identity of the expected cross-linked peptide (DWSFYLLYYTEFTPTEKDEYACR) (SNFLNCYVSGFHPSDIEVDLLK) (i.e. between Cys80 in the first peptide and Cys25 in the second peptide). The missed cleavage by trypsin at Lys75 in the first peptide is likely to be due to its close proximity to the cross-link site. Manual sequence analyses revealed that the MS/MS spectra displays high sequence coverage for both the y- and b-ions, with y4 and y10-y19 from peptide A (blue ions in Fig. 5C), as well as b11, b14 and b16 from peptide B (red ions in Fig. 5C) retaining the cross-link, thereby providing strong evidence for the Cys25-Cys 80 disulfide bond. The same Cys-Cys cross-linked peptide was detected as an ion with precursor m/z 1087.7031 (quintuply-charged, experimental mass 5433.5155 Da, mass error 5.78 ppm), but this showed a poorer fragment ion coverage of the candidate cross-linked peptides compared with the sextuply charged ion (Supplementary Fig. 2A and C).
Fig. 5.

Exposure to 1O2 results in loss of the native B2M disulfide bond. MS analysis of the sextuply charged Cys-Cys cross-linked peptide from B2M. Panel A: MS spectrum of the sextuply-charged Cys-Cys cross-linked peptide (m/z 906.59) displaying a +8 Da shift following trypsin digestion in H216O and H218O (1:1 ratio). Panel B: Illumination time-dependent loss of peak area for the Cys-Cys cross-linked peptide from B2M (based on the most intense isotope ion at m/z 907.09 ± 0.02) on photo-oxidation for 5, 15, 30, 60 or 90 min. Data was analyzed by one-way ANOVA with Dunnett's multiple comparison test, and are expressed as a ratio of the control data (native B2M). Panel C: MS/MS spectrum of the sextuply-charged cross-linked peptide showing fragment ions with intensity above 104. Blue b and y ions correspond to the peptide A, while red b and y fragments correspond to peptide B. Spectra were obtained from the software Data Analysis (Bruker), with the fragment ions annotated manually. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Photo-oxidation of B2M and subsequent MS analysis as described above revealed that the yield of the Cys-Cys cross-linked peptide decreased in a illumination time-dependent manner, with only ~36% and ~6% of the sextuply-charged disulfide-linked peptide detected after 5 and 90 min illumination, respectively, when compared with native B2M (CON; Fig. 5B); a similar trend was observed for the quintuply-charged disulfide peptide ion (Supplementary Fig. 1B). When B2M was illuminated in the absence of RB, or incubated with RB in the absence of illumination, no significant consumption of this cross-linked peptide was detected (Fig. 5B, Supplementary Fig. 1B). These data confirm that photo-oxidation rapidly modifies the single disulfide bond in B2M.
3.6. Identification of inter-protein disulfides between B2M and GAPDH using mass spectrometry
The potential formation of inter-protein disulfides between B2M and GAPDH was investigated by making use of the trypsin digestion and 18O labelling method described above. Analysis of the peptides arising from in-solution digestion of the B2M-GAPDH reaction mixtures (generated as described above) revealed two cross-linked peptides with the expected +8 Da mass shift in 18O-water (Fig. 6A, C). Strong evidence was obtained for a new Cys-Cys cross-linked peptide (SNFLNCYVSGFHPSDIEVDLLK) (IISNASCTTNCLAPLAK), formed between Cys25 in B2M (first peptide) and Cys149 in GAPDH (second peptide). This peptide (m/z 1054.2626) exhibited the characteristic mass difference of −2.016 Da of a cross-linked species (when compared to the sum of the two parent peptides), and an experimental mass (4213.0504 Da) very close to the expected theoretical mass (4213.0531 Da; mass error 0.64 ppm). The MS analysis shows a mass shift of +8 Da for the quadruply-charged cross-linked peptide ion with m/z 1054.2626 (Fig. 6A), and the fragment ions y17, y20 and b12 (indicated in blue) from the peptide SNFLNCYVSGFHPSDIEVDLLK, and y11, y12, y16 and b16 (indicated in red) from the peptide IISNASCTTNCLAPLAK, unambiguously locating the cross-link position to the indicated Cys residues (Fig. 6B).
Fig. 6.

New inter-protein disulfides are formed by oxidation of disulfide bonds and subsequent reaction with protein thiols. Panel A: MS spectrum of the quadruply charged Cys-Cys cross-linked peptide with m/z 1054.26 displaying a +8 Da shift following trypsin digestion in H218O versus H216O (1:1 ratio). Panel B: Assignment of a MS/MS spectrum of the cross-linked peptide (SNFLNCYVSGFHPSDIEVDLLK) (IISNASCTTNCLAPLAK) with the first peptide (peptide A) containing Cys25 from B2M, and the second peptide (peptide B) containing Cys149 from GAPDH. Panel C: MS spectrum of the quintuply charged Cys-Cys cross-linked peptide at m/z 799.58 displaying a +8 Da shift. Panel D: Assignment of a MS/MS spectrum of the cross-linked peptide (VPTPNVSVVDLTCR) (SNFLNCYVSGFHPSDIEVDLLK) with the first peptide (peptide A) containing Cys244 from GAPDH and the second peptide (peptide B) containing Cys25 from B2M. Blue b and y ions correspond to the peptide A, while red b and y fragments correspond to the peptide B. Spectra were obtained from the software Data Analysis, with the fragment ions annotated manually. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
A second Cys-Cys inter-protein cross-link was also identified with a quintuply-charged precursor ion at m/z 799.5808, consistent with the presence of the cross-linked peptide (VPTPNVSVVDLTCR) (SNFLNCYVSGFHPSDIEVDLLK) with a disulfide bond between Cys244 in GAPDH (peptide A) and Cys25 in B2M (peptide B). The mass loss (−2.016 Da) compared to the sum of the parent sequences is consistent with the Cys-Cys cross-link, and the theoretical mass (3992.9650 Da) is in good agreement with the observed mass (3992.9040 Da; mass error 15.28 ppm). The MS data showed a mass shift of +8 Da for the cross-linked peptide ion with m/z 799.5808 (quintuply charged, Fig. 6C), in agreement with the presence of an ion with two new C-termini. The assignment of the b- and y-fragment ions in the MS/MS spectra matches this Cys-Cys cross-link between B2M and GAPDH, with the ions from y2-y7 (in blue, Peptide A) and b12, b15 and b19 (in red, Peptide B) retaining the cross-link, providing compelling evidence for this linkage (Fig. 6D). Together these MS data provide compelling evidence for the generation of two different oxidation-induced cross-linked peptides between B2M and GAPDH; both cross-links involve the Cys25 residue of the (former) disulfide bond in B2M crosslinked to two different GAPDH Cys residues: Cys149 and Cys244. The first of these is the active site residue in GAPDH.
3.7. Generality of formation of cross-links between GAPDH and proteins containing oxidized disulfide bonds
The generality of the cross-linking process reported above was examined using three other proteins, each of which contains one or more disulfides but no free Cys residues: α-lactalbumin (aLA, four DSBs), lysozyme (LYSO, four DSBs) and C-reactive protein (CRP, one DSB). Photo-oxidation of each of the proteins (aLA, 100 μM; LYSO, 100 μM; CRP, 1.25 μM; all in 10 mM phosphate buffer, pH 7.4) was carried out as outlined in the Materials and methods using a Rose Bengal/visible light/O2 system, with a range of different illumination times. After cessation of illumination, any H2O2 formed was removed by addition of catalase, and the photo-oxidized protein samples then incubated with GAPDH (20 μM, in 10 mM phosphate buffer, pH 7.4) for 1 h. The protein samples were then separated using SDS-PAGE and either examined directly using Coomassie staining, or blotted to membranes and subjected to immunoblotting with appropriate antibodies.
SDS-PAGE gels of the reactions of photo-oxidized aLA and LYSO (Supplementary Fig. 3) provided clear evidence for the formation of aLA-GAPDH and LYSO-GAPDH protein cross-links with the detection of bands at ~50 kDa in each case (Supplementary Fig. 3A and C; lanes 6, 8, 10). In the case of aLA, bands assigned to dimeric aLA (band at ~25 kDa) and dimeric GAPDH were also detected. The yield of the aLA-GAPDH and LYSO-GAPDH protein cross-link bands showed a clear increase in intensity with an increasing time of photolysis of the aLA and LYSO, before incubation with the GAPDH (Supplementary Fig. 3A and C; lanes 6, 8, 10). Corresponding immunoblots with the anti-GAPDH antibody also revealed an increase in pixel intensity of the cross-linked protein band with longer light exposure (aLA-GAPDH: Fig. 7A, lanes 6, 8, 10; LYSO-GAPDH: Fig. 7C, lanes 4, 6, 8, 10). Corresponding studies were not carried out with anti-aLA or anti-LYSO antibodies as the cross-linked bands could be clearly identified on the Coomassie-stained gels, thereby allowing any problems arising from differential antibody recognition to be circumvented (see Discussion). Quantification of the immunoblots showed a significant increase in pixel density from the cross-link bands for both proteins when compared to control samples containing non-modified aLA or LYSO that had been incubated with GAPDH (Fig. 7D). For CRP, similar behavior was observed with CRP-GAPDH cross-links detected using both the anti-GAPDH antibody (Fig. 7B, lanes 6, 8, 10) and an anti-CRP antibody (Supplementary Fig. 3B; lanes 6, 8, 10). Quantification of the immunoblots showed significant increases with increasing initial photolysis time of the CRP, for both immunoblots (Fig. 7D, Supplementary Fig. 3D).
Fig. 7.

Photooxidation of aLA, Lyso and CRP and subsequent reaction with GAPDH induces protein cross-linking, and is dependent on the availability of free thiols on GAPDH. Panel A: representative immunoblot image of oxidized aLA reacted with GAPDH or GAPDH + NEM. Panel B: representative immunoblot image of oxidized CRP reacted with GAPDH or GAPDH + NEM. Panel C: representative immunoblot image of oxidized LYSO reacted with GAPDH or GAPDH + NEM. Panel D: optical density ratio (ODn min/OD0 min) of cross-linked bands from immunoblotting data presented in panels A–C, at the indicated photolysis times, for aLA, LYSO and CRP. Each blot is representative of one of three, obtained from three independent experiments. Quantitative data are presented as mean ± SD from three independent experiments. Statistical differences were examined using one-way analysis of variance (ANOVA) with Dunnett's multiple comparison test, and are indicated as follows: *P < 0.05 vs. lane 4 in panels A–C.
Pre-treatment of GAPDH with NEM (10-fold excess over GAPDH concentration) to block the free Cys groups on GAPDH, before incubation with the photo-oxidized proteins, decreased the intensity of the bands assigned to the protein-GAPDH cross-links (Fig. 7A, lanes 7, 9, 11; Fig. 7B, lanes 7, 9, 11; Fig. 7C, lanes 3, 5, 7, 9, 11; Supplementary Fig. 3, panels A–C). A low level of LYSO-GAPDH cross-linking was also detected in control samples where the LYSO was not subject to photo-oxidation (Fig. 7C, lanes 2, 4; Supplementary Fig. 3C, lanes 2, 4), which is ascribed to slow direct thiol-disulfide exchange reactions between two proteins; this was however at a lower level than detected with the photo-oxidized samples. For aLA and CRP, control samples which were illuminated in the absence of RB, or incubated with RB in the dark showed no cross-link formation on subsequent incubation with GAPDH (Fig. 7A and B; Supplementary Fig. 3A and B; lanes 2–5 in each case).
3.8. Mass spectrometric characterization of proteins detected in putative cross-link bands by SDS-PAGE, using in-gel digestion
The trypsin digestion/18O-labelling approach used with B2M to obtain information on the residues involved in the new disulfide bonds was unsuccessful with aLA and LYSO, due to the more complex nature of these proteins, due to the presence of 4 disulfide bonds and consequent poor digestion in the absence of reduction and alkylation. Therefore an alternative approach was employed to confirm the identity of the proteins present in the bands assigned to the novel cross-linked species detected on the SDS-PAGE gels. This involved in situ in-gel digestion of the putative protein cross-link bands with prior reduction and alkylation (although this destroys the new cross-link(s)), and subsequent mass spectrometric analysis, with the hypothesis being that detection of peptides from two different proteins in the cross-linked band, would provide evidence for the prior presence of covalent cross-links.
Initial examination of peptides arising from the SDS-PAGE cross-linked protein bands was conducted by nanoLC-MS/MS analysis, after reduction, alkylation and trypsin digestion. Using this approach consistent peptide data was obtained for the B2M − and LYSO-derived species. However, for the aLA species, despite the use of reduction and alkylation, and also the use of Lys-C in addition to trypsin, no peptides were obtained from the gel bands. In contrast, for the CRP-GAPDH species, additional digestion with Lys-C improved the efficiency significantly. The peptides observed from the various cross-link bands, and the parent protein identifications are summarized in Table 1, together with data on the peptides obtained from the native proteins (CRP, LYSO, B2M, GAPDH). In each case the extracted cross-link bands were determined to contain peptides from two proteins, consistent with the presence of reducible (likely to be disulfide) covalent linkages between each of the pairs of proteins.
Table 1.
Peptides detected by LC-MS after in-gel digestion of cross-linked and control protein bands.
| Protein ID |
Acc. No.a |
Peptide sequence |
m/z b |
Charge state |
Mass error (ppm) c |
Detected sampled |
|||
|---|---|---|---|---|---|---|---|---|---|
| CON protein band e | CRP-GAPDH band | LYSO-GAPDH band | B2M-GAPDH band | ||||||
| CRP | P02741 | ESDTSYVSLK | 564.77 | 2 | 0.05 | × | × | ||
| YEVQGEVFTK | 600.30 | 2 | 0.17 | × | × | ||||
| GYSIFSYATK | 568.78 | 2 | 0.17 | × | × | ||||
| APLTKPLK | 434.29 | 2 | 0.52 | × | × | ||||
| QDNEILIFWSK | 696.86 | 2 | 0.96 | × | × | ||||
| AFTVC36∗LHFYTELSSTR | 644.65 | 3 | 0.13 | × | × | ||||
| AFVFPK | 354.71 | 2 | 2.82 | × | |||||
| KAFVFPK | 418.76 | 2 | 0.46 | × | |||||
| PLKAFTVC36∗LHFYTELSSTR | 757.39 | 3 | 0.85 | × | |||||
| LYSO | P00698 | FESNFNTQATNR | 714.83 | 2 | 0.51 | × | × | ||
| GYSLGNWVC30∗AAK | 663.31 | 2 | 1.02 | × | × | ||||
| NTDGSTDYGILQINSR | 877.42 | 2 | 0.43 | × | × | ||||
| NLC76∗NIPC80∗SALLSSDITASVNC94∗AK | 1254.60 | 2 | 0.19 | × | × | ||||
| IVSDGNGMNAWVAWR | 838.40 | 2 | 0.77 | × | × | ||||
| GTDVQAWIR | 523.27 | 2 | 0.38 | × | × | ||||
| C6∗ELAAAM(ox)K | 455.21 | 2 | 2.31 | × | × | ||||
| C6∗ELAAAMK | 447.21 | 2 | 0.13 | × | × | ||||
| HGLDNYR | 437.71 | 2 | 1.72 | × | × | ||||
| RHGLDNYR | 515.76 | 2 | 0.19 | × | |||||
| WWC64∗NDGR | 497.20 | 2 | 0.06 | × | |||||
| B2M | P61769 | IQVYSR | 383.22 | 2 | 0.44 | × | × | ||
| VNHVTLSQPK | 561.82 | 2 | 0.18 | × | × | ||||
| SNFLNC25∗YVSGFHPSDIEVDLLK | 852.08 | 3 | 0.26 | × | × | ||||
| DWSFYLLYYTEFTPTEKDEYAC80∗R | 999.78 | 3 | 0.81 | × | × | ||||
| IEKVEHSDLSFSK | 506.93 | 3 | 0.85 | × | × | ||||
| IVKWDR | 408.74 | 2 | 2.08 | × | |||||
| TPKIQVYSR | 546.31 | 2 | 1.88 | × | × | ||||
| VEHSDLSFSK | 574.78 | 2 | 0.66 | × | |||||
| GAPDH | P46406 | AITIFQERDPANIK | 808.44 | 2 | 0.26 | × | × | × | × |
| VIISAPSADAPMFVMGVNHEK | 738.37 | 3 | 0.04 | × | × | × | × | ||
| VIHDHFGIVEGLMTTVHAITATQK | 655.35 | 4 | 0.17 | × | × | × | × | ||
| GAAQNIIPASTGAAK | 685.38 | 2 | 0.82 | × | × | × | × | ||
| LTGM(ox)AFR | 406.21 | 2 | 0.25 | × | × | × | × | ||
| VPTPNVSVVDLTC244∗R | 778.91 | 2 | 0.40 | × | × | × | × | ||
| LISWYDNEFGYSNR | 882.40 | 2 | 0.82 | × | × | × | × | ||
| VVDLMVHMASK | 615.32 | 2 | 0.76 | × | × | × | × | ||
| AITIFQER | 489.27 | 2 | 0.59 | × | × | × | × | ||
| IISNASC149∗TTNC153∗LAPLAK | 917.46 | 2 | 2.92 | × | × | × | × | ||
| QASEGPLK | 415.22 | 2 | 0.59 | × | × | × | × | ||
| VIPELNGK | 435.26 | 2 | 1.07 | × | × | × | × | ||
| VDVVAINDPFIDLHYMVYMFQYDSTHGK | 830.15 | 4 | 0.09 | × | × | × | × | ||
| YDDIKK | 391.21 | 2 | 0.93 | × | × | × | × | ||
| LVINGK | 322.21 | 2 | 2.21 | × | × | × | × | ||
| LTGMAFR | 398.21 | 2 | 1.00 | × | × | ||||
| FHGTVK | 344.69 | 2 | 0.72 | × | × | ||||
| YDNSLK | 370.18 | 2 | 1.74 | × | × | × | |||
Both control disulfide-containing protein (CRP, LYSO, B2M and GAPDH) and the cross-linking gel bands (CRP-GAPDH, LYSO-GAPDH and B2M-GAPDH) were analyzed by in-gel digestion. Proteins were obtained from 4 to 6 separate experiments. * indicates alkylation of Cys with a mass shift of +57 Da, M(ox) refers to methionine that was detected as oxidized species (+16 Da). Detailed analysis of peptide mass mapping data was performed using GPMAW 9.5 software.
Accession numbers correspond to the UniProt database numbering for these proteins.
Observed mass-to-charge ratio (m/z).
Mass error was calculated by Maxquant software.
Protein gel bands that contain the detected peptides.
Protein gel bands of the parent native protein.
For control (native) B2M, 8 peptides and a sequence coverage of 83.2% was determined, with 6 of these peptides (and a sequence coverage of 69.7%) also identified in the B2M-GAPDH cross-linked gel bands (Table 1). For native CRP, 9 peptides and a sequence coverage of 54.9% was detected, with 6 peptides from CRP and a sequence coverage of 39.3% detected in the CRP-GAPDH cross-linked band (Table 1). LYSO was identified with a sequence coverage percentage of 90.7% (11 peptides) and 79.1% (9 peptides) for control LYSO and LYSO-GAPDH cross-linked bands, respectively (Table 1). For GAPDH, 17 peptides were identified for the native protein (sequence coverage 64.0%), and the majority of these peptides (and a similar sequence coverage) were also detected in the protein-GAPDH cross-links (Table 1). Most of the peptides from these four proteins were also fragmented effectively in MS/MS experiments allowing confirmation of the protein identities in the cross-linked gel samples (Supplementary Figs. 4–7).
4. Discussion
Singlet oxygen (1O2) is an important reactive oxidant in biological systems and is known to induce modifications on biomolecules via type II photosensitization reactions (e.g. Refs. [16,[32], [33], [34]]). Rose Bengal generates high concentrations of 1O2 in the presence of molecular oxygen and visible light [19,35], and thus 1O2-mediated photo-oxidation primarily occurs in tissues that are routinely exposed to sunlight such as skin and eyes [[36], [37], [38]]. In proteins, the most reactive residues with 1O2 are the sulfur-containing (Cys, Met and cystine) and aromatic amino acids (His, Trp, and Tyr) [17,39].
The data obtained in this study demonstrate that photo-oxidation of B2M by the RB/visible light/O2 system induced a illumination-time dependent decrease in the parent B2M band, with increasing formation of dimer and trimer and higher aggregates of B2M, as detected using an anti-B2M antibody (Fig. 1A). Dimer formation was also detected with aLA and CRP (and previously for Lyso, e.g. Refs. [40,41]) on increasing exposure to 1O2 (cf. the SDS-PAGE and immunoblotting data in Supplementary Fig. 3). These aggregates were not formed in the absence of RB or visible light, consistent with 1O2 -mediated modification of specific amino acids in these proteins, which facilitate the formation of intra-protein cross-links. Similar data has been obtained previously for other proteins, though the mechanisms and nature of the amino acids involved have not always been fully elucidated. In some cases, data has been presented for the formation of non-reducible species involving strong carbon-carbon or carbon-heteroatom covalent bonds (e.g. di-tyrosine, di-tryptophan, tyrosine-tryptophan, histidine-lysine, histidine-arginine, histidine-histidine, tyrosine-lysine) [28,[40], [41], [42], [43], [44], [45], [46]]. However, in many cases, these species do not account quantitatively (see, e.g. Ref. [28]) for all the cross-linking detected, and there is abundant evidence for a major contribution from reducible species, and especially disulfide cross-links.
Whilst the formation of disulfide cross-links has often been ascribed to the oxidation of Cys residues (via either radical or non-radical reactions), such cross-links can also arise from thiol-disulfide exchange reactions [13]. This allows cross-links to be formed with proteins that do not contain any free Cys residues. The formation of such linkages via direct thiol-disulfide exchange is, however, very slow [13], and particularly with proteins where there are major steric and electronic barriers to the formation of these species, and little thermodynamic driving force due to the similar stabilities of the both the reactants and products. We show here that the formation of these types of cross-links can however be dramatically enhanced, both kinetically and in yield, if the disulfide bond is initially oxidized by 1O2: this provides a significant thermodynamic driving force for these reactions.
In this study, this mode of cross-linking was explored using multiple model proteins with different number of disulfides and no free Cys residues. The structurally-related proteins aLA and LYSO are stabilized by four disulfide bridges, and in both species one of these disulfides (Cys6-Cys120 in aLA, Cys6-Cys127 in LYSO) are known to be reactive due to their surface exposure (Supplementary Fig. 8) [47]. This may account for low level background thiol-disulfide exchange observed in this study (Fig. 7, Supplementary Fig. 3C), though this process is relatively slow. Here, we show that photo-oxidation of these two proteins and subsequent incubation with GAPDH (such that the GAPDH is not subject to direct oxidant exposure) results in formation of aLA-GAPDH and LYSO-GAPDH cross-links. With increasing photolysis time (and thus higher yields of 1O2), the extent of oxidation of these proteins increased, as evidenced by the time-dependent appearance of protein cross-links detected in SDS-PAGE and immunoblotting experiments (Fig. 7A, C; Supplementary Fig. 3). The formation of these cross-links has been confirmed in the case of LYSO by the concurrent detection of peptides from both LYSO and GAPDH in the cross-linked protein gel band by MS analysis.
Unlike aLA and LYSO, both B2M and monomeric CRP contain a single disulfide bond, with these buried within the structure [48,49]. Whilst this might be expected to inhibit cross-link formation, the data from the immunoblotting experiments, confirmed that cross-linking to GAPDH does occur, and that this increases with photolysis time (Fig. 1C and D; Fig. 7B and Supplementary Fig. 3B). This indicates that the initial exposure of these proteins to 1O2 results in significant, and sufficient, structural changes to allow reaction at the oxidized disulfide bond of B2M or CRP, with the one of the Cys residues on GAPDH, and consequent B2M-GAPDH and CRP-GAPDH cross-link formation. These data suggest that the time-dependent formation of such cross-links depends on initial modification of the protein structure and the formation of intermediate(s) with significant life-times as shown for the corresponding reactions with GSH [22,23,25].
It should be noted that interpretation of the quantitative comparisons between immunoblots, and particularly between native and modified systems (either cross-linked or oxidized) needs to be carried out with great care, as it is likely that recognition of the target protein epitope(s) by the antibody will vary as the protein is modified. Thus, there is unlikely to be a direct stoichiometric relationship between loss of the parent protein and the modified species (e.g. in terms of the pixel intensity), as recognition of the protein epitope may be increased (e.g. as a result of protein unfolding and greater exposure) or decreased (e.g. as a result of the epitope being buried within a dimer structure). It is therefore unwise to compare quantitatively (for example) the extent of formation of the B2M-GAPDH dimer depicted in Fig. 1, with the loss of functional activity of the GAPDH in Fig. 2. The latter suggests a much greater effect than the former, possibly as a result of a loss of GAPDH epitope recognition, or other effects of the modified B2M on the GAPDH activity. Correlation of these events requires accurate quantification of the extent of dimer formation, which is impossible to determine from the current data. However the GAPDH activity data, which may be more quantitatively accurate, do indicate disulfide-modified B2M has significant downstream functional effects on other proteins.
18O-labelling with MS analysis has allowed the specific characterization of a new disulfide bond formed between B2M and GAPDH after initial oxidation of B2M. The formation of B2M- , LYSO- and CRP- cross-links to GAPDH was also confirmed by in-gel digestion LC-MS/MS analysis. In these cases, the peptides identified from the SDS-PAGE bands (assigned to the cross-linked species) were in accordance with those expected on the basis of the protein sequences (Table 1), with good sequence coverage obtained in most cases (LYSO 79.1%, B2M 69.7%, CRP 39.3%, GAPDH 60.4–64.0%). The modest sequence coverage obtained with CRP, though definitive for the presence of this protein, probably arises from the compact hydrophobic core formed between the two β-sheets of this protein which may limit access to cleavage sites for trypsin (or other proteases) in the CRP sequence [50].
We propose that the formation of the protein cross-links involving GAPDH detected in this study occurs via initial oxidation of the disulfide bond in the photo-oxidized proteins (i.e. aLA, LYSO, B2M,CRP) by 1O2 to give either a zwitterionic peroxide, or possibly a thiosulfinate. The former is believed to be more likely, as the generation of a thiosulfinate requires further reactions. Both zwitterionic peroxides and thiosulfinates appear to have lifetimes of up to several hours [22,23,25], though this is structure dependent [51]. The oxidation of the disulfide bond as a result of these photo-oxidation reactions has been confirmed in the current study for B2M where a marked time-dependent loss of the cross-linked peptide containing the disulfide bond was detected by MS analysis. The zwitterionic peroxide (or thiosulfinate) species then reacts with a thiol group present on another protein, in this case one of the Cys residues of GAPDH, to give a new inter-protein disulfide bond (Fig. 8).
Fig. 8.

Proposed reaction pathway that yields new disulfide cross-linked proteins from initial photo-oxidation with 1O2 and subsequent reaction with GAPDH. Disulfide-containing proteins (A) undergo photo-oxidation to form a zwitterionic peroxide species (B), which serves as a possible precursor to thiosulfinates (C). The zwitterionic peroxide or thiosulfinates can react subsequently with the thiol-containing protein GAPDH (D) to yield inter-protein cross-links (E). Pretreatment of GAPDH with the thiol-blocking reagent NEM inhibits protein cross-linking formation. The disulfide reducing reagents DTT or TCEP can, at least partly, reverse protein cross-link formation.
Our previous studies on the reaction of peptides and proteins that contain disulfide bonds with oxidants (HOCl, ONOOH, H2O2, 1O2) have provided evidence for the incorporation of multiple oxygen atoms (as determined by the detection of a large series of ions with m/z increases of +16 in MS analyses) and for subsequent adduction of GSH (and other thiols) to peptide/protein-derived reactive intermediates [22,23,25]. Whilst some of the oxygen atom adduction is likely to occur at Met, His, Tyr and Trp residues (which are known targets for these oxidants [17,[52], [53], [54]]), data has also been obtained for oxygen atom addition at disulfide bonds to give intermediates that react with GSH [22,23,25]. The formation of these peptide-/protein-GSH adducts is (at least partially) reversible, and requires both the initial disulfide bond and an endogenous or added free thiol [22,23,25]. In the case of HOCl, ONOOH and H2O2, the intermediate species has been proposed to be a thiosulfinate [R–S(=O)–S-R’] species, whereas with 1O2, a zwitterionic peroxide [R–S+(OO−)-S-R’] may be the key species. The formation of such species has been reported previously in chemical systems [55,56].
The requirement for a free thiol on GAPDH for the formation of the new cross-link is illustrated by the experiments carried out with the thiol-blocking reagent NEM which significantly reduced the yield of each of the four protein-protein cross-links detected here (aLA-GAPDH, LYSO-GAPDH, B2M-GAPDH and CRP-GAPDH). Further evidence for a key role of the Cys residues on GAPDH in the formation of the B2M-GAPDH cross-links was obtained from the 18O MS analyses which identified two of the Cys residues on GAPDH (Cys149 and Cys244) as being directly involved in the cross-link, together with Cys25 from the former disulfide bond on B2M. Together these data provide strong evidence for a requirement for the initial disulfide bond, its oxidation to a reactive intermediate, and a free thiol on the second protein for the formation of the cross-links. As expected pre-treatment of the GAPDH with NEM also decreased the extent of the slow direct (control protein) thiol-disulfide exchange reaction detected between LYSO and GAPDH.
Incubation of the newly-formed inter-protein adducts with the disulfide reducing agents DTT and TCEP significantly reduced the yield of B2M-GAPDH cross-links, as detected by immunoblotting, consistent with the presence of a reducible inter-protein disulfide bonds. The extent of cross-link reduction was greater with TCEP than with DTT. This is in accord with previous reports of a greater efficiency in reduction by TCEP than DTT, at pH 7.5, although TCEP is a larger molecule, and for TCEP being significantly more effective in reducing surface-exposed protein disulfides [57,58]. Thus, the observed differences in reduction capacity of the protein cross-links between TCEP and DTT is likely to arise from both the reaction conditions as well as the location of new inter-protein disulfides.
Previous studies on the photo-oxidation of proteins, including some of those employed here (e.g LYSO), using Rose Bengal and other sensitizers, have provided evidence for the occurrence of both Type 1 and also Type 2 photochemistry (e.g. Refs. [40,41], reviewed [16,17,59]). The former process, which may be enhanced by sensitizer binding to the target protein [40], involves excited states formed on the sensitizer and subsequently the protein, and the generation of radicals from readily-oxidized sites such as tyrosine and tryptophan residues [16,17,59]. Some of these radicals undergo dimerization reactions to give di-tyrosine, di-tryptophan and tyrosine-tryptophan species [40,41]. However these species are typically of low yield, are not readily reversed by reducing agents such as TCEP and DTT (c.f. the reversal observed here) and require the two radicals to be present at the same time. As the GAPDH was added into these reaction systems after the cessation of illumination, no radicals should be present on this protein and therefore it is very unlikely that the cross-links involving GAPDH are formed via Type 1 photochemical processes and the generation of di-tyrosine, di-tryptophan, tyrosine-tryptophan or other carbon-carbon or carbon-heteroatom (e.g. iso-dityrosine) species. Such Type 1 processes may however account for some of the inter-molecular homo-dimeric cross-links detected on the photo-oxidized proteins (e.g. B2M-B2M, cf. data in Fig. 1A and B).
B2M levels have been reported to be elevated in the plasma of people with chronic uraemia due to impaired renal excretion, oxidative stress and inflammation. This elevation is associated with modifications to B2M including glycation and oxidation [60,61]. Native B2M is a 99-residue protein that forms the light-chain of the antigen class I major histocompatibility complex (MHC-I). Structurally, it has two β-sheets connected by the single Cys25-Cys80 disulfide bond, with this buried within the molecule [30,48] (Fig. 9). Interestingly, despite the buried nature of this disulfide, it appears to be readily modified by 1O2 (see above) and gives rise to a new disulfide bond with GAPDH. This ready reaction of the disulfide is however in agreement with previous hydrogen-deuterium exchange mass spectroscopy studies, that have shown that this protein has significant flexibility and can give rise to alternative ‘open’ conformations with significant lifetimes [62,63]. Thus the crystal structure rendered in Fig. 9 may be a misleading representation of the accessibility to the disulfide bond. Evidence was, however, only obtained for reaction with Cys25, and not Cys80 of B2M, as determined by the tryptic digestion and MS analysis; the reasons for this apparent selectivity are unclear and remain to be elucidated. Literature data indicate that aggregation of oxidized B2M is modulated by the disulfide bond in B2M [30], and that native (wild-type) B2M is resistant to aggregation without prior modification of the protein structure as observed here [30,64].
Fig. 9.

Rendering of the structure (Protein Data Bank, PDB ID: 1LDS) of B2M showing the site of the native disulfide, and of GAPDH (PDB ID: 1J0X) showing the sites of the free Cys residues. Panel A: The intra-disulfide (Cys25-Cys80) in B2M is shown as a van der Waals spheres representation: with the red colored atom being the sulfur atom of Cys25, and the green colored atom the sulfur atom of Cys80. Panel B: the Cys25-Cys80 disulfide in B2M is shown as a ball and stick representation and indicates the buried location of this species within the protein structure. Panel C: localization of the sulfur atoms of the free Cys residues in GAPDH: red colored atom - Cys149; blue colored atom - Cys153; green colored atom - Cys244; yellow colored atom - Cys281. Cys149 and Cys244 were identified as residues involved in the observed inter-protein disulfides between B2M and GAPDH, with Cys149 being highly exposed on the protein surface as indicated by the bright color of parts of this amino acid in the rendering of the protein structure. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
The partners with which the oxidized Cys25 forms a new linkage are also of interest with both Cys149 and Cys244 on GAPDH forming cross-links. The Cys149 residue of GAPDH is significantly exposed on the protein surface (Fig. 9), potentially allowing rapid and ready reaction with the oxidized disulfide on B2M, but Cys244 is less exposed though with significant surface accessibility (Fig. 9). The other Cys residues in GAPDH are less accessible. These data suggest that surface exposure is a significant factor in the generation of the new inter-protein linkages, but other factors may also play a significant role, such as the pKa of the thiol group in the Cys residues, as the thiolate form (RS−) is a much better nucleophile. This is a potentially important factor for GAPDH, as Cys149 is known to have a low pKa (5.5–6.0, due to the presence of a nearby His residue that acts as a proton acceptor [65,66]), and is part of the active site of this enzyme. Cys149 has been reported previously to contain an extremely reactive SH-group, and this residue is a major target for oxidative damage [67,68]. Cys 244 is also reported to have a low pKa, though the value is not known, as a result of electrostatic effects and formation of an ion-pair with neighboring basic amino acids [66,69]. Cys244 is a major site of covalent modification of GAPDH by electrophiles [65,70,71]. Modifications at either of these two Cys residues has been associated with GAPDH inactivation [70,71]. Modification of these Cys residues, by incorporation into a new disulfide would therefore also be expected to result in a loss of enzymatic activity, as detected here (Fig. 2). Thus, formation of these new disulfide bonds appears to have functional consequences. A loss of GAPDH activity, and aggregation of this protein, has been reported to be of pathophysiological relevance, and to be associated with neurodegenerative diseases such as Alzheimer disease (AD), Parkinson's disease (PD) and Huntington's Disease (HD) [72]. Additional studies to examine whether this loss of GAPDH activity, and aggregation of this protein, and also others, in diseased tissues (and models thereof) arises via the processes outlined above, would therefore be of great interest and potential importance.
Whilst thiol-disulfide exchange reactions, and oxidation of free Cys residues (by radicals or two-electron oxidants) have been previously postulated to be a major mechanism of novel disulfide bond formation, the studies described here illustrate a new pathway to protein cross-links via initial oxidation at disulfide bonds induced by singlet oxygen, and subsequent reaction with thiol groups from other proteins. Native protein cross-links are widespread (reviewed [31]), however, disturbances to native cross-linking and formation of new (non-native) cross-links has been proposed as both a contributing factor, and also a possible causative event, in a number of pathologies. The cross-linking pathway reported here significantly extends the potential repertoire of proteins that undergo such reactions as it provides a mechanism for facile aggregation of the (large number) of proteins that do not contain free Cys residues. Furthermore, the formation of these cross-links can also impact on the activity of the proteins with which they form cross-links as evidenced here for the loss of enzymatic activity of GAPDH.
It is well established that 1O2-mediated oxidation of proteins in cells can result in the formation of multiple reactive species and products, with Cys, cystine, Trp, His, and Tyr being major targets for 1O2, with the extent of reaction at each site dependent, at least in part, on their abundance, which clearly varies significantly between different proteins [16,52]. As 1O2 is a reactive species and has a limited diffusion radius, it is likely to react with a wide range of proteins within the cellular compartment where it is generated [16,18,52]. As a result of these two factors, it is difficult to make generalizations about which specific proteins will be affected, as this will depend on the site of 1O2 formation.
It is however already known that protein oxidation products, including those formed from 1O2 reactions, can have major biological effects, as some of these can induce further reactions (such as the peroxides formed from Trp, Tyr, and His residues, the sulfenic acids formed from Cys, and the zwitterion species formed from cystine reported in this work) [17,52]. It is therefore likely that these materials will have effects, and thereby may act as signals both within cells, and also between cells. However, this is an area that is poorly defined. The limited work that has been carried out in this area, clearly shows that some 1O2 -generated products, including a cis-hydroperoxide formed from Trp (cis-WOOH) have signaling functions, including on vascular tone [73]. The zwitterion peroxides, and the cystine oxidation products, that we report here, may act in a related manner, with the data reported here for GAPDH inactivation providing a first indication of the significance of these reactions. Taken together, this work opens the possibility of exploring the formation of disulfide cross-links as both biomarkers in diverse pathological conditions associated with oxidative stress, and also as a driver of disease initiation and progression. Understanding the full scope of this pathway requires further study.
Author contributions
SJ carried out most of the experimental work and the data analysis. MM and PH provided input into the LC-MS/MS experiments and subsequent analyses. LC helped supervise the project and contributed to the data analysis and implementation of the research. MJD conceived the study and supervised the project. SJ, PH, LC and MJD obtained funding to support the research. SJ wrote the first draft of the manuscript, and all authors contributed to its revision and final form.
Declaration of competing interest
The authors declare no conflicts of interest with regard to the data presented.
Acknowledgements
The authors are grateful for financial support from the Novo Nordisk Foundation (Laureate grant: NNF13OC0004294 to MJD), the China Scholarships Council (PhD scholarship to SJ: 201708340066), a WHRI International Fellowship (to LC) co-funded by the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (FP7/2007–2013) under REA grant agreement nº 608765, and an infrastructure grant from the Carlsberg Foundation (CF19-0451 to PH).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.101874.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Narayan M. Disulfide bonds: protein folding and subcellular protein trafficking. FEBS J. 2012;279:2272–2282. doi: 10.1111/j.1742-4658.2012.08636.x. [DOI] [PubMed] [Google Scholar]
- 2.Wong J.W.H., Hogg P.J. Analysis of disulfide bonds in protein structures. J. Thromb. Haemostasis. 2010;8 doi: 10.1111/j.1538-7836.2010.03894.x. 2345-2345. [DOI] [PubMed] [Google Scholar]
- 3.Chen V.M., Hogg P.J. Allosteric disulfide bonds in thrombosis and thrombolysis. J. Thromb. Haemostasis. 2006;4:2533–2541. doi: 10.1111/j.1538-7836.2006.02236.x. [DOI] [PubMed] [Google Scholar]
- 4.Okumura M., Kadokura H., Inaba K. Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radic. Biol. Med. 2015;83:314–322. doi: 10.1016/j.freeradbiomed.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Freedman R.B. The formation of protein disulphide bonds. Curr. Opin. Struct. Biol. 1995;5:85–91. doi: 10.1016/0959-440x(95)80013-q. [DOI] [PubMed] [Google Scholar]
- 6.Trujillo M., Alvarez B., Radi R. One-and two-electron oxidation of thiols: mechanisms, kinetics and biological fates. Free Radic. Res. 2016;50:150–171. doi: 10.3109/10715762.2015.1089988. [DOI] [PubMed] [Google Scholar]
- 7.Schoneich C. Thiyl radicals and induction of protein degradation. Free Radic. Res. 2016;50:143–149. doi: 10.3109/10715762.2015.1077385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang J., Carroll K.S., Liebler D.C. The expanding landscape of the thiol redox proteome. Mol. Cell. Proteomics. 2016;15:1–11. doi: 10.1074/mcp.O115.056051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poole L.B., Karplus P.A., Claiborne A. Protein sulfenic acids in redox signaling. Annu. Rev. Pharmacol. Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 10.Devarie-Baez N.O., Silva Lopez E.I., Furdui C.M. Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free Radic. Res. 2016;50:172–194. doi: 10.3109/10715762.2015.1090571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song J.J., Rhee J.G., Suntharalingam M., Walsh S.A., Spitz D.R., Lee Y.J. Role of glutaredoxin in metabolic oxidative stress. Glutaredoxin as a sensor of oxidative stress mediated by H2O2. J. Biol. Chem. 2002;277:46566–46575. doi: 10.1074/jbc.M206826200. [DOI] [PubMed] [Google Scholar]
- 12.Berndt C., Lillig C.H., Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H1227–H1236. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 13.Nagy P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxidants Redox Signal. 2013;18:1623–1641. doi: 10.1089/ars.2012.4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dasuri K., Zhang L., Keller J.N. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic. Biol. Med. 2013;62:170–185. doi: 10.1016/j.freeradbiomed.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 15.Andreu C.I., Woehlbier U., Torres M., Hetz C. Protein disulfide isomerases in neurodegeneration: from disease mechanisms to biomedical applications. FEBS Lett. 2012;586:2826–2834. doi: 10.1016/j.febslet.2012.07.023. [DOI] [PubMed] [Google Scholar]
- 16.Davies M.J. Singlet oxygen-mediated damage to proteins and its consequences. Biochem. Biophys. Res. Commun. 2003;305:761–770. doi: 10.1016/s0006-291x(03)00817-9. [DOI] [PubMed] [Google Scholar]
- 17.Pattison D.I., Rahmanto A.S., Davies M.J. Photo-oxidation of proteins. Photochem. Photobiol. Sci. 2012;11:38–53. doi: 10.1039/c1pp05164d. [DOI] [PubMed] [Google Scholar]
- 18.Ogilby P.R. Singlet oxygen: there is still something new under the sun, and it is better than ever. Photochem. Photobiol. Sci. 2010;9:1543–1560. doi: 10.1039/c0pp00213e. [DOI] [PubMed] [Google Scholar]
- 19.Redmond R.W., Gamlin J.N. A compilation of singlet oxygen yields from biologically relevant molecules. Photochem. Photobiol. 1999;70:391–475. [PubMed] [Google Scholar]
- 20.Clennan E.L., Wang D., Clifton C., Chen M.F. Geometry-dependent quenching of singlet oxygen by dialkyl disulfides. J. Am. Chem. Soc. 1997;119:9081–9082. [Google Scholar]
- 21.Clennan E.L. Persulfoxide: Key intermediate in reactions of singlet oxygen with sulfides. Acc. Chem. Res. 2001;34:875–884. doi: 10.1021/ar0100879. [DOI] [PubMed] [Google Scholar]
- 22.Carroll L., Jiang S., Irnstorfer J., Beneyto S., Ignasiak M.T., Rasmussen L.M., Rogowska-Wrzesinska A., Davies M.J. Oxidant-induced glutathionylation at protein disulfide bonds. Free Radic. Biol. Med. 2020;160:513–525. doi: 10.1016/j.freeradbiomed.2020.08.018. [DOI] [PubMed] [Google Scholar]
- 23.Jiang S., Carroll L., Rogowska-Wrzesinska A., Rasmussen L.M., Davies M.J. Oxidation of disulfide bonds in proteins by singlet oxygen gives rise to glutathionylated proteins. Redox Biol. 2021;38:101822. doi: 10.1016/j.redox.2020.101822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karimi M., Ignasiak M.T., Chan B., Croft A.K., Radom L., Schiesser C.H., Pattison D.I., Davies M.J. Reactivity of disulfide bonds is markedly affected by structure and environment: implications for protein modification and stability. Sci. Rep. 2016;6:38572. doi: 10.1038/srep38572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karimi M., Crossett B., Cordwell S.J., Pattison D.I., Davies M.J. Characterization of disulfide (cystine) oxidation by HOCl in a model peptide: evidence for oxygen addition, disulfide bond cleavage and adduct formation with thiols. Free Radic. Biol. Med. 2020;154:62–74. doi: 10.1016/j.freeradbiomed.2020.04.023. [DOI] [PubMed] [Google Scholar]
- 26.Wright A., Bubb W.A., Hawkins C.L., Davies M.J. Singlet oxygen–mediated protein oxidation: evidence for the formation of reactive side chain peroxides on tyrosine residues. Photochem. Photobiol. 2002;76:35–46. doi: 10.1562/0031-8655(2002)076<0035:sompoe>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 27.Shevchenko A., Tomas H., Havli J., Olsen J.V., Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006;1:2856. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 28.Leinisch F., Mariotti M., Rykaer M., Lopez-Alarcon C., Hägglund P., Davies M.J. Peroxyl radical-and photo-oxidation of glucose 6-phosphate dehydrogenase generates cross-links and functional changes via oxidation of tyrosine and tryptophan residues. Free Radic. Biol. Med. 2017;112:240–252. doi: 10.1016/j.freeradbiomed.2017.07.025. [DOI] [PubMed] [Google Scholar]
- 29.Mariotti M., Leinisch F., Leeming D.J., Svensson B., Davies M.J., Hägglund P. Mass-spectrometry-based identification of cross-links in proteins exposed to photo-oxidation and peroxyl radicals using 18o labeling and optimized tandem mass spectrometry fragmentation. J. Proteome Res. 2018;17:2017–2027. doi: 10.1021/acs.jproteome.7b00881. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y., Dokholyan N.V. A single disulfide bond differentiates aggregation pathways of ss2-microglobulin. J. Mol. Biol. 2005;354:473–482. doi: 10.1016/j.jmb.2005.09.075. [DOI] [PubMed] [Google Scholar]
- 31.Hägglund P., Mariotti M., Davies M.J. Identification and characterization of protein cross-links induced by oxidative reactions. Expert Rev. Proteomics. 2018;15:665–681. doi: 10.1080/14789450.2018.1509710. [DOI] [PubMed] [Google Scholar]
- 32.Cadet J., Douki T., Pouget J.P., Ravanat J.L., Sauvaigo S. Effects of UV and visible radiations on cellular DNA. Curr. Probl. Dermatol. 2001;29:62–73. doi: 10.1159/000060654. [DOI] [PubMed] [Google Scholar]
- 33.Girotti A.W. Photosensitized oxidation of cholesterol in biological systems: reaction pathways, cytotoxic effects and defense mechanisms. J. Photochem. Photobiol. B Biol. 1992;13:105–118. doi: 10.1016/1011-1344(92)85050-5. [DOI] [PubMed] [Google Scholar]
- 34.Clennan E.L. Overview of the chemical reactions of singlet oxygen. Compr. Ser. Photoch. 2016;13:353–367. [Google Scholar]
- 35.Wilkinson F., Helman W., Ross A. Quantum yields for the photosensitized formation of the lowest electronically excited singlet state of molecular oxygen in solution. J. Phys. Chem. 1993;22:113–262. [Google Scholar]
- 36.Davies M.J., Truscott R.J.W. Photo-oxidation of proteins and its role in cataractogenesis. J. Photochem. Photobiol. B Biol. 2001;63:114–125. doi: 10.1016/s1011-1344(01)00208-1. [DOI] [PubMed] [Google Scholar]
- 37.Baier J., Maisch T., Maier M., Landthaler M., Bäumler W. Direct detection of singlet oxygen generated by UVA irradiation in human cells and skin. J. Invest. Dermatol. 2007;127:1498–1506. doi: 10.1038/sj.jid.5700741. [DOI] [PubMed] [Google Scholar]
- 38.Giacomoni P.U., editor. Sun Protection in Man. Elsevier; Amsterdam: 2001. [Google Scholar]
- 39.Wilkinson F., Helman W.P., Ross A.B. Rate constants for the decay and reactions of the lowest electronically excited state of molecular oxygen in solution. An expanded and revised compilation. J. Phys. Chem. Ref. Data. 1995;24:663–1021. [Google Scholar]
- 40.Fuentes-Lemus E., Mariotti M., Hagglund P., Leinisch F., Fierro A., Silva E., Lopez-Alarcon C., Davies M.J. Binding of rose bengal to lysozyme modulates photooxidation and cross-linking reactions involving tyrosine and tryptophan. Free Radic. Biol. Med. 2019;143:375–386. doi: 10.1016/j.freeradbiomed.2019.08.023. [DOI] [PubMed] [Google Scholar]
- 41.Fuentes-Lemus E., Mariotti M., Reyes J., Leinisch F., Hägglund P., Silva E., Davies M.J., López-Alarcón C. Photo-oxidation of lysozyme triggered by riboflavin is O2-dependent, occurs via mixed type 1 and type 2 pathways, and results in inactivation, site-specific damage and intra- and inter-molecular crosslinks. Free Radic. Biol. Med. 2020;152:61–73. doi: 10.1016/j.freeradbiomed.2020.03.004. [DOI] [PubMed] [Google Scholar]
- 42.Leinisch F., Mariotti M., Hägglund P., Davies M.J. Structural and functional changes in RNAse A originating from tyrosine and histidine cross-linking and oxidation induced by singlet oxygen and peroxyl radicals. Free Radic. Biol. Med. 2018;126:73–86. doi: 10.1016/j.freeradbiomed.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 43.Fuentes-Lemus E., Silva E., Leinisch F., Dorta E., Lorentzen L., Davies M., López-Alarcón C. α-and β-casein aggregation induced by riboflavin-sensitized photo-oxidation occurs via di-tyrosine cross-links and is oxygen concentration dependent. Food Chem. 2018;256:119–128. doi: 10.1016/j.foodchem.2018.02.090. [DOI] [PubMed] [Google Scholar]
- 44.Xu C.F., Chen Y., Yi L., Brantley T., Stanley B., Sosic Z., Zang L. Discovery and characterization of histidine oxidation initiated cross-links in an IgG1 monoclonal antibody. Anal. Chem. 2017;89:7915–7923. doi: 10.1021/acs.analchem.7b00860. [DOI] [PubMed] [Google Scholar]
- 45.Shen H.-R., Spikes J.D., Smith C.J., Kopecek J. Photodynamic cross-linking of proteins IV. Nature of the His-His bond(s) formed in the rose bengal-photosensitized cross-linking of N-benzoyl-L-histidine. J. Photochem. Photobiol. Chem. 2000;130:1–6. [Google Scholar]
- 46.Shen H.R., Spikes J.D., Kopeckova P., Kopecek J. Photodynamic crosslinking of proteins. II. Photocrosslinking of a model protein-ribonuclease A. J. Photochem. Photobiol., B. 1996;35:213–219. doi: 10.1016/s1011-1344(96)07300-9. [DOI] [PubMed] [Google Scholar]
- 47.Gohda S., Shimizu A., Ikeguchi M., Sugai S. The superreactive disulfide bonds in α-lactalbumin and lysozyme. J. Protein Chem. 1995;14:731–737. doi: 10.1007/BF01886912. [DOI] [PubMed] [Google Scholar]
- 48.Smith D.P., Radford S.E. Role of the single disulphide bond of β2‐microglobulin in amyloidosis in vitro. Protein Sci. 2001;10:1775–1784. doi: 10.1110/ps.4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Braig D., Nero T.L., Koch H.-G., Kaiser B., Wang X., Thiele J.R., Morton C.J., Zeller J., Kiefer J., Potempa L.A. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat. Commun. 2017;8:14188. doi: 10.1038/ncomms14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szalai A.J., Agrawal A., Greenhough T.J., Volanakis J.E. C-reactive protein. Immunol. Res. 1997;16:127. doi: 10.1007/BF02786357. [DOI] [PubMed] [Google Scholar]
- 51.Clennan E.L., Hightower S.E. Natural bond orbital analyses of persulfoxide stabilization by remote functional groups. The conformationally induced electrostatic stabilization sulfide photooxygenation mechanism. J. Org. Chem. 2006;71:1247–1250. doi: 10.1021/jo052269c. [DOI] [PubMed] [Google Scholar]
- 52.Davies M.J. Protein oxidation and peroxidation. Biochem. J. 2016;473:805–825. doi: 10.1042/BJ20151227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davies M.J., Hawkins C.L. The role of myeloperoxidase (MPO) in biomolecule modification, chronic inflammation and disease. Antioxidants Redox Signal. 2020;32:957–981. doi: 10.1089/ars.2020.8030. [DOI] [PubMed] [Google Scholar]
- 54.Ferrer-Sueta G., Campolo N., Trujillo M., Bartesaghi S., Carballal S., Romero N., Alvarez B., Radi R. Biochemistry of peroxynitrite and protein tyrosine nitration. Chem. Rev. 2018;118:1338–1408. doi: 10.1021/acs.chemrev.7b00568. [DOI] [PubMed] [Google Scholar]
- 55.Clennan E.L. Persulfoxide: Key intermediate in reactions of singlet oxygen with sulfides. Acc. Chem. Res. 2001;34:875–884. doi: 10.1021/ar0100879. [DOI] [PubMed] [Google Scholar]
- 56.Clennan E.L., Hightower S.E., Greer A. Conformationally induced electrostatic stabilization of persulfoxides: a new suggestion for inhibition of physical quenching of singlet oxygen by remote functional groups. J. Am. Chem. Soc. 2005;127:11819–11826. doi: 10.1021/ja0525509. [DOI] [PubMed] [Google Scholar]
- 57.Cline D.J., Redding S.E., Brohawn S.G., Psathas J.N., Schneider J.P., Thorpe C. New water-soluble phosphines as reductants of peptide and protein disulfide bonds: reactivity and membrane permeability. Biochemistry. 2004;43:15195–15203. doi: 10.1021/bi048329a. [DOI] [PubMed] [Google Scholar]
- 58.Han J.C., Han G.Y. A procedure for quantitative determination of tris (2-carboxyethyl) phosphine, an odorless reducing agent more stable and effective than dithiothreitol. Anal. Biochem. 1994;220:5–10. doi: 10.1006/abio.1994.1290. [DOI] [PubMed] [Google Scholar]
- 59.Fuentes-Lemus E., Lopez-Alarcon C. Photo-induced protein oxidation: mechanisms, consequences and medical applications. Essays Biochem. 2020;64:33–44. doi: 10.1042/EBC20190044. [DOI] [PubMed] [Google Scholar]
- 60.Michelis R., Sela S., Kristal B. Intravenous iron-gluconate during haemodialysis modifies plasma β2-microglobulin properties and levels. Nephrol. Dial. Transplant. 2005;20:1963–1969. doi: 10.1093/ndt/gfh907. [DOI] [PubMed] [Google Scholar]
- 61.Wratten M.L., Tetta C., Ursini F., Sevanian A. Oxidant stress in hemodialysis: prevention and treatment strategies. Kidney Int. 2000;58:S126–S132. doi: 10.1046/j.1523-1755.2000.07616.x. [DOI] [PubMed] [Google Scholar]
- 62.Jørgensen T.J.D., Cheng L., Heegaard N.H.H. Mass spectrometric characterization of conformational preludes to beta 2-microglobulin aggregation. Int. J. Mass Spectrom. 2007;268:207–216. [Google Scholar]
- 63.Heegaard N.H.H., Jørgensen T.J.D., Cheng L., Schou C., Nissen M.H., Trapp O. Interconverting conformations of variants of the human amyloidogenic protein beta(2)-microglobulin quantitatively characterized by dynamic capillary electrophoresis and computer simulation. Anal. Chem. 2006;78:3667–3673. doi: 10.1021/ac060194m. [DOI] [PubMed] [Google Scholar]
- 64.Iadanza M.G., Silvers R., Boardman J., Smith H.I., Karamanos T.K., Debelouchina G.T., Su Y., Griffin R.G., Ranson N.A., Radford S.E. The structure of a β 2-microglobulin fibril suggests a molecular basis for its amyloid polymorphism. Nat. Commun. 2018;9:4517. doi: 10.1038/s41467-018-06761-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muronetz V.I., Barinova K.V., Stroylova Y.Y., Semenyuk P.I., Schmalhausen E.V. Glyceraldehyde-3-phosphate dehydrogenase: aggregation mechanisms and impact on amyloid neurodegenerative diseases. Int. J. Biol. Macromol. 2017;100:55–66. doi: 10.1016/j.ijbiomac.2016.05.066. [DOI] [PubMed] [Google Scholar]
- 66.Polgar L. Ion‐pair formation as a source of enhanced reactivity of the essential thiol group of D‐glyceraldehyde‐3‐phosphate dehydrogenase. Eur. J. Biochem. 1975;51:63–71. doi: 10.1111/j.1432-1033.1975.tb03907.x. [DOI] [PubMed] [Google Scholar]
- 67.Maller C., Schröder E., Eaton P. Glyceraldehyde 3-phosphate dehydrogenase is unlikely to mediate hydrogen peroxide signaling: studies with a novel anti-dimedone sulfenic acid antibody. Antioxidants Redox Signal. 2011;14:49–60. doi: 10.1089/ars.2010.3149. [DOI] [PubMed] [Google Scholar]
- 68.Souza J.M., Radi R. Glyceraldehyde-3-phosphate dehydrogenase inactivation by peroxynitrite. Arch. Biochem. Biophys. 1998;360:187–194. doi: 10.1006/abbi.1998.0932. [DOI] [PubMed] [Google Scholar]
- 69.Frizzell N., Lima M., Baynes J.W. Succination of proteins in diabetes. Free Radic. Res. 2011;45:101–109. doi: 10.3109/10715762.2010.524643. [DOI] [PubMed] [Google Scholar]
- 70.Blatnik M., Frizzell N., Thorpe S.R., Baynes J.W. Inactivation of glyceraldehyde-3-phosphate dehydrogenase by fumarate in diabetes: formation of S-(2-succinyl) cysteine, a novel chemical modification of protein and possible biomarker of mitochondrial stress. Diabetes. 2008;57:41–49. doi: 10.2337/db07-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blatnik M., Thorpe S.R., Baynes J.W. Succination of proteins by fumarate: mechanism of inactivation of glyceraldehyde-3-phosphate dehydrogenase in diabetes. Ann. N. Y. Acad. Sci. 2008;1126:272. doi: 10.1196/annals.1433.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chuang D., Hough C., Senatorov V.V. Glyceraldehyde-3-phosphate dehydrogenase, apoptosis, and neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 2005;45:269–290. doi: 10.1146/annurev.pharmtox.45.120403.095902. [DOI] [PubMed] [Google Scholar]
- 73.Stanley C.P., Maghzal G.J., Ayer A., Talib J., Giltrap A.M., Shengule S., Wolhuter K., Wang Y., Chadha P., Suarna C., Prysyazhna O., Scotcher J., Dunn L.L., Prado F.M., Nguyen N., Odiba J.O., Baell J.B., Stasch J.P., Yamamoto Y., Di Mascio P., Eaton P., Payne R.J., Stocker R. Singlet molecular oxygen regulates vascular tone and blood pressure in inflammation. Nature. 2019;566:548–552. doi: 10.1038/s41586-019-0947-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.