

Abstract
Coral gasdermin E is cleaved by activated caspase-3 to induce pyroptosis, a form of inflammatory programmed cell death, in response to a bacterial pathogen (see the related Research Article by Jiang et al.).
Invasive infection and signs of danger in mammals sound an immune alarm that culminates in pyroptosis, a highly inflammatory form of programmed necrotic cell death (1). Pyroptosis leads to the release of cellular alarmins, including inflammatory cytokines of the interleukin-1 family that are responsible for fever, ATP, and HMGB1, which recruit and activate white blood cells to sites of danger. Pyroptosis enables the immune system to respond to dangerous pathogens and avoid immune activation in response to nonthreatening commensal microbes. Although pyroptosis initially provides an alert that marshals protective immunity, under some circumstances, unconstrained inflammation can exacerbate disease. For example, pyroptosis lies at the root of the pathology of sepsis and cytokine storm, which can lead to acute organ failure and death. Recently, the molecular basis of pyroptosis in immune sentinel cells (monocytes, macrophages, and dendritic cells) and epithelial cells in the skin and mucosa, where the body often initially encounters a pathogen, was identified. Cytosolic sensors of danger, called inflammasomes, activate inflammatory caspases to cleave the cytosolic protein gasdermin D (GSDMD). GSDMD is composed of active N-terminal and inhibitory C-terminal domains separated by a flexible linker, whose cleavage liberates the N-terminal domain. The cleaved N-terminal domain then binds to the plasma membrane and assembles into pores that subsequently destroy cell membrane integrity through which cellular alarmins are released. GSDMD is a member of the gasdermin (GSDM) family consisting of six members in humans, GSDMA to GSDME and a more distant DFNB59, which, in mammals, is not known to form membrane pores like the other GSDMs. Cleavage of GSDMA to GSDME in the linker region causes pyroptosis, but the most important enzymes and pathways that activate each GSDM remain mostly unknown, except for GSDMD and GSDME (also known as DFNA5). GSDME is activated by the effector caspase of apoptosis (caspase-3), which converts noninflammatory apoptosis to inflammatory pyroptotic death with profound immune stimulatory consequences (2, 3). Recent studies have identified additional proteases (caspase-8, neutrophil, and killer lymphocyte granule serine proteases) that can cleave some of the GSDMs and trigger pyroptosis.
GSDME was originally identified as a mutated gene in patients with nonsyndromic hearing impairment, which is thought to be caused by autoactivation of GSDME to induce pyroptosis in cochlear hair cells (4). Besides cochlear hair cells, mammalian GSDME is also highly expressed in other tissues, including the brain, placenta, heart, and kidney. Stimuli that activate apoptosis and caspase-3, such as chemotherapy drugs, can trigger GSDME-mediated pyroptosis in those tissues, leading to tissue damage in mice (3). By contrast, GSDME is silenced in many cancers due to hypermethylation of its promoter region, preventing the tumor suppressive functions of GSDME (4). GSDME suppresses tumor growth by inducing pyroptosis and activating killer cell–mediated antitumor immunity (5).
GSDME and DFNB59 are the most evolutionarily ancient GSDM genes, which had previously been traced to the common ancestor of the chordates. In one species of fish, GSDME was shown to cause pyroptosis after linker cleavage by inflammatory caspase-1 and also, although less efficiently, by apoptotic caspases 3 and 7 (6). By contrast, GSDMA to GSDMD orthologs are largely restricted to mammals. In a study in this issue of Science Immunology, Jiang et al. (7) report that GSDME and pyroptosis go much further back in evolution to the Cnidaria, radially symmetric marine organisms that include sea anemones, jellyfish, and corals, and represent the oldest metazoan phylum. By searching available sequencing data, the authors identified a GSDM-like sequence in invertebrates, including corals, sea anemones, hydra, brachiopods, and mollusks. Phylogenetic analysis showed that those invertebrate GSDMs are evolutionarily closely related to mammalian GSDME and DFNB59 rather than GSDMA to GSDMD [Fig. 1A, modified based on fig. S3 of (7)]. Interestingly, invertebrate GSDMEs are more closely related to mammalian DFNB59 than GSDME. However, unlike mammalian DFNB59, which has an extremely short C terminus that is not believed to function as an N-terminal domain inhibitor, invertebrate GSDMEs contain a full-length C-terminal domain. The authors show that coral caspase-3 cleaves coral GSDME to cause pyroptosis (Fig. 1B). The pathway is evolutionarily conserved because human effector caspases 3 and 7 cleave GSDME derived from Orbicella faveolata (Of, a Caribbean reef-building coral) and overexpression of OfCASP3 and OfGSDME or the N-terminal fragment of OfGSDME in human HeLa cells triggered hallmarks of pyroptosis—a ballooning cell membrane displaying increased permeability and release of the large tetrameric protein lactate dehydrogenase (LDH). Consistent with a conserved GSDM function in pyroptosis, the tagged N-terminal fragment of OfGSDME concentrated on the plasma membrane, while full-length OfGSDME distributed throughout the cytosol of HeLa cells, like the mammalian proteins. Moreover, the authors identified two conserved sequences in the OfGSDME linker that correspond to canonical caspase-3 cleavage sites, 238DATD241 and 254DEPD257. Both N-terminal fragments produced by cleavage at these two sites caused pyroptosis. In human tumors, GSDME mutations cause loss of function and concentrate in the N-terminal domain and around the sequence encoding the caspase-3 cleavage site (5). In corals, the presence of two caspase-3 cleavage sites may serve as a safeguard to ensure that coral GSDME gets cleaved.
Fig. 1. Coral GSDME mediates infection-induced pyroptosis.
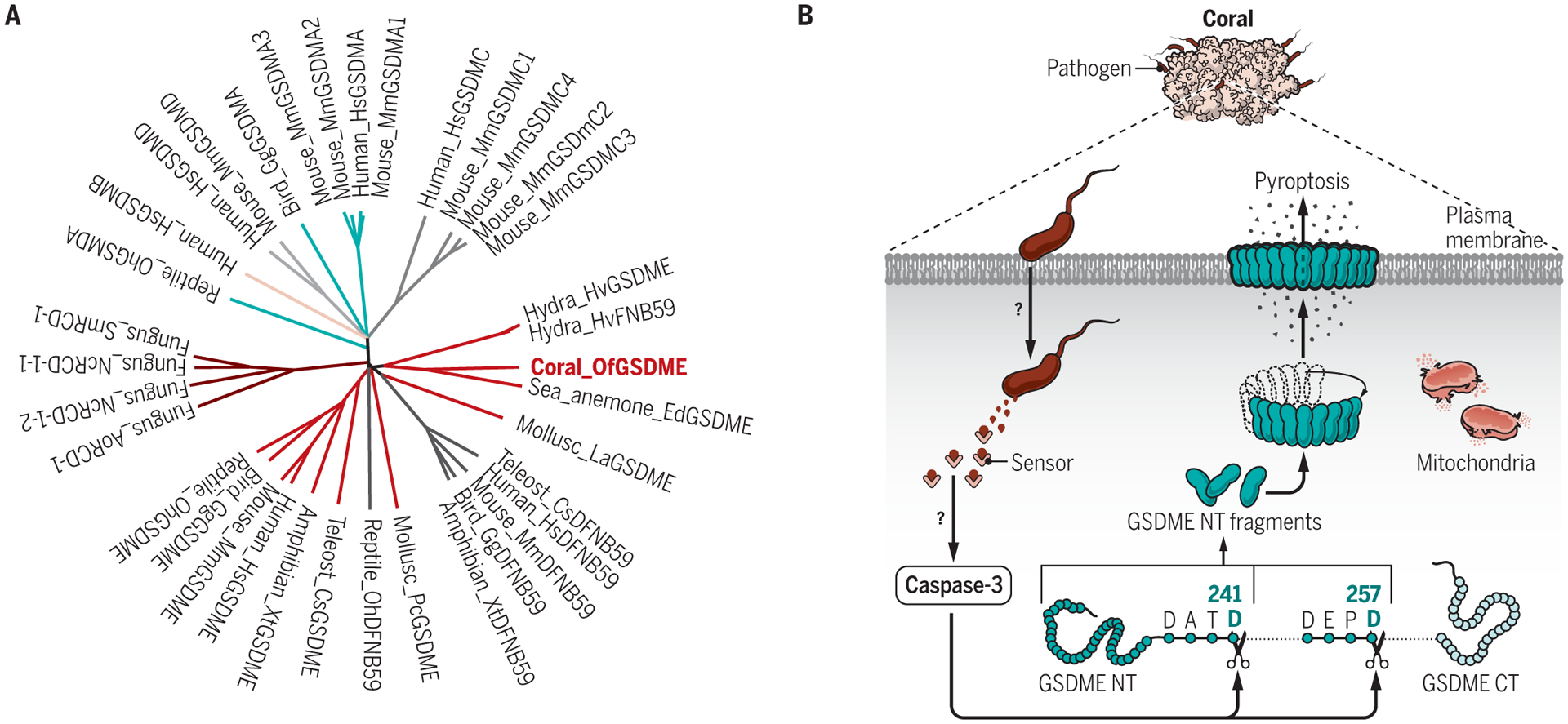
(A) Phylogenetic analysis of GSDME [modified based on fig. S3 of (7)]. The phylogenetic tree was generated using Clustal Omega (EMBL-EBI) and drawn using FigTree v1.4.4. (B) Pathogens in coral activate caspase-3 (CASP3) and GSDME-dependent pyroptosis. Invasive infection is recognized in corals by an unknown sensor and pathway to trigger activation of CASP3 to cleave GSDME after Asp241 and/or Asp257. The released GSDME N-terminal (NT) fragments oligomerize and form pores on the plasma membrane to induce pyroptosis. Accompanying mitochondrial damage was observed, but the underlying mechanism and its role in pyroptosis are unknown.
A recent study identified an even more distant ortholog of the GSDM N-terminal domain in the filamentous fungus Neurospora crassa, RCD-1 (8). This protein, which also binds to acidic phospholipids and triggers programmed cell death when coexpressed with another allele in human cells, triggers cell death when asexual spores from incompatible strains of the same species fuse in a defense mechanism known as heterokaryon incompatibility. Searches for proteins that shared sequence homology and a predicted structural similarity identified bacterial RCD-1 homologs. Thus, the molecular motif of GSDM pores may predate the metazoa and exist in prokaryotes and fungi.
The authors of the current study also show that infection of another coral species (Pocillopora damicornis) with Vibrio coralliilyticus, a bacterium that infects and causes destruction of reefs stressed by climate change, triggers caspase-3–dependent necrotic cell death. P. damicornis death was accompanied by cell membrane and mitochondrial damage but lacked hallmarks of apoptotic cell death such as chromatin condensation or other nuclear changes. Infection led to loss of polyps, necrosis, and destruction of the coral’s architecture, which was associated with GSDME cleavage and blocked by a caspase-3 inhibitor. These data strongly suggest that V. coralliilyticus infection of corals activates caspase-3, which, in turn, cleaves GSDME to cause pyroptosis and coral death. However, although the authors showed an association of GSDME cleavage with infection-induced necrotic death of corals, they were unable to definitively demonstrate that coral cell death was GSDME-dependent. These experiments would require—since there are no known GSDME inhibitors—manipulating GSDME expression by knockout or knockdown, or editing OfGSDME to mutate the caspase-3 cleavage sites. The authors also showed gross disruption of mitochondrial morphology after V. coralliilyticus infection; however, they did not examine whether mitochondrial damage directly caused or was just associated with cell death. Future studies could examine whether V. coralliilyticus infection induces biochemical changes within coral, such as generation of ROS or metabolic disruption that could have pathogenic effects, including “bleaching” caused by the loss of symbiotic algae that are highly sensitive to ROS.
This study raises many interesting questions, including: How does infection trigger pyroptosis in corals? Corals express Toll-like receptors, which monitor bacteria outside of the cell, but do they also have cytosolic sensors of invasive infection or sterile danger that resemble mammalian inflammasomes? Are LPS or the flagella of V. coralliilyticus recognized by coral cells as they are in mammals? The N-terminal fragment of GSDMs can form pores on bacterial membranes to kill bacteria. Do invertebrate GSDMs similarly target and help control the microbes that trigger pyroptosis? Do corals benefit from GSDME-triggered pyroptosis and does this pathway reduce bacterial load or the damage to the organism caused by V. coralliilyticus? If GSDME were not expressed, would caspase-triggered apoptosis of infected cells be less effective at controlling infection? In which cells is pyroptosis activated—in the infected cell or in the amoeboid migratory cells that are the primitive precursors of our own immune sentinel cells? The conversion of apoptosis to pyroptosis by mammalian GSDME in tumor cells activates protective killer cell–mediated antitumor immunity. Does pyroptosis similarly recruit amoebocytes to the site of infection and promote bacterial phagocytosis and killing and if so, by what mechanism?
This study provides evidence that pyroptosis is an evolutionarily conserved cell death mechanism across multiple phyla within the animal kingdom. Classical caspase-dependent apoptotic programmed cell death arose with multicellularity. Like the bacterial and fungal RCD-1 homologs of GSDME, structurally homologous caspase-like cysteine proteases (metacaspases) are expressed in bacteria and plants but are not thought to induce programmed cell death in these organisms. Apoptosis is an unselfish death mechanism by which cells that have outlived their usefulness sacrifice themselves for the overall benefit of the organism. This study suggests that pyroptosis developed around the same time evolutionarily as apoptosis and depends on the same critical protease. Generally, pyroptosis is responsible for cell death in response to pathogenic infection. This study provides evidence that pyroptosis may serve as an unselfish form of programmed cell death that benefits multicellular organisms by sacrificing infected cells to limit infection in the organism as a whole and sound an alarm to marshal innate immune defenses.
Given the extensive destruction of reef-building corals from increasing environmental stresses, disruption of favorable symbiosis, and secondary pathogen infection, this paper’s identification of an unexpected molecular mechanism of infection-induced coral death provides insight into how corals die and could lead to developing new approaches to protect coral reefs. A better understanding of the immune defenses activated by pyroptosis in invertebrates, how they are regulated, and the potential role of amoebocytes in protection and pathology is likely to provide insight into both the biological mechanisms contributing to coral destruction and the fundamental roles of pyroptosis in vertebrates.
Acknowledgments:
This work was supported by NIH R01CA240955 to J.L. J.L. discloses that she is a co-founder of Ventus Therapeutics in which she has an equity interest and receives consulting fees.
REFERENCES AND NOTES
- 1.Liu X, Lieberman J, A mechanistic understanding of pyroptosis: The fiery death triggered by invasive infection. Adv. Immunol 135, 81–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES, Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun 8, 14128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F, Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017). [DOI] [PubMed] [Google Scholar]
- 4.de Beeck KO, Van Laer L, Van Camp G, DFNA5, a gene involved in hearing loss and cancer: A review. Ann. Otol. Rhinol. Laryngol 121, 197–207 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, Junqueira C, Meza-Sosa KF, Mok TMY, Ansara J, Sengupta S, Yao Y, Wu H, Lieberman J, Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang S, Gu H, Zhao Y, Sun L, Teleost gasdermin E Is cleaved by caspase 1, 3, and 7 and induces pyroptosis. J. Immunol 203, 1369–1382 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Jiang S, Zhou Z, Sun Y, Zhang T, Sun L, Gasdermin-triggered pyroptosis in corals. Sci. Immunol 5, eabd2591 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Daskalov A, Mitchell PS, Sandstrom A, Vance RE, Glass NL, Molecular characterization of a fungal gasdermin-like protein. Proc. Natl. Acad. Sci. U.S.A 117, 18600–18607 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]