

Abstract
The novel SARS-CoV-2 coronavirus, which is responsible for COVID-19 disease, was first reported in Wuhan, China, in December of 2019. The virus rapidly spread, and the World Health Organization declared a pandemic by March 2020. With millions of confirmed cases worldwide, there is growing concern and considerable debate regarding the potential for coronavirus infection to contribute to an appreciable burden of chronic respiratory symptoms or fibrotic disease among recovered individuals. Because the first case of COVID-19 was documented less than one year ago, data regarding long-term clinical outcomes are not yet available, and predictions for long-term outcome are speculative at best. However, due to the staggering number of cases and the severity of disease in many individuals, there is a critical need to consider the potential long-term implications of COVID-19. This review examines current basic and clinical data regarding fibrogenic mechanisms of viral injury in the context of SARS-CoV-2. Several intersecting mechanisms between coronavirus infection and fibrotic pathways are discussed to highlight factors and processes that may be targetable to improve patient outcome. Reports of post-infection sequelae from previous coronavirus outbreaks are presented toward the goal of improved recognition of potential contributing risk factors for fibrotic disease.
Keywords: ARDS, COVID-19, lung injury, pulmonary fibrosis, SARS-CoV-2
INTRODUCTION
As an interface between the internal and external environment, the respiratory tract can be a considerable vulnerability for exposures and infection. The novel coronavirus SARS-CoV-2 is highly contagious, and the disease, COVID-19, resulting from infection, has proven to be more deadly than the flu. Yet COVID-19 may spread asymptomatically in many individuals (reviewed in Ref. 1). Although the factors that stratify patient risk for severe symptomatic infection remain unclear, given the relative severity of COVID-19 and the staggering number of infections worldwide, it is important to consider the potential long-term implications of viral infection on the lungs.
The first SARS-CoV-2 infections were reported in December of 2019 in Wuhan, China (2). Genetic analysis of the viral sequence suggests that this virus originated in bats and spread to humans via a currently unknown intermediate host (3, 4). SARS-CoV-2 is an enveloped virus. Like other coronaviruses, SARS-CoV-2 infects its host through a series of viral spike proteins that create the crown-like appearance for which the virus is named. A unique quality of the SARS-CoV-2 spike protein is that it can be cleaved by a number of ubiquitous serine proteases, contributing to its significant transmissibility and infectivity (4, 5). Following the binding of the spike proteins, a host protease, TMPRSS2, reveals the fusion domain of the spike protein allowing attachment to the angiotensin-converting enzyme-2 (ACE2) receptor on the host cell. Endocytosis ensues, and the viral genome is released into the host cell, where the virus hijacks the host cellular machinery to replicate and release viral particles extracellularly, which then infect neighboring cells. A comprehensive list of SARS-CoV-2 target cells and organ(s) is still under development; however, the virus is believed to infect several target cells, including type II pneumocytes, and alveolar macrophages (5) in the lungs, as well as enterocytes in the intestine (6), and potentially basal epithelial cells in the nasal passages, as supported by their known expression of the ACE2 receptor (7).
SARS-CoV-2 is rapidly spread through droplets and may also have a limited oral-fecal route of spread (8, 9). Although data are still being collected, SARS-CoV-2 has been reported to have an R0 of 1.4 or greater (reviewed in Ref. 10), with some reports as high as 5.7 (11), indicating the highly contagious nature of this strain of coronavirus. Symptomatic disease onset has been reported within a 14-day period following exposure to the virus, with most patients reporting symptoms within a median incubation period of ∼4–6 days postexposure (12). Current demographic data provided by the Centers for Disease Control (CDC) indicate that infection has been identified in patients of all ages, with the highest morbidity observed in those of advanced age (≥65 yr) and the highest mortality in those ≥85 years (13). According to a study by Lai et al. (14), an estimated 25.9% of COVID-19 patients have required intensive care hospitalization, and 11.5% of those hospitalized required ventilation support. An estimated 17.2 to 31% of COVID-19 patients suffer from viral-induced acute respiratory distress syndrome (ARDS) (14, 15).
There is evidence that viral infection may predispose or exacerbate existing respiratory conditions, including pulmonary fibrosis (PF) (reviewed in Ref. 16). PF is a disease characterized by scarring of the lungs. Fibrosis can present as stable disease in response to infection or injury, or it can be progressive and marked by periods of rapid exacerbation. Both stable and progressive fibrotic lung disease are associated with significant morbidity due to excessive deposition of extracellular matrix (ECM) molecules, such as collagen, laminin, and fibronectin, in the parenchymal lung tissue. This results in thickened alveolar walls, which hinders gas exchange and contributes to decreased and/or declining lung function, dyspnea, fatigue, and exercise intolerance. Alveolar epithelial injury is thought to be one of the primary initiating mechanisms of the disease, whereas the activated fibroblast is believed to be the primary effector of the disease. A direct pathogenic relationship between viral infection and stable or progressive fibrosis is unclear, although there are data indicating the potential for viral-induced fibrosis (17), including the “two-hit” hypothesis, as a potential explanation of origin for some cases of PF (reviewed in Ref. 16). We do not yet know whether recovered COVID-19 patients will be at increased risk of fibrotic disease. Therefore, it is important to consider the virally activated pathways, cellular mechanisms of viral injury, and current data on recovery from coronavirus infection to begin to identify intersecting processes that may be targeted to promote resolution, rather than fibrosis, as a long-term outcome for COVID-19 patients.
MECHANISMS OF VIRAL INJURY
Virally Mediated Profibrotic Pathways
Viruses, through direct engagement, or through activation of host signaling can upregulate expression of critical host cell surface receptors, signaling pathways, and production of growth factors (summarized in Fig. 1A, a). The ACE2 receptor, which is engaged by the S1 subunit of the SARS-CoV-2 spike protein for entry, is one such example. ACE2 acts as a regulator of the renin-angiotensin system (RAS). The RAS is responsible for hemostatic balance and fluid homeostasis through a balance of ACE and ACE2 activity. ACE cleaves angiotensin I to produce angiotensin II, which activates a broad range of signaling pathways as well as the release of aldosterone. In addition to inducing vasoconstriction and sodium and fluid retention, this arm of the RAS can have proinflammatory and profibrotic effects. As a counterbalance, ACE2 cleaves angiotensin II into protective angiotensin 1–7 and activates the Mas receptor axis. This arm of the RAS has protective anti-inflammatory and anti-fibrotic effects. Host ACE2 receptors undergo endocytosis when the SARS-CoV-2 spike proteins are cleaved on viral entry, reducing expression of ACE2 and reducing the ability of the host to balance the RAS. The effects of increased angiotensin II include activation of interleukin (IL)-6, tumor necrosis factor-α (TNFα), and increased recruitment of neutrophils and macrophages as well as direct endothelial cell injury. Angiotensin II has also been shown to promote collagen I gene activation through MAPK/ERK and transforming growth factor-β (TGFβ) (18), which are critical factors in a fibrotic response. Thus, through the mechanism of viral entry, SARS-CoV-2 contributes to activation of host proinflammatory and profibrotic pathways. Whether the level of activation and duration of signaling activity induced by the process of viral entry is sufficient to promote a fibrotic outcome or predispose infected individuals to fibrosis remains to be seen. However, increased angiotensin II and activation of angiotensin II-mediated downstream pathways during SARS-CoV-2 infection should be considered in evaluation of patient outcome following COVID-19.
Figure 1.
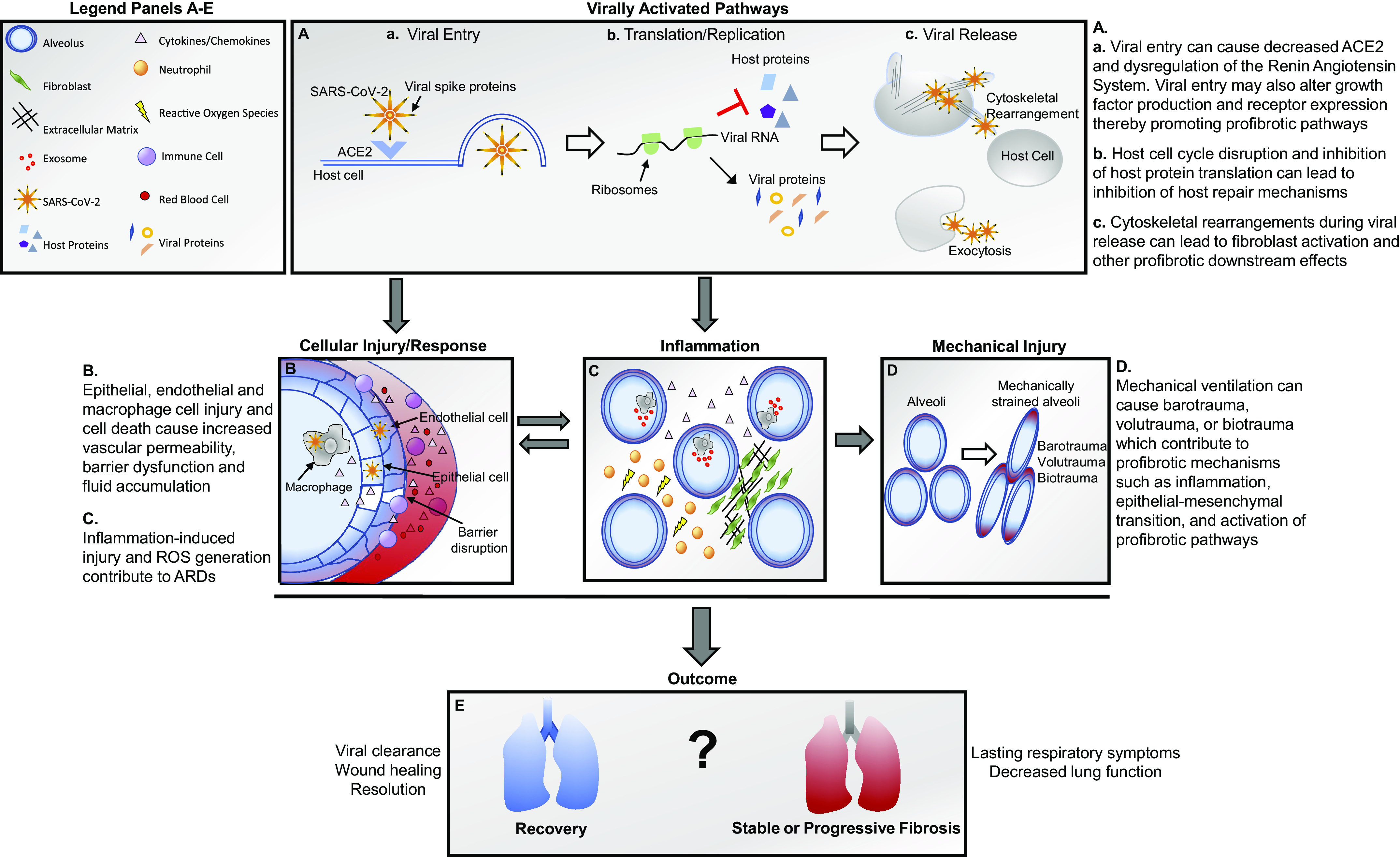
Summary of cellular and molecular mechanisms of viral injury.
Upon entry into the host cell, the virus promotes alterations in the host endoplasmic reticulum to promote synthesis of the viral RNA (Fig. 1A, b). The viral RNA and viral proteins are assembled, and the virus is transported to the cell surface and released by exocytosis (Fig. 1A, c). As this process ensues, the host machinery and cellular mechanisms are altered to promote the successful replication and spread of the viral particles. Using global phosphorylation analysis, Bouhaddou et al. (19) revealed that several host kinases were altered upon SARS-CoV-2 infection, including casein kinase II (CK2), cyclin-dependent kinase (CDK), and protein kinase C (PKC), suggesting an important role for these kinases in replication of the virus. Such kinases play a significant role in cell cycle regulation and cell cycle arrest. By arresting the cell cycle, the available proteins for viral particle production are increased. In the same study, analysis of phosphorylation sites in the structural coronavirus N protein, which plays a role in packaging of the viral RNA, suggested that the NH2-terminal portion of the protein may be responsible for inhibition of host translation (19). Inhibition of host translation allows for prioritization of viral protein synthesis over host proteins. Alterations in these processes can induce endoplasmic reticulum stress and an unfolded protein response (UPR) and integrated stress response (ISR), which, if sustained, may activate apoptotic and profibrotic pathways (reviewed in Ref. 20).
In addition to kinases, host growth factor signaling and growth factor receptors may also be altered to promote viral entry, replication, and evasion of the host immune response (reviewed in Ref. 21). Several pathways of interest include fibroblast growth factor (FGF), TGFβ, and epidermal growth factor receptor (EGFR). In a nonhuman primate model examining Middle East Respiratory Syndrome (MERS), increasing MERS viral load was correlated with increased SMAD7 and FGF-2 in lung tissue, which contributed to lung epithelial cell apoptosis based on colocalization of caspase-3 expression with SMAD-7, and FGF-2 in MERS infected cells in lung tissue (22). In vitro inhibition of SMAD7 in Calu-3 human lung epithelial cells increased cell viability and reduced viral load, further supporting an important role for this signaling pathway in host cell apoptosis and lytic viral release. In SARS-CoV infection, EGFR was upregulated, and its overexpression contributed to enhanced lung disease in a mouse model (23). In another study, the SARS-CoV RNA-binding protein (nucleocapsid N protein), a structural protein for viral nucleocapsid assembly that is also involved in regulation of host cell translation, was shown to promote SMAD3-mediated TGFβ signaling in human lung epithelial cells (24). The N protein also promoted PAI-1 and Col1A2 expression in human lung fibroblasts (24), showing direct profibrotic impact of SARS-CoV.
Cytoskeletal rearrangement, a process important for both cellular replication through mitosis and for cellular migration and invasion, is also impacted upon SARS-CoV-2 infection (Fig. 1A, c). Similar to other viruses, it appears that SARS-CoV-2 uses host cytoskeletal rearrangements to promote development of filapodia and for trafficking of the virus along actin fibrils to membrane extensions, allowing for increased efficiency in cell-cell spread of the virus (19). Cytoskeletal rearrangements exert both internal and external mechanical forces. These forces are sensed primarily by integrins and are transduced through activation of mechanosensitive pathways. As connectors between the internal cytoskeleton and the ECM, integrins allow for mechanical sensing of matrix stiffness and contraction as well as provide a biochemical mechanism by which a functional cellular response to cytoskeletal and matrix changes can occur. This functional response can include downstream effects such as fibroblast activation, macrophage phagocytosis, and changes in endothelial barrier function (reviewed in Ref. 25). Integrins can also directly activate important profibrotic pathways in response to mechanical stimuli. For example, integrins, including αvβ3, αvβ5, and αvβ6, can directly activate latent TGFβ by binding to the latency-associated peptide of TGFβ and inducing a conformational change that liberates active TGFβ (reviewed in Refs. 26 and 27). This can then contribute to a feed-forward loop of fibroblast activation, cellular contraction, increased collagen deposition, and consequent increases in matrix stiffness (reviewed in Refs. 27). In this way, cytoskeletal alterations, such as those induced by viral infection, may promote fibrogenic mechanisms in the host.
Cellular Level Host Response
In addition to alterations in host signaling and function, viral infection also leads to cellular injury through direct viral infection of cells (summarized in Fig. 1B). This can result in altered cell function and secretory profile as well as cell death. Although multiple cell types may become infected, in terms of lung damage, infection and death of type II alveolar epithelial cells (AEC) are significant, as these cells stabilize and repair the epithelial barrier, secrete vital prosurfactant (28), and are not efficiently replaced (29). In addition, type II AEC damage is thought to be an initiating event in the development of pulmonary fibrosis (reviewed in Refs. 30 and 31). Using an in vitro air-liquid interface model with human AECs, Qian et al. (32) demonstrated that infection of the cells with SARS-CoV occurred within 24 h of exposure and was detected by the appearance of viral antigens in the cytoplasm. These authors noted variability in infection of the cells, which they suggested may be due to varied expression levels of the ACE2 receptor among individuals. Following infection of AECs, significant increases in gene expression of inflammatory cytokines occurred, including interferon-β (IFN-β), IL-6, IL-29, CXCL10, CXCL11, and several others. Infection of type II AECs with SARS-CoV has also been shown to cause diffuse alveolar damage and promote macrophage, lymphocyte, and neutrophil recruitment (reviewed in Ref. 33).
Expression of ACE2 on alveolar macrophages and their role in phagocytosis of infected cells suggested that they may also be susceptible to direct infection by SARS-CoV-2. This possibility was examined by Chu et al. (5), who demonstrated direct SARS-CoV-2 infection of alveolar macrophages in ex vivo human lung explants. Activation of macrophages in response to viral infection results in production of inflammatory cytokines and subsequent recruitment of additional immune cells. Activated myeloid populations such as monocytes and macrophages have been shown to play a causal role in development of PF (34, 35) and in modulating inflammatory pathways and immune cell phenotypes (36, 37). Although the impact of macrophage activation in response to SARS-CoV-2 infection on future development of PF is unknown, given the critical role of these cells in both pathologies, examination of the myeloid response may provide insight for future studies.
Disruption of the alveolar epithelial surface and loss of alveolar macrophage populations can also leave endothelial cells susceptible to direct infection. Direct endothelial cell infection has been observed in COVID-19 patients in postmortem lung and bowel sections (38). In lung specimens from these patients, evidence of apoptotic endothelial cells was observed, as was increased mononuclear and neutrophilic infiltrate and thickened lung septa. Endothelial cell death is significant in that it leads to disruption of barrier function and increased vascular permeability (Fig. 1B), which substantially increases the extent of pulmonary injury. This type of damage to the lung microvascular architecture may contribute to development of PF (reviewed in Ref. 39). In an animal model, low-dose chemical injury to the lung (bleomycin) combined with an endothelial barrier disruption agent (FTY720, a nonselective sphingosine 1-phosphate receptor modulator) to increase vascular permeability was sufficient to induce PF (40). In this model, thrombin inhibition reduced collagen deposition, attenuated disruption of the alveolar-capillary barrier, and decreased extravascular fibrin deposition. Thrombin inhibition also decreased expression of the αvβ6 integrin and inhibited activation of TGFβ signaling through SMAD2. This suggests that thrombin may play an essential role in development of PF resulting from increased vascular permeability. In addition to local impact on the lungs, altered vascular permeability and hemostatic imbalance due to endothelial damage can also lead to coagulopathy and thromboembolism, which cause damage to distant organs. Similar thrombolytic events have been observed as a considerable complication of COVID-19 in some patients (41).
Inflammatory Response
Inflammation promotes viral clearance; however, the balance of chemokines and cytokines versus interferons and the control of the intensity of the inflammatory response are essential to eliminating infection without inducing significant injury. Although excessive cytokine response can be damaging (summarized in Fig. 1C and reviewed in Ref. 42), cytokines and chemokines play an important role in the wound-healing response. They promote activation of immune populations that clear infection and promote immunity through T cell and B cell recruitment. They also lead to activation of macrophage populations that clear apoptotic cellular debris. However, in acute lung injury, activated macrophages may also contribute to induction of neutrophil recruitment and activation through exosome-mediated crosstalk (43). Neutrophilic infiltrate, in turn, contributes to generation of reactive oxygen species (ROS; Fig. 1C). Although neutrophilic infiltrate and ROS may promote viral clearance, in abundance it may contribute to tissue injury (44, 45). Ultimately, fibroblasts are recruited and activated to mediate this injury. Activated fibroblasts deposit collagen and other ECM molecules to repair the damaged tissue (Fig. 1C). However, when this wound-healing response is dysregulated, it can result in reduced tissue function due to increased thickness, stiffening and altered vascularization, and PF.
In COVID-19, numerous inflammatory cytokines have been reported as increased in COVID-19 patients versus uninfected patients (46) (reviewed in Refs. 42, 47, and 48). In one study, among COVID-19 study participants requiring intensive care unit (ICU) level care, plasma levels of cytokines, including IL-2, IL-7, IL-10, IP10, MCP-1, MIP1A, and TNFα, were elevated versus non-ICU COVID-19 patients (46) (reviewed in Refs. 47 and 48). In another study, Blanco-Melo et al. (49) have demonstrated that SARS-CoV-2 infection induces a unique inflammatory pattern not commonly seen in other viral infections. This pattern is characterized by significant chemokine activation but only mild interferon I and III response. These authors speculate that the mild interferon response may be a possible explanation for the pronounced morbidity and mortality found among the elderly with COVID-19, as the aging immune system produces significantly less interferon response than seen in the young.
Mechanical Injury
Mechanical ventilation may induce stretch force injury and alveolar injury and may contribute to ARDS (Fig. 1D). In a study of 1,591 COVID-19 patients, 99% of patients in intensive care required respiratory support, and 88% of those required assisted ventilation by mechanical ventilator, according to a report published in the Journal of the American Medical Association (JAMA) (50). Increased lung stretch can induce oxidative injury, increase cytokine production, increase epithelial-mesenchymal transition (EMT) (51, 52), and increase collagen deposition in the lungs (51, 53). Mechanical ventilation can cause pulmonary injury through several mechanisms, including barotrauma, due to complications of positive pressure ventilation, or through volutrauma or atelectrauma due to hyperinflation. Biotrauma, which is the biological response to mechanical injury, may also contribute to the injury process by promoting inflammatory response (reviewed in Ref. 54). One example of biotrauma is the transmission of mechanosensory signals. In the alveolar epithelium, this occurs, at least in part, through protein kinase R-like endoplasmic reticulum-Kinase (PERK)-mediated integrated stress response (ISR) (55). Using a rat model of ventilator-induced lung injury, Dolinay et al. (55) demonstrated that inhibition of PERK reduced cell count in bronchoalveolar lavage, pulmonary edema, and neutrophil recruitment, suggesting that PERK activation may contribute to development of ventilation-induced lung injury.
In a murine acid aspiration model, Yang et al. (56) demonstrated that mechanical ventilation induced expression of EMT markers, which contributed to development of PF. This was mediated in part by Smad2/3 signaling and increased TGFβ. The effect was ameliorated by Resolvin-D1 and was associated with suppression of Smad2/3 phosphorylation, suggesting that TGFβ pathway activation is another contributor to ventilation-induced lung injury. This finding is supported by a study by Froese et al. (57), who showed that mechanical ventilation challenge of fibrotic lungs at high inspiratory pressures increased phosphorylation of Smad2/3, indicating activation of TGFβ. This contrasted with mechanical ventilation challenge of normal lungs, which did not activate TGFβ. Using inhibitors of protease activity and protein secretion, these authors demonstrated that mechanical strain was sufficient to induce release of TGFβ from the extracellular matrix of fibrotic lung, but not from normal lung, suggesting that stretch-induced TGFβ release is related to matrix stiffness. Reduction of cell contraction and integrin binding by pharmacological agents indicated that this stretch-induced TGFβ activation was likely integrin mediated (57; and reviewed in Ref. 26). Together, these studies suggest that careful ventilation of injured lungs, or lungs that may have increased stiffness, could potentially prevent stretch-induced TGFβ activation and thereby help to minimize ventilator-induced profibrotic signaling.
POTENTIAL FOR FIBROTIC RESPONSE
Given the recent appearance of SARS-CoV-2, data are not available on the long-term impact of COVID-19 on lung health. There is considerable debate as to whether survivors of COVID-19 will fully recover or whether they will experience stable lung damage or progressive fibrosis post-infection (Fig. 1E). Experience from other coronaviruses, including MERS and SARS-CoV, suggest that fibrotic disease as an outcome of COVID-19 is a concern (reviewed in Ref. 58). A study of MERS noted that 33% of patients with abnormal chest radiographs had lung fibrosis (59). These patients had longer ICU stays, were older, and had higher chest radiographic scores as well as higher peak lactate dehydrogenase levels versus patients without pulmonary fibrosis. Studies from SARS-CoV indicated that 27.8% to 62% of patients infected with SARS-CoV exhibited decreased lung function and increased fibrosis (60–64). In a 15-year follow-up from the 2003 SARS outbreak, ∼9% of study participants experienced fibrosis following infection, and that percentage diminished within one year and remained stable until a 15-year follow-up in 2018 (65). The study found that most patients recovered from interstitial damage and functional decline within 2 years after rehabilitation (65). The factors mediating a profibrotic response to coronavirus infection are not fully known, but studies suggest that the innate immune response (66), altered gene expression profile in myeloid populations (67), hyperactivation of alternatively activated macrophages (68), and proinflammatory and profibrotic factor production (69, 70) may contribute. Age and complicating comorbidities have also been implicated as contributing factors in severe and fibrotic outcomes (reviewed in Ref. 16).
Age
PF is predominantly a disease of the aged, indicating that the aging lung is particularly susceptible to development of fibrotic response. The most severe COVID-19 disease is reported among the elderly, and this population may be at increased risk for development of postinfection fibrosis, as was the case with SARS-CoV infection (71). Age is associated with ECM dysregulation, altered intracellular communication, and stem cell exhaustion (72). Delayed recovery following viral infection may occur due to other age-related changes such as cellular senescence, telomere shortening, or epigenetic changes, which can contribute to homeostatic imbalance and altered intracellular communication. Repair of the lung may also be impacted with age-related changes to stem-like cellular populations. The stem populations that moderate repair are still under investigation (reviewed in Ref. 73); however, perhaps the most recognized example of stem cell-like function in the lung is renewal of the alveolar epithelial population. Type II AECs have been shown to self-renew and have capacity to differentiate into type I AECs either directly (29) or through a progenitor population (74). In the aging lung, these and other stem-like populations may have reduced capacity to differentiate and mediate effective regeneration of depleted cellular populations. This, in turn, may contribute to the continued inflammation and dysregulated wound-healing response that often characterize fibrotic disease.
Experience from Other Viral Infections
In general, there is conflicting evidence regarding the possibility that viral infection may predispose one to development of fibrosis. A recent meta-analysis by Sheng et al. (75) found that acute viral infection did not correlate with fibrotic development, but data supported increased risk of idiopathic pulmonary fibrosis (IPF) with chronic viral infection. It is thought that chronic viral infection may contribute to fibrotic response through promoting a state of mild but chronic inflammation, which disrupts homeostasis and healing, thereby leading to increased susceptibility to secondary insult. Likewise, the two-hit hypothesis proposes that an initiating insult causes alterations in the lung microenvironment, increasing susceptibility to fibrotic development following a secondary insult. Coronavirus infection tends to have an acute duration; however, there is evidence from ARDS that even a duration of less than one week can lead to fibrosis (76). Therefore, severe cases leading to ARDS and those with extended recovery may be at increased risk for fibrosis rather than full recovery.
ARDS-Induced Fibrosis
PF subsequent to ARDS is well-recognized, and given the relatively high incidence of ARDS among COVID-19 patients (14), PF as a potential long-term outcome of COVID-19 is concerning. Distinct from the idiopathic form of PF or other progressive interstitial lung diseases, fibrosis resulting from ARDs is largely stable. However, whereas some patients with fibrosis post-ARDS may fully recover, some may have lasting symptoms of decreased lung function. Underlying pathology of ARDS is complex (77), and the inflammatory response and immune system play a critical role. As research efforts continue, identifying patients at greatest risk for ARDS resulting from severe COVID-19 may be possible and could lead to positive patient outcome. In addition, there is discussion regarding the potential for early intervention with anti-fibrotic agents to provide benefit for COVID-19 and the possibility that these agents may prevent potential post-infection lung damage (reviewed in Refs. 78–80).
PERSPECTIVES
There is much to be explored and understood about the lasting effects of severe viral respiratory infection and treatment and prevention of post-infection or post-injury PF, especially in the context of COVID-19 (reviewed in Refs. 58, 80, and 81). The SARS-CoV-2 virus appears unique among coronaviruses in its high transmission rate, atypical pattern of inflammatory response, range of disease severity from infection, and the distribution of those infected, with the young only mildly impacted. It remains unknown whether COVID-19 will contribute to any lasting respiratory symptoms or disease; however, it is possible that these unique features of SARS-CoV-2 infection may convey specific physiological impacts that contribute to a profibrotic environment. In this review, several fibrogenic molecular pathways engaged by coronaviruses and mechanisms of pulmonary injury related to viral infection are summarized (Fig. 1 and Table 1). Clinical data presented in this review regarding patient outcomes from respiratory infections with a longer case history demonstrate a range from full recovery to stable fibrosis or lasting disease. This varied response is perhaps one reason for the uncertainty among physicians and scientists in predicting the course of recovery from COVID-19. As the number of clinical trials increase, and basic bench studies are completed, the cellular mechanisms underlying pathophysiological response to SARS-CoV-2 infection will begin to be elucidated and will refine current understanding of the risks of COVID-19 and strategy for its treatment. Mortality rates have declined since the outset of this pandemic, suggesting that early detection, improved intervention, and application of repurposed drugs are beginning to have a positive impact, although it is unknown whether this will translate to decreased lasting effects on lung or decreased potential for post-infection fibrosis. The increased knowledge gained through the rapid deployment of scientific research and state-of-the-art healthcare provides hope that early and improved intervention will lead to positive outcomes, but we must remain vigilant in consideration of the potential for lasting lung damage and post-COVID-19 fibrosis.
Table 1.
Summary of fibrogenic mechanisms associated with viral infection
| Mechanism | Summary |
|---|---|
| Viral activation of profibrotic pathways | Altered renin-angiotensin system balance |
| Inhibition of host translation and altered cell cycle | |
| Activation of growth factors (e.g., FGF, EGF, and TGFβ) | |
| Cytoskeletal rearrangement | |
| Direct cellular injury | Type II alveolar epithelial cells |
| Macrophages | |
| Endothelial cells | |
| Cytokine-induced injury | Acute respiratory distress syndrome |
| Immune recruitment | |
| Neutrophil reactive oxygen species | |
| Macrophage exosomes | |
| Aberrant wound-healing response | |
| Mechanical Injury | Volutrauma/atelectrauma |
| Barotrauma | |
| Biotrauma | |
| Age | Altered cellular communication |
| Stem cell exhaustion | |
| Extracellular matrix dysregulation |
EGF, epidermal growth factor; FGF, fibroblast growth factor; TGFβ, transforming growth factor-β.
GRANTS
This work was supported by U.S. Department of Veterans Affairs Grant IK2 BX004072 (L.T.M).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author.
AUTHOR CONTRIBUTIONS
L.T.M. drafted manuscript; edited and revised manuscript; and approved final version of manuscript.
ACKNOWLEDGMENTS
The Author thanks Dr. Amanda C. LaRue, PhD, for helpful discussion in preparation of this article.
REFERENCES
- 1.Furukawa NW, Brooks JT, Sobel J. Evidence supporting transmission of severe acute respiratory syndrome coronavirus 2 while presymptomatic or asymptomatic. Emerg Infect Dis 26, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. Pneumonia of unknown cause – China (Online), 2020; https://www.who.int/csr/don/05-january-2020-pneumonia-of-unkown-cause-china/en/. [Google Scholar]
- 3.Benvenuto D, Giovanetti M, Salemi M, Prosperi M, De Flora C, Junior Alcantara LC, Angeletti S, Ciccozzi M. The global spread of 2019-nCoV: a molecular evolutionary analysis. Pathog Glob Health 114: 64–67, 2020. doi: 10.1080/20477724.2020.1725339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181: 281–292.e6, 2020. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu H, Chan JF, Wang Y, Yuen TT, Chai Y, Hou Y, Shuai H, Yang D, Hu B, Huang X, Zhang X, Cai JP, Zhou J, Yuan S, Kok KH, To KK, Chan IH, Zhang AJ, Sit KY, Au WK, Yuen KY. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin Infect Dis 71: 1400–1409, 2020. doi: 10.1093/cid/ciaa410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamers MM, Beumer J, van der Vaart J, Knoops K, Puschhof J, Breugem TI, Ravelli RBG, Paul van Schayck J, Mykytyn AZ, Duimel HQ, van Donselaar E, Riesebosch S, Kuijpers HJH, Schipper D, van de Wetering WJ, de Graaf M, Koopmans M, Cuppen E, Peters PJ, Haagmans BL, Clevers H. SARS-CoV-2 productively infects human gut enterocytes. Science 369: 50–54, 2020. . doi: 10.1126/science.abc1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203: 631–637, 2004. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Y, Guo C, Tang L, Hong Z, Zhou J, Dong X, Yin H, Xiao Q, Tang Y, Qu X, Kuang L, Fang X, Mishra N, Lu J, Shan H, Jiang G, Huang X. Prolonged presence of SARS-CoV-2 viral RNA in faecal samples. Lancet Gastroenterol Hepatol 5: 434–435, 2020. doi: 10.1016/S2468-1253(20)30083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao F, Tang M, Zheng X, Liu Y, Li X, Shan H. Evidence for Gastrointestinal Infection of SARS-CoV-2. Gastroenterology 158: 1831–1833.e3, 2020. . doi: 10.1053/j.gastro.2020.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Viceconte G, Petrosillo N. COVID-19 R0: Magic number or conundrum? Infect Dis Rep 12: 8516, 2020. doi: 10.4081/idr.2020.8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanche S, Lin YT, Xu C, Romero-Severson E, Hengartner N, Ke R. High contagiousness and rapid spread of severe acute respiratory syndrome coronavirus 2. Emerg Infect Dis 26: 1470–1477, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lauer SA, Grantz KH, Bi Q, Jones FK, Zheng Q, Meredith HR, Azman AS, Reich NG, Lessler J. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Ann Intern Med 172: 577–582, 2020. doi: 10.7326/M20-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.CDC COVID-19 Response Team. Severe outcomes among patients with coronavirus disease 2019 (COVID-19) - United States. MMWR Morb Mortal Wkly Rep 69: 343–346, 2020. doi: 10.15585/mmwr.mm6912e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int J Antimicrob Agents 55: 105924, 2020. doi: 10.1016/j.ijantimicag.2020.105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, Guan L, Wei Y, Li H, Wu X, Xu J, Tu S, Zhang Y, Chen H, Cao B. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395: 1054–1062, 2020. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naik PK, Moore BB. Viral infection and aging as cofactors for the development of pulmonary fibrosis. Expert Rev Respir Med 4: 759–771, 2010. doi: 10.1586/ers.10.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiao J, Zhang M, Bi J, Wang X, Deng G, He G, Luan Z, Lv N, Xu T, Zhao L. Pulmonary fibrosis induced by H5N1 viral infection in mice. Respir Res 10: 107, 2009. doi: 10.1186/1465-9921-10-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tharaux PL, Chatziantoniou C, Fakhouri F, Dussaule JC. Angiotensin II activates collagen I gene through a mechanism involving the MAP/ER kinase pathway. Hypertension 36: 330–336, 2000. doi: 10.1161/01.HYP.36.3.330. [DOI] [PubMed] [Google Scholar]
- 19.Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M, , et al. The Global phosphorylation landscape of SARS-CoV-2 infection. Cell 182: 685–712, 2020. e619 doi: 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, Wang Y, Pandupuspitasari NS, Wu G, Xiang X, Gong Q, Xiong W, Wang CY, Yang P, Ren B. Endoplasmic reticulum stress, a new wrestler, in the pathogenesis of idiopathic pulmonary fibrosis. Am J Transl Res 9: 722–735, 2017. [PMC free article] [PubMed] [Google Scholar]
- 21.Hondermarck H, Bartlett NW, Nurcombe V. The role of growth factor receptors in viral infections: An opportunity for drug repurposing against emerging viral diseases such as COVID-19? FASEB Bioadv 2: 296–303, 2020. doi: 10.1096/fba.2020-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeung ML, Yao Y, Jia L, Chan JF, Chan KH, Cheung KF, Chen H, Poon VK, Tsang AK, To KK, Yiu MK, Teng JL, Chu H, Zhou J, Zhang Q, Deng W, Lau SK, Lau JY, Woo PC, Chan TM, Yung S, Zheng BJ, Jin DY, Mathieson PW, Qin C, Yuen KY. MERS coronavirus induces apoptosis in kidney and lung by upregulating Smad7 and FGF2. Nat Microbiol 1: 16004, 2016. doi: 10.1038/nmicrobiol.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venkataraman T, Coleman CM, Frieman MB. Overactive Epidermal Growth Factor Receptor Signaling Leads to Increased Fibrosis after Severe Acute Respiratory Syndrome Coronavirus Infection. J Virol 91, 2017. doi: 10.1128/JVI.00182-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao X, Nicholls JM, Chen YG. Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-beta signaling. J Biol Chem 283: 3272–3280, 2008. doi: 10.1074/jbc.M708033200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burgstaller G, Oehrle B, Gerckens M, White ES, Schiller HB, Eickelberg O. The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J 50: 1601805, 2017. doi: 10.1183/13993003.01805-2016. [DOI] [PubMed] [Google Scholar]
- 26.Hinz B, Suki B. Does breathing amplify fibrosis? Am J Respir Crit Care Med 194: 9–11, 2016. doi: 10.1164/rccm.201601-0149ED. [DOI] [PubMed] [Google Scholar]
- 27.Freeberg MAT, Pereleas A, Rebman JK, Phipps RP, Thatcher TH, Sime PJ. Mechanical feed-forward loops contribute to idiopathic pulmonary fibrosis. Am J Pathol 191: 18–25, 2021. doi: 10.1016/j.ajpath.2020.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respir Res 2: 33–46, 2001. doi: 10.1186/rr36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BL. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest 123: 3025–3036, 2013. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parimon T, Yao C, Stripp BR, Noble PW, Chen P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int J Mol Sci 21, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winters NI, Burman A, Kropski JA, Blackwell TS. Epithelial Injury and Dysfunction in the Pathogenesis of Idiopathic PulmonaryFibrosis. Am J Med Sci 357: 374–378, 2019. doi: 10.1016/j.amjms.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qian Z, Travanty EA, Oko L, Edeen K, Berglund A, Wang J, Ito Y, Holmes KV, Mason RJ. Innate immune response of human alveolar type II cells infected with severe acute respiratory syndrome-coronavirus. Am J Respir Cell Mol Biol 48: 742–748, 2013. doi: 10.1165/rcmb.2012-0339OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miura TA, Holmes KV. Host-pathogen interactions during coronavirus infection of primary alveolar epithelial cells. J Leukoc Biol 86: 1145–1151, 2009. doi: 10.1189/jlb.0209078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, , et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 214: 2387–2404, 2017. doi: 10.1084/jem.20162152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang N, Yang K, Bai J, Yi J, Gao C, Zhao J, Liang S, Wei T, Feng L, Song L, Han H, Qin H. Myeloid-specific blockade of Notch signaling alleviates murine pulmonary fibrosis through regulating monocyte-derived Ly6c(lo) MHCII(hi) alveolar macrophages recruitment and TGF-beta secretion. FASEB J 34: 11168–11184, 2020. [DOI] [PubMed] [Google Scholar]
- 36.Lebrun A, Lo Re S, Chantry M, Izquierdo Carerra X, Uwambayinema F, Ricci D, Devosse R, Ibouraadaten S, Brombin L, Palmai-Pallag M, Yakoub Y, Pasparakis M, Lison D, Huaux F. CCR2(+) monocytic myeloid-derived suppressor cells (M-MDSCs) inhibit collagen degradation and promote lung fibrosis by producing transforming growth factor-beta1. J Pathol 243: 320–330, 2017. doi: 10.1002/path.4956. [DOI] [PubMed] [Google Scholar]
- 37.McDonald LT, Johnson SD, Russell DL, Young MRI, LaRue AC. Role of a novel immune modulating DDR2-expressing population in silica-induced pulmonary fibrosis. PLoS One 12: e0180724, 2017. doi: 10.1371/journal.pone.0180724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 395: 1417–1418, 2020. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Probst CK, Montesi SB, Medoff BD, Shea BS, Knipe RS. Vascular Permeability in the Fibrotic Lung. Eur Respir J 56: 1900100, 2020., doi: 10.1183/13993003.00100-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shea BS, Probst CK, Brazee PL, Rotile NJ, Blasi F, Weinreb PH, Black KE, Sosnovik DE, Van Cott EM, Violette SM, Caravan P, Tager AM. Uncoupling of the profibrotic and hemostatic effects of thrombin in lung fibrosis. JCI Insight 2, 2017. doi: 10.1172/jci.insight.86608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Middeldorp S, Coppens M, van Haaps TF, Foppen M, Vlaar AP, Müller MCA, Bouman CCS, Beenen LFM, Kootte RS, Heijmans J, Smits LP, Bonta PI, van Es N. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost 18: 1995–2002, 2020. . doi: 10.1111/jth.14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science 368: 473–474, 2020. doi: 10.1126/science.abb8925. [DOI] [PubMed] [Google Scholar]
- 43.Ye C, Li H, Bao M, Zhuo R, Jiang G, Wang W. Alveolar macrophage - derived exosomes modulate severity and outcome of acute lung injury. Aging (Albany NY) 12: 6120–6128, 2020. doi: 10.18632/aging.103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng Y, Herbert JA, Robinson E, Ren L, Smyth RL, Smith CM. Neutrophil: airway epithelial interactions result in increased epithelial damage and viral clearance during RSV infection. J Virol, 94, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herbert JA, Deng Y, Hardelid P, Robinson E, Ren L, Moulding D, Smyth RL, Smith CM. beta2 integrin LFA1 mediates airway damage following neutrophil trans-epithelial migration during RSV infection. Eur Respir J 56: 1902216, 2020. doi: 10.1183/13993003.02216-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395: 497–506, 2020. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Costela-Ruiz VJ, Illescas-Montes R, Puerta-Puerta JM, Ruiz C, Melguizo-Rodriguez L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ragab D, Salah Eldin H, Taeimah M, Khattab R, Salem R. The COVID-19 Cytokine Storm; What We Know So Far. Front Immunol 11: 1446, 2020. doi: 10.3389/fimmu.2020.01446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, tenOever BR. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181: 1036–1045, 2020. e1039 doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A, Cereda D, Coluccello A, Foti G, Fumagalli R, Iotti G, Latronico N, Lorini L, Merler S, Natalini G, Piatti A, Ranieri MV, Scandroglio AM, Storti E, Cecconi M, Pesenti A; COVID-19 Lombardy ICU Network. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA 323: 1574, 2020. doi: 10.1001/jama.2020.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cabrera-Benítez NE, Parotto M, Post M, Han B, Spieth PM, Cheng WE, Valladares F, Villar J, Liu M, Sato M, Zhang H, Slutsky AS. Mechanical stress induces lung fibrosis by epithelial-mesenchymal transition. Crit Care Med 40: 510–517, 2012. doi: 10.1097/CCM.0b013e31822f09d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang R, Pan Y, Fanelli V, Wu S, Luo AA, Islam D, Han B, Mao P, Ghazarian M, Zeng W, Spieth PM, Wang D, Khang J, Mo H, Liu X, Uhlig S, Liu M, Laffey J, Slutsky AS, Li Y, Zhang H. Mechanical Stress and the Induction of Lung Fibrosis via the Midkine Signaling Pathway. Am J Respir Crit Care Med 192: 315–323, 2015. doi: 10.1164/rccm.201412-2326OC. [DOI] [PubMed] [Google Scholar]
- 53.Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med 157: 294–323, 1998. doi: 10.1164/ajrccm.157.1.9604014. [DOI] [PubMed] [Google Scholar]
- 54.Madahar P, Beitler JR. Emerging concepts in ventilation-induced lung injury. F1000Res 9: 222, 2020. doi: 10.12688/f1000research.20576.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dolinay T, Aonbangkhen C, Zacharias W, Cantu E, Pogoriler J, Stablow A, Lawrence GG, Suzuki Y, Chenoweth DM, Morrisey E, Christie JD, Beers MF, Margulies SS. Protein kinase R-like endoplasmatic reticulum kinase is a mediator of stretch in ventilator-induced lung injury. Respir Res 19: 157, 2018. doi: 10.1186/s12931-018-0856-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Y, Hu L, Xia H, Chen L, Cui S, Wang Y, Zhou T, Xiong W, Song L, Li S, Pan S, Xu J, Liu M, Xiao H, Qin L, Shang Y, Yao S. Resolvin D1 attenuates mechanical stretch-induced pulmonary fibrosis via epithelial-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol 316: L1013–L1024, 2019. doi: 10.1152/ajplung.00415.2018. [DOI] [PubMed] [Google Scholar]
- 57.Froese AR, Shimbori C, Bellaye PS, Inman M, Obex S, Fatima S, Jenkins G, Gauldie J, Ask K, Kolb M. Stretch-induced activation of transforming growth factor-beta1 in pulmonary fibrosis. Am J Respir Crit Care Med 194: 84–96, 2016. doi: 10.1164/rccm.201508-1638OC. [DOI] [PubMed] [Google Scholar]
- 58.Spagnolo P, Balestro E, Aliberti S, Cocconcelli E, Biondini D, Casa GD, Sverzellati N, Maher TM. Pulmonary fibrosis secondary to COVID-19: a call to arms? Lancet Respir Med 8: 750–752, 2020. doi: 10.1016/S2213-2600(20)30222-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Das KM, Lee EY, Singh R, Enani MA, Al Dossari K, Van Gorkom K, Larsson SG, Langer RD. Follow-up chest radiographic findings in patients with MERS-CoV after recovery. Indian J Radiol Imaging 27: 342–349, 2017. doi: 10.4103/ijri.IJRI_469_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Antonio GE, Wong KT, Hui DS, Wu A, Lee N, Yuen EH, Leung CB, Rainer TH, Cameron P, Chung SS, Sung JJ, Ahuja AT. Thin-section CT in patients with severe acute respiratory syndrome following hospital discharge: preliminary experience. Radiology 228: 810–815, 2003. doi: 10.1148/radiol.2283030726. [DOI] [PubMed] [Google Scholar]
- 61.Chan KS, Zheng JP, Mok YW, Li YM, Liu YN, Chu CM, Ip MS. SARS: prognosis, outcome and sequelae. Respirology 8 Suppl: S36–40, 2003. doi: 10.1046/j.1440-1843.2003.00522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hui DS, Joynt GM, Wong KT, Gomersall CD, Li TS, Antonio G, Ko FW, Chan MC, Chan DP, Tong MW, Rainer TH, Ahuja AT, Cockram CS, Sung JJ. Impact of severe acute respiratory syndrome (SARS) on pulmonary function, functional capacity and quality of life in a cohort of survivors. Thorax 60: 401–409, 2005. doi: 10.1136/thx.2004.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hui DS, Wong KT, Ko FW, Tam LS, Chan DP, Woo J, Sung JJ. The 1-year impact of severe acute respiratory syndrome on pulmonary function, exercise capacity, and quality of life in a cohort of survivors. Chest 128: 2247–2261, 2005. doi: 10.1378/chest.128.4.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ngai JC, Ko FW, Ng SS, To KW, Tong M, Hui DS. The long-term impact of severe acute respiratory syndrome on pulmonary function, exercise capacity and health status. Respirology 15: 543–550, 2010. doi: 10.1111/j.1440-1843.2010.01720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang P, Li J, Liu H, Han N, Ju J, Kou Y, Chen L, Jiang M, Pan F, Zheng Y, Gao Z, Jiang B. Long-term bone and lung consequences associated with hospital-acquired severe acute respiratory syndrome: a 15-year follow-up from a prospective cohort study. Bone Res 8, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sheahan T, Morrison TE, Funkhouser W, Uematsu S, Akira S, Baric RS, Heise MT. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog 4: e1000240, 2008. doi: 10.1371/journal.ppat.1000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hu W, Yen YT, Singh S, Kao CL, Wu-Hsieh BA. SARS-CoV regulates immune function-related gene expression in human monocytic cells. Viral Immunol 25: 277–288, 2012. doi: 10.1089/vim.2011.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Page C, Goicochea L, Matthews K, Zhang Y, Klover P, Holtzman MJ, Hennighausen L, Frieman M. Induction of alternatively activated macrophages enhances pathogenesis during severe acute respiratory syndrome coronavirus infection. J Virol 86: 13334–13349, 2012. doi: 10.1128/JVI.01689-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huang KJ, Su IJ, Theron M, Wu YC, Lai SK, Liu CC, Lei HY. An interferon-gamma-related cytokine storm in SARS patients. J Med Virol 75: 185–194, 2005. doi: 10.1002/jmv.20255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wong CK, Lam CW, Wu AK, Ip WK, Lee NL, Chan IH, Lit LC, Hui DS, Chan MH, Chung SS, Sung JJ. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol 136: 95–103, 2004. doi: 10.1111/j.1365-2249.2004.02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chan TY, Miu KY, Tsui CK, Yee KS, Chan MH. A comparative study of clinical features and outcomes in young and older adults with severe acute respiratory syndrome. J Am Geriatr Soc 52: 1321–1325, 2004. doi: 10.1111/j.1532-5415.2004.52362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meiners S, Eickelberg O, Königshoff M. Hallmarks of the ageing lung. Eur Respir J 45: 807–827, 2015. doi: 10.1183/09031936.00186914. [DOI] [PubMed] [Google Scholar]
- 73.Basil MC, Katzen J, Engler AE, Guo M, Herriges MJ, Kathiriya JJ, Windmueller R, Ysasi AB, Zacharias WJ, Chapman HA, Kotton DN, Rock JR, Snoeck HW, Vunjak-Novakovic G, Whitsett JA, Morrisey EE. The cellular and physiological basis for lung repair and regeneration: past, present, and future. Cell Stem Cell 26: 482–502, 2020. doi: 10.1016/j.stem.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, Zhou S, Cantu E, Morrisey EE. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 555: 251–255, 2018. doi: 10.1038/nature25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sheng G, Chen P, Wei Y, Yue H, Chu J, Zhao J, Wang Y, Zhang W, Zhang HL. Viral Infection Increases the Risk of Idiopathic Pulmonary Fibrosis: A Meta-Analysis. Chest 157: 1175–1187, 2020. doi: 10.1016/j.chest.2019.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thille AW, Esteban A, Fernández-Segoviano P, Rodriguez JM, Aramburu JA, Vargas-Errázuriz P, Martín-Pellicer A, Lorente JA, Frutos-Vivar F. Chronology of histological lesions in acute respiratory distress syndrome with diffuse alveolar damage: a prospective cohort study of clinical autopsies. Lancet Respir Med 1: 395–401, 2013. doi: 10.1016/S2213-2600(13)70053-5. [DOI] [PubMed] [Google Scholar]
- 77.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 78.George PM, Wells AU, Jenkins RG. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir Med 8: 807–815, 2020. doi: 10.1016/S2213-2600(20)30225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seifirad S. Pirfenidone: A novel hypothetical treatment for COVID-19. Med Hypotheses 144: 110005, 2020. doi: 10.1016/j.mehy.2020.110005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vasarmidi E, Tsitoura E, Spandidos DA, Tzanakis N, Antoniou KM. Pulmonary fibrosis in the aftermath of the COVID-19 era (Review). Exp Ther Med 20: 2557–2560, 2020. doi: 10.3892/etm.2020.8980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ojo AS, Balogun SA, Williams OT, Ojo OS. Pulmonary fibrosis in COVID-19 survivors: predictive factors and risk reduction strategies. Pulm Med 2020: 6175964, 2020., doi: 10.1155/2020/6175964. [DOI] [PMC free article] [PubMed] [Google Scholar]