

Benefits from biologic therapy for inflammatory bowel disease (IBD) come at a cost of side effects and antidrug antibodies.1 This necessitates novel anti-inflammatory therapies.
Histone acetylation is an important epigenetic gene expression regulator. Acetylation of histone-3 lysine-27 (H3K27ac), H3K9ac, and trimethylation of H3K4 (H3K4me3) mark active enhancers. H3K27ac can differentiate active from poised enhancers and is linked to gene expression.2 Trimethylation of H3K27 leads to gene repression. IBD-associated single-nucleotide polymorphisms overlap with regulatory elements marked by H3K27ac.3 H3K27ac is reduced in dextran sulfate sodium (DSS)-induced colitis in mice and broad-acting histone deacetylases (HDAC) inhibitors (eg, SAHA and valproic acid [VPA]) restore H3K27ac levels and inhibit inflammatory cytokine production.4,5
We extend findings from models to patients with IBD to evaluate the therapeutic potential of HDAC inhibitors. We assessed H3K27ac in patients with IBD and tested whether VPA could inhibit inflammatory cytokine production in IBD biopsies cultured ex vivo. VPA was selected because of its safety record in treating neurologic disorders. Methods and patient characteristics are given in the Supplementary Material.
To validate our approach, we confirmed a reduction in the percentage of H3K27ac positive (H3K27ac+) cells in conjunction with the development of inflammation in a DSS model (Figure 1A–C).4 Moreover, after removal of DSS, H3K27ac+ cells significantly increased during resolution of inflammation (Figure 1C). No significant changes in HDAC mRNA were detected during the inflammatory phase, despite an increase in interleukin (IL) 6 mRNA levels (positive control for DSS-treatment) (Figure 1D). During the resolution of inflammation (Day 17), expression of 4 Hdacs differed from baseline (downregulated, Hdac1, 5, and 10; upregulated, Hdac2) (Figure 1D).
Figure 1.
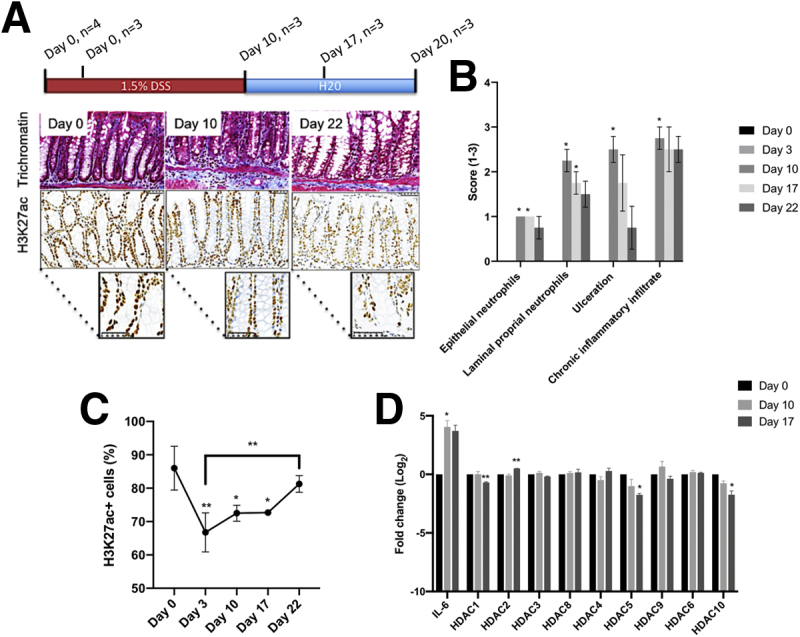
(A) H3K27ac in mouse DSS colitis. Representative Masson trichrome and H3K27ac stains. (B) Assessment of trichromatee stains confirmed an increase in inflammation markers post–DSS treatment, which lowered on DSS removal. (C) Conversely, percentage of H3K27ac+ cells decreased following DSS treatment and increased following DSS removal. (D) DSS treatment associated with increased IL6 mRNA levels. DSS did not alter HDAC mRNA levels, although these decreased during recovery following DSS removal. Significant changes: ∗< .05, ∗∗< .01.
A reduction in H3K27ac+ cells was confirmed in actively inflamed IBD biopsies relative to inactive IBD control subjects (Figure 2A and B) and was maintained in a paired subanalysis of the same samples between patient matched active and inactive segments (-16.25%; P = .0243; n = 11). This indicated that confounding factors, including disease duration and medications, could not account for the observed differences. A similar pattern of change was observed in patients with Crohn’s disease (CD) and ulcerative colitis (UC), although the reduction in H3K27ac+ cells in active biopsies was significant only in patients with UC (Figure 2B). As in the DSS model, active disease in patients with IBD was marked by increased IL6 mRNA but was not associated with changes in HDAC mRNA levels (Figure 2C). A subanalysis of patients with UC and CD (Supplementary Figure 1) found increased HDAC 9 in UC consistent with its inflammatory role.6
Figure 2.

(A, B) Fluorescent protein analysis of the percentage of H3K27ac+ cells in inactive and active patients with IBD confirmed a reduction in active relative to inactive IBD control subjects. (C) No changes in HDAC mRNA were observed in IBD biopsies with active disease. (D) Culturing biopsies with an HDAC inhibitor (5 mM VPA) increased the percentage H3K27ac+ cells in IBD biopsies relative to patient-matched biopsies treated with a vehicle control (RPMI medium), and (E–G) decreased inflammatory cytokine mRNA levels and protein production. Significant changes: ∗< .05, ∗∗< .01, ∗∗∗< .001, and ∗∗∗∗< .0001. ELISA, enzyme-linked immunosorbent assay; IFNG, interferon gamma; NS, not significan; TNF, tumor necrosis factor.
Supplementary Figure 1.

(A) A table comparing IL6 and HDAC mRNA levels between IBD biopsies with inactive (n = 8) and active disease (n = 11); with a separate analysis for patients with CD (CD inactive and CD active, n = 4 and 5, respectively) and patients with UC (UC inactive and UC active, n = 4 and 6, respectively). Data are expressed as log2 fold-change in active biopsies relative to the mean level of expression in inactive biopsies. Differences between groups were determined using Student t test, assuming equal variance, and the P values for each comparison are given in the table. (B) The expression levels for IL6 and HDAC9 are also graphically represented (∗< .05, ∗∗< .01).
Inhibiting HDAC activity in mucosal IBD biopsies cultured ex vivo with 5 mM VPA (Figure 2D) increased H3K27ac levels and reduced expression of IL6, IL10, IL1B, and IL23 mRNA significantly relative to patient-matched biopsies treated with control media (Figure 2E). Reduced production of IL6, IL10, and IL23 protein into culture media was confirmed (Figure 2F), and a reduction in tumor necrosis factor and interferon gamma was found (Figure 2F). Analysis of IL6 in an expanded cohort again indicated that VPA effects were greater in biopsies from areas with active disease, and in patients with UC (Figure 2G); namely, groups that had the most pronounced reductions in H3K27ac+ cells (Figure 2B).
In biopsy cultures, we cannot discern the primary cellular source of IL6 or other inflammatory cytokines. VPA can suppress inflammation by inducing apoptosis of laminal propria mononuclear cells,7 and we observed a decrease in BLC3 mRNA and an increase in CASP9 expression in VPA-treated IBD biopsies (Supplementary Figure 2). Although not significant, there was an increase in the percentage of CASP+ cells in VPA-treated biopsies and the proportion of CASP+/CD3+ T cells (Supplementary Figure 2).
Supplementary Figure 2.

(A) The mRNA levels of BLC3, which protects against apoptosis, and (B) CASP9 and (C) CASP3, which promote apoptosis, were quantified by quantitative polymerase chain reaction in paired IBD biopsies treated with or without 5 mM VPA. Paired Student t tests showed a significant decrease in BCL3 expression, coupled with an increase in CASP9 mRNA levels (D–G). There was also a small increase in the percentage of CASP9+ cells measured using immunofluorescence. This increase was relatively larger in CD3+ T cells. However, these differences did not reach statistical significance. Significant changes: ∗∗∗∗< .0001. N.S, not significant.
Other potentially relevant mechanisms of action for VPA include modulation of acetylation at inflammatory gene promoters and regulation of microRNAs. For example, in a model of colitis-accelerated colon carcinogenesis, DSS treatment increased HDAC activity and decreased H3K27ac levels in the intestine. However, promoters of inflammatory genes, including the IL6 promoter, were hyperacetylated. Conversely, anti-inflammatory treatment with aspirin increased global acetylation levels but reduced H3K27ac levels at the promoter regions of proinflammatory genes.4 Others have reported that in T cells, HDAC inhibitors promote FOXP3 expression by acetylating its promoter and inducing microRNA signatures associated with regulatory T cells. Thus, reducing IL6/STAT3/IL17 signaling in naive CD4+ T cells and blocking the polarization of Th17 cells.8 HDAC inhibitors also acetylate nonhistone proteins including STAT1, blocking their phosphorylation and inhibiting proinflammatory signalling.9 Increasing histone acetylation may alter other histone modifications (eg, H3K27me3), which is altered by DSS treatment.
VPA can have HDAC-independent effects, including regulating DNA methylation leading to gene repression and metabolic changes. Because chemically diverse HDAC inhibitors have similar effects, an HDAC-dependent mechanism seems likely. Indeed, SAHA (vorinostat), which is more potent than VPA in IBD models but currently only used for cutaneous T-cell lymphoma, is being trailed in patients with CD.10
Our data identify H3K27ac levels in the mucosa as a potential biomarker of response to HDAC therapy and establish ex vivo biopsy cultures as an alternative screen for HDAC inhibitors. Although further work is needed to elucidate VPA mechanism of action, we highlight the potential to repurpose VPA, a widely used and inexpensive drug, to treat inflammation in IBD.
Footnotes
Conflicts of interest The authors disclose no conflicts.
Funding Supported by European Crohn’s and Colitis Organisation (Fellowship award 2015 to CF), Rosetrees Trust (research grant CM550 to CF and AS), and The Barts Charity (research grant MGU0399 to AS and JOL).
Supplementary Material
Materials and Methods
Dextran Sulphate Sodium Exposed Mice
Animal experiments were approved by the Ethics Committee for Animal Experimentation of the Fondazione IRCCS Istituto Nazionale dei Tumori of Milan. C57BL/6 wild-type mice (4–6 weeks) were purchased from Charles River and housed in filter-top cages held under pathogen-free conditions. Mice were administered 1.5% DSS in drinking water for 10 days. Recovery was evaluated 10 days after DSS withdrawal. Mice were sacrificed at Day 0 (n = 3), Day 3 (n = 4), Day 10 (n = 4), Day 17 (n = 3), and Day 22 (n = 3). The colon was removed, washed in phosphate-buffered saline (PBS) and formalin fixed and paraffin embedded (FFPE). Other mice were sacrificed at Day 0 (n = 3), Day 10 (n = 3), and Day 17 (n = 3) and the colon homogenized in Trizol reagent (Invitrogen, Waltham, MA) before RNA extraction.
Robarts histopathology index score of trichromatin-stained FFPE sections monitored disease activity.1 The Robarts histopathology index score correlated well with previously published measures in these mice.2,3
Immunohistochemistry for Mouse Formalin Fixed and Paraffin Embedded Tissue
H3K27ac+ nuclei were quantified using standard immunohistochemistry protocols. Antigen retrieval was performed in citrate buffer pH 6.0 for 20 minutes in a microwave. Slides were cooled in a 1x PBS solution for 2 minutes, before blocking in goat serum for 15 minutes (Dake, diluted 1:25). Primary anti-H3K27ac (ab4729, Abcam, Cambridge, MA; 1:1000) was added and incubated for 45 minutes at room temperature, followed by a PBS wash (5 minutes under agitation). The secondary antibody (goat antirabbit horseradish peroxidase secondary antibody, 1:250; Dako) was added and incubated for 45 minutes. Next, slides were washed in 1x PBS, before incubation with 3,3’-dianminobenzidine solution for 10 minutes, washed in distilled water and counterstained with hematoxylin. Finally, slides were dehydrated and mounted from xylene using distyrene plasticizer reagent. All slides were scanned using the NanoZoomer Digital Pathology (NDP.view2) system (Hamamatsu Corp, Bridgewater, NJ).
For H3K27ac quantification 3 high-power fields of the mucosa were taken from each slide, processed on ImageJ, and each field counted to assess positively stained (brown) and total nuclei (brown and purple). H3K27ac+ cells were expressed as a percentage of the total number of cells. Between 600 and 1800 nuclei were analyzed per field.
Immunofluorescence
Surgically resected intestinal FFPE tissue sections from patients with IBD were obtained from the Royal London Hospital Pathology Archive. Specimens with dysplasia or established malignancy were excluded. IBD FFPE tissues analyzed (n = 28) included 11 inactive IBD samples (CD, n = 8; UC, n = 3) and 17 active IBD samples (CD, n = 9; UC, n = 8). The cohort included 11 inactive and active patient matched pairs. Disease activity for each block was assessed using pathology reports. Cohort characteristics are shown in Supplementary Table 1.
For H3K27ac analysis antigen retrieval and primary antibody incubations were performed as described in the immunohistochemistry section. However, H3K27ac staining was visualized using a fluorescent secondary antibody (Ab150077, diluted 1:250 in PBS), and Vectashield HardSet (Vector Biosystems, Malvern, PA) mounting medium with DAPI was used for counterstaining. Imaging used ×40 magnification. H3K27ac+ nuclei were scored manually from 3 independent mucosal images. H3K27ac+ nuclei were expressed as a percentage of the total number of cells in that field. Typically, 200–400 nuclei were scored per field. Data were normally distributed and differences in the percentage of H3K27ac+ cells between active and inactive IBD tissue determined using a 2-tailed Student t test assuming equal variance. A subanalysis of the paired patient-matched inactive and active tissue was performed using a paired Student t test.
Inflammatory Bowel Disease Biopsy Culture
IBD biopsies from the same area of the intestine were collected during routine endoscopies at the Royal London Hospital. For each patient the disease activity, age, date of diagnosis, extension and behavior of disease (Montreal classification), clinical activity of disease (Harvey-Bradshaw Index for CD and Mayo score for UC), comorbidities, and all previous and current medications were recorded. Exclusion criteria included a diagnosis of infective colitis and prior use of VPA. In total 29 patients with IBD were recruited, but not all patients had sufficient biopsies taken to be included in all analyses. The numbers for each analysis are listed individually under the appropriate methods sections. The cohort characteristics as a whole are in Supplementary Table 2.
Biopsies were placed immediately into ice-cold RPMI 1640 medium and then washed in Hank’s balanced salt solution with dithiothreitol 0.01% to eliminate mucus and debris before culturing. Biopsies were culture with either control medium (RPMI 164 supplemented with fetal bovine serum [5%], L-glutamine [0.01%], penicillin/streptomycin [0.01%], and gentamycin [0.0025%]) or medium containing VPA (5 mM) at 37°C in a humidified incubator at 5% CO2 for 24 hours. The dose of VPA used corresponded to that used to reduce inflammatory cytokine production and treat in DSS-exposed mice.4 VPA was diluted from a stock 50-mM solution dissolved in RPMI medium.
Following culture, the supernatant was removed and stored at -80°C, and the biopsies stored either in RNALater at -80°C or FFPE embedded. As a positive control for treatment (n = 6 pairs), the percentage of H3K27ac cells was determined as in the immunofluorescence protocol. Difference between patient-matched biopsies treated with either VPA or a vehicle control (RPMI media) was determined using a paired Student t test.
RNA Extraction and Quantitative Polymerase Chain Reaction
IBD biopsies stored in RNALater were homogenized in 700 μL of Qiazol reagent and RNA extracted using the miRNeasy Kit (Qiagen, Hilden, Germany) and RNA quality determined using a bioanalyzer. RNA was converted to cDNA using the High Capacity RNA-to-cDNA Kit (Applied Biosystems, Wilmington, DE). The cDNAs were diluted 1:10 before incubation with commercial Taqman gene expression probes and Universal Mastermix (Applied Biosystems) on a 7500 System RealTime PCR cycler (Applied Biosystems). Resultant cycle threshold (Ct) values were exported and normalized to control genes RPLPO using the 2-ΔCt method.
All normalized data were log2 transformed before statistical analysis and normally distributed. IL6 and HDAC mRNA levels in the DSS mouse at Day 10 (n = 3) and Day 17 (n = 3) were compared with baseline at Day 0 (n = 3) using a 2-tailed Student t test assuming equal variance. Differences in IL6 mRNA and HDAC mRNA between inactive (n = 8) and active IBD biopsies (n = 11) were similarly analyzed.
Luminex and Enzyme-Linked Immunosorbent Assays
Supernatants from cultured IBD biopsies (n = 21 pairs) were assessed using a customized 6-plex Luminex panel according to manufacturer’s instructions (EMD Millipore, Billerica, MA). Changes in IL6 were further validated by enzyme-linked immunosorbent assays (DY206-05; n = 17 pairs). Data were not normally distributed and differences between VPA-treated biopsies and biopsies cultured with the vehicle control (RPMI media) were determined using a Wilcoxon matched-pairs signed rank test.
Supplementary Table 1.
Clinical Characteristics of the FFPE Cohort
| Patient ID | Age | Gender | CD/UC | Disease extent/Montreal | CRP | Endoscopic evidence active disease | Medications |
|---|---|---|---|---|---|---|---|
| 1 | 26 | F | CD | A2L3B2p | <5 | Yes | Steroids |
| 2 | 37 | F | CD | A?L3B3p | 9 | Yes | None |
| 3 | 26 | F | CD | A1L1B3 | 52 | Yes | Thiopurine |
| 4 | 38 | F | CD | A?L3B3p | 25 | Yes | Adalimumab and thiopurine |
| 5 | 19 | M | CD | A1L1B2 | 7 | Yes | Adalimumab |
| 9 | 18 | F | CD | A1L3B3 | <5 | Yes | Steroids |
| 10 | 23 | M | CD | A2L3B2p | 35 | Yes | Adalimumab and thiopurine |
| 11 | 36 | M | CD | A1L3B2p | Yes | Adalimumab and thiopurine | |
| 12 | 64 | F | CD | A3L3B3 | 53 | Yes | Adalimumab and steroids |
| 6 | 35 | F | UC | E3 | Yes | None | |
| 7 | 50 | F | UC | E3 | 37 | Yes | None |
| 8 | 34 | M | UC | E3 | <5 | Yes | Infliximab |
| 13 | 47 | M | UC | E1 | 34 | Yes | None |
| 14 | 14 | F | UC | E3 | 31 | Yes | Steroids |
| 15 | 28 | M | UC | E3 | 9 | Yes | Biologics and steroids |
| 16 | 16 | M | UC | E3 | 10 | Yes | Immunomodulator and steroids |
| 17 | 28 | F | UC | E3 | 32 | Yes | Tacrolimus |
CD, Crohn’s disease; CRP, C-reactive protein; FFPE, formalin fixed and paraffin embedded; UC, ulcerative colitis.
Supplementary Table 2.
Clinical Characteristics of Patients Used for Biopsy Cultures
| Sample | Patient | Age | Gender | CD/UC | Disease extent UC/Montreal | CRP | Endoscopic evidence active disease | Medications |
|---|---|---|---|---|---|---|---|---|
| cf01 | 1 | 44 | M | CD | A1L1L4B2 | 13 | Yes | Thiopurine and steroids |
| cf02 | 2 | 20 | F | CD | A1L2L4B1 | 6 | Yes | Adalimumab and thiopurine |
| cf03 | 3 | 41 | F | CD | A2L2B1p | No | None | |
| cf04 | 4 | 25 | M | CD | A2L2B1p | <5 | Yes | Thiopurine and steroids |
| cf05 | 5 | 55 | M | CD | A2L3B1P | <5 | Yes | Thiopurine |
| cf06 | 6 | 59 | F | UC | E2 | <5 | Yes | Steroids |
| cf07 | 7 | 59 | F | UC | E1 | <5 | No | None |
| cf08 | 8 | 58 | M | UC | E3 | <5 | Yes | None |
| cf09 | 9 | 68 | M | UC | E3 | <5 | No | None |
| cf10 | 10 | 41 | M | UC | E3 | <5 | Yes | None |
| cf11 | 11 | 35 | M | UC | E2 | 19 | Yes | None |
| cf12 | 12 | 37 | M | CD | A1L3B2 | <5 | Yes | None |
| cf13 | 13 | 17 | F | CD | A1L3B2 | <5 | Yes | Thiopurine |
| cf14 | 14 | 40 | F | UC | E3 | <5 | Yes | None |
| cf15 | 15 | 39 | F | UC | E3 | <5 | No | None |
| cf16 | 16 | 21 | M | UC | E3 | <5 | No | Thiopurine and steroids |
| cf17 | 17 | 21 | M | UC | E3 | <5 | Yes | Methotrexate and steroids |
| cf21 | 18 | 50 | F | UC | E2 | <5 | Yes | None |
| cf23 | 19 | 41 | M | UC | E3 | <5 | Yes | Thiopurine |
| cf24 | 20 | 51 | F | UC | E1 | <5 | Yes | None |
| cf25 | 21 | 20 | M | CD | 16 | Yes | Infliximab | |
| cf26 | 22 | 21 | M | UC | E3 | <5 | No | None |
| cf28 | 23 | 27 | F | CD | A2L3B2p | <5 | No | Steroids |
| cf32 | 24 | 59 | M | CD | A3L2B1 | <5 | No | None |
| cf33 | 25 | 40 | F | UC | E3 | <5 | No | None |
| cf34 | 26 | 29 | M | CD | A2L1 | <5 | Yes | Methotrexate |
| cf36 | 27 | 46 | F | CD | A1L3B2 | 7 | Yes | None |
| cf37 | 28 | 46 | F | CD | A1L3B2 | 7 | Yes | None |
| cf38 | 29 | 43 | F | CD | A2L3B2p | <5 | Yes | Adalimumab and methotrexate |
CD, Crohn’s disease; CRP, C-reactive protein; UC, ulcerative colitis.
References
- 1.Gareb B. Pharmaceutics. 2020;12:E539. doi: 10.3390/pharmaceutics12060539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Creyghton M. PNAS. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mokry M. Gastroenterology. 2014;146:1040–1047. doi: 10.1053/j.gastro.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Guo Y. Carcinogenesis. 2016;37:616–624. doi: 10.1093/carcin/bgw042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Felice C. Aliment Pharmacol Ther. 2015;41:26–38. doi: 10.1111/apt.13008. [DOI] [PubMed] [Google Scholar]
- 6.De Zoeten E.F. Gastroenterology. 2010;138:583–594. doi: 10.1053/j.gastro.2009.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glauben R. J Immunol. 2006;176:5015–5022. doi: 10.4049/jimmunol.176.8.5015. [DOI] [PubMed] [Google Scholar]
- 8.Glauben R. J Biol Chem. 2014;289:6142–6151. doi: 10.1074/jbc.M113.517599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dahllöf M.S.J. Interferon Cytokine Res. 2015;35:63–70. doi: 10.1089/jir.2014.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Available at: https://clinicalcenter.nih.gov/recruit/protocols/17_crohns.html Refer to study #17-I-0101. Accessed June 2020.
References
- 1.Mosli M.H. Gut. 2017;66:50–58. doi: 10.1136/gutjnl-2015-310393. [DOI] [PubMed] [Google Scholar]
- 2.Rigoni A. Cancer Res. 2015;75:3760–3770. doi: 10.1158/0008-5472.CAN-14-3767. [DOI] [PubMed] [Google Scholar]
- 3.Rigoni A. Inflamm Bowel Dis. 2017;24:136–148. doi: 10.1093/ibd/izx024. [DOI] [PubMed] [Google Scholar]
- 4.Glauben R. J Immunol. 2006;176:5015–5022. doi: 10.4049/jimmunol.176.8.5015. [DOI] [PubMed] [Google Scholar]