

Abstract
Tyrosyl-DNA phosphodiesterase 1 (TDP1) is an enzyme crucial for cleavage of the covalent topoisomerase 1-DNA complex, an intermediate in DNA repair. TDP1 plays a role in reversing inhibition of topoisomerase I by camptothecins, a series of potent and effective inhibitors used in the treatment of colorectal, ovarian, and small-cell lung cancers. It is hypothesized that inhibition of TDP1 activity may enhance camptothecin sensitivity in tumors. Here, we describe the design, development, and execution of a novel assay to identify inhibitors of TDP1 present in natural product extracts. The assay was designed to address issues with fluorescent “nuisance” molecules and to minimize the detection of false-positives caused by polyphenolic molecules known to nonspecifically inhibit enzyme activity. A total of 227,905 purified molecules, prefractionated extracts, and crude natural product extracts were screened. This yielded 534 initial positives (0.23%). Secondary prioritization reduced this number to 117 (0.05% final hit rate). Several novel inhibitors have been identified showing micromolar affinity for human TDP1, including halenaquinol sulfate, a pentacyclic hydroquinone from the sponge Xestospongia sp.
Keywords: cancer and cancer drugs, enzyme assays or enzyme kinetics, natural products screening, tyrosyl-DNA phosphodiesterase
Introduction
DNA topoisomerases play a fundamental role in DNA metabolism and chromatin structure by removing DNA knots and catenanes and relaxing DNA supercoiling in a manner conducive to further processing by additional enzymes.1 Without the constant intervention of DNA topoisomerases, the accumulation of positive DNA supercoiling leads to fork stalling while the accumulation of negative supercoiling contributes to the formation of abnormal DNA structures. Therefore, topoisomerases are key pharmacological targets for the treatment of cancers. Topoisomerase I (TOP1) inhibitors are among the most potent anticancer drugs with clinical utility against lymphomas, cervical, ovarian, breast, small-cell lung, and colorectal cancer, among others.1–3
The TOP1-DNA covalent complex is the therapeutic target of irinotecan and topotecan, which are water-soluble derivatives of camptothecin (CPT), a natural product inhibitor originally isolated from the bark of Camptotheca acuminate.4 As part of its normal catalytic cycle, TOP1 binds to supercoiled DNA, conducts strand breakage, religation, and departs from the relaxed strand. In the presence of external intervention from an inhibitor, TOP1 release from the DNA substrate is not possible, and TOP1 stalls midprocess,5 resulting in a covalently attached complex. Formation of these complexes prevents further processing until the complex is removed. To reverse this occurrence, several mechanisms to remove covalently linked TOP1 from the damaged DNA strand have evolved using additional DNA repair enzymes, including tyrosyl-DNA phosphodiesterase 1 (TDP1).6–8
TDP1 is a member of the phospholipase D family of enzymes7 that catalyzes cleavage of the 3′-phospho-tyrosyl bond linking the active site tyrosine in TOP1 and the phosphate of the DNA substrate backbone in the stalled complex.8 TDP1 removes the tyrosyl moiety, allowing subsequent repair of the strand break by polynucleotide kinase phosphatase and DNA ligase III. TDP1 was originally shown to play a role in DNA repair in Saccharomyces cerevisae9 and later implicated in spinocerebellar ataxia with axonal neuropathy (SCAN1), a recessive hereditary disorder caused by an H493R active site mutation in the human enzyme.10 Cell lines derived from SCAN1 patients show enhanced sensitivity to treatment with CPT due to severely decreased levels of TDP1 catalytic activity,11 suggesting a potential opportunity for pharmacological intervention to enhance the activity of CPTs.
There has been increasing interest in identifying inhibitors of TDP1 as a means of sensitizing cells to TOP1 inhibitors, such as topotecan and irinotecan.12 One report showed that the synthetic TDP1 inhibitor furamidine is synergistic with irinotecan in an in vivo model of murine lupus nephritis.13 Further utility for TDP1 inhibitor adjunct therapy was also found with other chemotherapeutic agents such as bleomycin, which causes oxidative DNA damage that can be cleared by TDP1,14,15 and anticancer nucleoside analogs such as cytarabine, which cause chain-terminating deoxyribo- or ribonucleotides, which can be removed by TDP1’s nucleosidase activity.16
Natural products have been developed into a large number of pharmacologically important drugs. Since the 1980s, approximately 40% of all approved antitumor drugs worldwide have been either natural products or derived from natural product pharmacophores.17 To date, a number of efforts have been made to identify synthetic chemical inhibitors of TDP1 activity in vitro with varying degrees of success.18–20 As TDP1 is conserved throughout much of evolution,10 we sought to exploit the possibility that natural molecules might have evolved to interact with this protein. Here, we describe the first reported attempt to develop a suitable assay platform to identify, isolate, and characterize natural product inhibitors of TDP1 catalytic activity from crude natural product extracts. The assay was used to identify several natural TDP1 inhibitors, including the marine sponge metabolite halenaquinol sulfate, which is a covalent inhibitor of TDP1.
Materials and Methods
Reagents
Both fluorophore-labeled 14Y oligo substrates used in this study were purchased from The Midland Certified Reagent Company Inc. (Midland, TX).
Purified human TDP1 proteins (both full-length and truncated missing the first 149 amino acids) were provided by the Protein Expression Laboratory (James Hartley, Dominic Esposito, and William Gillette, Leidos Biomedical Research Inc., Frederick, MD). Full-length TDP1 as reported by Champoux1 was used for all assay procedures. TDP1 proteins (>95% pure) were stored in 20 mM HEPES, 50 mM KCl, 2 mM DTT, 1 mM EDTA, and 50% glycerol at pH 8.0 and at a concentration of 40 μM. Unless otherwise stated, all assays were carried out in 1X phosphate-buffered saline (PBS) at pH 8.0 containing 80 mM KCl, 1 mM tris(2-carboxyethyl)phosphine, 10% DMSO, 0.05% Tween 20, and 0.01% bovine serum albumin (BSA).
Characterization of Substrate Molecules prior to Use
Prior to use of rTDP1 for the screening and characterization of new inhibitor molecules, the Km for both Förster resonant energy transfer (FRET) and biotinylated TDP1 substrates was measured by establishing a twofold dilution series from 0 to 2 μM (screening substrate) and 0 to 5 μM (FRET substrate) over 11 dilution steps. Experiments were carried out in appropriate assay plates depending on the substrate immobilization requirement, in the presence of 500 pM TDP1, at a final volume of 100 μL. For the biotinylated screening substrate, separate control samples were set up lacking TDP1 to allow a measure of the fluorescence of the fully intact eosin fluorophore. To ensure linear kinetics during the course of this experiment, all reactions using the screening substrate were run for 30 min at 25 °C prior to quenching. For the FRET oligo substrate, reaction rates were measured directly from linear time course data collected at each concentration of substrate, with inner filter correction carried out prior to data analysis. Data were collected using a Tecan Safire plate reader (Tecan US, Durham, NC) at excitation and emission wavelengths of 525 nm and 545 nm, respectively. The Km and kcat values for both substrates were estimated by fitting a variant of the Michaelis-Menten equation (Eq. 1) to both data sets using Graphpad Prism (GraphPad Software, San Diego, CA).
| (1) |
where V is the reaction velocity, [E] the total enzyme concentration, [S] the concentration of substrate, kcat the turnover number, and Km the Michaelis constant. The cleavage of the eosin tag from the biotinylated screening assay substrate was confirmed by high-resolution liquid chromatography/mass spectrometry (LC/MS), which clearly showed the TDP1-dependent loss of 868Da from the substrate (corresponding to the mass of the eosin tag).
Preparation of Natural Product Extracts
All crude natural product source material was obtained from the Natural Products Branch of the National Cancer Institute (NCI) Developmental Therapeutics Program. Crude extract material was collected and prepared using a standardized procedure21 prior to delivery to the Molecular Targets Laboratory. A selection of ~12,000 crude natural product extracts from the NCI Natural Products Repository were subjected to a prefractionation process in an effort to reduce the level of assay false-positives resulting from interference by common nonspecific nuisance compounds as previously described.22
The natural products halenaquinol sulfate, halenaquinone, and xestoquinone were isolated from an organic extract of the sponge Xestospongia sp. The natural product 2,3-epoxy-8-hydroxy-lapachol was isolated from the stems of the Madagascaran plant Barleria alluaudii. Compound identifications for all natural products were determined by comparison of nuclear magnetic resonance spectroscopy and mass spectrometry data for the isolated molecules and results published previously for the halenaquinone series23 and 2,3-epoxy-8-hydroxy-lapachol.24
Endpoint HTS Assay for Detection of TDP1 Inhibitors
The screening assay was carried out in 384-well polypropylene plates (Greiner No. 781280 from Sigma-Aldrich, St. Louis, MO) at a volume of 25 μL. The assay used a 5′-biotinylated substrate oligo that had been labeled with a 3′-eosin fluorescent group. Eosin has absorption and emission peaks at 525 nm and 545 nm, respectively. TDP1 enzyme and substrate were present at 500 pM and 500 nM, respectively. Sample screening was carried out at differing concentrations of inhibitor, depending on the source material being screened. For purified molecules, a final concentration of 10 μM was used. For prefractionated and crude natural product extracts, a final concentration of 50 μg/mL total material was used in all cases. Positive and negative control wells were included on each screening plate containing 10 mM sodium vanadate quencher or assay buffer, respectively. For all wells, DMSO was present at 10% v/v. All compounds and extracts were initially tested at a single point during primary screening.
To each well of a 384-well plate, 10 μL of a 1.25 nM TDP1 stock was added, followed by 5 μL of a 50 μM or 250 μg/mL inhibitor stock (or relevant control solutions). To start the reaction, 10 μL of a 1.25 μM substrate solution was added to each well and mixed. The plates were incubated for 50 min at 25 °C, followed by addition of 10 μL per well of a 50 mM sodium vanadate quencher solution. The contents of each well were then transferred to a black polystyrene streptavidin-coated 384-well microtiter plate (Pierce No. 15506) and allowed to incubate at 25 °C for 5 min. The wells were washed three times using a BioTek ELx405 plate washer (BioTek, Winooski, VT) to remove cleaved fluorescent tag and other nonimmobilized reaction constituents, and the remaining nonutilized fluorescent substrate was measured at 550 nm using a BMG PHERAstar Plus plate reader (BMG LABTECH Inc., Durham, NC) equipped with a fluorescence intensity 520/550 optical module. Samples displaying at least 50% inhibition or crude extracts displaying inhibition greater than three times the standard deviation of the mean of all extracts from similar source materials were identified for further confirmation.
Confirmation of Hits from Primary Screening
Compounds and extracts displaying inhibitory activity in the primary screening assay (>3 standard deviations away from the mean of all samples tested from that source group) were subjected to a confirmation protocol involving three assay systems. Using the primary screening assay itself, initial positives were primarily confirmed by quadruplicate dose response at concentrations spanning 100 μg/mL to 0.78 μg/mL for natural product extracts or 20 μM to 0.16 μM for purified source material across eight dilutions. Where source material was scarce, confirmation was carried out via quadruplicate single dose at the original screening concentration of 50 μg/mL.
Compounds and extracts confirmed in the primary screening assay were then subjected to further evaluation using additional gel-based TDP1 activity assays displaying an orthogonal radiometric readout. In this assay, 5′−32P–labeled TDP1 DNA substrate (1 nmol/L; N14Y; 5′-GATCTAAAAGACTT-pY-3′) was incubated with 20 pM of recombinant full length or 3 pM of truncated (Δ1–149) TDP1 in the absence or presence of inhibitor for 15 min at 25 °C in an assay buffer containing 50 mM Tris HCl, pH 7.5, 80 mM KCl, 2 mM EDTA, 1 mM DTT, 40 μg/mL BSA, and 0.01% Tween-20. Reactions were terminated by the addition of two volumes of gel loading buffer (99.5% [v/v] formamide, 5 mmol/L EDTA, 0.01% [w/v] xylene cyanol, and 0.01% [w/v] bromophenol blue). Samples were subjected to a 16% denaturing polyacrylamide gel electrophoresis, and gels were exposed after drying to a PhosphorImager screen (GE Healthcare). Gel images were scanned using a Typhoon 8600, and densitometric analyses were performed using the ImageQuant software package (GE Healthcare).
A second gel-based assay measured inhibition of TDP1 in the presence of cellular constituents. In this assay, 1 × 107 DT40 knock-out cells complemented with full-length human TDP1 were collected and washed once with PBS. The supernatant was removed, and the pellet was resuspended with 50 μL of CellLytic M Cell Lysis Reagent (SIGMA C2978), pipetting several times over ice. After 15 min, the lysate was centrifuged at a 12,000 g for 10 min, and the supernatant was transferred to a new tube. Protein concentration was determined using the Protein Assay Dye Reagent Concentrate (Bio-Rad No. 500–0006). 5′−32P–labeled DNA substrate (1 nmol/L; N14Y; 5′-GATCTAAAAGACTT-pY-3′) was incubated with 125 μg/mL of cell extract in the absence or presence of inhibitor for 15 min at 25 °C in the same assay buffer as above. Reactions were terminated and visualized as with the gel-based assay described above.
In a final assay to determine the presence of false-positives inhibiting the assay by interacting with the fluorescent screening substrate, confirmed hits were incubated at their screening concentration with 2 μM substrate for 30 min at 25 °C in nonbinding black fluorescent plates (Corning Costar No. 3915) at a final volume of 100 μL. The emission fluorescence of the eosin fluorphore was then measured using a Tecan Safire plate reader (Tecan US) at excitation and emission wavelengths of 525 nm and 545 nm, respectively. Comparative emission levels for wells containing inhibitor only, substrate only, and mixed inhibitor and substrate were measured. Inhibitorcontaining wells displaying an enhancement of fluorescence were tagged as working via a substrate-binding mechanism.
Continuous FRET Assay for Mechanistic Characterization of Isolated Inhibitors
A variation of the screening assay employing FRET was developed for mechanistic characterization of isolated hit molecules. To measure time course rate data in solution, a substrate was designed containing eosin and TAMRA fluorophores at opposite ends of the oligo molecule. TAMRA absorbs and emits at 546 nm and 574 nm, respectively. Absorption at 546 nm overlaps with the emission of eosin at 545 nm, quenching the emission signal at this wavelength. Cleavage of the substrate by TDP1 yields an increase in eosin fluorescence as quenching by TAMRA is mitigated. Use of this substrate facilitated continuous measurement of reaction rates. All assays using the FRET substrate were carried out in nonbinding, black fluorescent assay plates at 28 °C, as this was the constant temperature capability of the plate reader (room temperature + 3 °C). Prior to final analysis, FRET assay data were corrected for inner filter signal quenching using a standardized method.25 More specific details on the individual uses of this assay are described below.
IC50 Determination of Inhibitors of TDP1
To determine the potency of compounds inhibiting TDP1, a series of 12-point, two-fold serial dilutions were used to generate dose-response curves at inhibitor concentrations ranging from 200 μM down to 195 nM plus a no-inhibitor control. Final concentrations of TDP1 and FRET substrate were 500 pM and 1.5 μM, respectively, in all assay wells. Dose-response experiments were carried out in a final assay volume of 15 μL.
Under experimental conditions, 5 μL each of 1.5 nM TDP1 and 3X inhibitor were added to wells on the plate. After incubation at 4 °C for 1 h, 5 μL of a 4.5 μM FRET substrate solution was added to start the reaction. Fluorescence time course data were collected using a Tecan Safire plate reader (Tecan US) at excitation and emission wavelengths of 525 nm and 545 nm, respectively, for 1 h after addition of substrate at 28 °C. Rate data were normalized as percentage enzyme activity relative to the no-inhibitor control rate and analyzed using an appropriate equation (Eq. 2) in GraphPad Prism (GraphPad Software, San Diego, CA) as shown below.
| (2) |
where IC50 is the concentration of inhibitor giving 50% inhibition of enzyme activity, [I] is the concentration of inhibitor, and Hillslope is the slope factor of the dose-response curve.
Reversibility
Measurement of the reversibility of compound inhibition was carried out using a combination of TDP1 preequilibration at high inhibitor concentration, followed by rapid dilution to induce recovery of enzyme activity.26 Incubation samples were set up in assay buffer by combining 50 nM TDP1 with inhibitor at 10X IC50 concentration. The samples incubated on ice for 1 h to allow for equilibrium between both binding partners. Under these concentration conditions, approximately 90% of TDP1 activity is inhibited. To induce recovery of activity, samples were then diluted 100-fold into assay buffer containing 6 μM FRET substrate, reducing the concentrations of TDP1 and inhibitor to 500 pM and 0.1X IC50, respectively. Time course emission data were collected using a Tecan Safire plate reader as described previously. All reversibility experiments were carried out in 96-well, black fluorescent assay plates (Corning Costar No. 3915) at a final volume of 100 μL.
Determination of Compound Mechanism of Inhibition
Compound mechanism of inhibition experiments were carried out by measuring the differing reaction rates produced by TDP1 under a matrix of varying substrate and inhibitor concentrations. FRET substrate concentration was varied from 2.25 μM down to 0.035 μM over eight twofold dilution steps. Inhibitor was present at three concentrations representing 0.33X IC50, 1.0X IC50, and 3.0X IC50. A no-inhibitor sample was also included to allow measurement of the rate of reaction in the absence of inhibition.
For each experiment, separate dilution series of substrate and compounds were created at 3X their final required concentration. Five microliters each of 1.5 nM TDP1 and 3X inhibitor were transferred to the appropriate wells on the assay plate and allowed to incubate on ice for 1 h. After incubation, 5 μL of the 3X substrate solution was added to the plate to initiate the reaction. The plates were placed in a Tecan Safire plate reader, and time course data were collected for approximately 1 h at 28 °C. TDP1 was present at a final fixed concentration of 500 pM for all wells on the plate.
Experimental data were plotted as reaction rate versus substrate concentration for each inhibitor concentration and analyzed using models for competitive, noncompetitive (pure and mixed), and uncompetitive inhibition Eq. 3A–D using GraphPad Prism. To identify the most appropriate mechanistic model describing the inhibition data for each inhibitor, Akaike’s Information Criterion27 was employed.
Competitive Inhibition
| (3A) |
where Vmax is the maximum reaction velocity and Ki is the inhibition constant.
Pure Noncompetitive Inhibition
| (3B) |
where Ki is the inhibition constant in the presence or absence of substrate.
Mixed Noncompetitive Inhibition
| (3C) |
where Kie is the inhibition constant for binding to the enzyme in the absence of substrate and Kies the inhibition constant for binding to the ES complex.
Uncompetitive Inhibition
| (3D) |
where Kies is the inhibition constant for binding to the ES complex.
Measurement of Direct Binding of Compounds to TDP1 by Intrinsic Tryptophan Quenching
Measurement of compound binding was conducted by observing the decrease in tryptophan fluorescence emission intensity as compound was titrated into a cuvette containing a fixed concentration of TDP1. Binding experiments were carried out in a quartz cuvette containing 3 mL of standard assay buffer lacking BSA. TDP1 enzyme was present at a concentration of 100 nM. To this, a total of 15X 0.6 μl aliquots of 20 mM compound stock (in 100% DMSO) were titrated into the cuvette. The cuvette contents were allowed to equilibrate for 5 min between addition of each aliquot and measurement of the resultant fluorescence intensity level. Fluorescence measurements were carried out at 25 °C using an AMINCO-Bowman Series 2 (AB2) spectrofluorometer (Thermo Fisher Scientific, Waltham, MA) at excitation and emission wavelengths of 295 nm (slit width 2 nm) and 340 nm (slit width 16 nm), respectively. The data were corrected for inner filter effect signal quenching via an established correction method.28 Corrected data were analyzed using GraphPad Prism software by first calculating the concentration of the enzyme-ligand (EL) complex using a quadratic root
| (4) |
where Et is the total concentration of TDP1 enzyme, Lt is the concentration of compound, and Kd is the equilibrium dissociation constant. This permitted calculation of fluorescent values at each concentration of compound using the following expression:
| (5) |
where F is the calculated fluorescence value, Fo is the starting fluorescence of TDP1 in the absence of compound, and Famp is the total fluorescence amplitude decrease observed in the presence of infinite compound.
Results
Design of the Fluorescent 14Y Oligo Substrates
The biotinylated, fluorescent substrate used for primary inhibitor screening (Fig. 1A) was similar to a substrate previously used to screen synthetic compounds for TDP1 inhibition.18 The necessity to reduce false-positives caused by lower-wavelength fluorescing nuisance compounds in natural product extracts directed the selection of eosin (λex = 525nm, λem = 545nm) as a fluorophore (Fig. 1B). The addition of a 5′-biotin moiety allowed for attachment of the substrate and product to streptavidin-coated assay plates, which enabled a wash step to be added. The combination of the longer wavelength of visualization and the wash step reduced the occurrence of fluorescent interference on the assay results (data not shown).
Figure 1.

Design of tyrosyl-DNA phosphodiesterase 1 (TDP1) substrates used in this study. (A) The natural cellular substrate of TDP1 was used as a model for the design of two fluorescent substrates. (B) The presence of a 5′-biotin moiety on the screening substrate allowed for immobilization on streptavidin-coated plates. This facilitated the inclusion of a postreaction wash step to remove cleaved 3′-eosin fluorescent product prior to measuring residual fluorescent signal from the remaining intact substrate. (C) By replacing the 5′-biotin moiety with a TAMRA fluorphore, a Förster resonant energy transfer substrate was designed for use during inhibitor characterization. TAMRA quenches the fluorescence emission signal in the intact substrate molecule. Cleavage of the substrate by TDP1 removes this interference, producing an increase in eosin emission signal that can be monitored continuously.
The biotinylated substrate is cleaved in solution in the presence of TDP1 and a natural product extract. After transferring the quenched reaction mixture to a streptavidin-coated plate, the intact substrate and the nonfluorescent 5′-end of the reaction product adhere to the plate. Immobilization facilitates removal of the cleaved 3′-fluorescent end of the reaction product, allowing measure of the remaining fluorescence derived from residual unreacted substrate. The wash step also plays a crucial role in the removal of fluorescent or quenching natural molecules from the well that might interfere with active compound determination.
For additional kinetic analysis of active molecules, a second substrate was developed by replacing the 5′-biotin with a TAMRA fluorophore (Fig. 1C). TAMRA (λex = 546 nm, λem = 574 nm) absorbs strongly around the maximum emission wavelength for eosin and quenches the eosin emission signal when the substrate molecule is intact. Cleavage of the phospho-tyrosyl bond allows the opposing fluorescent groups to separate, reducing interference by TAMRA and producing a sharp increase in the eosin emission signal. Use of this substrate facilitated continuous observation of TDP1 catalytic activity in solution.
Kinetic analysis of both assay substrates (Fig. 2) with TDP1 resulted in Km values of 3.7 ± 0.1 μM and 1.3 ± 0.1 μM for screening (Fig. 2A) and FRET substrates (Fig. 2B), respectively. Turnover numbers (kcat) were 181.6 ± 6.8 s−1 (screening) and 120.5 ± 2.2 s−1 (FRET), producing kcat/Km ratios of 5.0 × 107 s−1.M–1 and 9.4 × 107 s−1.M−1, respectively. Under the finalized screening concentrations of 500 pM TDP1 and 500 nM screening substrate, a greater than 50% reduction in absolute fluorescent signal was routinely observed over the course of a 50-min incubation at 25 °C (Fig. 3A, B) while yielding acceptable linearity and signal to noise. When tested in 384-well plates under assay conditions, Z′ values of 0.6 to 0.8 were routinely achieved (Fig. 3C).
Figure 2.

Tyrosyl-DNA phosphodiesterase 1 (TDP1) assay substrate kinetics. (A) Data were generated for the eosin-labeled screening substrate by measuring the linear decrease in residual fluorescence after incubation with 500 pM TDP1 for 30 min. Concentration of substrate ranged from 1.95 nM to 2 μM. A series of matching control dilution wells lacking TDP1 were used to measure the fluorescent signal derived from fully intact substrate. These values were used to calculate the rate of product formation over the assay time frame. (B) Data for the FRET substrate were generated by measuring the linear reaction rate from time course data in the presence of 1 nM TDP1 over a period of up to 1 h at 28 °C. The concentration of FRET substrate ranged from 9.76 nM to 5 μM. The rate of product formation was calculated using a standard curve of product fluorescence.
Figure 3.

Tyrosyl-DNA phosphodiesterase 1 (TDP1) screening assay performance. (A) The screening assay was initially examined for suitability in the detection of inhibitors of TDP1 catalytic activity. Representative data show residual fluorescence values for wells containing 500 nM substrate and 500 pM TDP1 in the absence (■) and presence (▲) of 10 mM vanadate inhibitor after incubation for 50 min. The Z′ value for these data is 0.74, with coefficient of variation (CV) values of 4.48% and 3.08% for uninhibited and inhibited wells, respectively. (B) In the presence of natural product extracts, the assay was well behaved, yielding a low number of extracts displaying significant inhibition (>50%) of TDP1 activity. The example data shown above are from a single 384-well plate containing 352 individual extract sample wells (◆), 16 fully inhibited control wells (●), and 16 uninhibited control wells (■). The Z′ value for this data set was 0.64, with CV values of 6.34% and 4.80% for uninhibited and inhibited wells, respectively. (C) Overall screening data quality was monitored via routine calculation of the Z′ value for each assay plate. The data displayed above shows Z′ values for all 387 natural product extract plates screened over the course of several months. The average Z′ value is 0.62, with the majority of the plates displaying Z′ values significantly above the lower limit of 0.4.
Development, Optimization, and Validation of an Assay to Detect TDP1 Inhibitors in Crude Natural Product Extracts
For the TDP1 assay, several features were included to reduce interference by common nuisance molecules found in natural product extracts: (1) A fluorescent label that absorbed and emitted light toward the red end of the spectrum was used, as this has been reported to be beneficial in removing interference from nuisance organic compounds.29 (2) Immobilization of the substrate and 5′-end of the product on a streptavidin-coated plate facilitated a wash step to selectively remove complicating fluorescent or quenching molecules, and reaction product, from each well. (3) Biochemical assays, where the active assay protein is the only proteinaceous component present, are vulnerable to inactivation by nonspecific protein-binding molecules such as tannins. It has been reported that the presence of an excipient protein can reduce the level of this interference.30 Bovine serum albumin was added to the screening assay buffer at a concentration of 0.01% w/v to reduce the effect of these compounds on TDP1. (4) Compound-induced aggregation/sequestration of active enzyme can lead to the identification of nuisance compounds in biochemical assays; here, we added the detergent Tween 20 (0.05%) to reduce the likelihood of identifying aggregating compounds as hits. These targeted adaptations for natural product extracts enabled screening at a crude extract concentration of 50 μg/mL total material, a concentration higher than initially thought possible. At this concentration, the rate of initial positive identification remained less than 1% (Table 1), indicating some success in suppressing the typical sources of false-positive hit rates observed during natural product extract screening.
Table 1.
Results of Screen for Inhibitors of Tyrosyl-DNA Phosphodiesterase 1.
| Total Screened | Initial Positives | Confirmed Hits | Final Hit Rate (%) | |
|---|---|---|---|---|
| Synthetic compounds | 8258 | 30 | 9 | 0.1 |
| Pure natural products | 4043 | 43 | 10 | 0.25 |
| Prefractionated extracts | 71,642 | 113 | 59 | 0.1 |
| Crude extracts | 143,962 | 348 | 39 | 0.03 |
| Plants: 106,738 | Plants: 149 | Plants: 3 | Plants: 0.003 | |
| Algal: 2464 | Algal: 34 | Algal: 9 | Algal: 0.36 | |
| Fungal: 19,272 | Fungal: 87 | Fungal: 17 | Fungal: 0.09 | |
| Marine: 15,488 | Marine: 78 | Marine: 10 | Marine: 0.06 | |
| Total | 227,905 | 534 | 117 | 0.05 |
The resulting assay produced reliable activity data (Fig. 3A). A selection of representative crude natural product extracts was tested to uncover any potential issues remaining with the assay that might affect the ability to detect natural product inhibitors (Fig. 3B). These data confirmed that the assay was suitable for crude extract screening, yielding a Z′ value >0.5 and an acceptable hit rate.
High-Throughput Screening for Inhibitors of Human TDP1
A total of 227,905 samples (Table 1) were screened, including 143,962 crude natural product samples, 71,642 prefractionated natural product samples, and 12,301 purified single compounds, with a hit rate of 0.05% across all libraries. The assay maintained an average Z′ of 0.62 for crude natural product extract plates. The use of a rigorous confirmation process involving both fluorescent and nonfluorescent assay readouts was vital in lowering the final number of confirmed hits significantly. Step 1 was a radioactive gel assay using TDP1 from recombinant sources to confirm sample activity (Suppl. Fig. S1A–C), which was followed by examination of the activity of test samples in whole-cell lysates containing TDP1 (Suppl. Fig. S2A–C). Samples active in these two assays were then considered confirmed hits for further analysis and compound isolation.
Biochemical Analysis of Isolated Molecules Showing Inhibition of TDP1 Catalytic Activity
A number of inhibitors of TDP1 catalytic activity have been identified in this screen. Table 2 lists some of the compounds identified from all libraries screened. Aurintricarboxylic acid (ATA) and daphnetin diacetate were identified from pure compound libraries. ATA is a well-established inhibitor of numerous DNA binding proteins31 and was recently shown to inhibit influenza neuraminidase.32 Daphnetin is a coumarin derivative isolated from the Daphne genus of plants and has previously been evaluated as a serine-threonine kinase inhibitor and in the treatment of malaria.33
Table 2.
Examples of Natural Product Inhibitors of Tyrosyl-DNA Phosphodiesterase 1.
| Name | Structure | Origin | IC50 (μM) |
|---|---|---|---|
| 2,3-epoxy-8-hydroxy-lapachol | 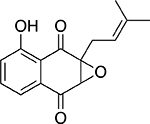 |
Plant Barleria alluaudii |
22 ± 1.0 |
| Minquartynoic acid | 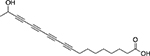 |
Plant Ochanostachys amentaceae |
52 ± 1.0 |
| Aurintricarboxylic acid | 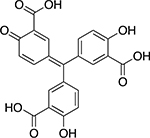 |
Synthetic | 8.0 ± 1.0 |
| Daphnetin diacetate | 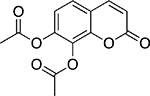 |
Plant Daphne sp. |
1.1 ± 0.2 |
| Halenaquinone | 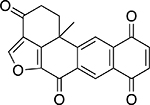 |
Marine sponge Xestospongia sp. |
26 ± 1.0 |
| Halenaquinol sulfate | 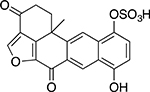 |
Marine sponge Xestospongia sp. |
24 ± 1.1 |
| Xestoquinone | 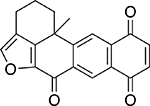 |
Marine sponge Xestospongia sp. |
93 ± 1.1 |
Screening of natural product extracts identified compounds from three very different structural groups. The first inhibitor, minquartynoic acid, is reported to have anti-HIV activity.34 We also identified 2,3-epoxy-8-hydroxy-lapachol (EHL) as an inhibitor of TDP1. EHL is a compound isolated from the organic extract of the stems of Barleria alluaudii, a plant from the Acanthaceae family native to Madagascar24 (Fig. 4A). Follow-up dose-response work on the purified compound suggested moderately potent inhibition of TDP1 catalytic activity, with an IC50 of 21.9 ± 0.32 μM (Fig. 4B). Dilution studies to check for reversibility of inhibition by this compound (Fig. 4C) show that EHL is fully reversible. When the mechanism of inhibition was probed, EHL displayed mixed-model noncompetitive inhibition kinetics (Fig. 4D). Under this model, differential affinity for TDP1 is observed in both the presence and absence of substrate. In this case, a higher affinity for the TDP1-substrate complex (Kies: 9.95 ± 1.14 μM) than for TDP1 alone (Kie: 40.93 ± 14.14 μM) is observed. Akaike’s Information Criterion27 was used to compare this model with fits to models for pure noncompetitive inhibition (where Kie = Kies) and uncompetitive inhibition (where inhibition takes place only against the ES complex). In both cases, the model for mixed-mode inhibition proved to best (>99.8% probability) describe the data (Suppl. Table S1).
Figure 4.

2,3-epoxy-8-hydroxy-lapachol as an inhibitor of human tyrosyl-DNA phosphodiesterase 1 (TDP1). (A) 2,3-epoxy-8-hydroxy-lapachol was isolated from the stems of Barleria alluaudii. (B) Dose-response analysis yields an IC50 of 21.9 ± 0.32 μM. (C) Reversibility studies of 2,3-epoxy-8-hydroxy-lapachol (◆) and aurintricarboxylic acid (■) show recovery of 90% of the uninhibited control rate (●) after preincubation with compound for 1 h. Halenoquinone shows no such recovery of activity (▼), indicating irreversible inhibition. (D) A series of substrate saturation curves carried out in the presence of 0 μM (◆), 6.6 μM (●), 20 μM (■), and 60 μM (▲) 2,3-epoxy-8-hydroxy-lapachol produced a pattern of inhibition best described by model of a mixed-type noncompetitive mechanism. (E) Binding of 2,3-epoxy-8-hydroxy-lapachol to the TDP1 apoprotein was observed by monitoring the decrease in intrinsic tryptophan fluorescence under increasing concentrations of compound (●). The equilibrium dissociation constant for the interaction (Kd) was estimated to be 19.1 ± 0.7 μM.
As a further test to confirm the direct interaction between EHL and TDP1, and also to fully exclude an uncompetitive model of inhibition, quenching of the intrinsic tryptophan fluorescence of TDP1 was used to monitor binding of this compound to the TDP128 (Fig. 4E). The comparatively high number of tryptophan residues in the TDP1 amino acid sequence yielded a high fluorescent signal from the unbound enzyme, allowing experiments to be conducted at protein concentrations of 50 to 100 nM. Titrating EHL up to 60 μM produced a fluorescence plateau suggesting complete saturation of TDP1 by the compound and resulted in a decrease in tryptophan emission intensity of ~75%. Analyzing these data with a model describing a single binding interaction produced an estimate of the Kd of 19.1 ± 0.008 μM approximately twofold lower than the estimate of Kie produced from fitting the data in a model for mixed inhibition. The relatively large error produced from this fit further suggests that the values of Kie and Kd might be somewhat closer than estimated from the kinetic mechanistic studies. This observation would appear to support the mixed-model mode of inhibition for EHL inhibiting TDP1.
Another distinct group of natural TDP1 inhibitors was a series of marine sponge metabolites including halenaquinone, halenaquinol sulfate, and xestaquinone (Table 2). Past studies have established these molecules as possessing activity against cancer.35 Here, we found that, unlike EHL, both halenaquinone and halenaquinol sulfate irreversibly inhibited TDP1 (Suppl. Fig. S3). Previously, halenaquinone and its analogs have been reported to covalently bind to proteins through either cysteine orlysine residues.36 Studies have also detailed their binding to the active site of enzymes requiring adenosine triphosphate.23,35 Because of the sulfate group on its D ring, halenaquinol sulfate is unable to react with Cys residues and instead shows a preference for reacting with Lys residues.37 When TDP1 was incubated with 5 μM halenaquinol sulfate and then analyzed by LC/MS, the data showed that only one residue of TDP1 had been modified (out of 37 possible lysine residues; Suppl. Fig. S4). Halenaquinol sulfate shows structural similarities to the compound wortmannin (Suppl. Fig. S5A), a microbial natural product also reported to competitively inhibit kinases such as phosphatidylinositol 3-kinase,38 via its reactive keto-furan ring. To see if this structural motif alone was responsible for the activity of halenaquinol sulfate, we tested wortmannin in the TDP1 assay. At concentrations up to 500 μM, wortmannin was unable to inhibit TDP1 (Suppl. Fig. S5B), indicating that additional structural elements (outside of the reactive keto-furan) are responsible for the selective inhibition of TDP1 by halenaquinol sulfate.
Discussion
Natural product compounds, and those derived from natural pharmacophores, have had success in specifically disrupting many of the critical processes involved in the pathogenesis of numerous important cancers.39–41 Although advances in medicinal chemistry have led to a more recent shift away from reliance on natural product extracts as primary sources of new pharmacophores, there is still untapped potential in this source of chemical diversity.41 New advances in how extracts are screened, and in how primary screening hits are evaluated to identify active molecules, are sparking a positive reassessment of the feasibility of reliably obtaining new drug molecules from natural sources.42
Screening crude natural product extracts presents special challenges to the investigator that are generally not encountered when dealing with synthetic chemical libraries. The work described here aimed to develop a robust screening assay to selectively identify specific natural product inhibitors of human TDP1 catalytic activity, a first for this target, and to employ this system to screen the NCI repository of crude natural product extracts. As mentioned earlier, a number of previous screening efforts have sought to identify synthetic inhibitors of TDP1 using largely proprietary screening technologies. Although successful at working with refined compound libraries, these assay platforms were found to be less than ideal for use with our crude extract library (data not shown). We addressed this by developing a simple fluorescence-based assay designed to confront the main challenges encountered when screening extracts, principally interference from optically active molecules, and nonspecific inhibition of the enzyme target by promiscuous polyphenolic protein binders. By using a redshifted fluorophore, we sought to remove many of the sources of optical interference exhibited by crude extracts in the ultraviolet end of the spectrum. The presence of a low concentration of BSA in our screening assay buffers aimed to limit the activity of nonspecific protein binding compounds. In addition, a low concentration of the detergent Tween-20 was also added to the assay buffer to reduce the nonspecific effects of aggregating agents within the extracts. Lastly, as a final step to remove any remaining sources of interference possibly affecting the fluorescent assay readout, a wash step was included to eliminate everything but the residual intact substrate from the final detection plates. These simple features resulted in a significantly improved assay that greatly reduced the abundance of errant positive samples detected during primary screening operations.
The second stage of the screening process implemented a rigorous confirmation regime to triage the number of extracts to those specifically and repeatedly inhibiting TDP1 catalytic activity. This was achieved by using a three-step approach involving initial confirmation in the screening assay by dose response or replicate points, detection of extracts interacting with the substrate via an enhancement of fluorescence, and measurement of activity in a nonfluorescent orthogonal assay format. As seen from Table 1, this process produced an almost fivefold reduction in the number of compounds and extracts between primary screening and the end of confirmation, which increased confidence in final outcomes significantly.
The prefractionation of crude natural product extracts aims to separate out nuisance molecules while concentrating other low-percentage molecules from the extract based on their polarity. The process has the effect of simplifying the extract from a screening perspective and lowering the hit rate for nonspecific inhibitors of the enzyme, while amplifying the level of inhibition observed from more specific compounds now present at higher concentration within the extract samples. The data presented in Table 1 appears to supports this trend, where screening of the Molecular Targets Laboratory (Center for Cancer Research/NCI) library of prefractionated natural product extracts22 produced fewer initial positives yet maintained a significantly higher confirmation rate than seen for the originating library of crude extracts. Although the preparation of prefractionated extracts is both expensive and time-consuming, it does appear to produce a far superior product with which to screen, confirming data previously published on a cell-based screen using this library.22
Screening of all available libraries ultimately produced a diverse collection of confirmed hit molecules, including the first natural product inhibitors of TDP1 catalytic activity. As expected from a screen of natural product extracts, a number of unusual pharmacophores were identified showing inhibition of TDP1 catalytic activity (Table 2). The compounds initially isolated represented three structural groups, with the largest group populated by the halenaquinone series of marine metabolites. The individual compounds within this particular group have all been previously studied for their role as inhibitors of other drug targets, including their activity as kinase inhibitors.23,35 The three marine quinone molecules (halenaquinone, halenaquinol sulfate, and xestoquinone) were identified as inhibitors of TDP1, with halenaquinone and halenaquinol sulfate irreversibly inhibiting TDP1 (Suppl. Fig. S3). Halenaquinol sulfate shares significant structural similarity to published compounds including wortmannin, a fungal metabolite with potent anti-kinase activity,38 and NSC 88915, an inhibitor of TDP1 recently discovered in a screen of synthetic drug compounds.20 We established, however, that wortmannin did not inhibit TDP1 (Suppl. Fig. S5B), which suggests that other aspects of the structure of the halenaquinol molecule must be responsible for some level of specificity in inhibiting this enzyme. As we determined by mass spectrometry (Suppl. Fig. S4) that a single halenaquinol sulfate molecule appears to be bound to TDP1, further analysis of the structural basis for inhibition by this molecule to identify this binding site may provide some valuable insights into potential avenues for the development of less promiscuous molecules as novel inhibitors of TDP1 activity.
Supplementary Material
Acknowledgment
We thank Drs Brice Wilson and James McMahon (MTL/NCI) for helpful discussions and review of the manuscript. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN26120080001E, and supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (BC006150). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health (NIH), under contract HHSN26120080001E and supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (BC006150).
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ Note
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Supplementary material is available online with this article.
References
- 1.Champoux JJ DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem 2001, 70, 369–413. [DOI] [PubMed] [Google Scholar]
- 2.Morham SG; Kluckman KD; Voulomanos N; et al. Targeted Disruption of the Mouse Topoisomerase I Gene by Camptothecin Selection. Mol. Cell Biol 1996, 16, 6804–6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holden JA DNA Topoisomerases as Anticancer Drug Targets: From the Laboratory to the Clinic. Curr. Med. Chem. Anticancer Agents 2001, 1, 1–25. [DOI] [PubMed] [Google Scholar]
- 4.Wall ME; Wani MC Camptothecin and Taxol:Discovery to Clinic—Thirteenth Bruce F. Cain Memorial Award Lecture. Cancer Res 1995, 55, 753–760. [PubMed] [Google Scholar]
- 5.Kingma PS; Osheroff N The Response of Eukaryotic Topoisomerases to DNA Damage. Biochim. Biophys. Acta 1998, 1400, 223–232. [DOI] [PubMed] [Google Scholar]
- 6.Interthal H; Pouliot JJ; Champoux JJ The Tyrosyl-DNA Phosphodiesterase TDP1 Is a Member of the Phospholipase D Superfamily. Proc. Natl. Acad. Sci. U.S.A 2001, 98, 12009–12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pommier Y; Huang SY; Gao R; et al. Tyrosyl-DNA-Phosphodiesterases (TDP1 and TDP2). DNA Repair (Amst.) 2014, 19, 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang SW; Burgin AB Jr.; Huizenga BN; et al. A Eukaryotic Enzyme That Can Disjoin Dead-End Covalent Complexes between DNA and Type I Topoisomerases. Proc. Natl. Acad Sci. U.S.A 1996, 93, 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pouliot JJ; Yao KC; Robertson CA; et al. Yeast Gene for a Tyr-DNA Phosphodiesterase That Repairs Topoisomerase I Complexes. Science 1999, 286, 552–555. [DOI] [PubMed] [Google Scholar]
- 10.Takashima H; Boerkoel CF; John J; et al. Mutation of TDP1, Encoding a Topoisomerase I-Dependent DNA Damage Repair Enzyme, in Spinocerebellar Ataxia with Axonal Neuropathy. Nat. Genet 2002, 32, 267–272. [DOI] [PubMed] [Google Scholar]
- 11.Interthal H; Chen HJ; Kehl-Fie TE; et al. SCAN1 Mutant TDP1 Accumulates the Enzyme—DNA Intermediate and Causes Camptothecin Hypersensitivity. EMBO J 2005, 24, 2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen TX; Abdelmalak M; Marchand C; et al. Synthesis and Biological Evaluation of Nitrated 7-,8-,9-, and 10-Hydroxyindenoisoquinolines as Potential Dual Topoisomerase I (TOP1)-Tyrosyl-DNA Phosphodiesterase I (TDP1) Inhibitors. J. Med. Chem 2005, 58, 3188–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keil A; Frese-Schaper M; Steiner SK; et al. The Topoisomerase I Inhibitor Irinotecan and the Tyrosyl-DNA Phosphodiesterase 1 Inhibitor Furamidine Synergistically Suppress Murine Lupus Nephritis. Arthritis Rheumatol 2015, 67, 1858–1867. [DOI] [PubMed] [Google Scholar]
- 14.Inamdar KV; Pouliot JJ; Zhou T; et al. Conversion of Phosphoglycolate to Phosphate Termini on 3′ Overhangs of DNA Double Strand Breaks by the Human Tyrosyl-DNA Phosphodiesterase hTDP1. J. Biol. Chem 2002, 277, 27162–27168. [DOI] [PubMed] [Google Scholar]
- 15.Murai J; Huang SY; Das BB; et al. Tyrosyl-DNA Phosphodiesterase 1 (TDP1) Repairs DNA Damage Induced by Topoisomerases I and II and Base Alkylation in Vertebrate Cells. J. Biol. Chem 2012, 287, 12848–12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang SY; Murai J; Dalla Rosa I; et al. TDP1 Repairs Nuclear and Mitochondrial DNA Damage Induced by Chain-Terminating Anticancer and Antiviral Nucleoside Analogs. Nucleic Acids Res 2013, 41, 7793–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newman DJ; Cragg GM Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- 18.Antony S; Marchand C; Stephen AG; et al. Novel High-Throughput Electrochemiluminescent Assay for Identification of Human Tyrosyl-DNA Phosphodiesterase (TDP1) Inhibitors and Characterization of Furamidine (NSC 305831) as an Inhibitor of TDP1. Nucleic Acids Res 2007, 35, 4474–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchand C; Lea WA; Jadhav A; et al. Identification of Phosphotyrosine Mimetic Inhibitors of Human Tyrosyl-DNA Phosphodiesterase I by a Novel AlphaScreen High-Throughput Assay. Mol. Cancer Ther 2009, 8, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dexheimer TS; Gediya LK; Stephen AG; et al. 4-Pregnen-21-ol-3,20-Dione-21-(4-Bromobenzenesulfonate) (NSC 88915) and Related Novel Steroid Derivatives as Tyrosyl-DNA Phosphodiesterase (TDP1) Inhibitors. J. Med. Chem 2009, 52, 7122–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCloud TG High Throughput Extraction of Plant, Marine and Fungal Specimens for Preservation of Biologically Active Molecules. Molecules 2010, 15, 4526–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henrich CJ; Cartner LK; Wilson JA; et al. Deguelins, Natural Product Modulators of NF1-Defective Astrocytoma Cell Growth Identified by High-Throughput Screening of Partially Purified Natural Product Extracts. J. Nat. Prod 2015, 78, 2776–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee RH; Slate DL; Moretti R; et al. Marine Sponge Polyketide Inhibitors of Protein Tyrosine Kinase. Biochem. Biophys. Res. Commun 1992, 184, 765–772. [DOI] [PubMed] [Google Scholar]
- 24.Whitson EL; Sun H; Thomas CL; et al. Synergistic TRAIL Sensitizers from Barleria alluaudii and Diospyros maritima. J. Nat. Prod 2012, 75, 394–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lakowicz JR Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, 2006. [Google Scholar]
- 26.Copeland RA Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, Wiley-Interscience: Hoboken, NJ, 2005. [PubMed] [Google Scholar]
- 27.Akaike H Information Theory and an Extension of the Maximum Likelihood Principle In Second International Symposium on Information Theory; Csaki B. N. P. a. F., Ed.; Akademiai Kiado: Budapest, 1973. [Google Scholar]
- 28.Gauthier TD; Shane EC; Guerin WF; et al. Fluorescence Quenching Method for Determining Equilibrium Constants for Polycyclic Aromatic Hydrocarbons Binding to Dissolved Humic Materials. Environ. Sci. Technol 1986, 20, 1162–1166. [Google Scholar]
- 29.Grant SK; Sklar JG; Cummings RT Development of Novel Assays for Proteolytic Enzymes Using RhodamineBased Fluorogenic Substrates. J. Biomol. Screen. 2002, 7, 531–540. [DOI] [PubMed] [Google Scholar]
- 30.Sasiela CA; Stewart DH; Kitagaki J; et al. Identification of Inhibitors for MDM2 Ubiquitin Ligase Activity from Natural Product Extracts by a Novel High-Throughput Electrochemiluminescent Screen. J. Biomol. Screen. 2008, 13, 229–237. [DOI] [PubMed] [Google Scholar]
- 31.Ghosh U; Giri K; Bhattacharyya NP Interaction of Aurintricarboxylic Acid (ATA) with Four Nucleic Acid Binding Proteins DNase I, RNase A, Reverse Transcriptase and Taq Polymerase. Spectrochim. Acta. A Mol. Biomol. Spectrosc 2009, 74, 1145–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hashem AM; Flaman AS; Farnsworth A; et al. Aurintricarboxylic Acid Is a Potent Inhibitor of Influenza A and B Virus Neuraminidases. PLoS One 2009, 4, e8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang YZ; Ranz A; Pan HZ; et al. Daphnetin: A Novel Antimalarial Agent with In Vitro and In Vivo Activity. Am. J. Trop. Med. Hyg 1992, 46, 15–20. [DOI] [PubMed] [Google Scholar]
- 34.Rashid MA; Gustafson KR; Cardellina JH II; et al. Absolute Stereochemistry and Anti-HIV Activity of Minquartynoic Acid, a Polyacetylene from Ochanostachys amentacea. Nat. Prod. Lett 2001, 15, 21–26. [DOI] [PubMed] [Google Scholar]
- 35.Fujiwara H; Matsunaga K; Saito M; et al. Halenaquinone, a Novel Phosphatidylinositol 3-Kinase Inhibitor from a Marine Sponge, Induces Apoptosis in PC12 Cells. Eur. J. Pharmacol 2001, 413, 37–45. [DOI] [PubMed] [Google Scholar]
- 36.Drahl C; Cravatt BF; Sorensen EJ Protein-Reactive Natural Products. Agnew. Chem. Int. Ed 2005, 44, 5788–5809. [DOI] [PubMed] [Google Scholar]
- 37.Wang J; Bourguet-Kondracki ML; Longeon A; et al. Chemical and Biological Explorations of the Electrophilic Reactivity of the Bioactive Marine Natural Product Halenaquinone with Biomimetic Nucleophiles. Bioorg. Med. Chem. Lett 2011, 21, 1261–1264. [DOI] [PubMed] [Google Scholar]
- 38.Ui M; Okada T; Hazeki K; et al. Wortmannin as a Unique Probe for an Intracellular Signalling Protein, Phosphoinositide 3-Kinase. Trends Biochem. Sci 1995, 20, 303–307. [DOI] [PubMed] [Google Scholar]
- 39.Cragg GM; Grothaus PG; Newman DJ Impact of Natural Products on Developing New Anti-Cancer Agents. Chem. Rev 2009, 109, 3012–3043. [DOI] [PubMed] [Google Scholar]
- 40.Cassinelli G The Roots of Modern Oncology: From Discovery of New Antitumor Anthracyclines to their Clinical Use. Tumori 2016, 2016, 226–235. [DOI] [PubMed] [Google Scholar]
- 41.Li JW; Vederas JC Drug Discovery and Natural Products: End of an Era or an Endless Frontier? Science 2009, 325, 161–165. [DOI] [PubMed] [Google Scholar]
- 42.Bailly C Ready for a Comeback of Natural Products in Oncology. Biochem. Pharmacol 2009, 77, 1447–1457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.