

Abstract
Methods for highly multiplexed RNA imaging are limited in spatial resolution, and thus in their ability to localize transcripts to nanoscale and subcellular compartments. We adapt expansion microscopy, which physically expands biological specimens, for long-read untargeted and targeted in situ RNA sequencing. We applied untargeted expansion sequencing (ExSeq) to mouse brain, yielding readout of thousands of genes, including splice variants. Targeted ExSeq yielded nanoscale-resolution maps of RNAs throughout dendrites and spines in neurons of the mouse hippocampus, revealing patterns across multiple cell types; layer-specific cell types across mouse visual cortex; and the organization and position-dependent states of tumor and immune cells in a human metastatic breast cancer biopsy. Thus ExSeq enables highly multiplexed mapping of RNAs, from nanoscale to system scale.
One Sentence Summary:
In situ sequencing of physically expanded specimens enables multiplexed mapping of RNAs at nanoscale, subcellular resolution.
Tissues are made of cells of many different types and states that are regulated by, and contribute to, the cells’ spatial organization. Multiplexed measurements of the locations and identities of RNA molecules within cells has been useful for exploring these relationships (1–13). Furthermore, mapping the subcellular locations of RNAs is important for understanding diverse biological processes (14, 15), such as how RNAs in dendritic spines help regulate synaptic function (16–19).
Imaging RNAs within such compartments, and throughout detailed cellular morphologies, requires nanoscale precision. Such precision is not easily achieved within tissues with current multiplexed optical methods to image RNA. Indeed, no method can currently perform multiplexed imaging of RNA within tissues in the context of nanoscale cellular morphology. Even though seqFISH+ allows high resolution imaging of RNA molecules, it cannot resolve the detailed cellular and tissue context with nanoscale precision (20).
Ideally one would be able to perform the enzymatic reactions of sequencing in situ with high multiplexing capacity, while providing for fast nanoscale imaging of cellular and tissue context. We here present a toolbox for the untargeted (i.e., not restricted to a pre-defined list of gene targets) and targeted in situ sequencing of RNAs within intact tissues, in the context of nanoscale cellular morphology.
Adapting expansion microscopy to improve in situ sequencing
We created an untargeted in situ sequencing technology that enables the sequencing of arbitrary RNAs within detailed cellular and tissue contexts. Untargeted approaches have the potential to discover spatially localized sequence variants, such as splice variants and retained introns (21). Fluorescent in situ sequencing (FISSEQ) enables such data to be acquired from cultured cells (22), but was not fully demonstrated in tissues (22). Therefore, we adapted the chemistry of expansion microscopy (ExM; (23, 24)) to separate RNAs from nearby molecules. We reasoned that this may facilitate the chemical access needed for in situ sequencing within tissues. We also expected that the resolution boost from ExM would enable high spatial resolution mapping of RNAs and their cellular and tissue context on conventional microscopes.
In FISSEQ, untargeted in situ sequencing of RNA is performed to amplify RNA into ‘nanoballs’ of cDNA (or ‘amplicons’), containing many copies of an RNA sequence (22, 25). These sequences are interrogated in situ with standard next-generation sequencing chemistries on a fluorescence microscope. In ExM (23) we isotropically separate gel-anchored biomolecules of interest by a ~4x linear expansion factor, which facilitates both nanoscale imaging with conventional optics, and better chemical access to the separated biomolecules (24). ExM enables better resolution of normally densely packed RNA transcripts for in situ hybridization imaging (26, 27).
Expanding specimens is expected to benefit FISSEQ by dividing the effective size of the FISSEQ amplicon (200-400 nm; (22)) by the expansion factor. This reduces the packing density of amplicons and facilitates their tracking over many rounds of sequencing. We adapted ExM chemistry to enable FISSEQ in expanded tissues. In particular, the anchoring (Fig. 1Ai), polymerization (Fig. 1Aii), and expansion (Fig. 1Aiii) steps, which separate RNAs for nanoscale imaging (26), result in charged carboxylic acid groups throughout the swellable gel. This suppresses the enzymatic reactions required for FISSEQ (Fig. S1). We thus stabilized expanded specimens by re-embedding them in uncharged gels (26), and then chemically treated samples to result in a neutral charge environment (Fig. S1). We reasoned that this would allow FISSEQ signal amplification (Fig. 1Aiv) and readout (Figs. 1Av–vi and 1B) steps to proceed.
Fig. 1. Untargeted expansion sequencing (ExSeq) concept and workflow.
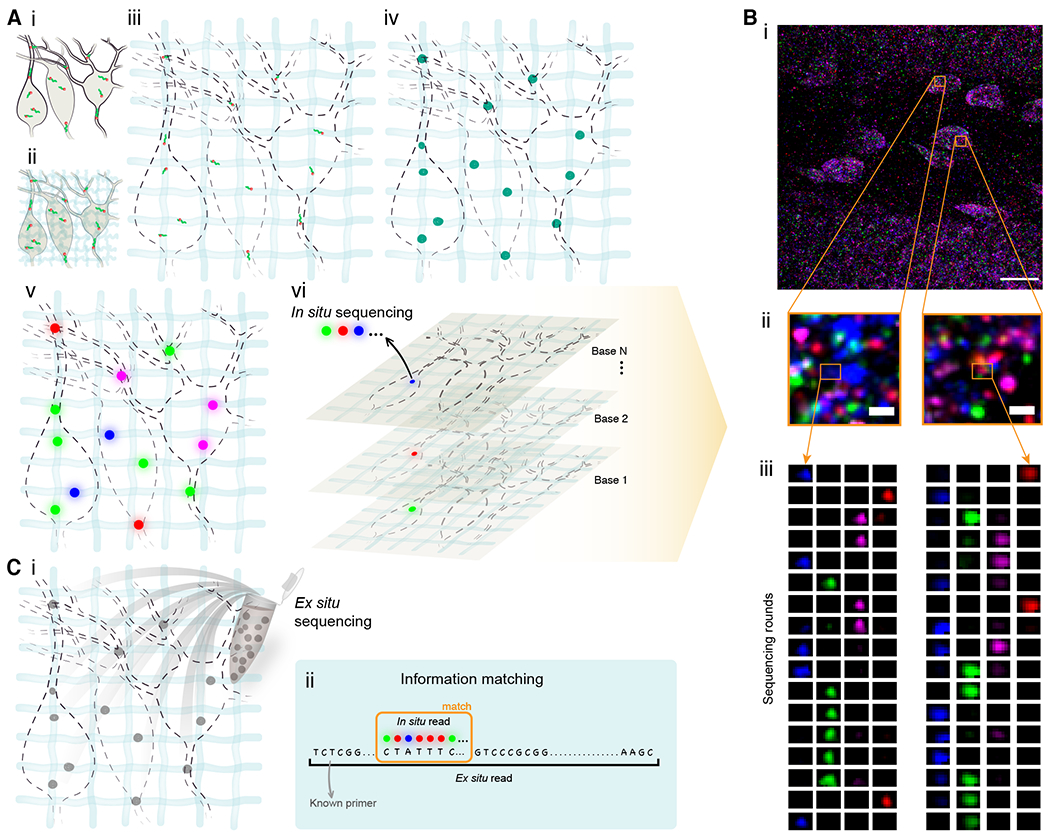
(A) ExSeq schematic. (i) A specimen is fixed, and RNA molecules (green) bound by an anchor (orange). (ii) The specimen is embedded in a swellable gel material (light blue, not to scale), mechanically softened, and then expanded with water (iii). RNA molecules are anchored to the gel. (iv) RNA molecules are reverse transcribed and amplified using FISSEQ (v) in situ sequencing. Colored dots indicate the colors used in the sequencing chemistry. (vi) In each sequencing round colors (blue, magenta, green, and red) reveal the current base of the cDNA. (B) Example of ExSeq from a 50 micron thick slice of mouse dentate gyrus. (i) One sequencing round, with two zoomed-in regions (ii), and puncta histories obtained over 17 rounds of in situ sequencing (iii). (C) Ex situ sequencing. (i) After in situ sequencing, cDNA amplicons are eluted from the sample, and resequenced ex situ with next-gen sequencing. (ii) In situ reads are matched to their longer ex situ counterparts, focusing on unique matches, augmenting the effective in situ read length. Scale bars: Bi, 17 microns (in biological, i.e. pre-expansion, units used throughout, unless otherwise indicated), Bii, 700 nanometers.
In situ sequencing involves many rounds of adding fluorescent oligonucleotides (22). Accordingly, we established an automated sequencing system (28). Because the resultant datasets consist of a series of 3D images, one for each successive base sequenced, we created a software pipeline (Fig. S2; https://doi.org/10.5281/zenodo.4075515, ref (93)). This software can align, across images from many rounds, the puncta for each expressed gene to within 1 pixel (validated in Fig. S3 and S4). Finally, puncta are segmented and bases called (Fig. 1Biii).
In situ sequencing has previously been limited to short reads of 5-30 bases (10, 11, 22). This limitation reflects laser-induced damage during imaging (25) and dependence of the signal for a given cycle on signals from previous cycles (known as “phasing”), caused by incomplete enzymatic reactions (29). Alignment of such short reads to the genome is challenging (30). Moreover, short reads do not easily capture mRNA complexity, such as alternative splicing.
Accordingly, we added a follow-on round of ex situ classical “next-gen” sequencing (Fig. 1Ci, (28)). Importantly, the random nature of untargeted sequencing (28) results in the creation of unique molecular identifiers from the in situ sequenced region of the amplified cDNA (Fig. S5). This allows us to use ex situ information as a dictionary to align and directly interpret the in situ reads (Figs. 1Cii and S5A, bottom panel).
92% of all matches, and 97% of the matches aligned against non-rRNA, were strictly unique. We removed the handful of in situ reads that matched to more than one ex situ library entry (28). Thus, one in situ read matched one ex situ library entry (Fig. S5C). This allowed us to explore sequence variations in mRNA, such as alternative splicing, using the longer ex situ matched reads (Fig. S6).
Biological validations of ExSeq
Expansion sequencing (ExSeq) produced data from a variety of specimens (Tables S1–S5), including mouse brain (Fig. 1B), C. elegans (Fig. S7A), Drosophila embryos (Fig. S7B), and HeLa cells (Fig. S7C). To validate ExSeq, we used the following mouse specimens: cultured hippocampal neurons (Figs. 2A–B and S8), a 15 micron thick hippocampal slice (Fig. 2C–D), and a 50 micron thick hippocampal slice (Fig. 2E–F). To improve the efficiency of cDNA circularization, we restricted the size of cDNA fragments to ~100 bases long, so ex situ Illumina reads typically contained several repeats of a given cDNA fragment that were, on average, 76 bases long (Fig. S6).
Fig. 2. In situ sequencing in cells and tissues with untargeted ExSeq.
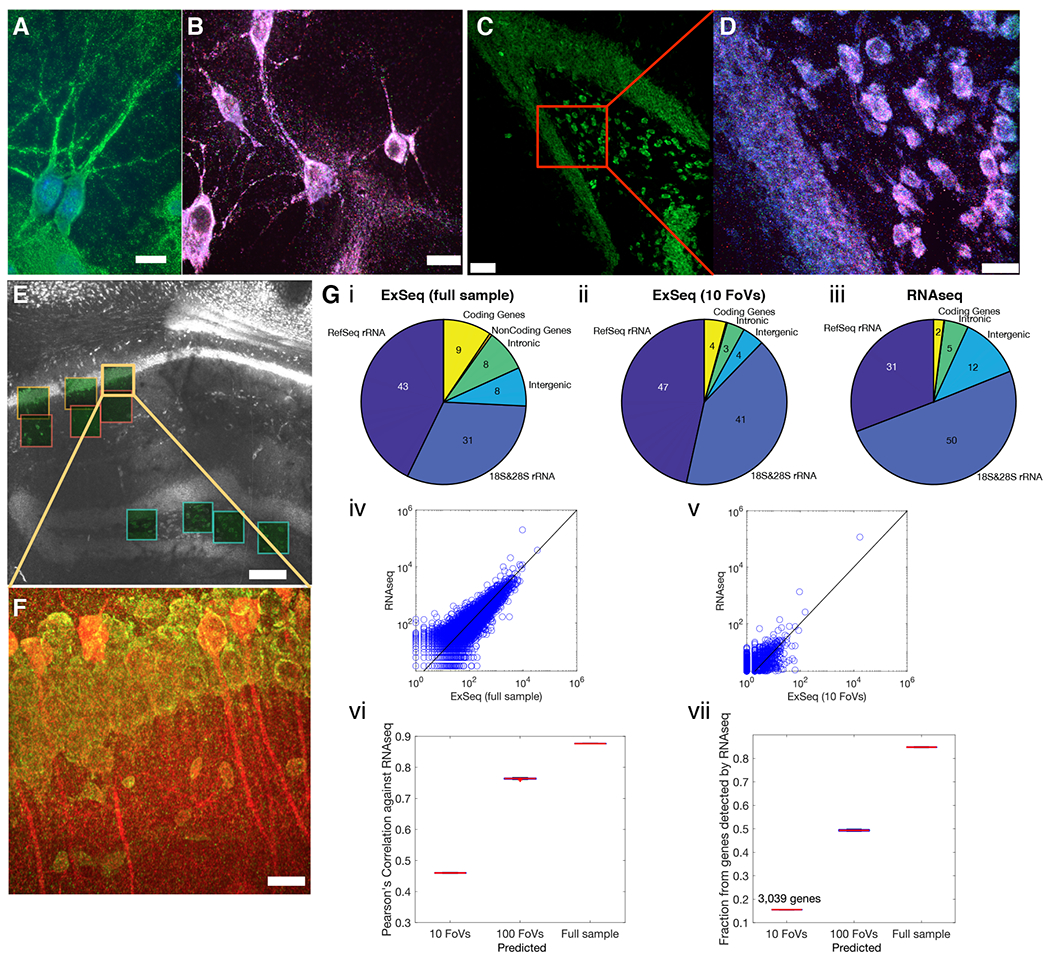
(A) Example of ExSeq library preparation in hippocampal culture (green, hybridization probe against amplified cDNA; blue, DAPI). (B) Maximum intensity projection of one sequencing round in hippocampal culture; color scheme as in Fig. 1B. (C) Low magnification image of ExSeq library preparation in a 15 𝜇m slice of mouse hippocampus (green, hybridization probe against amplified cDNA). (D) Maximum intensity projection of a higher magnification image of the specimen of C, focusing on one sequencing round; color scheme as in Fig. 1B. (E) Low magnification image of ExSeq library preparation in a 50 𝜇m slice of mouse hippocampus. Fields of view (FoVs) acquired with a higher magnification objective are shown as green squares. White, hybridization probe against amplified cDNA. (F) Maximum intensity projection of one FoV of panel E, with antibody staining post in situ sequencing (red, antibody against YFP; specimen from a Thy1-YFP mouse; green, hybridization probe against amplified cDNA). (G) Sequence analysis of ExSeq specimen shown in (E). (i-iii) RNA content obtained with ExSeq, either using ex situ sequencing data from the entire slice (i) or using ex situ data that correspond to in situ reads observed within the FoVs of panel E (ii), is comparable to the RNA content of an adjacent slice obtained with standard RNAseq (iii). Numbers inside the pie chart represent percentage of the total. (iv) Agreement between the normalized expression levels of all well-annotated genes (RefSeq genes) using RNAseq and ExSeq with full ex situ sequencing data as in i. (v) As in (iv), but using the 10 acquired FoVs, as in ii. (vi) Pearson’s correlation between the log-transformed expression of RefSeq genes using ExSeq and using RNAseq, as a function of the number of acquired FoVs (estimated by sampling from the full ex situ sequencing data to simulate the number of expected reads for 100 FoVs; (28)). The value for the 100 FoVs is plotted using the MATLAB boxplot function; central mark, median; bottom and top edges of the box, 25th and 75th percentiles, respectively. (vii) Fraction of RefSeq genes detected using ExSeq vs. RNAseq, as a function of the number of acquired FoVs (estimated by sampling from the full ex situ sequencing data to simulate the number of expected reads for 100 FoVs). Scale bars: A-D&F, 13μm E, 130μm. Note that deconvolution was used in panels D and F (28).
Antibody staining after in situ sequencing, as with previous ExM-related protocols (31), enabled visualization of specific proteins. This was demonstrated by staining with antibodies against YFP in a Thy1-YFP mouse (32) to visualize in situ sequencing reads within neural morphology (Fig. 2F).
As a validation of ExSeq, we performed RNA sequencing (RNAseq), with random primers, on a 50 micron thick hippocampal slice adjacent to the 50 micron thick ExSeq specimen (Fig. 2E–F). As expected for total RNA analysis, most of the RNA detected in both cases was ribosomal. We observed overall agreement between the RNA types obtained with both methods (Fig. 2Gi–iii); although ExSeq exhibited a slightly higher percentage of coding RNA (4-9% with ExSeq vs 2% with RNAseq). Gene ontology analysis revealed expected functional enrichments for this specimen, including ‘synapse’, ‘neuron projection’ and ‘hippocampus’ (Fig. S9 and Table S6).
In FISSEQ highly abundant genes were underrepresented, for example genes involved in translation and splicing (22). In contrast, we did not observe this detection bias with ExSeq (28). The expression levels of well-annotated genes (RefSeq genes) using RNAseq and ExSeq were highly correlated (Pearson’s r = 0.89) (Fig. 2Giv and Fig. S8C).
The correlation between ExSeq and RNAseq increased with the ExSeq volume imaged. For instance, 10 microscope volumes (each 350x350x100 microns in size, post-expansion, and ~100x100x28 microns pre-expansion) resulted in a Pearson’s r = 0.47 (Fig. 2Gv; Student’s t, p-value 9 x 10−164) comparable to recent targeted in situ sequencing methods (33). Larger volumes, simulated by sampling (28), yielded higher correlations (Fig. 2Gvi). With 10 volumes, 3,039 genes were detected, comprising ~16% of all the genes detected in the sample via RNAseq, again increasing with the volume sampled (Fig. 2Gvii). Thus, ExSeq is able to report on genome-wide expression in situ, in an untargeted, highly multiplexed way.
Subcellular pinpointing of transcript locations in neurons
We next sought to utilize the improved spatial resolution of ExSeq to pinpoint RNAs relative to antibody-stained morphology. We traced 13 hippocampal CA1 pyramidal neurons (28). We analyzed the locations of RNAs inside identified neurons with a custom 3D viewer (Figs. 3 and S10, (28)). The number of sequencing reads per neuron was 229 ± 74 (mean ± standard deviation used throughout) including rRNA, and 30 ± 14 for non-ribosomal RNA, for cell bodies and dendrites imaged up to ~100 microns from the cell body. Not including rRNAs, 326 RefSeq genes were observed in these imaged volumes. These numbers are comparable to those obtained by the original FISSEQ protocol (25), applied to cultured cell lines.
Fig. 3. Untargeted ExSeq enables mapping of RNAs and their variants in dendrites of neurons.
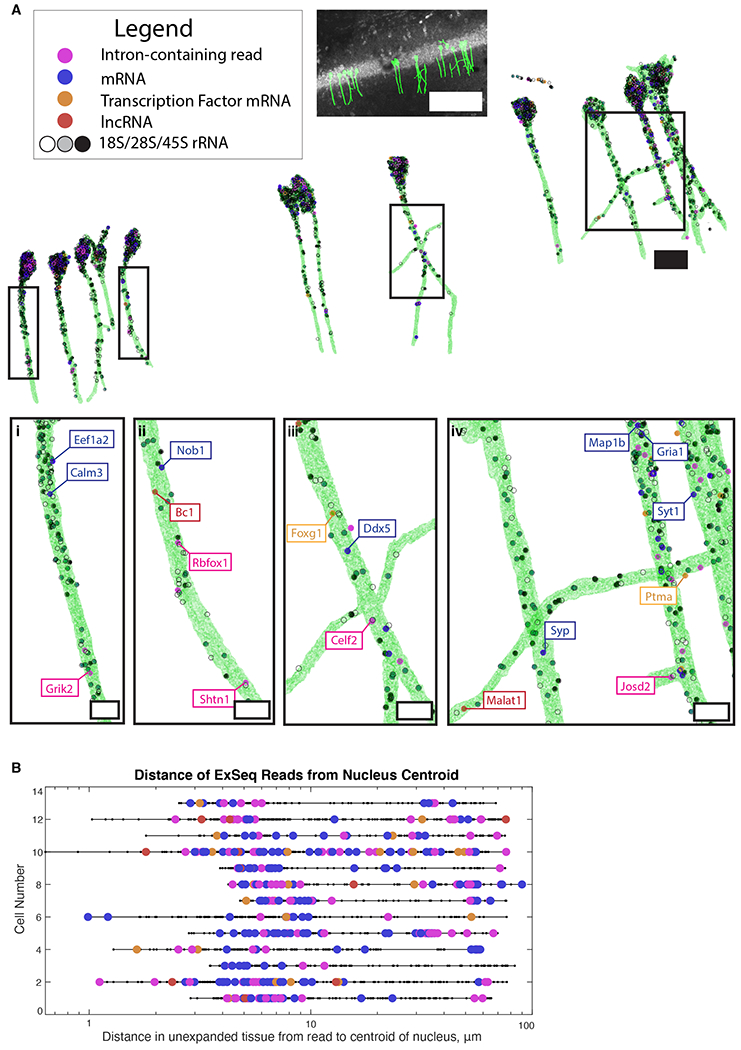
(A) 3D render of Thy1-YFP CA1 neuronal morphology as determined by YFP antibody staining, containing RNA types as indicated. (i-iv), zoomed-in dendritic regions (boxed above). Scale bars: top, middle and bottom, 100, 20, and 5 microns, respectively. (B) Euclidean distance, relative to the center of the cell body, of sequencing reads for neurons in A. Color code, as in A.
Neurons contain one nucleus vs. thousands of synapses. This raises the question of whether splicing of mRNAs, such as those that contribute to synaptic function, is regulated in a spatially dependent manner along dendritic trees (34). We examined reads that corresponded to intronic regions and observed that, while 70% of such reads were located at the soma, introns in YFP-containing dendritic projections could be found as far down the dendrite as we looked, consistent with previous reports (35, 36). For example, glutamate ionotropic receptor kainate type subunit 2 (Grik2), which encodes a receptor subunit involved in excitatory glutamatergic neurotransmission, appears in our data in dendrites with a retained intron (Fig. 3Ai). The Grik1 subunit had been identified earlier as a dendritically-targeted intron-retaining sequence (35, 37). Dendritic splicing of glutamate receptor subunit RNAs may contribute to the regulation of the state or plasticity of excitatory synapses. Indeed, splicing in dendrites has been characterized previously in cultured neurons (38).
The long sequencing reads (Fig. S6) and untargeted nature of ExSeq also allowed for mapping of alternative splicing in situ. We quantified the expression of known alternative splicing isoforms with ExSeq vs. RNAseq. The two methods were highly correlated (Pearson’s r = 0.944; Fig. S11A). Using only 10 confocal microscope fields of view, of sizes described above, we detected 112 sequencing reads that corresponded to known alternative splicing events. 67% of these sequencing reads revealed the expressed alternative splicing isoforms, including ribosomal protein S24 (Rps24) and microtubule-associated protein 2 (Map2) (Fig. S11B). We also identified possibly new isoforms, for example for the gene spectrin beta (Sptbn1) (Fig. S11B).
Importantly, ExSeq provides the ability to locate these alternative splicing events in space. As an example, isoforms of Map2, a key dendritic protein (39), and the transcription factor Cux1, which is involved in dendrite and spine formation (40), could be localized to the neuronal soma, outside of the nucleus (Figs. S10, Neurons 9 and 7, respectively, and S11B).
Many genes may have unappreciated connections to neuronal signaling inside dendritic trees. mRNAs for specific transcription factors have been identified inside dendrites (41), for example MAX dimerization protein (Mga; (42); Fig. S10, Neuron 6). However, the full complement of dendritically localized transcription factors in any neuron type is unknown.
In our hippocampus sample (Table S2), 914 of the known 1675 mouse transcription factors (RIKEN transcription factor database) were detected by ExSeq. This included 32 reads localized within YFP-expressing cells, and 11 reads in the dendrites of these cells. These reads include forkhead box protein G1 (Foxg1), involved with neural development (43), and prothymosin alpha (Ptma), involved in learning and memory and neurogenesis (44) (Fig. 3Aiii and 3Aiv). We also found long non-coding RNAs (lncRNAs) and protein coding genes with unknown function in dendrites (Fig. 3A). For example, BC1 (Fig. 3Aii) is a lncRNA from an RNA polymerase III transcript that complexes with proteins to form a ribonucleoprotein particle. BC1 is dendritically localized (45) and involved with activity-dependent synaptic regulation (46). Additionally, MALAT1 (Fig. 3Aiv) has roles in neural growth and synaptogenesis, but its localization had not been determined in hippocampal tissue (47, 48).
We localized genes that had been found in dendrites of CA1 pyramidal cells at the protein level, but had not been mapped at the mRNA level; such as gamma-aminobutyric acid (GABA) type A receptor gamma2 subunit (Gabrg2; Fig. S10, Neuron 2) (49). Thus, ExSeq allows us to expand our knowledge of dendritically localized genes of known function, which may point to new regulatory mechanisms for their gene products. Furthermore, we identified transcripts encoding genes of unknown function in the hippocampus (e.g, Nob1, Fig. 3Aii) (50), which may contribute to their functional analysis.
To more systematically understand how the types and identities of transcripts varied with location along a dendrite, we measured the distance from each read to the centroid of its corresponding neuron’s cell body (Fig. 3B). These measurements reveal the positions of RNAs encoding for transcription factors, intron-containing reads, and lncRNAs up to 100 microns from the soma. To follow up with a more in-depth examination of specific genes, we next generated a targeted form of ExSeq.
Targeted ExSeq
Untargeted sequencing enables transcriptome-wide exploration of localized RNAs, including rare variants and those of unknown function. However, the diversity of possible reads generated by untargeted methods lead to a lower per-gene copy number of detected molecules, and a larger number of biochemical and imaging cycles to distinguish among reads. Targeted methods, in contrast, detect a smaller predefined set of genes and are applicable to mapping cell types and states, their spatial relationships in situ, and visualizing subcellular gene regulation.
An ideal technology for targeted multiplexed RNA mapping would satisfy the following list of criteria. First, it should possess sufficient yield (probability of detecting a present molecule) to detect low copy number transcripts such as transcription factors or sparse RNA molecules. Second, the technology should have resolution below the diffraction limit both laterally and axially to resolve nanoscale morphological features, such as dendritic spines in neurons. Third, the method should provide the ability to image both RNAs and proteins, and to work with 3D tissues, to localize RNAs in biological contexts. Finally, the method should work with various tissue types, including human tissues. We thus developed a targeted version of ExSeq to match these specifications (Tables S7–S8).
In targeted ExSeq, oligonucleotide padlock probes bearing barcodes hybridize to transcripts (11, 51). Amplicons are then generated for readout through in situ sequencing of the barcodes (Figs. 4A and S12). The inefficient (22) reverse transcription step required by untargeted in situ sequencing (11, 22, 52) is circumvented by the binding and ligation of padlock probes on each targeted transcript using PBCV-1 DNA ligase (also known as SplintR ligase). This enzyme can ligate DNA on an RNA template ~100x faster than T4 DNA Ligase (51, 53–56). After circularization and rolling circle amplification, the barcodes are sequenced in situ. As barcodes are sequenced across multiple rounds of imaging, the number of identifiable molecular targets scales exponentially with the number of imaging rounds.
Fig. 4. Targeted ExSeq of transcripts specifying neuron types of mouse primary visual cortex.
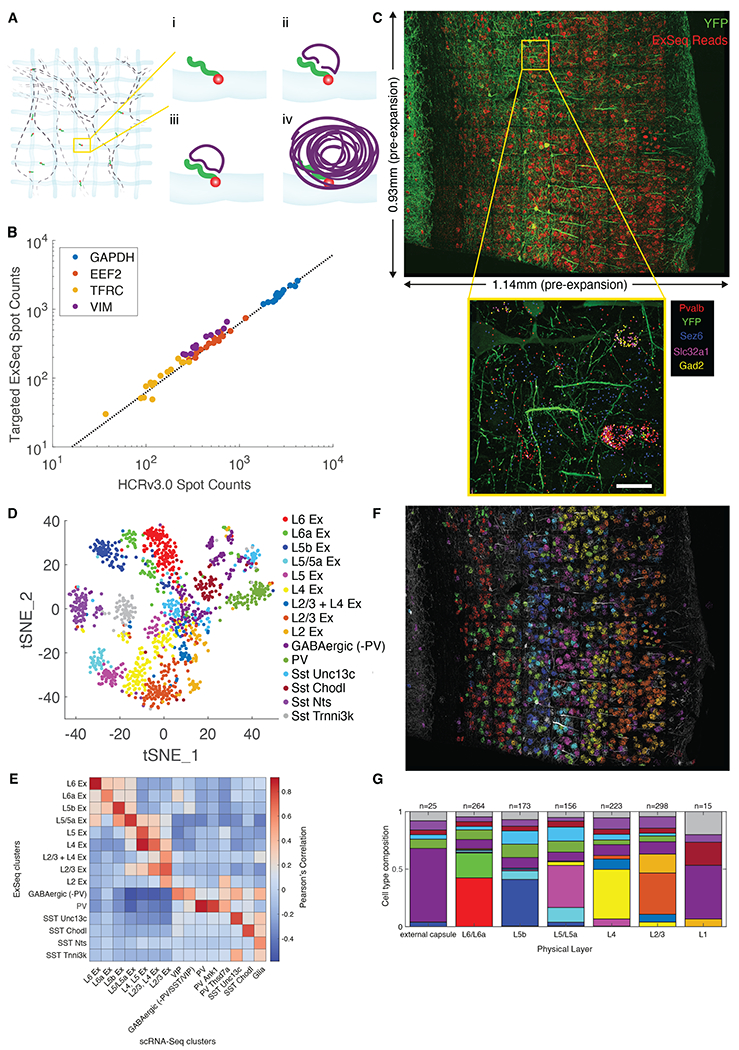
(A) Targeted ExSeq library preparation: (i) RNA anchoring and expansion; (ii) padlock probe hybridization; (iii) probe ligation; (iv) rolling circle amplification. (B) Amplicon counts for targeted ExSeq vs. HCRv3.0-amplified ExFISH for the same transcript in the same HeLa cell (60 cells); slope, 0.62 (Pearson’s r = 0.991). (C) Targeted ExSeq of 42 cell type marker genes in Thy1-YFP mouse visual cortex. Top, maximum intensity projection image showing targeted ExSeq reads (red) and YFP (green). Bottom, localization of marker genes Pvalb (red), Sez6 (cyan), Slc32a1 (magenta), and Gad2 (yellow), with YFP (green). (D) Targeted ExSeq gene expression profiles of 1154 cells clustered into 15 cell types. Cluster legend and colors apply to panels D, F, and G. (E) Heatmap showing Pearson’s correlation between clusters identified in targeted ExSeq vs. a prior scRNA-seq study (60). (F) Spatial organization of cell types identified in (D). Cell-segmented reads are shown, colored by cluster assignment, and overlaid on YFP (white). (G) Layer-by-layer cell-type composition across segmented cortical layers. Scale bars: (C) bottom, 20 microns (pre-expansion).
We explored the performance of targeted ExSeq in a variety of contexts (Table S5). To validate the yield, hybridization chain reaction (HCR) v3.0-amplified expansion FISH (ExFISH) and targeted ExSeq were sequentially performed for the same genes in expanded HeLa cells (26, 57). Targeted ExSeq exhibited an mRNA detection yield of ~62% (Pearson’s r = 0.991) relative to HCRv3.0-ExFISH (Fig. 4B, Tables S9–S11), which has a detection efficiency of ~70% in tissue (26). For comparison, single-cell RNA sequencing captures ~10% of mRNA (58, 59).
Cell type mapping with spatial context in the visual cortex
We mapped the cell types of the mouse primary visual cortex, for which single cell RNA sequencing (scRNA-Seq) data-based classification of cell types has been performed (60). We designed a panel of probes targeting 42 genes (Tables S9–S10) that mark key excitatory and inhibitory neuron types. We performed targeted ExSeq of these 42 genes across a coronal section of the primary visual cortex of a Thy1-YFP mouse over a volume of 0.933 mm x 1.140 mm x 0.02 mm, sequencing 265,347 reads (Fig. 4C, top and Table S12).
The spatial distribution of ExSeq reads recapitulated spatial distributions in the Allen in situ hybridization (ISH) atlas (Fig. S13). Transcripts known to express in the same cell type appeared in similar positions -- for example, in parvalbumin-positive (Pvalb+) interneurons (PV interneurons), parvalbumin (Pvalb), vesicular inhibitory amino acid transporter (Slc32a1), and glutamate decarboxylase 2 (Gad2) transcripts co-localized (Fig. 4C, inset). In contrast, seizure protein 6 homolog (Sez6) transcripts, associated with excitatory neurons in deep cortical layers (as well as vasoactive intestinal peptide (VIP+) interneurons) was not co-localized with Pvalb, Slc32a1, and Gad2 transcripts (Fig. 4C, inset).
Segmenting cells (Fig. S14, (28)) yielded a total of 1915 cells containing a total of 220,783 reads, out of which 1154 cells with at least 50 reads each (177 ± 127 reads/cell) were analyzed. We k-means clustered expression profiles, and embedded them into a low-dimensional space using t-Stochastic Neighbor Embedding (t-SNE) (61) (Fig. 4D). Clusters were identified with known markers (28), such as those corresponding to excitatory neurons (“Ex”, and sub-annotated by their layer location) and inhibitory neurons (annotated with relevant cell type markers). Clusters expressed marker genes consistent with prior studies (60) (Fig. S15).
We compared our results to a previous study of scRNA-Seq of the mouse primary visual cortex (28, 60) (Fig. 4E). We observed the canonical layer-by-layer stratification of excitatory neurons in the visual cortex (Figs. 4F and S16). The 9 ExSeq clusters of excitatory neurons corresponded, with slightly different groupings, to 7 scRNA-Seq clusters of excitatory neurons (Fig. 4E). We found inhibitory neuron ExSeq clusters that matched 1-to-1 to scRNA-Seq clusters. For example, two somatostatin interneuron clusters found across the layers of the cortex, the SST cluster expressing Unc-13 homolog C (cluster SST Unc13c) and the SST cluster expressing Chondrolectin (cluster SST Chodl), appeared prominently in both datasets (Fig. 4F–G). Some ExSeq clusters of inhibitory neurons mapped onto multiple scRNA-Seq clusters. For example, two ExSeq clusters, which we denoted PV and GABAergic (-PV), mapped onto multiple scRNA-Seq clusters (Fig. 4E).
Such poolings of scRNA-Seq clusters into ExSeq clusters (and vice versa) are likely due to the smaller number of cells analyzed with ExSeq vs. scRNA-seq, the small number of markers interrogated, and the use of a simple k-means algorithm for clustering. Some substructure is visible in the t-SNE plot for the cluster GABAergic (−PV) (Fig. 4D). This suggests that alternative clustering approaches, for instance utilizing morphological criteria or protein markers, could be devised in the future to yield more precise delineations of cell types.
We varied the parameters used for cell segmentation of the ExSeq dataset and for clustering of the single-cell dataset, and found the above conclusions to be robust (Figs. S17–S18). Non-neuronal cells (e.g., glial cells) did not highly express the interrogated markers, and were likely non-specifically clustered with other cell types.
As described (60, 62), the layer-specific excitatory neuron transcription factor marker genes homeobox protein cut-like 2 (Cux2), RAR-related orphan receptor beta (Rorb), fasciculation and elongation protein zeta-2 (Fezf2) and forkhead box protein P2 (Foxp2) were expressed in L2/3, L4, L5b, and L6 respectively (Figs. 4E–F and S15). We used the clusters featuring these markers to segment the cortex into layers (Fig. S14D), so that the cell types within each layer could be quantified (Fig. 4G; raw counts, Fig. S19). Each cluster of inhibitory neurons was dispersed across layers (Fig. 4G and S19), consistent with earlier work (60, 62). Thus, targeted ExSeq enables sensitive RNA detection across circuit-relevant volumes of tissue, and enables cell types to be analyzed in spatial context.
Nanoscale RNA compartmentalization in mouse hippocampal neurons
We next used targeted ExSeq to explore nanoscale RNA compartmentalization within neurons of the mouse hippocampus, where dendritic RNAs are implicated in synaptic plasticity and learning (63–65). We traced YFP in neurons to identify dendrites and spines, and targeted for sequencing 34 transcripts previously found in CA1 neuron dendrites (66). We note that spines were not observed in the untargeted ExSeq hippocampus data because the antibody staining was performed post-sequencing, resulting in weaker staining, whereas here antibody staining was performed pre-expansion (28).
We performed four rounds of in situ sequencing to localize these transcripts on 170 fields of view (1.7mm x 1mm x 0.02mm total, Table S5). This sequencing spanned a coronal section containing subfields of the hippocampus, yielding 1.2 million reads, 90,000 of which localized within YFP expressing neurons (Fig. 5A and Table S13). The distributions of expressed genes were similar to those reported in the Allen Brain Atlas in situ hybridization dataset (Fig. S20).
Fig. 5. Targeted ExSeq characterization of nanoscale transcriptomic compartmentalization in mouse hippocampal neuron dendrites and spines.
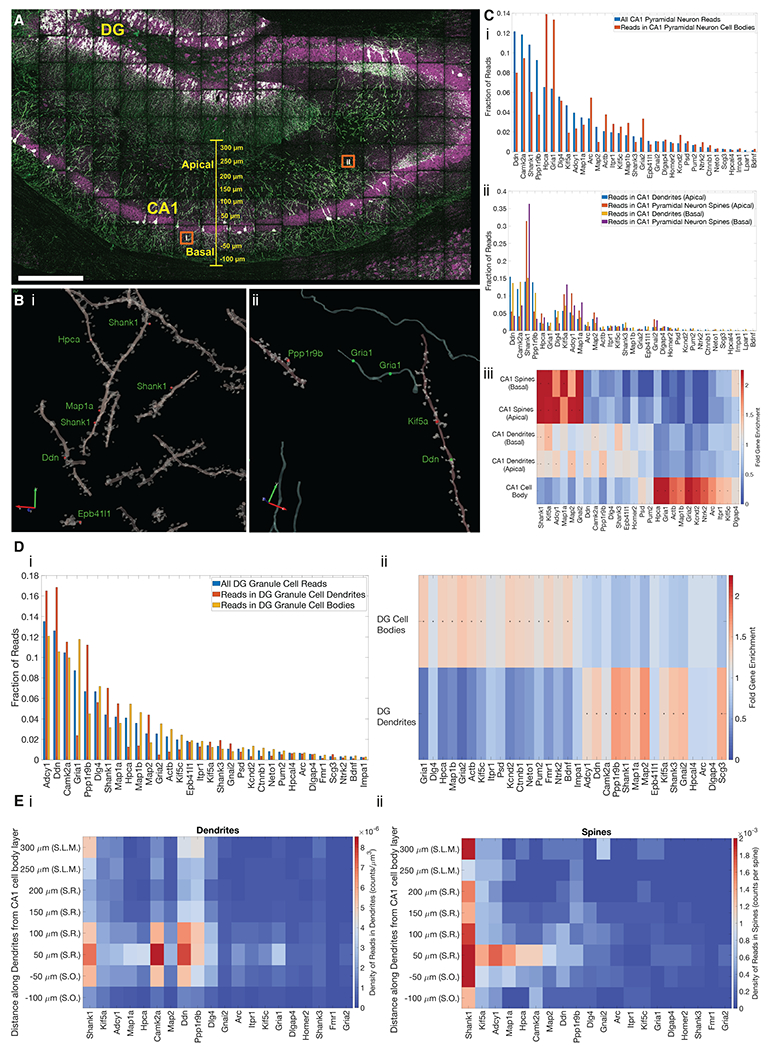
(A) Confocal image showing targeted ExSeq of a 34-panel gene set across slice of mouse hippocampus. Green, YFP; magenta, reads identified via ExSeq; white, reads localized within YFP-expressing cells. DG, dentate gyrus; CA1, CA1 region of hippocampus. (B) 3D reconstruction of dendrites, spines, and axons showing reads localized in spines (red dots) and processes (green dots) for regions indicated by orange boxes in A. (C) The abundance of transcripts in cellular compartments of CA1 pyramidal neurons: (i) abundance of transcripts in all cellular compartments vs. cell bodies; (ii) abundance of transcripts in apical and basal dendrites and spines; (iii) heat map showing the enrichment of transcripts in apical and basal dendritic and spine compartments of CA1 pyramidal neurons, vs. cell bodies; (*) indicates statistically significant enrichment (bootstrapped p-value <0.001). (D) The abundance of transcripts in cellular compartments of dentate gyrus (DG) granule cells; (i) abundance of transcripts in the cell bodies and dendrites of DG granule cells; (ii) heat map showing enrichment of transcripts in compartments of DG granule cells; (*) indicates statistically significant enrichment (bootstrapped p-value <0.001). (E) Plots showing the density of transcripts in the dendrites (i) and spines (ii) of CA1 pyramidal neurons along the apical-basal axis (Euclidean distance) of CA1, including regions S.R. (stratum radiatum), S.O. (stratum oriens), and S.L.M. (stratum lacunosum moleculare). Scale bars: A, 300 microns, Bi and Bii, 2 and 3 microns respectively, red and green arrows (pre-expansion).
Using the YFP signal, we segmented the CA1 pyramidal neurons and dentate gyrus granule cells (although the spines and axons of the latter exhibited low signal-to-noise and were not analyzed further). We found transcripts within dendrites (CA1, DG), spines (CA1), and to a much smaller extent, in axons (CA1) (Fig. 5B). In 106,000 spines examined, we found 730 reads in dendritic spines (each spine had one RNA, except for one that had two). Through simulations (Fig. S21), we concluded that it was unlikely that this sparsity of distribution was due to chemical artifacts of the ExSeq procedure.
In CA1 neurons, as expected, genes such as the postsynaptic density protein dendrin, the synaptic plasticity-associated gene Camk2a, and the postsynaptic scaffolding protein SH3 and multiple ankyrin repeat domains 1 (Shank1) were prominent in dendrites. The neuronal calcium sensor Hpca and the synaptic glutamate receptor Gria1 were amongst the most abundant in cell bodies (Fig. 5C). In spines, we found Shank1, Adenylyl cyclase 1 (Adcy1) and kinesin family member 5a (Kif5a) to be amongst the most abundant transcripts. We found that the distribution of reads in cell bodies, apical dendrites, basal dendrites, apical dendritic spines, and basal dendritic spines was each statistically different from the others (bootstrapped 2-sample Kolmogorov-Smirnov test p-value < 0.001), except for apical versus basal spines which were not different from each other (Fig. 5Ciii). This suggests a common set of spine RNAs and spine RNA trafficking principles throughout these neurons.
We validated these observations through bulk RNA sequencing from hippocampal slices adjacent (+/− 100 μm coronally) to the section used for targeted ExSeq. We observed a high level of correlation between in situ sequencing results and bulk RNA sequencing results (Pearson’s r = 0.85, Fig. S22). For the genes studied through both untargeted and targeted versions of ExSeq, we observed high correlation between the read counts (Pearson’s r = 0.68, Fig. S22 and Table S13). Using these genes, we estimated the yield of untargeted ExSeq to be 0.6% vs. targeted ExSeq (Table S13).
Specific genes were significantly (using bootstrapping, p-value <0.001) enriched in specific CA1 neuronal compartments (Fig. 5C). Transcripts for Shank1, Kif5a, Adcy1, Map1a, Map2, and Gnai2, were highly enriched in spines, and to a smaller extent in apical and basal dendrites, compared to cell bodies, perhaps pointing to a process through which these transcripts are enriched the closer they get to synapses. Many of these genes serve structural roles in spines and dendrites (67–69). On the other hand, a distinct set of genes, including Hpca, Gria1, ActB, and Map1b, among others, were highly enriched in cell bodies as compared to dendrites or spines, consistent with an earlier study (66). Interestingly, Arc, whose RNA is known to be dendritically targeted in plasticity contexts, was enriched in cell bodies, consistent with the highly regulated nature of its presence in dendrites (70, 71). In addition, a few genes, such as Camk2a and Ddn, were enriched in dendrites as compared to both spines and cell bodies, consistent with earlier work (66).
In dentate gyrus dendrites, we found transcripts similar to those found in CA1 apical and basal dendrites, such as Shank1, Map2, and Pppr1r9b (Fig. 5D). Across the entire 34 gene set, we observed similar dendritic localizations of RNAs in dentate gyrus granule cells vs. CA1 pyramidal neurons (Pearson’s r = 0.91, Fig. S23). This similarity raises the possibility that there may be general rules, applicable to multiple neuron types, governing the dendritic transport of specific RNAs.
Transcripts exhibit varied distributions along dendrites (66). We found that most transcripts within dendrites were close (+/− 50 μm) to the cell body layer, and their density decayed rapidly towards distal regions of dendrites, similar to previous observations for these genes (66) (Figs. 5Ei and S24).
Some transcripts, such as Shank1, Ddn, and Ppp1r9b were present in distal regions of dendrites. When we quantified the presence of transcripts within spines along dendrites, however, we observed a markedly different distribution (Figs. 5Eii and S24). For most transcripts found in spines among those in our probe set, their highest density occurred close to the cell body layer. However, spine-localized Shank1 transcripts exhibited a strong presence throughput spines in both proximal and distal regions of dendrites in both apical and basal directions. Kif5a and Adcy1, to a lesser extent, were also found in the spines of distal dendrites. Thus, although spines are directly connected to dendritic branches, they can exhibit strikingly different mRNA distributions.
ExSeq mapping of cell type relationships in cancer
We next explored how ExSeq might reveal spatial patterns of gene expression in the context of cancer biology and immunology. One key question is to understand how tumor microenvironments, including the state of immune cells, govern tumor growth, metastasis, and treatment resistance (72). Multiplexed spatial mapping of RNA performed in human tissues to date has not achieved high enough resolution for single cell quantification, let alone subcellular resolution (33, 73, 74) (Table S7).
A core biopsy was taken from a patient with metastatic breast cancer infiltration into the liver, and 297 tumor-related genes of interest (28) were profiled. We resolved 1.15 million reads, including 771,904 reads in 2,395 DAPI-segmented cells (Fig. 6A, counts in Table S14). The high 3D spatial resolution of ExSeq allowed the detection of 516 RNA reads inside nuclear structures <1 micron in size, possibly nucleoplasmic bridges, challenging structures to resolve in tissue (75) (Fig. S25).
Fig. 6. Targeted ExSeq resolves maps of cell types and states in cancer.
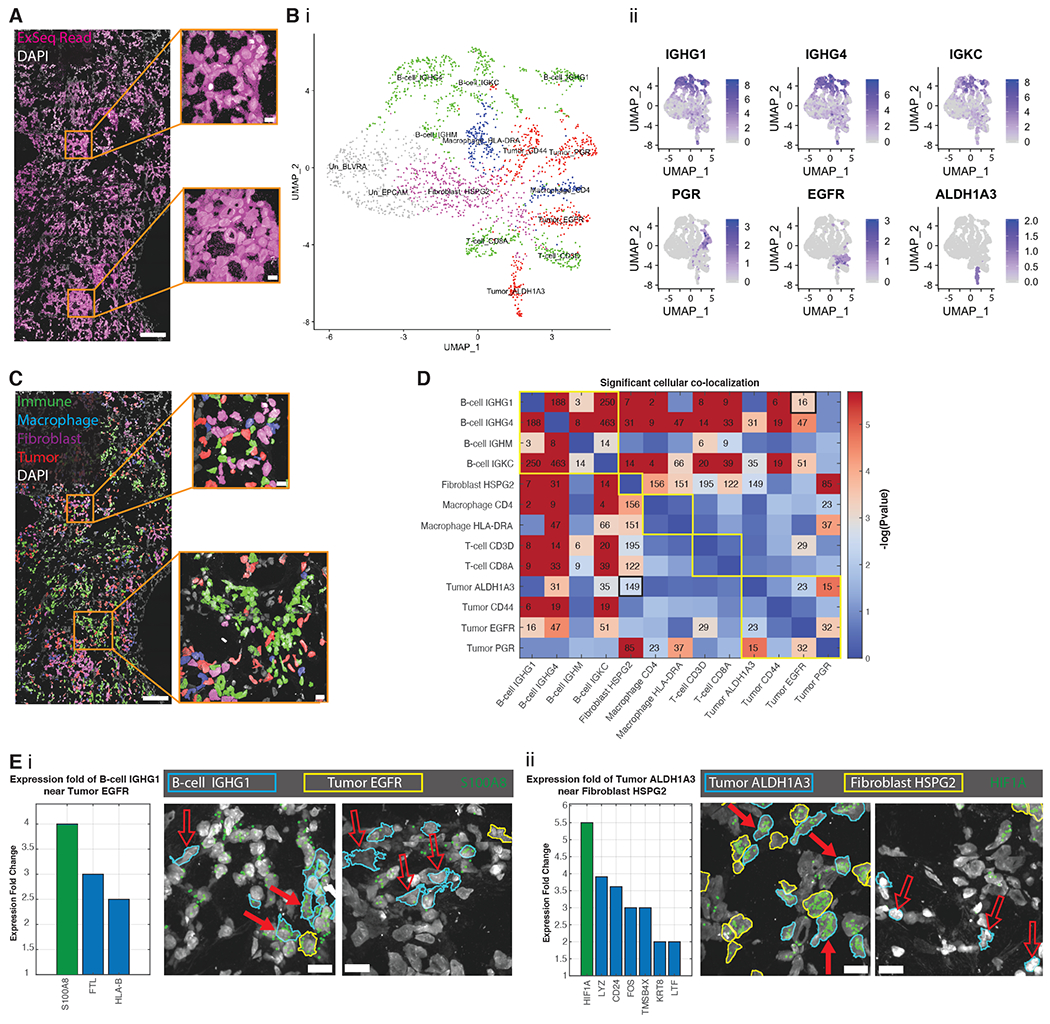
(A) ExSeq resolves 771,904 reads in 2,395 cells (with >100 reads/cell) of 297 genes in a metastatic breast cancer biopsy. (B) Uniform manifold approximation and projection (UMAP) representation of PCA-based expression clustering reveals immune and tumor cell clusters, indicated by different colors: green (T-cells, B-cells), red (tumor cells), blue (macrophages), magenta (fibroblasts) and gray (un-annotated clusters, (28)) (i), which express known cell markers for immune cells (ii, top row) and tumor cells (ii, bottom row); expression projected onto UMAP as log2(1+counts). (C) Transcriptionally-defined cell clusters mapped onto tissue context (colors as in B(i)). (D) Spatial colocalization analysis of cell clusters. Adjacency matrix text values, number of cell pairs of indicated type that are in close proximity (nucleus centroid distance of <20 ¼m; robustness analysis in Fig. S26). Adjacency matrix heatmap, p-value (500,000 bootstrapping iterations) relative to obtaining the same or higher number of cells in close proximity by chance. Adjacency matrix entries with text values are statistically significant (Benjamini Hochberg false-discovery rate of 1.5%). Yellow borders along the diagonal illustrate major cell type categories (B-cell, fibroblast, macrophage, T-cell, Tumor); two black-bordered entries correspond to pairs shown in (E). (E) ExSeq analysis of cell state as a function of physical proximity, measured by calculating differential expression when cells of different kinds are spatially adjacent (<20¼m) vs. far apart. The gene with the largest fold change in a specific cell type when adjacent versus non-adjacent to another specific cell type is shown in green in the histogram (p-value = 1e-4 using 100,000 bootstrapping iterations, all other genes shown in the histogram have p-value < 0.05), as well as in the image showing the gene’s read locations in the original sample. (i), fold-change of gene expression in IGHG1-positive B cells when in proximity to EGFR-positive tumor cells (B cells and tumor cells shown with blue and yellow boundaries, respectively). Solid arrows, cells in close proximity; hollow arrows, cells not in close proximity. (ii), fold-change of gene expression in ALDH1A3-positive tumor cells when in proximity to HSPG2-positive fibroblasts (tumor and fibroblast cells shown with blue and yellow boundaries, respectively). Scale bars: (A) and (C) 100 microns, and 10 microns for the insets, (E) 10 microns (pre-expansion).
Expression clustering of DAPI-segmented cells (28, 76) revealed the expected mixture of cell types, including tumor, immune (T-cell, B-cell, and macrophage), and fibroblast cell clusters, characterized with known biomarkers. These biomarkers include members of the immunoglobulin family (IGHG1, IGHG4, IGKC) found in B-cells, and genes known to be expressed in metastatic breast cancer (progesterone receptor, PGR (77); epidermal growth factor receptor, EGFR (78); aldehyde dehydrogenase 1 family member A3, ALDH1A3 (79)) (Fig. 6B).
Tumor and non-tumor cells were highly intermixed (Fig. 6C). We examined spatial colocalizations (proximity within 20 microns) between cell types (Fig. 6D; results were robust to distance parameter value (Fig. S26)). Different B-cell clusters tended to co-localize in space, consistent with previous observations (80). B-cell clusters exhibited statistically significant (using bootstrapping (28)) co-localizations to all the other cell clusters (Fig. 6D) except for one tumor cluster expressing the gene marker PGR (Tumor PGR). This is consistent with B-cells directly interacting with tumor cells and macrophages, with such interactions contributing to humoral responses in the microenvironment (80, 81). Our analysis also indicates other cell type co-localizations, for example between fibroblast clusters and macrophage, T-cell and tumor clusters (Fig. 6D). Such mappings thus may help illuminate the role of fibroblasts in supporting leukocyte aggregation at sites of cancer (82) or the spatial distributions of fibroblast cell types in cancers (83).
We finally analyzed whether one cell type could express genes differently as a function of physical proximity to another cell type. For example, one cell might change state depending on physical contact or close proximity to another cell. For each pair of cell clusters that exhibited colocalization, we searched gene expression differences between specific cells that were close (i.e, within 20 microns) vs. not close, using bootstrapping (28). Hypoxia-inducible factor (HIF1A) was >5-fold overexpressed in ALDH1A3-positive tumor cells when they were in close proximity to HSPG2-positive fibroblasts (Fig. 6Eii). Given that HIF1A serves as a proxy of hypoxic environments, and is a microenvironmental cue for tumor cell maintenance (84), ExSeq maps may be helpful for further probing such relationships. The mRNA level of HIF1A may also indicate tumor radiotherapy resistance (85). As a second example, the gene S100A8, a regulator of inflammatory processes and immune responses that may be a biomarker for relapse or progression in breast cancer patients (86–88), was 4-fold overexpressed in IGHG1-positive B cells when they were close to EGFR-positive tumor cells (Fig. 6Ei).
Discussion
ExSeq adapts two techniques -- expansion microscopy and in situ sequencing -- to enable spatially precise, highly multiplexed imaging of RNAs in cells and tissues. ExSeq, in both untargeted and targeted forms, facilitates the investigation of scientific questions involving subcellular, and even nanoscale, RNA localization in intact cellular and tissue contexts (e.g., as indicated by antibody staining of proteins or DAPI staining of nuclei). It can be applied to specimens of multiple organ systems and species, ranging from the mouse brain to human cancer biopsies, to reveal spatial relationships within, and between, cells. Such data may reveal principles of cellular organization and function, and provide insights into potential mechanisms of how cells interact, or are coordinated, in complex tissues and multicellular systems. We anticipate that beyond neuroscience and cancer biology, ExSeq will find uses in other fields where many cell types are operating within a complex tissue context, ranging from developmental biology, to immunology, to aging.
Beyond spatial genomics, we expect ExSeq to be useful for in situ sequencing of lineage (89) and/or connectome (90–92) indexing RNA barcodes, which incorporate designed or randomized base-level variation that is not naturally addressed by a FISH approach with a fixed set of tags and targets. More generally, the approaches for re-embedding, passivation, many-round sequential probing, image analysis and ex situ sequence matching in expanded samples that we have developed for ExSeq should be broadly applicable to other kinds of in situ enzymatic readouts, such as for the multiplexed readout of endogenous DNA or of antibody-attached tags, which may benefit from nanoscale spatial resolution in intact tissues.
Materials and methods
All tissues were fixed, optionally immunostained, and treated with the RNA anchoring reagent LabelX. The tissues were then gelled, digested, and expanded (23, 26). Next, the tissues were re-embedded and passivated, enabling enzymatic reactions to be performed in situ. For untargeted ExSeq, the in situ sequencing library was generated by performing reverse transcription with random primers, circularization of cDNA, and rolling circle amplification (RCA). For targeted ExSeq, padlock probes bearing barcodes were hybridized to transcripts of interest, circularized, and RCA-amplified. In situ sequencing of the cDNA amplicons was then performed through iterative rounds of sequencing chemistry and imaging. The imaging data was converted to nucleotide reads localized in 3D space by a custom image processing pipeline. Reads were ascribed to cells by using immunostaining or other morphological markers. For untargeted ExSeq, the reference for alignment of in situ reads was generated by extracting and sequencing the cDNA amplicons from the sample, enabling augmentation of the in situ read length. Full methods are available in (28).
Supplementary Material
Acknowledgments:
We acknowledge A. Lin for the help with Illumina sequencing, D. Park for providing cultured neurons, E. Murray for providing cultured HeLa cells, K. Piatkevich for performing transcardial perfusions, R. Kalhor for helpful discussions, and E. A. Pnevmatikakis for helpful discussions on image processing. We also acknowledge the SpaceTx analysis working group, for help in clustering: T. Bakken, Z. Yao, P. Kharchenko, and in gene selection: E. D. Vaishnav, B. Aevermann, R. Scheuermann, K. Harris.
Funding: R.E.K. and A.H.M. acknowledge IARPA MICrONS (D16PC0008); E.R.D. acknowledges government support under HG005550 and HG008525 awarded by the National Institutes of Health and under DGE1144152 awarded by the National Science Foundation; S.A. was supported by the HHMI fellowship of the LSRF; D.G. is NSF GRFP fellow; F.C. is supported by the Schmidt Fellows Program at the Broad Institute; A.T.W. was supported by the Hertz Foundation Fellowship and the Siebel Scholarship; A.S. was supported by the NIH Neuroimaging Training Program T32 grant 5T32EB001680; J.M. and E.L. thank the Chan Zuckerberg Initiative, an advised fund of Silicon Valley Community Foundation for their support (Award No.: 2017-0525). E.S.B. was supported by Lisa Yang, John Doerr, the Open Philanthropy Project, Cancer Research UK Grand Challenge grant C31893/A25050, U. S. Army Research Laboratory and the U. S. Army Research Office under contract/grant number W911NF1510548, NSF Grant 1734870, NIH UF1NS107697, NIH IDIQ17X149, NIH 1RM1HG008525, NIH 1R01MH103910, NIH 1R01MH114031, NIH 1R01MH110932, NIH 1R01EB024261, NIH 1R01DA045549, IARPA D16PC00008, NIH 1U19MH114821, NIH 1R01NS102727, the Chan Zuckerberg Initiative Human Cell Atlas pilot program, the Ludwig Foundation, the HHMI-Simons Faculty Scholars Program, and the HHMI investigator program. This project has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, Task Order No. HHSN261100039 under Contract No. HHSN261201500003I.
Footnotes
Competing interests: S.A., D.G., A.T.W., A.S., F.C., E.R.D., A.C.P., P.T., P.R., G.M.C., A.H.M. and E.S.B. are inventors on several patents relating to ExSeq. E.S.B. is a co-founder of Expansion Technologies, which has commercial interests in the space of expansion microscopy. F.C is a paid consultant of Celsius Therapeutics, and is a member of Celsius Thinklab. E.R.D. is a co-founder of ReadCoor, part of 10X Genomics, which has commercial interests in the space of expansion microscopy and FISSEQ.
Data and materials availability: All the raw Illumina sequencing data was deposited to NCBI Sequence Read Archive (SRA), BioProject PRJNA663046. The entire MATLAB pipeline to process ExSeq datasets from the microscope to spatial analysis of gene expression is publicly accessible at Zenodo (93). The linked wiki includes a tutorial with step-by-step instructions on how to run the pipeline, and a tutorial set of targeted ExSeq data from the mouse visual cortex. The 3D visualization tool is hosted at Zenodo (94).
References and Notes:
- 1.Crosetto N, Bienko M, van Oudenaarden A, Spatially resolved transcriptomics and beyond. Nat. Rev. Genet 16, 57–66 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Gregor T, Garcia HG, Little SC, The embryo as a laboratory: quantifying transcription in Drosophila. Trends Genet. 30, 364–375 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alon S, Huynh GH, Boyden ES, Expansion microscopy: enabling single cell analysis in intact biological systems. FEBS J. 286, 1482–1494 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moor AE et al. , Global mRNA polarization regulates translation efficiency in the intestinal epithelium. Science. 357, 1299–1303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halpern KB et al. , Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature. 542, 352–356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lein E, Borm LE, Linnarsson S, The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science. 358, 64–69 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Regev A et al. , The Human Cell Atlas. Elife. 6 (2017), doi: 10.7554/eLife.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moffitt JR et al. , Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science. 362 (2018), doi: 10.1126/science.aau5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shah S, Lubeck E, Zhou W, Cai L, In Situ Transcription Profiling of Single Cells Reveals Spatial Organization of Cells in the Mouse Hippocampus. Neuron. 92, 342–357 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X et al. , Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 361 (2018), doi: 10.1126/science.aat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ke R et al. , In situ sequencing for RNA analysis in preserved tissue and cells. Nature Methods. 10 (2013), pp. 857–860. [DOI] [PubMed] [Google Scholar]
- 12.Femino AM, Fay FS, Fogarty K, Singer RH, Visualization of single RNA transcripts in situ. Science. 280, 585–590 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Levsky JM, Shenoy SM, Pezo RC, Singer RH, Single-cell gene expression profiling. Science. 297, 836–840 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Franchini D-M et al. , Microtubule-Driven Stress Granule Dynamics Regulate Inhibitory Immune Checkpoint Expression in T Cells. Cell Rep. 26, 94–107.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Balagopal V, Parker R, Polysomes P bodies and stress granules: states and fates of eukaryotic mRNAs. Current Opinion in Cell Biology. 21 (2009), pp. 403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holt CE, Martin KC, Schuman EM, Local translation in neurons: visualization and function. Nat. Struct. Mol. Biol 26, 557–566 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Schuman EM, mRNA Trafficking and Local Protein Synthesis at the Synapse. Neuron. 23 (1999), pp. 645–648. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez AJ, Czaplinski K, Condeelis JS, Singer RH, Mechanisms and cellular roles of local protein synthesis in mammalian cells. Current Opinion in Cell Biology. 20 (2008), pp. 144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hafner A-S, Donlin-Asp PG, Leitch B, Herzog E, Schuman EM, Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science. 364 (2019), doi: 10.1126/science.aau3644. [DOI] [PubMed] [Google Scholar]
- 20.Eng C-HL et al. , Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature. 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan JT, Fink GR, Bartel DP, Excised linear introns regulate growth in yeast. Nature. 565, 606–611 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JH et al. , Highly multiplexed subcellular RNA sequencing in situ. Science. 343, 1360–1363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen F, Tillberg PW, Boyden ES, Expansion microscopy. Science. 347 (2015), pp. 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wassie AT, Zhao Y, Boyden ES, Expansion microscopy: principles and uses in biological research. Nature Methods. 16 (2019), pp. 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JH et al. , Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc 10, 442–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen F et al. , Nanoscale imaging of RNA with expansion microscopy. Nat. Methods. 13, 679–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang G, Moffitt JR, Zhuang X, Multiplexed imaging of high-density libraries of RNAs with MERFISH and expansion microscopy. Sci. Rep 8, 4847 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Detailed materials and methods are available as supplementary materials.
- 29.Kircher M, Heyn P, Kelso J, Addressing challenges in the production and analysis of illumina sequencing data. BMC Genomics. 12 (2011), doi: 10.1186/1471-2164-12-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li W, Freudenberg J, Miramontes P, Diminishing return for increased Mappability with longer sequencing reads: implications of the k-mer distributions in the human genome. BMC Bioinformatics. 15, 2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tillberg PW et al. , Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat. Biotechnol 34, 987–992 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng G et al. , Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 28, 41–51 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Svedlund J et al. , Generation of in situ sequencing based OncoMaps to spatially resolve gene expression profiles of diagnostic and prognostic markers in breast cancer. EBioMedicine. 48, 212–223 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kosik KS, Life at Low Copy Number: How Dendrites Manage with So Few mRNAs. Neuron. 92, 1168–1180 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Khaladkar M et al. , Subcellular RNA sequencing reveals broad presence of cytoplasmic intron-sequence retaining transcripts in mouse and rat neurons. PLoS One. 8, e76194 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saini H, Bicknell AA, Eddy SR, Moore MJ, Free circular introns with an unusual branchpoint in neuronal projections. Elife. 8 (2019), doi: 10.7554/eLife.47809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buckley PT et al. , Cytoplasmic Intron Sequence-Retaining Transcripts Can Be Dendritically Targeted via ID Element Retrotransposons. Neuron. 69, 877–884 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glanzer J et al. , RNA splicing capability of live neuronal dendrites. Proc. Natl. Acad. Sci. U. S. A 102, 16859–16864 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harada A, Teng J, Takei Y, Oguchi K, Hirokawa N, MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J. Cell Biol 158, 541–549 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cubelos B et al. , Cux1 and Cux2 regulate dendritic branching, spine morphology, and synapses of the upper layer neurons of the cortex. Neuron. 66, 523–535 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crino P et al. , Presence and phosphorylation of transcription factors in developing dendrites. Proc. Natl. Acad. Sci. U. S. A 95, 2313–2318 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Middleton SA, Eberwine J, Kim J, Comprehensive catalog of dendritically localized mRNA isoforms from sub-cellular sequencing of single mouse neurons. BMC Biol. 17, 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pancrazi L et al. , Foxg1 localizes to mitochondria and coordinates cell differentiation and bioenergetics. Proc. Natl. Acad. Sci. U. S. A 112, 13910–13915 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ueda H et al. , Prothymosin alpha-deficiency enhances anxiety-like behaviors and impairs learning/memory functions and neurogenesis. J. Neurochem 141, 124–136 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Tiedge H, Fremeau RT Jr, Weinstock PH, Arancio O, Brosius J, Dendritic location of neural BC1 RNA. Proc. Natl. Acad. Sci. U. S. A 88, 2093–2097 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muslimov IA, Banker G, Brosius J, Tiedge H, Activity-dependent regulation of dendritic BC1 RNA in hippocampal neurons in culture. J. Cell Biol 141, 1601–1611 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Hamblin MH, Yin K-J, The long noncoding RNA Malat1: Its physiological and pathophysiological functions. RNA Biol. 14, 1705–1714 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Briggs JA, Wolvetang EJ, Mattick JS, Rinn JL, Barry G, Mechanisms of Long Non-coding RNAs in Mammalian Nervous System Development, Plasticity, Disease, and Evolution. Neuron. 88, 861–877 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Hörtnagl H et al. , Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience. 236, 345–372 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han Y et al. , Up-regulation of Nob1 in the rat auditory system with noise-induced hearing loss. Neurosci. Lett 491, 79–82 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Schneider N, Meier M, Efficient in situ detection of mRNAs using the Chlorella virus DNA ligase for padlock probe ligation. RNA. 23, 250–256 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fürth D, Hatini V, Lee JH, In Situ Transcriptome Accessibility Sequencing (INSTA-seq). bioRxiv (2019), p. 722819. [Google Scholar]
- 53.Cartier AE et al. , Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J. Neurosci 29, 7857–7868 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lohman GJS, Zhang Y, Zhelkovsky AM, Cantor EJ, Evans TC Jr, Efficient DNA ligation in DNA-RNA hybrid helices by Chlorella virus DNA ligase. Nucleic Acids Res. 42, 1831–1844 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deng R, Zhang K, Sun Y, Ren X, Li J, Highly specific imaging of mRNA in single cells by target RNA-initiated rolling circle amplification. Chem. Sci 8, 3668–3675 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iyer EPR et al. , Barcoded oligonucleotides ligated on RNA amplified for multiplex and parallel in-situ analyses. bioRxiv (2018), p. 281121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi HMT et al. , Third-generation hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development. 145 (2018), doi: 10.1242/dev.165753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marinov GK et al. , From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res. 24, 496–510 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Torre E et al. , Rare Cell Detection by Single-Cell RNA Sequencing as Guided by Single-Molecule RNA FISH. Cell Syst. 6, 171–179.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tasic B et al. , Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci 19, 335–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van der Maaten L, Hinton G, Visualizing Data using t-SNE. J. Mach. Learn. Res 9, 2579–2605 (2008). [Google Scholar]
- 62.Lein ES et al. , Genome-wide atlas of gene expression in the adult mouse brain. Nature. 445, 168–176 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Ostroff LE, Fiala JC, Allwardt B, Harris KM, Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron. 35, 535–545 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Steward O, Worley P, Local synthesis of proteins at synaptic sites on dendrites: role in synaptic plasticity and memory consolidation? Neurobiol. Learn. Mem 78, 508–527 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Yoon YJ et al. , Glutamate-induced RNA localization and translation in neurons. Proc. Natl. Acad. Sci. U. S. A 113, E6877–E6886 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cajigas IJ et al. , The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron. 74, 453–466 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hung AY et al. , Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci 28, 1697–1708 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hirokawa N, Niwa S, Tanaka Y, Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 68, 610–638 (2010). [DOI] [PubMed] [Google Scholar]
- 69.Koleske AJ, Molecular mechanisms of dendrite stability. Nature Reviews Neuroscience. 14 (2013), pp. 536–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Steward O, Wallace CS, Lyford GL, Worley PF, Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron. 21, 741–751 (1998). [DOI] [PubMed] [Google Scholar]
- 71.Steward O, Worley PF, Selective targeting of newly synthesized Arc mRNA to active synapses requires NMDA receptor activation. Neuron. 30, 227–240 (2001). [DOI] [PubMed] [Google Scholar]
- 72.Chen DS, Mellman I, Elements of cancer immunity and the cancer–immune set point. Nature. 541 (2017), pp. 321–330. [DOI] [PubMed] [Google Scholar]
- 73.Ståhl PL et al. , Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 353, 78–82 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Rodriques SG et al. , Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. 363, 1463–1467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fenech M et al. , Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis. 26, 125–132 (2011). [DOI] [PubMed] [Google Scholar]
- 76.Stuart T et al. , Comprehensive Integration of Single-Cell Data. Cell. 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kitahara M et al. , HER2-Positive Conversion in a Metastatic Liver Focus in Late Recurrent Breast Cancer. Case Reports in Oncology. 12 (2019), pp. 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Masuda H et al. , Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat 136, 331–345 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marcato P et al. , Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. 29, 32–45 (2011). [DOI] [PubMed] [Google Scholar]
- 80.Garaud S et al. , Tumor infiltrating B-cells signal functional humoral immune responses in breast cancer. JCI Insight. 5 (2019), doi: 10.1172/jci.insight.129641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pipi E et al. , Tertiary Lymphoid Structures: Autoimmunity Goes Local. Front. Immunol 9, 1952 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nayar S et al. , Immunofibroblasts are pivotal drivers of tertiary lymphoid structure formation and local pathology. Proc. Natl. Acad. Sci. U. S. A 116, 13490–13497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bartoschek M et al. , Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun 9, 5150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Colwell N et al. , Hypoxia in the glioblastoma microenvironment: shaping the phenotype of cancer stem-like cells. Neuro. Oncol 19, 887–896 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cimmino F et al. , HIF-1 transcription activity: HIF1A driven response in normoxia and in hypoxia. BMC Med. Genet 20, 37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gebhardt C, Németh J, Angel P, Hess J, S100A8 and S100A9 in inflammation and cancer. Biochem. Pharmacol 72, 1622–1631 (2006). [DOI] [PubMed] [Google Scholar]
- 87.Zhong J-M et al. , Protein S100-A8: A potential metastasis-associated protein for breast cancer determined via iTRAQ quantitative proteomic and clinicopathological analysis. Oncol. Lett 15, 5285–5293 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang D et al. , Clinical Significance of Elevated S100A8 Expression in Breast Cancer Patients. Front. Oncol 8, 496 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kalhor R et al. , Developmental barcoding of whole mouse via homing CRISPR. Science. 361 (2018), 10.1126/science.aat9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen X, Sun Y-C, Church GM, Lee JH, Zador AM, Efficient in situ barcode sequencing using padlock probe-based BaristaSeq. Nucleic Acids Research. 46 (2018), pp. e22–e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peikon ID et al. , Using high-throughput barcode sequencing to efficiently map connectomes. Nucleic Acids Res. 45, e115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zador AM et al. , Sequencing the Connectome. PLoS Biology. 10 (2012), p. e1001411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Publicly accessible at Zenodo (2020). 10.5281/zenodo.4075515 [DOI]
- 94.Publicly accessible at Zenodo (2020). 10.5281/zenodo.4075649. [DOI] [Google Scholar]
- 95.Dell RB, Holleran S, Ramakrishnan R, Sample size determination. ILAR J. 43, 207–213 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Klapoetke NC et al. , Independent optical excitation of distinct neural populations. Nat. Methods. 11, 338–346 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chow BY et al. , High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 463, 98–102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen F, Tillberg PW, Boyden ES, Expansion microscopy. Science. 347, 543–548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Karmakar S et al. , Organocatalytic removal of formaldehyde adducts from RNA and DNA bases. Nat. Chem 7, 752–758 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hermanson GT, Bioconjugate Techniques (Academic Press, 2013). [Google Scholar]
- 101.Mitchell D 3rd et al. , In vivo RNA structural probing of uracil and guanine base-pairing by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC). RNA. 25, 147–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rouhanifard SH et al. , ClampFISH detects individual nucleic acid molecules using click chemistry-based amplification. Nat. Biotechnol (2018), doi: 10.1038/nbt.4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Harris C, Stephens M, in Alvey vision conference (Citeseer, 1988), vol. 15, pp. 10–5244. [Google Scholar]
- 104.Murray E et al. , Simple, Scalable Proteomic Imaging for High-Dimensional Profiling of Intact Systems. Cell. 163, 1500–1514 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lowe DG, Distinctive Image Features from Scale-Invariant Keypoints. Int. J. Comput. Vis 60, 91–110 (2004). [Google Scholar]
- 106.Scovanner P, Ali S, Shah M, in Proceedings of the 15th ACM international conference on Multimedia (ACM, 2007), pp. 357–360. [Google Scholar]
- 107.Bookstein FL, Principal warps: thin-plate splines and the decomposition of deformations. IEEE Trans. Pattern Anal. Mach. Intell 11, 567–585 (1989). [Google Scholar]
- 108.Schindelin J et al. , Fiji: an open-source platform for biological-image analysis. Nat. Methods. 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S, Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods. 5, 877–879 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li H et al. , The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Morgulis A, Gertz EM, Schäffer AA, Agarwala R, WindowMasker: window-based masker for sequenced genomes. Bioinformatics. 22, 134–141 (2006). [DOI] [PubMed] [Google Scholar]
- 113.Quinlan AR, Hall IM, BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ondov BD, Varadarajan A, Passalacqua KD, Bergman NH, Efficient mapping of Applied Biosystems SOLiD sequence data to a reference genome for functional genomic applications. Bioinformatics. 24, 2776–2777 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huang DW, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 116.Zhang J, Kobert K, Flouri T, Stamatakis A, PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 30, 614–620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Duerr JS, Immunohistochemistry. WormBook, 1–61 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stiernagle T, Maintenance of C. elegans. WormBook, 1–11 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Berger DR, Seung HS, Lichtman JW, VAST (Volume Annotation and Segmentation Tool): Efficient Manual and Semi-Automatic Labeling of Large 3D Image Stacks. Front. Neural Circuits. 12, 88 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.OpenFrameworks Community, OpenFrameworks (https://openframeworks.cc). [Google Scholar]
- 121.Nicovich PR et al. , Multimodal cell type correspondence by intersectional mFISH in intact tissues. bioRxiv (2019), p. 525451. [Google Scholar]
- 122.Tasic B et al. , Shared and distinct transcriptomic cell types across neocortical areas. Nature. 563, 72–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fang FC, Frawley ER, Tapscott T, Vázquez-Torres A, Bacterial Stress Responses during Host Infection. Cell Host & Microbe. 20 (2016), pp. 133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xu Q, Schlabach MR, Hannon GJ, Elledge SJ, Design of 240,000 orthogonal 25mer DNA barcode probes. Proc. Natl. Acad. Sci. U. S. A 106, 2289–2294 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Buschmann T, DNABarcodes: an R package for the systematic construction of DNA sample tags. Bioinformatics (2017), p. btw759. [DOI] [PubMed] [Google Scholar]
- 126.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Keren L et al. , A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell. 174, 1373–1387.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Qian X et al. , Probabilistic cell typing enables fine mapping of closely related cell types in situ. Nat. Methods. 17, 101–106 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ke R et al. , In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods. 10, 857–860 (2013). [DOI] [PubMed] [Google Scholar]
- 130.Codeluppi S et al. , Spatial organization of the somatosensory cortex revealed by osmFISH. Nat. Methods. 15, 932–935 (2018). [DOI] [PubMed] [Google Scholar]
- 131.Xia C, Babcock HP, Moffitt JR, Zhuang X, Multiplexed detection of RNA using MERFISH and branched DNA amplification. Sci. Rep 9, 7721 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Moffitt JR et al. , High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl. Acad. Sci. U. S. A 113, 11046–11051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vickovic S et al. , High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods. 16, 987–990 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kast RJ, Lanjewar AL, Smith CD, Levitt P, FOXP2 exhibits projection neuron class specific expression, but is not required for multiple aspects of cortical histogenesis. eLife. 8 (2019), , doi: 10.7554/elife.42012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gunnersen JM et al. , Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron. 56, 621–639 (2007). [DOI] [PubMed] [Google Scholar]
- 136.Tantirigama MLS et al. , Fezf2 expression in layer 5 projection neurons of mature mouse motor cortex. J. Comp. Neurol 524, 829–845 (2016). [DOI] [PubMed] [Google Scholar]
- 137.Zeng H et al. , Large-scale cellular-resolution gene profiling in human neocortex reveals species-specific molecular signatures. Cell. 149, 483–496 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Daigle TL et al. , A Suite of Transgenic Driver and Reporter Mouse Lines with Enhanced Brain-Cell-Type Targeting and Functionality. Cell. 174, 465–480.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rudy B, Fishell G, Lee S, Hjerling-Leffler J, Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol 71, 45–61 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.