

Abstract
The Paramecium aurelia complex, a group of morphologically similar but sexually incompatible sibling species, is a unique example of the evolutionary plasticity of mating-type systems. Each species has two mating types, O (Odd) and E (Even). Although O and E types are homologous in all species, three different modes of determination and inheritance have been described: genetic determination by Mendelian alleles, stochastic developmental determination, and maternally inherited developmental determination. Previous work in three species of the latter kind has revealed the key roles of the E-specific transmembrane protein mtA and its highly specific transcription factor mtB: type O clones are produced by maternally inherited genome rearrangements that inactivate either mtA or mtB during development. Here we show, through transcriptome analyses in five additional species representing the three determination systems, that mtA expression specifies type E in all cases. We further show that the Mendelian system depends on functional and nonfunctional mtA alleles, and identify novel developmental rearrangements in mtA and mtB which now explain all cases of maternally inherited mating-type determination. Epistasis between these genes likely evolved from less specific interactions between paralogs in the P. aurelia common ancestor, after a whole-genome duplication, but the mtB gene was subsequently lost in three P. aurelia species which appear to have returned to an ancestral regulation mechanism. These results suggest a model accounting for evolutionary transitions between determination systems, and highlight the diversity of molecular solutions explored among sibling species to maintain an essential mating-type polymorphism in cell populations.
Keywords: self-incompatibility systems, programmed genome rearrangements, evolutionary genomics, ciliates
Significance
Sibling species of the Paramecium aurelia complex have long been known to use one of three different systems to determine the two complementary mating types (genetic, stochastic, or maternally inherited determination), but the mechanisms involved have been elucidated only for three species of the latter kind. This study shows that expression or nonexpression of the transmembrane protein mtA specifies mating types in all three systems, and identifies five different molecular switches that turn off mtA expression in one of the mating types. The evolutionary history of the genes involved illustrates the fascinating diversity of molecular solutions explored among sibling species to maintain an essential polymorphism in cell populations.
Introduction
Sex, defined by meiosis and fertilization and the alternation of haploid and diploid phases of the life cycle, is an ancient evolutionary innovation that facilitates the evolution of eukaryotic genomes through the shuffling of alleles within populations. Likely present in the last common ancestor of all eukaryotes, it is almost universally conserved across phyla. In contrast, self-incompatibility systems, such as male/female sexual dimorphism or mating types are thought to have evolved multiple times independently, possibly to limit inbreeding though other ideas have been proposed (Billiard et al. 2011; Perrin 2012; Heitman 2015; Hadjivasiliou and Pomiankowski 2016; Umen and Coelho 2019). The mechanisms of sex or mating-type determination, and the genes involved, are highly diverse and appear to be continuously diversifying, even among related species.
The Paramecium aurelia complex, a group of morphologically indistinguishable but sexually isolated sibling species, offers an interesting example of this evolutionary lability (Phadke and Zufall 2009). The complex was initially described as a single species containing 14 different “varieties” numbered from 1 to 14, each possessing two mating types called I and II for variety 1, III and IV for variety 2, etc.; but as gene flow appeared to be impossible between varieties, they were recognized as distinct species and received binomial names reflecting the variety numbers, Paramecium primaurelia through Paramecium quadecaurelia (reviewed in Sonneborn 1975a). Paramecium sonneborni was later admitted as a 15th member of the P. aurelia complex (Aufderheide et al. 1983; Przyboś et al. 2015), and the newly described Paramecium quindecaurelia is a 16th (Potekhin and Mayén-Estrada 2020). Despite being closely related to P. sonneborni, four other species were not included because of differences in nuclear morphology: Paramecium jenningsi (Diller and Earl 1958), Paramecium schewiakoffi (Fokin et al. 2004), and two groups of strains initially assigned to P. jenningsi but later shown to be distinct species (Sonneborn 1958; Przyboś and Tarcz 2016; Przyboś and Tarcz 2019). As argued in supplementary figure S1, Supplementary Material online, however, molecular phylogenies suggest that all 20 species should now be considered bona fide members of the P. aurelia complex.
As in all ciliates, nuclear dimorphism underlies the separation of germline and somatic functions (Cheng et al. 2020; see Sonneborn 1975b for this and other general features of nuclear organization and life cycle in P. aurelia species). Each cell contains diploid germline micronuclei (MICs) that only serve during sexual events, undergoing meiosis to produce gametic nuclei, and a highly polyploid somatic macronucleus (MAC) which is responsible for all gene expression but is not passed on to sexual progeny. In each generation, both types of nuclei develop from mitotic products of the diploid zygotic nucleus formed by karyogamy. Sexual genetic exchange occurs by conjugation, the reciprocal fertilization of cells of different mating types. When mildly starved, young vegetative clones become sexually reactive, a physiological state in which cells of different mating types will adhere to each other through antero-ventral cilia upon contact, resulting in the agglutination of many cells in large clumps within minutes after the mixing of mating types. After 60–90 min, individual pairs of cells form “holdfast unions,” that is, pairs firmly united at the antero-ventral surface after local resorption of cilia (Watanabe 1978). These pairs are then set loose from the clumps, and the two conjugants proceed with meiosis and cross-fertilization.
The two mating types of each P. aurelia species were renamed O (Odd) and E (Even) so that these would be homologous across the entire complex. Identification of homologous O and E types was based in part on interspecies agglutination reactions that can be observed between some species, often closely related ones (fig. 1), but only between type O of one species and type E of another (Sonneborn 1975a). In some cases, this results in the formation of holdfast unions and successful fertilization can even yield viable F1 hybrids, but these are usually sterile and thus cannot mediate gene flow between species. Identification of O and E types also relied on a fundamental asymmetry uncovered by genetic analyses. Mutagenesis screens only yielded recessive mutations restricting homozygotes to the expression of mating type O, but never to type E, indicating that O is a “default” state and that expression of E requires additional genetic functions (Butzel 1955; Taub 1963; Sonneborn 1977). In the most thoroughly studied species Paramecium tetraurelia, complementation tests defined three unlinked loci (mtA, mtB, and mtC) that are specifically required for E expression: mutant homozygotes at any of these loci always express type O (Byrne 1973). Similarly, a few natural isolates were found in several species to be genetically restricted to one type, and in all cases this was O (Butzel 1955; Sonneborn 1977). The fact that no mutant was ever found to be restricted to the expression of type E suggests that no gene is specifically required for type O expression. In other words, all of the genes involved in type O expression may also be involved in type E expression; the phenotype that would be expected for mutations in such genes is a complete lack of sexual reactivity. In species with no reported mating-type mutant or cross-reaction with other species, the asymmetrical phenomenon called “phenomic lag” (Sonneborn 1975b, 1977), in essence a dominance test in cells containing several macronuclei or macronuclear fragments determined for opposite mating types, was also used to define E as the “dominant” type.
Fig. 1.

Phylogeny of the 14 classical Paramecium aurelia species and interspecies mating reactions. (A) COII phylogeny. The phylogenetic tree of the mitochondrial COII gene identifies groups of closely related species among the 14 “classical” species (also identified by the original “variety” numbers after binomial names). Bootstrap support values are shown only when ≥50%; these nodes are confirmed in the multi-gene tree of sequenced species shown in supplementary figure S1, Supplementary Material online. Together with the mtA gene tree in figure 2 and the mtB gene tree in supplementary file S1, Supplementary Material online the COII tree clarifies the correct phylogenetic position of Paramecium septaurelia, which was erroneous in many previous publications due to wrong species assignments (see Extended Data figs. 6–8 in Singh et al. 2014). The strains used are: Paramecium primaurelia AZ9-3, Paramecium biaurelia V1-4, Paramecium triaurelia 325, Paramecium tetraurelia 51, Paramecium pentaurelia 87, Paramecium sexaurelia AZ8-4, Paramecium septaurelia 227, Paramecium octaurelia 138, Paramecium novaurelia TE, Paramecium decaurelia 223, Paramecium undecaurelia 219, Paramecium dodecaurelia 274, Paramecium tredecaurelia 321, Paramecium quadecaurelia N1A. Color shadings indicate the mechanism of mating-type determination in each species: orange, maternal inheritance; blue, stochastic determination; green, Mendelian. (B) Mating reactions between species. The 14 species are designated by numbers, as in (A). The diagonal (red) represents intraspecies reactions between O and E clones, where both agglutination and pair formation show the best efficiency. The table reports the efficiency of all observed interspecies agglutination reactions, always between opposite mating types: +++ (orange), ∼90–95% of cells in clumps; ++ (yellow), ∼40% of cells in clumps; + (green), only a few % of cells in clumps; +/– (blue), brief and weak contacts one pair at a time; ? (gray), dubious. Whenever observed, the formation of conjugating pairs is also quantified: C3, ∼90% of clumped cells; C2, 40–50% of clumped cells; C1, a few % of clumped cells. The data are from Sonneborn (1975a).
The recent identification of the mtA, mtB, and mtC genes in P. tetraurelia provided molecular insight into an E-specific biochemical pathway (reviewed in Orias et al. 2017). mtA was shown to encode a transmembrane protein that is specifically expressed in sexually reactive cells of type E and localizes to cilia of the antero-ventral surface, whereas mtB and mtC encode factors that are required for mtA transcription in E reactive cells (Singh et al. 2014). Importantly, RNA-seq of a null mtB mutant revealed that mtA is the only gene that requires mtB for transcription during sexual reactivity (Singh et al. 2014). These genes are conserved in the sibling species Paramecium septaurelia, where mtB-dependent mtA transcription also characterizes type E; cross-species transformation experiments provided evidence that the mtA protein is directly involved in the species-specific recognition of O reactive cells (Singh et al. 2014).
Despite the apparent conservation of a basic biochemical pathway with “on” and “off” states corresponding to E and O phenotypes, respectively, three different systems of mating-type determination have been described among P. aurelia species (reviewed in Sonneborn 1975b, 1977). The simplest of these systems, Mendelian determination, is represented by a single species, Paramecium tredecaurelia. In that species, mating types are genetically determined in the MIC (and in the derived MAC) by a pair of alleles at a single locus named mt, with a dominant allele Mt specifying type E and a recessive allele mt determining type O (Sonneborn 1975b). Mating types thus simply follow Mendelian inheritance after conjugation or after autogamy, an alternative sexual process through which single cells of P. aurelia species can fertilize themselves.
In all other species examined, mating types are not genetically determined in the germline MIC, which is totipotent; instead, mating type becomes irreversibly fixed as O or E in each new MAC as it develops, and thereafter remains unchanged during vegetative divisions of the resulting clone. Species with nongenetic mating-type determination can be further subdivided into two groups.
In seven of them (figs. 1A and 2), developmental determination of the new MAC for O or E is not random but controlled through the cytoplasm (Sonneborn 1947) by the parental MAC (Sonneborn 1954; Nanney 1957), still present at that stage in the form of transcriptionally active fragments, so that the zygotic MAC usually becomes determined for the same mating type. Because little cytoplasm is exchanged between the two mates during conjugation, this determination system results in the maternal inheritance of mating types: each F1 progeny acquires the mating type of its cytoplasmic parent in 90–99% of cases, depending on the species and strain (Taub 1963; Brygoo et al. 1980). Such transgenerational epigenetic inheritance is even stronger after autogamy, and sexual progeny become determined for the same mating type as the parental clone in the vast majority of cases. In P. tetraurelia strain d4-2 for instance, <1/1,000 postautogamous progeny switch to the other type (Brygoo 1977).
Fig. 2.

Conservation of mating-type genes in the P. aurelia complex. Check marks in the mtA, mtB, and mtC columns on the right indicate the presence of a potentially functional gene in the 16 species examined; red crosses indicate that the gene is absent or present as a pseudogene. The phylogenetic tree of mtA proteins, fully consistent with the multi-gene tree of sequenced species in supplementary figure S1, Supplementary Material online, is used to show the most parsimonious scenario for evolutionary changes in the three genes. The blue triangle in the P. aurelia common ancestor branch indicates the appearance of mtB and mtC and the insertion of a T in the mtA promoter motif; red triangles indicate the most likely branches where mtB was lost or pseudogenized and where the mtA promoter insertion was reversed; the orange triangles indicate the most likely branches where mtC was lost. The strains used for mtA protein alignment are the same as in figure 1A for the 14 classical species, strain 30995 (ATCC) for Paramecium sonneborni, strain M for Paramecium jenningsi (later renamed Paramecium trijenningsi, see supplementary fig. S1, Supplementary Material online; mating-type determination system is unclear), and strain MO3c4 of Paramecium multimicronucleatum. Color shadings indicate the mechanism of mating-type determination in each species: orange, maternal inheritance; blue, stochastic determination; green, Mendelian.
In six other species (figs. 1A and 2), there is no influence of the maternal MAC and each new MAC develops as O or E in a stochastic manner, whether the zygote is formed by conjugation or by autogamy; as in the similar stochastic system observed in some distantly related Tetrahymena species (Orias et al. 2017), the probability of each mating type can be influenced by environmental factors, such as temperature (Sonneborn 1977). The distribution of these three determination systems (Mendelian, maternal inheritance, stochastic) in the P. aurelia phylogenetic tree implies multiple changes from one system to another during the evolution of these species (figs. 1A and 2, and supplementary fig. S1, Supplementary Material online).
So far, mating-type determination mechanisms have been elucidated for three of the species showing maternal inheritance (Singh et al. 2014). They rely on the programmed genome rearrangements that occur during MAC development (reviewed in Bétermier and Duharcourt 2014; Rzeszutek et al. 2020), a process which probably evolved to remove Transposable Elements (TEs) from the expressed somatic genome (Coyne et al. 2012; Dubois et al. 2012), but here appears to have been co-opted to inactivate E-specific genes in type O clones (Singh et al. 2014). In P. aurelia species, rearrangements include the excision of numerous single-copy Internal Eliminated Sequences (IESs), which are thought to be the degenerate relics of ancient TE insertions and must be precisely excised from coding sequences to reconstitute functional genes in the MAC (Arnaiz et al. 2012; Bétermier and Duharcourt 2014; Sellis et al. 2020). IESs are invariably bounded by two TA dinucleotides that are required for cleavage by the endonuclease Pgm (Baudry et al. 2009); a single TA is left at the MAC junction after excision and repair by the Non Homologous End Joining pathway (Marmignon et al. 2014; Abello et al. 2020). Despite the frequent occurrence of a short degenerate consensus at IES ends (Arnaiz et al. 2012), the excision machinery appears to have little additional sequence specificity, and its targeting to the correct sites often depends on epigenetic marks (Lhuillier-Akakpo et al. 2014; Frapporti et al. 2019).
In P. tetraurelia, type O clones are produced by excision of the mtA promoter during MAC development, which prevents mtA expression during sexual reactivity (Singh et al. 2014). This excision event depends on the IES excision machinery and is regulated by scnRNAs (Singh et al. 2014), a class of piRNA-like small RNAs that is essential for elimination of TEs and of a fraction of IESs. Produced from the entire MIC genome during meiosis, scnRNAs are thought to mediate a genomic subtraction with the maternal MAC genome which selects the scnRNAs corresponding to MIC-specific sequences and licenses them to later target, in the developing MAC, the elimination of the same sequences (reviewed in Duharcourt et al. 2009; Chalker et al. 2013; Bétermier and Duharcourt 2014; Rzeszutek et al. 2020). The mechanism thus enables the zygotic MAC to reproduce the deletion of any germline sequence that had been eliminated in the MAC of the previous generation, explaining the maternal inheritance of mtA promoter excision or retention as well as that of many other alternative rearrangements unrelated to mating types (Meyer and Garnier 2002; Garnier et al. 2004).
This determination system was shown to be entirely conserved in the closely related Paramecium octaurelia (Singh et al. 2014). In P. septaurelia, a more divergent species which also shows maternal inheritance but groups with stochastic-determination species (figs. 1A and 2), mtA is always rearranged into a functional MAC form and type O clones are produced by inactivation of mtB through the excision of part of its coding sequence, which similarly prevents mtA expression (Singh et al. 2014). Thus similar switches have evolved independently in different genes to control the “on” and “off” states of the same pathway.
In this study, we have examined the conservation, expression and developmental rearrangements of mating-type genes in additional species of the P. aurelia complex. The results support the hypothesis that type E is specified by mtA expression in all species. Novel polymorphisms and rearrangements are uncovered in mtA and mtB, which together explain all cases of genetic and maternally inherited mating-type determination.
Materials and Methods
Paramecium Strains and Cultivation
Unless otherwise stated, all experiments were carried out with entirely homozygous strains from the strain collection of the Institute of Systematics and Evolution of Animals, Polish Academy of Sciences, Cracow, or from the RC CCM strain collection of Saint Petersburg State University, St Petersburg. Cultivation and autogamy were carried out at 27 °C as described in Beisson et al. (2010a, 2010b). Cells were grown in wheat grass powder (WGP, Pines International, USA) medium bacterized the day before with Klebsiella pneumoniae and supplemented with 0.8 mg/l β-sitosterol (Merck, Germany).
Mating-Type Tests
For species with genetic or maternally inherited determination, testers were prepared from cell lines of known mating types by refeeding ∼1,000 autogamous cells in tubes with 4 ml of 0.2× WGP medium bacterized with K. pneumoniae (standard medium) and incubating overnight at 27 °C. The next day, tubes were refed with 8 ml of standard medium and again incubated overnight at 27 °C. The following day, reactive cells concentrated near the top of the tube were collected (∼1.5 ml per tube), checked by mixing aliquots with reactive cells of the complementary type, and used in mating-type tests. Mass postautogamous progenies to be tested were made reactive in the same way. For species with stochastic determination or to test individual clones, single karyonides (out of autogamy or conjugation) were isolated in 250 μl of standard medium and incubated until starved. They were then refed with 250 μl and tested the next day.
DNA and RNA Extraction
Small-scale DNA samples were prepared from around 1,000 Paramecium cells in 1-ml cultures using the NucleoSpin Tissue kit (Macherey-Nagel, Germany) according to manufacturer’s instructions (protocol for blood, buffy coat or body fluids). Large-scale DNA samples were prepared from 800 ml of exponentially growing cultures at 1000–1500 cell/ml as described in (Baudry et al. 2009). RNA extraction was performed from 400-ml reactive cell cultures using TRI Reagent (MRC, USA).
RNA-Seq Analysis
Illumina libraries were prepared by the “mRNA stranded” protocol and sequenced (paired-end, 2× 80 nt) using the NextSeq 500/550 High Output Kit v2. Reads were mapped on Paramecium references (supplementary tables S1 and S2, Supplementary Material online) with Tophat (v2.1.1 –min-intron-length 15 –max-intron-length 100 –coverage-search –read-mismatches 1 –library-type fr-firststrand) (Kim et al. 2013). The number of fragments mapped on each gene were then normalized by the total number of fragments mapped on all genes (Million) and by the length of the coding sequence (kb) to express results in FPKMs, using Cufflinks (v2.2.1 –min-intron-length 15) (Trapnell et al. 2010).
PCR and Sequencing
PCR and agarose gel electrophoresis were carried out using standard procedures. A list of the PCR primers used in this study is given in supplementary table S3, Supplementary Material online.
Phylogeny of Paramecium Species
To reconstruct the species phylogeny shown in supplementary figure S1, Supplementary Material online, we searched for single-copy gene families in Paramecium genomes. We selected 19 species (15 Paramecium and four Tetrahymena species, to be used as outgroups) for which genome sequences and annotations are available (supplementary table S1, Supplementary Material online). All protein sequences were compared with each other with diamond (Buchfink et al. 2015). The diamond output was then processed with SiLiX (Miele et al. 2011) to cluster homologous proteins sharing at least 35% identity over 80% of their length. We selected all families with a single member in each of the 15 Paramecium species (N = 473 single-copy genes, presumably orthologous). Prior to alignment, we applied PREQUAL (Whelan et al. 2018) on protein sequences to mask nonhomologous segments. Protein sequences were aligned with ClustalOmega (Sievers and Higgins 2018); multiple alignments included a total of 305,634 sites. We then used IQTREE (Nguyen et al. 2015) (version 2.0.5 for Linux 64-bit built May 15, 2020) to calculate a species tree using each protein alignment as a partition (“LG” model with substitution rate heterogeneity among sites following a discrete gamma model with four parameters and invariable sites). Branch support values were calculated with SH-aLRT and ultrafast bootstrap with a value of 5,000.
iqtree2 -redo -nt 10 -m LG+I+G -bb 5000 -alrt 5000 -p partition.nexus
Links:
diamond: https://github.com/bbuchfink/diamond
SiLiX: http://lbbe.univ-lyon1.fr/SiLiX
PREQUAL: https://github.com/simonwhelan/prequal
ClustalOmega: http://www.clustal.org/omega/
IQ-TREE: http://www.iqtree.org/
Other Phylogenetic Analyses
Sequences were aligned using MAFFT at https://mafft.cbrc.jp/alignment/server/ (Katoh et al. 2019) and phylogenetic trees were computed using the Phylogeny.fr server (Dereeper et al. 2008), by the bootstrapping procedure for the trees shown in figures 1 and 2 (500 bootstraps), or by the approximate likelihood ratio test method PhyML 3.1/3.0 aLRT with default parameters for the trees shown in supplementary figures S2 and S3, Supplementary Material online and in supplementary files S1 and S2, Supplementary Material online.
Plasmids Used
The plasmid containing the mtB227 gene has been described (Singh et al. 2014). The mtA223 plasmid contains the entire gene as well as 329 bp of sequences upstream of the ATG translation initiation codon and 191 bp of sequences downstream of the TGA stop codon (complete sequence available upon request).
Microinjection
Paramecium cells were microinjected as described in Beisson et al. (2010c). Briefly, cells were microinjected in Dryl’s solution or Volvic water (France) containing 0.2% bovine serum albumin, under paraffin oil, whereas they were visualized with a phase-contrast inverted microscope.
Results
Conservation of Mating-Type Genes and Paralogs in P. aurelia Species and Outgroups
The search for mtA, mtB, and mtC orthologs and paralogs was facilitated by high synteny conservation in the MAC genomes of the 13 P. aurelia species sequenced so far, and to a lesser extent in the two outgroup species Paramecium multimicronucleatum and Paramecium caudatum (supplementary table S1, Supplementary Material online) (Aury et al. 2006; McGrath, Gout, Johri, et al. 2014; McGrath, Gout, Doak, et al. 2014; Gout et al. 2019; Sellis et al. 2020). PCR primers based on multiple alignments further allowed us to sequence orthologous genes in the three unsequenced species Paramecium undecaurelia, Paramecium triaurelia, and Paramecium septaurelia. In P. tetraurelia, mtA and mtB both belong to small multigene families, albeit with different evolutionary histories. Four genes (mtAL1–mtAL4) encode transmembrane proteins that are structurally similar to mtA (and to the MTA and MTB proteins involved in conjugation in Tetrahymena, Cervantes et al. 2013; Orias et al. 2017): five C-terminal transmembrane helices, a couple of cysteine-rich, furin-like repeats, a large N-terminal domain exposed outside the membrane), though they share only limited sequence similarity. None of these five genes was found to retain any duplicate (ohnolog) from the two successive Whole-Genome Duplications (WGDs) thought to have occurred in a common ancestor of P. aurelia species, after its divergence from outgroup species (Aury et al. 2006; McGrath, Gout, Johri, et al. 2014; McGrath, Gout, Doak, et al. 2014; Yi et al. 2014; Johri et al. 2017; Gout et al. 2019; Sellis et al. 2020). All five are conserved in all examined P. aurelia species as well as in the pre-WGD outgroups (supplementary fig. S2, Supplementary Material online), with the possible exceptions of mtAL3 in P. quadecaurelia, mtAL1 and mtAL2 in P. jenningsi, and mtA in P. caudatum, which could not be found in the current MAC genome assemblies. The mtA-like gene family thus probably results from very ancient segmental duplications.
In contrast, at least three of the five mtB-like genes in P. tetraurelia are more recent WGD duplicates. Although mtB is missing its ohnolog from the last WGD, the two most similar genes, mtBL1 and mtBL2, were identified as its ohnologs from the previous WGD. Consistently, only a single homolog of this group is present in pre-WGD outgroup species (supplementary fig. S3, Supplementary Material online). Thus mtB itself only occurs in the P. aurelia complex, and it is present in all but threee of the 16 species examined (fig. 2): in P. jenningsi and P. tredecaurelia it has decayed into a nonfunctional pseudogene (supplementary file S1, Supplementary Material online), whereas in P. sexaurelia it is completely absent from the homologous locus. Given the phylogenetic tree of P. aurelia species (supplementary fig. S1, Supplementary Material online), the most parsimonious hypothesis is that mtB was lost on three independent occasions, after each speciation event (fig. 2). The alternative hypothesis that mtB did not exist in the last common ancestor of the Paramecium sexaurelia/Paramecium sonneborni/P. jenningsi subclade but only appeared in the other subclade, and was later horizontally transferred to P. sonneborni, is not consistent with 1) the hypothesis that the last two WGDs preceded any P. aurelia speciation, 2) the presence of a recognizable mtB pseudogene in P. jenningsi, and 3) the phylogeny of mtB genes, where the P. sonneborni gene and P. jenningsi pseudogene form a sister clade to other P. aurelia species (supplementary file S1, Supplementary Material online), like the rest of their genomes (supplementary fig. S1, Supplementary Material online).
Like P. tetraurelia, all sequenced species have lost the mtB ohnolog from the last WGD. They all have two homologs of the mtBL1/2 type, except for P. jenningsi where none could be found (supplementary fig. S3, Supplementary Material online). The two more distant mtB-like members, mtBL3 and mtBL4, have no identified ohnolog in P. tetraurelia; each has one or two homologs in other P. aurelia species. In pre-WGD outgroups, a single homolog branches off before the mtBL3/mtBL4 divergence (supplementary fig. S3, Supplementary Material online), suggesting these two genes may be ohnologs from the previous WGD that escaped detection in the P. tetraurelia analysis.
Like mtB, mtC is found only in P. aurelia species, but it does not have any close homolog or reported ohnolog. It is missing in P. sexaurelia and has become a pseudogene in P. jenningsi (supplementary file S2, Supplementary Material online), but is present in all other species including P. tredecaurelia (fig. 2). Phylogenetic analyses place the P. sonneborni mtC and P. jenningsi pseudogene in a sister clade to all other P. aurelia species (supplementary file S2, Supplementary Material online), indicating that a functional gene was present in the last common ancestor of the P. sexaurelia/P. sonneborni/P. jenningsi subclade and was then lost twice independently.
Expression of Mating-Type Genes and Paralogs in P. tetraurelia
Previous RNA-Seq studies have determined relative expression levels of P. tetraurelia genes at different stages of the life cycle (Arnaiz et al. 2017; Singh et al. 2014). Most of the mtA-like genes (mtA and mtAL1/2/3) are silent in exponentially growing vegetative cells and are turned on specifically in sexually reactive cells, at similar and relatively high levels (supplementary fig. S4, Supplementary Material online). The only difference is that mtA cannot be expressed in mating type O cells, where its promoter is excised, whereas mtAL1/2/3 are expressed in both mating types. However, unlike mtA the transcription of mtAL1/2/3 requires neither mtB nor mtC, implying a different regulation mechanism. The last mtA-like gene, mtAL4, is unique in that it is not expressed in sexually reactive cells, but at an early autogamy stage which best coincides with meiosis (supplementary fig. S4, Supplementary Material online).
As previously reported, mtB and mtC are expressed throughout the life cycle, though at very low levels that make read counts quite variable (supplementary figs. S5 and S6, Supplementary Material online). The four mtB-like genes are expressed at much higher levels, and show one of two distinct profiles: expression of mtBL1 and mtBL3 is limited to meiosis, like that of mtAL4, whereas mtBL2 and mtBL4 resemble mtAL1/2/3 in being expressed almost exclusively in sexually reactive cells of both mating types (supplementary fig. S5, Supplementary Material online).
mtA Expression Specifies Mating Type E in Other P. aurelia Species
To test whether mating type E is associated with mtA expression in other species, we carried out RNA-Seq analyses of sexually reactive cells of both mating types for five additional species, representing the three determination systems (supplementary table S2, Supplementary Material online). For the “Mendelian” species P. tredecaurelia, we used strains 209 (type O) and 321 (type E). Random-determination species included Paramecium pentaurelia (strain 87), P. sonneborni (ATCC 30995), and P. undecaurelia (strain 219). In all four cases, mtA was expressed in sexually reactive cells of one mating type, but not of the other (supplementary figs. S7 and S8, Supplementary Material online). We also analyzed two different strains of the maternal-inheritance species P. sexaurelia (AZ8-4 and 159), and in each of them we found that mtA is transcribed in both mating types. However, in one of the mating types (the same in both strains, as determined by cross-testing) the transcripts do not include the last 372 bp of the coding sequence and therefore cannot produce the wild-type mtA protein (supplementary fig. S7, Supplementary Material online). We also quantified RNA-Seq reads mapping to mtA-like genes mtAL1–3 in these species, except for P. undecaurelia where their sequence is unknown. As in P. tetraurelia, these genes are expressed in sexually reactive cells, but no significant difference in relative expression levels is observed between the two mating types (supplementary fig. S7, Supplementary Material online); the mapping of reads also did not reveal any difference in mRNA structure.
Thus in all five tested species, only one of the two mating types expresses full-length mtA mRNAs during sexual reactivity. That type is known to be E in P. tredecaurelia, where strain 321 is homozygous for the E-determining allele Mt (Sonneborn 1975b), and it can also be formally identified as E in P. pentaurelia, where analyzed clones could be typed using cross-reactions with strain P of P. primaurelia, a natural mutant genetically restricted to type O (Butzel 1955). Although genetically fixed tester strains were not available for the other three species, the mtA-expressing type can be identified as E in all cases, based on the more general definition of mating type E as the type requiring the expression of additional genes.
mtA Alleles Determine Mating Types in P. tredecaurelia
We then looked for genomic polymorphisms that may explain mating-type determination in different species by resequencing the mtA, mtB, and mtC genes in MACs of both mating types. Starting with the search for the O and E mt alleles in P. tredecaurelia, we found that the mtA alleles of strains 209 and 321 are 100% identical over the coding sequence, introns, and downstream intergenic region, and only differ by three point mutations in the upstream intergenic region: a 1-bp deletion located 69 bp upstream of the ATG initiation codon, and two substitutions 417 and 515 bp upstream. The point deletion falls in a 10-bp motif (GGTTGAAAAA) that is highly conserved among P. aurelia species (supplementary fig. S9, Supplementary Material online), within the ∼200-bp segment that was shown to contain essential elements of the mtA promoter in P. tetraurelia, and could thus conceivably affect mtA transcription. Resequencing of the mtB genomic region showed that strains 209 and 321 have exactly the same allele of the mtB pseudogene, which therefore cannot be the mt locus. As for mtC, the 209 and 321 alleles differ by a single substitution resulting in an Asp/His amino acid change outside of the protein’s zinc finger, which may or may not affect function.
To test whether the mtA or mtC genetic differences might determine mating types in P. tredecaurelia, we analyzed the cosegregation of 209 and 321 alleles with mating types in a cross between these two strains. After conjugation, F1 clones were confirmed to be true heterozygotes by testing their mating types (expected to be E for both heterozygous exconjugants from a single pair) and resequencing the mtA promoter, and then allowed to undergo autogamy. This self-fertilization process always results in entirely homozygous F2 progeny clones, which have a 50% chance to inherit either of the parental F1 alleles. As previously reported for P. tredecaurelia crosses (Sonneborn 1975b; Przyboś et al. 2013; Przyboś and Tarcz 2018), F2 lethality was very high: out of 180 independent F2 clones, only 13 survived and only one was healthy enough to be tested for mating types and genotyped. Although such F2 lethality precluded segregation analysis, the healthy clone, which was type E, was homozygous for the 321 allele of mtA and for the 209 allele of mtC, indicating that the latter does not prevent mating type E expression. That clone was back-crossed to the 209 strain and the procedure was repeated, resulting in a much lower lethality rate among second-round F2s. A total of 23 F2 clones could be tested for mating types and genotyped for the mtA allele: 14 were E and had the 321 allele, and nine were O and had the 209 allele. The perfect cosegregation demonstrates that the mt locus is mtA, or closely linked to it.
Thus the 209 allele of mtA, which is not expressed during sexual reactivity, appears to be intrinsically nonfunctional in P. tredecaurelia. It may seem surprising that this is the allele in which the ancestral 10-bp promoter motif is conserved, whereas the functional 321 allele shows the derived 9-bp variant GGTGAAAAA. However, this is not inconsistent with the possibility that the 10-bp ancestral motif is the mtB binding site in other species, since mtA transcription obviously does not depend on mtB in P. tredecaurelia. Furthermore, the same 9-bp variant (GGTGAAAAA) is also observed in P. sexaurelia and P. jenningsi, the other two species that lack the mtB gene. Like the loss of mtB, the point deletion in the mtA promoter motif appears to have occurred three times independently in these species, after their speciation (fig. 2). This suggests that the 9-bp variant may be required, in the absence of the mtB gene, for mtA transcription to be driven by some other factor. Interestingly, similar motifs are found in the promoter regions of mtAL1, mtAL2, and mtAL3 (supplementary figs. S10–S12, Supplementary Material online), which in P. tetraurelia are expressed during sexual reactivity of both mating types in an mtB-independent manner, but not in that of mtAL4 (supplementary fig. S13, Supplementary Material online), which has a distinct expression profile. These motifs are all found at similar distances from the ATG initiation codons and share the common core GTGAAA, which can be extended to the degenerate consensus RGTGAAAWWWG (supplementary fig. S14, Supplementary Material online). This raises the possibility that mtA expression may be regulated, in the three species lacking mtB, by the same factor as mtAL1–3.
The above observations suggest that the only reason why the 209 allele of mtA is not expressed in P. tredecaurelia is the lack of a functional mtB. The previous demonstration (Singh et al. 2014) that the mtA promoter of one species (P. septaurelia) can be activated by the mtB factor of another (P. tetraurelia) prompted us to directly test this point by microinjecting the functional mtB gene from P. septaurelia strain 227 into the MAC of P. tredecaurelia strain 209 cells. Transformed clones were identified by a plasmid-specific PCR, and all clones were mildly starved to induce sexual reactivity. Mating type tests revealed that three of four transformed clones expressed mating type E (one was not reactive and could not be tested), whereas nontransformed clones that could be tested remained mating type O (fig. 3). Thus strain 209 is indeed restricted to mating type O by the loss of mtB, and mating type E expression appears to be rescued in strain 321 by the point deletion in the mtA promoter.
Fig. 3.

Transformation of Paramecium tredecaurelia strain 209 with mtB227 results in mating type E expression. Lanes 1–8: eight type O cells from strain 209 were microinjected with a plasmid containing the mtB227 gene from Paramecium septaurelia. Successfully transformed clones were identified by a plasmid-specific PCR and tested for mating types with type O (strain 209) and type E (strain 321) testers. The number of + signs below the lanes indicates the strength of the agglutination reaction; – sign, no reaction. Clones 5 and 7 were not sexually reactive and their mating types could therefore not be determined. Lane c1, noninjected control clone (strain 209); lane c2, positive PCR control on the mtB227 plasmid.
Paramecium tredecaurelia is a rare species and for a long time only strains 209 (France, 1953) and 321 (Mexico, 1956) were known, raising the question of the generality of the genetic mating-type determinant in that species. We resequenced the mtA promoter in three recently described natural isolates from different continents (supplementary fig. S9, Supplementary Material online); all three alleles differ from both 209 and 321 by at least one substitution. Strain WO from southern Ural (Potekhin A and Lanzoni O, unpublished data) conjugates with strain 321 but not with strain 209, indicating it is mating type O, and shows the ancestral 10-bp motif. Strains ET-3a from Ethiopia (Przyboś and Tarcz 2018) and TaB from Thailand (Przyboś et al. 2013) (Note that P. tredecaurelia mating types were inverted in that study due to the swapping of 209 and 321 testers.) have the 9-bp derived variant and 10-bp ancestral motif, respectively. Although these strains could not be retested with 209 and 321, they could conjugate together and are therefore of opposite mating types. Thus at least two different P. tredecaurelia alleles show the 9-bp derived variant, and the available evidence suggests that mating type determination by this genetic polymorphism may be general in that species.
Paramecium decaurelia and P. dodecaurelia Share the P. tetraurelia/octaurelia Mechanism
Among species with maternal inheritance of mating types, P. tetraurelia (strain 51) and P. octaurelia (strains 138 and GFg-1) were previously shown to share a common determination mechanism, based on excision or retention of the mtA promoter. We first looked at the two closest species P. decaurelia (strain 223) and P. dodecaurelia (strains 274 and J4), which form a specific subclade with the former two (fig. 2). By resequencing the mtA promoter region in clones of both mating types, we found that in both species the mtA promoter was similarly retained in type E MACs but excised in type O MACs (as determined by cross-reactions with P. tetraurelia testers), with precisely the same excision boundaries (supplementary fig. S9, Supplementary Material online). To test whether this deletion actually determines type O, we microinjected the promoter-containing, E form of the MAC mtA gene from P. decaurelia strain 223 into the MAC of O cells of the same strain. Transformed clones were identified by a plasmid-specific PCR, and all clones were mildly starved to induce sexual reactivity. Mating type tests revealed that three of five transformed clones expressed mating type E (two were not reactive and could not be tested), whereas nontransformed clones remained type O (fig. 4). Thus, as in P. tetraurelia, excision of the mtA promoter is the only feature in the MAC genome of O cells that prevents them to express type E specificity. We conclude that this determination system was inherited from the common ancestor of the four species.
Fig. 4.

Transformation of type O cells from Paramecium decaurelia strain 223 with full-length mtA223 results in mating type E expression. Lanes 1–7: seven type O cells from strain 223 were microinjected with a plasmid containing the full-length mtA223 gene. Successfully transformed clones were identified by a plasmid-specific PCR and tested for mating types with type O and type E testers from strain 223. The number of + signs below the lanes indicates the strength of the agglutination reaction; – sign, no reaction. Clones 5 and 7 were not sexually reactive and their mating types could therefore not be determined. Lane c1, noninjected control clone (strain 223); lane c2, positive PCR control on the mtA223 plasmid.
Paramecium biaurelia: An mtB-Based Mechanism, Distinct from That of P. septaurelia
We then turned to P. biaurelia (strain Rieff), a maternal-inheritance species not closely related to any other (fig. 2 and supplementary fig. S1, Supplementary Material online). Resequencing the mtA and mtC MAC genes did not reveal any difference between the two mating types, but the MAC mtB gene was found to be rearranged differently: one mating type shows a 168-bp deletion in the coding sequence, whereas the other has a full-length, functional gene. The latter can be identified as E, according to the general definition of mating type E as the type requiring the expression of additional genes. Like the previously documented mtB deletion in type O of P. septaurelia, the deletion occurs between two TA dinucleotides flanked by good matches to the IES end consensus sequence (Arnaiz et al. 2012; Swart et al. 2014), although it affects a different part of the gene (fig. 5A). To complement the mtB deficiency, we microinjected the functional mtB gene from P. septaurelia into the MACs of type O cells. Transformed clones were identified by PCR, and mating type tests revealed that they expressed type E, whereas nontransformed clones remained type O (fig. 5B). Thus the presence or absence of a functional mtB gene in the MAC is what determines mating types in that species, as in P. septaurelia; here again, successful mtB complementation excludes the possibility that any additional feature of the MACs of O cells prevents type E expression. However, the excised mtB segments are distinct in these two species and appear to be determined in each ortholog solely by the position of sites that happen to be possible substrates for the IES excision machinery, indicating that the two systems evolved independently.
Fig. 5.
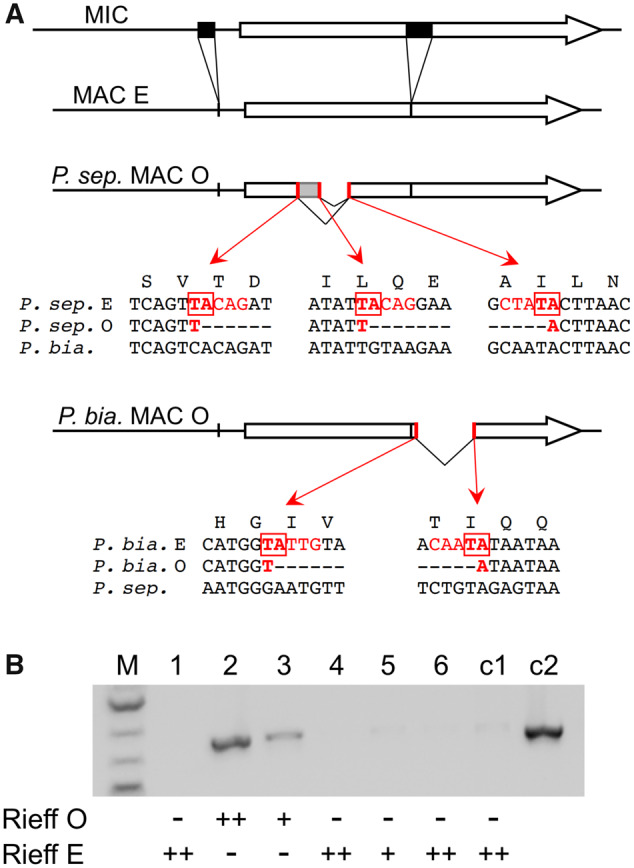
mtB rearrangements determine mating types in Paramecium biaurelia. (A) Structure of the mtB gene in the MIC and MAC genomes of Paramecium septaurelia and P. biaurelia. In both species the MIC gene (top drawing) contains two IESs (black boxes) that are precisely excised in the MACs of type E clones (second drawing). In the MACs of P. septaurelia (strain 227) type O clones (third drawing), two alternative deletions further remove parts of the coding sequence. Red bars indicate the two left boundaries and the common right boundary. The local sequences are given below, together with the homologous sequences from P. biaurelia (strain Rieff); the IES-like end sequences are shown in red, and the TAs used for recombination are boxed (data from Singh et al. 2014). In the MACs of P. biaurelia (strain Rieff) type O clones (fourth drawing), a different part of the coding sequence is excised, again between IES-like boundaries that are conserved in other strains (supplementary figs. S15 and S16, Supplementary Material online). (B) Transformation of type O cells from P. biaurelia strain Rieff with mtB227 results in mating type E expression. Lanes 1–6: six type O cells were microinjected with a plasmid containing the mtB227 gene from P. septaurelia. Successfully transformed clones were identified by a plasmid-specific PCR and tested for mating types with type O and type E testers from strain Rieff. The number of + signs below the lanes indicates the strength of the agglutination reaction; – sign, no reaction. Lane c1, noninjected control clone; lane c2, positive PCR control on the mtB227 plasmid.
To test whether the same alternative rearrangement occurs in other strains of P. biaurelia, individual cells from 10 different natural isolates were grown and taken through autogamy. Because mating types are maternally inherited, most of the postautogamous progenies should be determined for the same mating type as their parental clone. Mass progenies (∼1,000 cells) were grown, made sexually reactive and tested against each other to identify opposite mating types, and the structure of the MAC mtB gene was tested by PCR. Four cell populations showed the deleted form and were of the same mating type (O), and two had the full-length form and were the other type (E). The remaining four populations contained both forms and showed selfing (S), that is, intrapopulation conjugation (supplementary fig. S15A, Supplementary Material online). Sequencing of the PCR product from each strain revealed low allelic diversity (three SNPs defining at least four different alleles), and showed that the coding-sequence deletion had the same boundaries in all cases (supplementary fig. S15B, Supplementary Material online). Further support was provided by the mapping of whole-genome sequencing reads from nine additional P. biaurelia strains (Johri et al. 2017): four of them showed precisely the same deletion, suggesting these genomes were sequenced from type O clones (supplementary fig. S16, Supplementary Material online). We conclude that this excision event is widely used among P. biaurelia strains to determine mating type O.
Paramecium sexaurelia: A Novel mtA-Based Mechanism Not Affecting Transcription
The last maternal-inheritance species, P. sexaurelia, does not contain the mtB and mtC genes. We probed the structure of the MAC mtA gene in two different strains, AZ8-4 and 159, which happened to be of opposite mating types in our stocks. Consistent with the transcriptome data indicating that mtA is transcribed in both mating types, its promoter was found to be retained in both strains. We then checked the maternal inheritance of mating types in a cross between these strains. After conjugation, the two F1 exconjugants were checked for heterozygosity by resequencing SNPs, and the maternal (cytoplasmic) origin of each was ascertained by typing a mitochondrial polymorphism. F1s were then tested for mating types; each of them was indeed of the same mating type as its maternal parent. F1 samples were used to test the structure of the complete mtA gene by PCR, which revealed a deletion at the 3′ end of the gene in one of the mating types (fig. 6A).
Fig. 6.
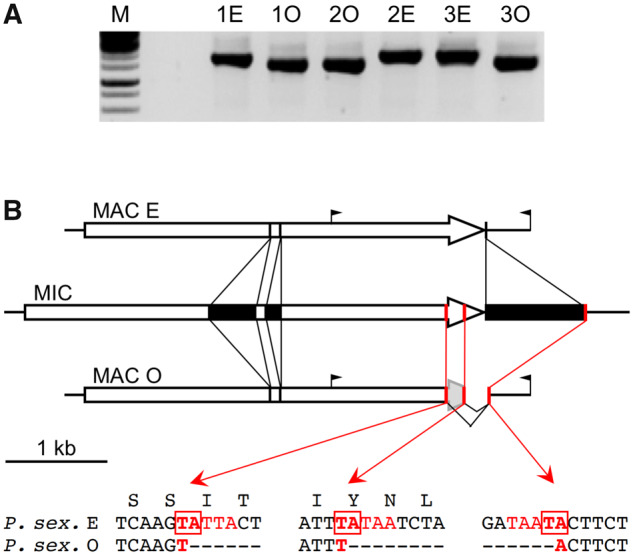
Novel mtA rearrangements determine mating types in Paramecium sexaurelia. (A) The 3′ end of mtA differs between mating types. PCR amplification of the 3′ end of mtA on three pairs of heterozygous F1s (1–3) from the AZ8-4 × 159 cross; in each pair the type E F1 derives from the AZ8-4 parent, and the type O F1 from the 159 parent. The faster-migrating band in type O F1s is in fact a poorly resolved doublet. The positions of PCR primers are indicated in (B). (B) Structure of the mtA gene in the MIC and MAC genomes. The MIC mtA gene in P. sexaurelia strain AZ8-4 (middle drawing) has two IESs (black boxes) inserted within the coding sequence, and a third one 23 bp downstream of the stop codon. All three IESs are precisely excised in type E MACs, resulting in a functional gene (top drawing). The black flags indicate the positions of the PCR primers used in (A). In type O MACs, the third IES is excised using alternative boundaries (red bars) located outside of the correct ones, resulting in the deletion of the 3′ end of the coding sequence. The same two alternative left boundaries and common right boundary are used in strain 159 and other P. sexaurelia strains (supplementary fig. S17, Supplementary Material online); local sequences around the TAs used for recombination are indicated below.
To determine whether this deletion correlates with mating types in each of the homozygous parental strains, we first sought to establish clones of both mating types for each of them, by screening large-scale mass postautogamous progenies for rare mating-type revertants that would form pairs with nonrevertants upon sexual reactivity. Pairs were isolated and allowed to proceed with conjugation; the two F1 exconjugants were then grown separately and tested for mating types. Revertants can be expected to transmit the new mating type to their cytoplasmic progeny, and in 11 out of 15 F1 pairs (both strains) the two homozygous F1s were indeed of opposite mating types. One such pair was then tested by PCR for each strain: for both AZ8-4 and 159, one mating type (the same in both strains, as determined by cross-testing) was associated with a deletion at the 3′ end of mtA, but not the other.
Sequencing of the PCR products showed that the deletions were precisely the same in the two strains, and revealed their developmental origin. In one of the mating types, a 939-bp IES located 23 bp downstream of the mtA stop codon appears to be excised using alternative excision boundaries further apart in the flanking sequences. On the 5′ side these alternative boundaries are located within the coding sequence, 197 and 372 bp upstream of the stop codon (fig. 6B). As a result, the end of the coding sequence is absent from the MAC genome, explaining the defective mtA transcripts observed in that mating type. If translated, these transcripts would produce a protein lacking the last two transmembrane helices, which may be essential for function given their complete conservation in mtA and mtA-like proteins; that mating type is therefore most likely O. The O-specific alternative excision boundaries all coincide with TA dinucleotides flanked by good matches to the IES end consensus (Arnaiz et al. 2012; Swart et al. 2014). These TAs are conserved in the recently sequenced MAC genomes of additional P. sexaurelia strains (Johri et al. 2017) and appear to be used to remove the 3′ end of the mtA gene in seven out of 13 of these genomes (supplementary fig. S17, Supplementary Material online), presumably reflecting the mating type of the particular clone sequenced for each strain. Thus, this alternative rearrangement appears to be widely used among P. sexaurelia strains to determine type O by preventing production of the full-length mtA protein.
Discussion
A previous study of P. tetraurelia and P. septaurelia concluded that the key difference between mating types is the expression of the transmembrane protein mtA in type E, but not type O, reactive cells. The transcriptome analyses presented in this work show that in five additional P. aurelia species representing the three determination systems, full-length mtA mRNAs are produced in only one of the mating types, which by definition must be type E. In four of these species (P. tredecaurelia, P. pentaurelia, P. sonneborni, P. undecaurelia), mtA transcription appears to be abolished in O cells, whereas in P. sexaurelia, one of the earliest diverging species, mtA is transcribed in both mating types but full-length transcripts are only observed in type E. Thus expression of the full-length mtA protein may well be what specifies mating type E in all P. aurelia species. That ciliary protein was previously shown to be directly involved in the recognition of conspecific O cells, but the nature of the putative mtA receptor remains unknown; the available evidence suggests that it may be produced in both mating types, but made capable of binding mtA only in O cells (i.e., in the absence of endogenous mtA) through posttranslational processes (Singh et al. 2014; Orias et al. 2017).
A functional mtB gene was shown to be required for mtA transcription during sexual reactivity in P. tetraurelia and P. septaurelia (Singh et al. 2014). Although this point was not directly tested here in any other species, we showed that the full-length mtB gene is required in P. biaurelia for expression of mating type E in sexually reactive cells. Because mating type E is specified by mtA expression in all seven of the other species tested, this strongly suggests that mtB is required for mtA transcription in P. biaurelia as well. Similar indirect evidence is provided in P. primaurelia by the finding that strain P, a natural mutant unable to express type E (Butzel 1955), has a nonfunctional mtB allele: a mutation of the 3′ splice site of the first intron is predicted to prevent splicing and cause a frameshift (supplementary file S1, Supplementary Material online), suggesting that mtA transcription may also depend on mtB in that species. However, this cannot be true of all P. aurelia species, since a functional mtB gene is missing in three of them: P. tredecaurelia, P. jenningsi, and P. sexaurelia. The phylogenetic tree suggests mtB was present in the last common ancestor of all P. aurelia species, and then was lost three times independently (fig. 2). Consistent with this conclusion, mtB owes its evolutionary origin to the duplication of an mtB-like gene during a WGD thought to have occurred in the P. aurelia ancestor, before its radiation into extant species but after its divergence from outgroups P. caudatum and P. multimicronucleatum (Aury et al. 2006; McGrath, Gout, Johri, et al. 2014; McGrath, Gout, Doak, et al. 2014; Yi et al. 2014; Johri et al. 2017; Gout et al. 2019; Sellis et al. 2020). Thus the mtA orthologs of outgroup species, like the three P. aurelia species that later lost the mtB function, must be regulated by some other transcription factor.
After its emergence in the P. aurelia ancestor, mtB appears to have taken up the exclusive role of driving transcription from the mtA promoter. In P. tetraurelia at least, transcriptome analyses showed that mtA is the only gene whose transcription is affected by the null mutation mtBO (Singh et al. 2014). This may be true in all species with a functional mtB, as suggested by the fact that P. septaurelia and P. biaurelia use mtB inactivation to determine type O (mating types are not known to differ in any other phenotype). Transcription of the mtA-like genes mtAL1–3, which are expressed during sexual reactivity in both mating types, would then be driven by another factor in all P. aurelia species. A candidate regulatory motif, GTGAAA, was found to be conserved upstream of the coding sequences in mtAL1–3, but not in mtAL4 which has a distinct expression profile.
At a similar distance upstream of the mtA coding sequence, a similar motif, GTTGAAA, appears to result from the insertion of a T in most P. aurelia species. Strikingly, the only exceptions are the three species that have lost the mtB function, suggesting that GTTGAAA may be the mtB binding site (supplementary fig. S14, Supplementary Material online). These three species show the GTGAAA motif instead, though this does not appear to reflect continuous conservation of the ancestral motif found in outgroup species (supplementary fig. S14, Supplementary Material online). Rather, the most parsimonious hypothesis is here again that the T insertion occurred in the P. aurelia common ancestor, and was later reversed independently in the three species. Among them, P. tredecaurelia is a special case in that both variants are found, depending on the mtA allele in each strain; our results indicate that GTGAAA alleles determine mating type E, and GTTGAAA alleles type O. We further showed that transforming the type O strain 209 with a functional mtB gene from another species results in type E expression, consistent with the idea that GTTGAAA may be the mtB binding site.
Together these results suggest a possible evolutionary scenario in which an ancestral mtB-like factor was responsible for the co-induction of mtA and mtAL1–3 during sexual reactivity, through the binding of a common GTGAAA motif in their promoters. This P. aurelia ancestor could have produced type O clones through genetic or developmental inactivation of the mtA gene; like all extant species, it was probably equipped with all of the factors known to be involved in IES excision and in the scnRNA-based mechanism for epigenetic inheritance of alternative rearrangements (Chalker et al. 2013; Bétermier and Duharcourt 2014; Rzeszutek et al. 2020), but it is impossible to guess which determination system it may have used. After the first WGD, one of the mtB-like duplicates became free to evolve randomly into a variant recognizing GTTGAAA, that is, mtB. This would allow a second random mutation to modify the motif in the mtA promoter and enable its activation by the mtB variant, in effect disconnecting the regulation of mtA from that of mtAL1–3. That step may also have involved the appearance of the P. aurelia-specific mtC factor, although its essential role in mtA transcription was only demonstrated in P. tetraurelia. What selective pressures (if any) may have led to the fixation of these mutations in the population is not obvious, but the high specificity of the new mtB-mtA interaction would make mtB a second possible target for developmental rearrangements that prevent mtA expression in type O clones, as observed in P. septaurelia and P. biaurelia.
In this scenario, reversion of the new GTTGAAA motif in the mtA promoter to the original GTGAAA would be tolerated, because this would still allow mtA transcription to be properly induced by the other mtB-like duplicate (or after the second WGD, by at least one of its two descendants, mtBL1 and mtBL2). This would make mtB useless and set the stage for its decay and eventual disappearance, here shown to have occurred in three species. mtC might also have become dispensable since it was jointly lost in two of these species. Paramecium tredecaurelia would represent a stage where mtB is no longer functional but the two variants of the mtA promoter motif still coexist in the population, making it the only extant species known to use genetic determination of mating types. P. sexaurelia and P. jenningsi would represent more advanced stages in which reversion of the mtA promoter motif to GTGAAA has become fixed; the lack of functional mtB and mtC genes would restrict possible type O determination mechanisms to developmental rearrangements inactivating mtA (or unidentified genes that may be required for mtA transcription), which could be either retained from the ancestor or newly evolved.
This model makes a number of predictions that remain to be tested, such as the putative roles of mtB-like paralogs in the regulation of mtA-like genes, and, in species lacking mtB, of mtA itself. Further work will also be required to assess the possible roles of mtA-like genes in sexual reactivity, to identify the putative mtA receptor in type O cells, and to unravel the mechanisms of stochastic mating-type determination in other P. aurelia species. Nevertheless the present work has elucidated the various mechanisms at work in all P. aurelia species with genetic or maternally inherited determination (fig. 7), illustrating the fascinating diversity of the molecular solutions that can evolve among closely related sibling species to regulate a single conserved gene and maintain mating-type polymorphism in cell populations.
Fig. 7.
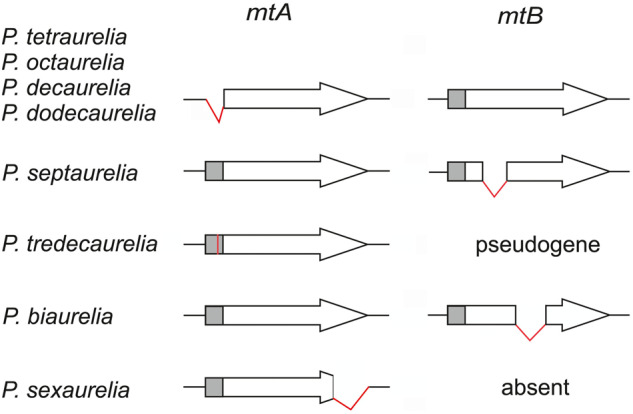
Five distinct mechanisms preventing mtA expression in mating type O. The schematic drawings summarize the state of the MAC mtA and mtB genes in mating type O clones for the seven maternal-inheritance species, and for the Mendelian species Paramecium tredecaurelia. In the former, developmental rearrangements disrupt either mtA or mtB, preventing the production of the wild-type mtA protein. Excision of the mtA promoter (gray box) is shared between Paramecium tetraurelia and the three most closely related species, but other rearrangements are unique to each species. In P. tredecaurelia and Paramecium sexaurelia, the mtB transcription factor was lost and the mtA promoter must be activated by some other factor; in P. tredecaurelia this is prevented by a point mutation (red bar) in the O-determining alleles of mtA.
Supplementary Material
Acknowledgments
Experiments and sequencing were partly performed thanks to the Core Facility Centers “Cultivation of Microorganisms” and “Molecular and Cell Technologies” of St. Petersburg State University. We thank the French National Sequencing Center (Genoscope CEA) and the France Génomique Paramecium Sequencing Project coordinated by S. Duharcourt, as well as M. Lynch and collaborators, for permission to use MAC and MIC genomes prior to publication. RNA sequencing benefited from the facilities and expertise of the high-throughput sequencing platform of I2BC. This study was supported by National Science Centre (Grant No. 2013/09/N/NZ8/03198) (Paramecium tredecaurelia study) to N.S.-G. and S.T. and fellowship 2013/08/T/NZ1/00306 to N.S.-G. (Paramecium biaurelia study), by the Agence Nationale de la Recherche (Grant No. ANR 18-CE12-0005-01) to E.M., and by the Russian Foundation for Basic Research (Grant No. 19-04-00710a) to I.N. (Paramecium sexaurelia, Paramecium dodecaurelia, and Paramecium tredecaurelia studies). It received support under the program “Investissements d’Avenir” launched by the French Government and implemented by ANR with the references ANR-10-LABX-54 MEMOLIFE and ANR-10-IDEX-0001-02 PSL Research. Mobility to Paris was supported by St. Petersburg State University grants 1.42.1436.2015 to I.N. and COLLAB2020_1 50414369 to A.P.
Data Availability
The RNA-seq data underlying this article are available in the European Nucleotide Archive, at http://www.ebi.ac.uk/ena/data/view/PRJEB38593, and accession numbers for individual sequencing data sets are listed in supplementary table S2, Supplementary Material online. The mtA and mtA-like, mtB and mtB-like, and mtC protein sequences used are listed in supplementary files S3–S5, Supplementary Material online, respectively; individual gene sequences determined in this study are shown in supplementary file S6, Supplementary Material online.
Literature Cited
- Abello A, et al. 2020. Functional diversification of Paramecium Ku80 paralogs safeguards genome integrity during precise programmed DNA elimination. PLoS Genet. 16(4):e1008723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaiz O, et al. 2012. The Paramecium germline genome provides a niche for intragenic parasitic DNA: evolutionary dynamics of internal eliminated sequences. PLoS Genet. 2012;8(10):e1002984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaiz O, et al. 2017. Improved methods and resources for Paramecium genomics: transcription units, gene annotation and gene expression. BMC Genomics 18(1):483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aufderheide KJ, Daggett PM, Nerad TA.. 1983. Paramecium sonneborni n. sp., a new member of the Paramecium aurelia species-complex. J Protozool. 30(1):128–131. [Google Scholar]
- Aury JM, et al. 2006. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 444(7116):171–178. [DOI] [PubMed] [Google Scholar]
- Baudry C, et al. 2009. PiggyMac, a domesticated piggyBac transposase involved in programmed genome rearrangements in the ciliate Paramecium tetraurelia. Genes Dev. 23(21):2478–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisson J. et al. 2010a. Maintaining clonal Paramecium tetraurelia cell lines of controlled age through daily reisolation. Cold Spring Harb Protoc. 2010(1):pdb.prot5361. [DOI] [PubMed] [Google Scholar]
- Beisson J. et al. 2010b. Mass culture of Paramecium tetraurelia. Cold Spring Harb Protoc. 2010(1):pdb.prot5362. [DOI] [PubMed] [Google Scholar]
- Beisson J. et al. 2010c. DNA microinjection into the macronucleus of Paramecium. Cold Spring Harb Protoc. 2010(1):pdb.prot5364. [DOI] [PubMed] [Google Scholar]
- Bétermier M, Duharcourt S.. 2014. Programmed rearrangement in ciliates: paramecium. Microbiol Spectrum. 2(6):MDNA3-0035-2014. [DOI] [PubMed] [Google Scholar]
- Billiard S, et al. 2011. Having sex, yes, but with whom? Inferences from fungi on the evolution of anisogamy and mating types. Biol Rev Camb Philos Soc. 86(2):421–442. [DOI] [PubMed] [Google Scholar]
- Brygoo Y. 1977. Genetic analysis of mating type differentiation in Paramecium tetraurelia. Genetics 87(4):633–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brygoo Y, Sonneborn TM, Keller AM, Dippell RV, Schneller MV.. 1980. Genetic analysis of mating type differentiation in Paramecium tetraurelia. II. Role of the micronuclei in mating-type determination. Genetics 94(4):951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, Huson DH.. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 12(1):59–60. [DOI] [PubMed] [Google Scholar]
- Butzel HM. 1955. Mating type mutations in variety 1 of Paramecium aurelia, and their bearing upon the problem of mating type determination. Genetics 40(3):321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne BC. 1973. Mutational analysis of mating type inheritance in Syngen 4 of Paramecium aurelia. Genetics 74(1):63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes M.D. et al. 2013. Selecting one of several mating types through gene segment joining and deletion in Tetrahymena thermophila. PLoS Biol. 2013;11(3):e1001518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker DL, Meyer E,, Mochizuki K.. 2013. Epigenetics of ciliates. Cold Spring Harb Perspect Biol. 5(12):a017764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Orias E, Leu JY, Turkewitz AP.. 2020. The evolution of germ-soma nuclear differentiation in eukaryotic unicells. Curr Biol. 30(10):R502–R510. [DOI] [PubMed] [Google Scholar]
- Coyne R, Lhuillier-Akakpo M, Duharcourt S.. 2012. RNA-guided DNA rearrangements in ciliates: is the best genome defence a good offence? Biol Cell. 104(6):309–325. [DOI] [PubMed] [Google Scholar]
- Dereeper A, et al. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36(Web Server):W465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diller WF, Earl PR.. 1958. Paramecium jenningsi, n. sp. J Protozool. 5(2):155–158. [Google Scholar]
- Dubois E, et al. 2012. Transposon invasion of the Paramecium germline genome countered by a domesticated piggyBac transposase and the NHEJ pathway. Int J Evol Biol. 2012:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duharcourt S, Lepere G, Meyer E.. 2009. Developmental genome rearrangements in ciliates: a natural genomic subtraction mediated by non-coding transcripts. Trends Genet. 25(8):344–350. [DOI] [PubMed] [Google Scholar]
- Fokin S, et al. 2004. Morphological and molecular investigations of Paramecium schewiakoffi sp. nov. (Ciliophora, Oligohymenophorea) and current status of distribution and taxonomy of Paramecium spp. Eur J Protistol. 40(3):225–243. [Google Scholar]
- Frapporti A, et al. 2019. The Polycomb protein Ezl1 mediates H3K9 and H3K27 methylation to repress transposable elements in Paramecium. Nat Commun. 10(1):2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier O, Serrano V, Duharcourt S, Meyer E.. 2004. RNA-mediated programming of developmental genome rearrangements in Paramecium tetraurelia. Mol Cell Biol. 24(17):7370–7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout JF, et al. 2019. Universal trends of post-duplication evolution revealed by the genomes of 13 Paramecium species sharing an ancestral whole-genome duplication. bioRxiv 2019:573576. [Google Scholar]
- Hadjivasiliou Z,, Pomiankowski A.. 2016. Gamete signalling underlies the evolution of mating types and their number. Philos Trans R Soc Lond B Biol Sci. 371(1706):20150531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman J. 2015. Evolution of sexual reproduction: a view from the Fungal Kingdom supports an evolutionary epoch with sex before sexes. Fungal Biol Rev. 29(3–4):108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri P, et al. 2017. Population genomics of Paramecium species. Mol Biol Evol. 34(5):1194–1216. [DOI] [PubMed] [Google Scholar]
- Katoh K, Rozewicki J, Yamada KD.. 2019. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinformatics 20(4):1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, et al. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14(4):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhuillier-Akakpo M, et al. 2014. Local effect of enhancer of zeste-like reveals cooperation of epigenetic and cis-acting determinants for zygotic genome rearrangements. PLoS Genet. 10(9):e1004665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmignon A, et al. 2014. Ku-mediated coupling of DNA cleavage and repair during programmed genome rearrangements in the ciliate Paramecium tetraurelia. PLoS Genet. 10(8):e1004552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath CL, Gout JF, Doak TG, Yanagi A, Lynch M.. 2014. Insights into three whole-genome duplications gleaned from the Paramecium caudatum genome sequence. Genetics 197(4):1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath CL, Gout JF, Johri P, Doak TG, Lynch M.. 2014. Differential retention and divergent resolution of duplicate genes following whole-genome duplication. Genome Res. 24(10):1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer E, Garnier O.. 2002. Non-Mendelian inheritance and homology-dependent effects in ciliates. Adv Genet. 46:305–337. [DOI] [PubMed] [Google Scholar]
- Miele V, Penel S, Duret L.. 2011. Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinformatics 12(1):116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanney DL. 1957. Mating type inheritance at conjugation in variety 4 of Paramecium aurelia. J Protozool. 4(2):89–95. [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol Biol Evol. 32(1):268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orias E, Singh DP, Meyer E.. 2017. Genetics and epigenetics of mating type determination in Paramecium and Tetrahymena. Annu Rev Microbiol. 71(1):133–156. [DOI] [PubMed] [Google Scholar]
- Perrin N. 2012. What uses are mating types? The ``developmental switch” model. Evolution 66(4):947–956. [DOI] [PubMed] [Google Scholar]
- Phadke SS, Zufall RA.. 2009. Rapid diversification of mating systems in ciliates. Biol J Linn Soc. 98(1):187–197. [Google Scholar]
- Potekhin A, Mayén-Estrada R.. 2020. Paramecium diversity and a new member of the Paramecium aurelia species complex described from Mexico. Diversity 12(5):197. [Google Scholar]
- Przyboś E, Tarcz S.. 2016. Paramecium jenningsi complex: existence of three cryptic species confirmed by multi-locus analysis and strain crosses. Syst Biodivers. 14(2):140–154. [Google Scholar]
- Przyboś E, Tarcz S.. 2018. New stands of the Paramecium aurelia spp. complex (Protista, Oligohymenophorea) in Ethiopia, Madagascar, Taiwan, and Romania. Folia Biol (Krakow)). 66(3):111–119. [Google Scholar]
- Przyboś E, Tarcz S.. 2019. Global molecular variation of Paramecium jenningsi complex (Ciliophora, Protista): a starting point for further, detailed biogeography surveys. Syst Biodivers. 17(5):527–539. [Google Scholar]
- Przyboś E, Tarcz S, Rautian M, Sawka N.. 2015. Delimiting species boundaries within a paraphyletic species complex: insights from morphological, genetic, and molecular data on Paramecium sonneborni (Paramecium aurelia species complex, Ciliophora, Protozoa) . Protist 166(4):438–456. [DOI] [PubMed] [Google Scholar]
- Przyboś E, Tarcz S, Surmacz M, Sawka N, Fokin S.. 2013. Paramecium tredecaurelia, a unique non-polymorphic species of the P. aurelia spp. complex (Oligohymenophorea, Ciliophora). Acta Protozool. 52:257–266. [Google Scholar]
- Rzeszutek I, Maurer-Alcalá XX, Nowacki M.. 2020. Programmed genome rearrangements in ciliates. Cell Mol Life Sci. 77(22):4615–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellis D, et al. 2020. Massive colonization of protein-coding exons by selfish genetic elements in Paramecium germline genomes. bioRxiv 2020. doi: 10.1101/2020.12.23.424184. [DOI] [PMC free article] [PubMed]
- Sievers F, Higgins DG.. 2018. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 27(1):135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DP, et al. 2014. Genome-defence small RNAs exapted for epigenetic mating-type inheritance. Nature 509(7501):447–452. [DOI] [PubMed] [Google Scholar]
- Sonneborn TM. 1947. Recent advances in the genetics of Paramecium and Euplotes. Adv Genet. 1:263–358. [DOI] [PubMed] [Google Scholar]
- Sonneborn TM. 1954. Patterns of nucleo-cytoplasmic integration in Paramecium. Caryologia 6(Suppl):307–325. [Google Scholar]
- Sonneborn TM. 1957. Breeding systems, reproductive methods and species problems in protozoa. In: Mayr E, editor. The species problem. Washington (DC: ): American. Association for the Advancement of Science. p. 155–324. [Google Scholar]
- Sonneborn TM. 1958. Classification of syngens of the Paramecium aurelia-multimicronucleatum complex. J Protozool. 5(Suppl):17–18. [Google Scholar]
- Sonneborn TM. 1975a. The Paramecium aurelia complex of 14 sibling species. Trans Am Microsc Soc. 94(2):155–178. [Google Scholar]
- Sonneborn TM. 1975b. Paramecium aurelia. In: King CR, editor. Handbook of genetics: plants, plant viruses and protists. Vol. 2. New York: Plenum Press, p. 469–594. [Google Scholar]
- Sonneborn TM. 1977. Genetics of cellular differentiation: stable nuclear differentiation in eucaryotic unicells. Annu Rev Genet. 11(1):349–367. [DOI] [PubMed] [Google Scholar]
- Swart EC, et al. 2014. Genome-wide analysis of genetic and epigenetic control of programmed DNA deletion. Nucleic Acids Res. 42(14):8970–8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taub SR. 1963. The genetic control of mating type differentiation in Paramecium. Genetics 48(6):815–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28(5):511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umen J, Coelho S.. 2019. Algal sex determination and the evolution of anisogamy. Annu Rev Microbiol. 73(1):267–291. [DOI] [PubMed] [Google Scholar]
- Watanabe T. 1978. A scanning electron-microscopic study of the local degeneration of cilia during sexual reproduction in Paramecium. J Cell Sci. 32:55–66. [DOI] [PubMed] [Google Scholar]
- Whelan S, Irisarri I, Burki F.. 2018. PREQUAL: detecting non-homologous characters in sets of unaligned homologous sequences. Bioinformatics 34:3929–3930. [DOI] [PubMed] [Google Scholar]
- Yi Z, Strüder-Kypke M, Hu X, Lin X, Song W.. 2014. Sampling strategies for improving tree accuracy and phylogenetic analyses: a case study in ciliate protists, with notes on the genus Paramecium. Mol Phylogenet Evol. 71:142–148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data underlying this article are available in the European Nucleotide Archive, at http://www.ebi.ac.uk/ena/data/view/PRJEB38593, and accession numbers for individual sequencing data sets are listed in supplementary table S2, Supplementary Material online. The mtA and mtA-like, mtB and mtB-like, and mtC protein sequences used are listed in supplementary files S3–S5, Supplementary Material online, respectively; individual gene sequences determined in this study are shown in supplementary file S6, Supplementary Material online.