

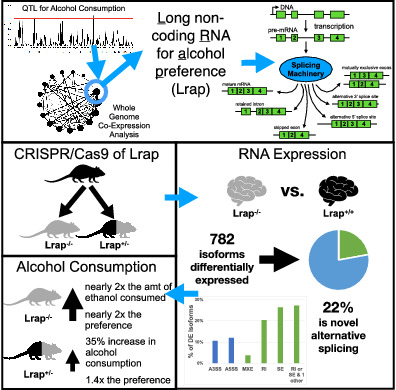
Abstract
LncRNAs are important regulators of quantitative and qualitative features of the transcriptome. We have used QTL and other statistical analyses to identify a gene coexpression module associated with alcohol consumption. The “hub gene” of this module, Lrap (Long non‐coding RNA for alcohol preference), was an unannotated transcript resembling a lncRNA. We used partial correlation analyses to establish that Lrap is a major contributor to the integrity of the coexpression module. Using CRISPR/Cas9 technology, we disrupted an exon of Lrap in Wistar rats. Measures of alcohol consumption in wild type, heterozygous and knockout rats showed that disruption of Lrap produced increases in alcohol consumption/alcohol preference. The disruption of Lrap also produced changes in expression of over 700 other transcripts. Furthermore, it became apparent that Lrap may have a function in alternative splicing of the affected transcripts. The GO category of “Response to Ethanol” emerged as one of the top candidates in an enrichment analysis of the differentially expressed transcripts. We validate the role of Lrap as a mediator of alcohol consumption by rats, and also implicate Lrap as a modifier of the expression and splicing of a large number of brain transcripts. A defined subset of these transcripts significantly impacts alcohol consumption by rats (and possibly humans). Our work shows the pleiotropic nature of non‐coding elements of the genome, the power of network analysis in identifying the critical elements influencing phenotypes, and the fact that not all changes produced by genetic editing are critical for the concomitant changes in phenotype.
Keywords: brain RNA expression networks, long non‐coding RNA, predisposing factors, genetic modification, CRISPR/Cas, quantitative genetics, recombinant inbred rat strains, systems genetics, transcriptome, voluntary alcohol consumption
Lrap, a novel lncRNA, is a mediator of alcohol consumption and a modifier of brain RNA expression and splicing. Categories from GO and KEGG pathways that are enriched within the group of differentially expressed transcripts in brains of the Lrap +/+ versus Lrap −/− rats (A). B and C depict heat maps illustrating the direction and magnitude of difference in expression of the genes included in the category “response to ethanol” (C) and “tight junction” between Lrap +/+, Lrap +/− and Lrap −/− rats. Relations between these genes and differential alcohol consumption of genetically, Lrap modified rats are discussed in the manuscript.
Abbreviations
- A3SS
alternative 3′ splice site
- A5SS
alternative 5′ splice site
- API
application programming interface
- AS
alternative splicing
- BN
Brown Norway
- CNS
central nervous system
- Ct
cycle threshold
- DE
differentially expressed
- FDR
false discovery rate
- GO
gene ontology
- HISAT2
hierarchical indexing for spliced alignment of transcripts 2
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- lncRNA
long non‐coding RNA
- LOD
logarithm of the odds
- Lrap
long non‐coding RNA for alcohol preference
- MXE
mutually exclusive exons
- ORF
open reading frame
- PCR
polymerase chain reaction
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- QTL
quantitative trait loci
- ReI
retained intron
- RI
recombinant inbred
- rMATS
replicate multivariate analysis of transcript splicing
- RNASeq
RNA sequencing
- RSEM
RNA‐seq by expectation–maximization
- SE
skipped exon
- sgRNA
single guide RNA
- SHR
spontaneously hypertensive rat
- SNP
single nucleotide polymorphism
- SOI
sum of isoforms
- WGCNA
weighted gene co‐expression network analysis
1. INTRODUCTION
Long non‐coding RNA (lncRNA) is a class of transcribed elements that is present in living cells from an organism's conception to death. 1 , 2 These >200 bp transcripts have been shown to influence transcription, translation and splicing events, 3 , 4 , 5 and to play a critical role in the development and senescence of an organism. 6 , 7 During adulthood, the mammalian brain has the largest assortment of lncRNAs, compared with other organs, and several have been shown to participate in neuronal and glial function. 8 , 9
Alcohol (ethanol) consumption is evident in phyla from insects (Drosophila) 10 to mammals (including Homo sapiens). Alcohol is consumed for a number of reasons, including being a source of calories, for gustatory reward, and for its psychoactive properties. 11 , 12 The consumption of alcohol is also a sine qua non for development of alcohol addiction (Alcohol Use Disorder), and both alcohol consumption and Alcohol Use Disorder have been shown to have a major genetic component. 13
In exploring the genetic and neural determinants of levels of alcohol consumption by animals, we previously applied a series of systems genetics analyses to microarray data that included measures of brain RNA expression levels across a panel of recombinant inbred (RI) and selectively bred rats that differed significantly in their levels of voluntary alcohol consumption. 14 Our results identified a gene co‐expression module, consisting of 17 gene products, that was implicated in influencing the levels of alcohol consumption. The “hub gene” of the module (the most connected transcript within the module) was identified as an unannotated transcript that showed all of the characteristics of an lncRNA, and we refer to this transcript as Lrap (Long non‐coding RNA for alcohol preference).
We now extend our work by determining, through statistical analysis, whether the expression of one of the genes in the module influences the association between other genes in the module. 15 Based on the results of this analysis we find that the “hub gene” of the module, Lrap, is critical for cohesion of the co‐expression module. We generate further information on the sequence of Lrap and illustrate its presence in mouse and human as well as rat.
We then produce rats that are genetically modified by deletion of a portion of the Lrap gene using CRISPR/Cas9 techniques. Using these animals and their wild type controls, we show that this genomic deletion produces a significant change in the quantity of alcohol consumption in a two‐bottle choice paradigm. We show that disruption of Lrap produces correlated changes in expression of another transcript that is a member of the module for which Lrap is the “hub gene”, and also produces broad changes in expression and splicing of other transcripts in brain. In all, we show that the constitutive disruption of the Lrap gene sequence produces a broad spectrum of changes in brain gene expression, and that subsets of the Lrap‐affected transcriptional events can significantly alter alcohol consumption.
2. MATERIALS AND METHODS
All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh or the Institutional Animal Care and Use Committee of the University of Colorado Anschutz Medical Campus and were performed in accordance with the guidelines in the NIH Guide for the Care and Use of Laboratory Animals.
2.1. Identification of transcripts responsible for network cohesiveness
To prioritize which transcript should be disrupted within the previously‐identified candidate module for alcohol consumption, 14 we assessed the individual association (correlation) of each transcript with alcohol consumption, and the association of the expression levels of each transcript with the alcohol consumption QTL on rat chromosome 12. 14 We also evaluated each transcript's connectivity within the module, and the changes in network cohesion when we “mimicked” a scenario where the expression level of one of the transcripts does not vary among samples (partial correlation analysis).
2.1.1. Use of data from previously published work for further analysis
Gene expression analysis
Expressed transcripts were identified and their expression was quantitated (log base 2), by use of the exon‐specific probes on Affymetrix Rat Exon Arrays. 14 In the current work, the individual association of each transcript in the module with alcohol consumption was assessed using Pearson correlation.
Measurement of alcohol consumption in RI rats
The alcohol consumption values were the measured average daily alcohol consumption by animals of a particular recombinant inbred strain during the second week of a two‐bottle choice paradigm. 14 The alcohol consumption for each animal was expressed in grams of alcohol per kilogram of body weight. 14
QTL analysis and genetic association analysis
The alcohol consumption QTL that was originally shown to be associated with the candidate brain coexpression module is located on chromosome 12. 14 In the current work, the SNP with the highest LOD score in the genetic region of the alcohol consumption QTL was used to represent the QTL in an association with individual transcript expression levels. Associations were determined using a Welch 2‐sample t test that assumes unequal variances. For this test, the mean expression level of strains with the BN‐Lx genotype at that marker was compared with the mean expression level of strains with the SHR genotype at that marker. Association of individual transcript expression levels with the candidate module eigengene were evaluated using Pearson correlation.
2.1.2. Current work
Intramodular connectivity analysis to ascertain a target for genetic disruption
Intramodular connectivity was calculated in two ways. First, it was defined for an individual transcript as the sum of the absolute value of its correlation coefficients with all other transcripts within the module. Second, it was defined for an individual transcript as the sum of its topological overlap with each other transcript within the module. Topological overlap values are described as part of the weighted gene co‐expression network analysis (WGCNA) 16 and are derived by first transforming pairwise correlation coefficients to reflect a scale‐free network and then quantitating both direct and indirect associations between a pair of transcripts.
Assessment of module cohesion
To examine the influence of an individual transcript, or the module eigengene QTL, on module cohesion, we compared the number of edges among the other transcripts in the module to the number of edges that remain in an 'adjusted' module after controlling for the effect of the transcript or QTL. For this analysis, an edge in the original module is defined as a correlation coefficient with an absolute value greater than 0.30. An edge that is retained in the 'adjusted' network is defined as a partial correlation coefficient with an absolute value greater than 0.30. In the context of the transcript effect, partial correlation is used to examine the association between expression levels of two transcripts after accounting for the variation in both transcripts that may be due to a “third” transcript. A large decrease in the number of edges between the original and the adjusted networks may indicate that the “third” transcript influences the relationships among other transcripts in the module, and that eliminating the variation in expression of this transcript disrupts the network structure.
2.2. Molecular disruption of Lrap
2.2.1. sgRNA/Cas9 production
CRISPR/Cas9 was used to create Lrap knockout (−/−) rats using techniques previously described. 17 , 18 Briefly, two sgRNAs targeting Lrap in exon 3 surrounding a suspected open reading frame 14 were identified using the CRISPR Design Tool. 19 sgRNA#1 was designed to cut Lrap immediately 5′ of the putative start codon. This sgRNA was created from 2 overlapping PCR primers (F:GAAATTAATACGACTCACTATAGGCT GTCTCCACATCTTTGGGCGTTTTAGAGCTAGAAATAGC; R:AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC) to generate a T7 promoter‐containing sgRNA template as described. 20 sgRNA#2 was designed to cut immediately downstream of the putative translation termination codon. This sgRNA was similarly created using the same reverse primer described above and an overlapping forward primer (GAAATTAATACGACTCACTAT AGGAGCCCTTTTGTGAGCATATGTTTTAGAGCTAGAAATAGC). These templates were transcribed in vitro from the T7 promoter using a MEGAshortscript Kit (Ambion/Thermo Fisher Scientific, Austin, TX). The Cas9 coding sequence was amplified from pX330 21 using a T7 promoter‐containing forward primer (tattacgactcactataggGAGAATGGACTATAAGGACCACGAC) and reverse primer (GCGAGCTCTAGGAATTCTTAC) and subcloned into pCR2.1‐TOPO. This plasmid was linearized with EcoRI, in vitro transcribed and polyA tailed using the mMessage mMachine T7 Ultra Kit (Ambion). Following synthesis, the sgRNAs and Cas9 mRNA were purified using the MEGAclear Kit (Ambion), ethanol precipitated, and resuspended in DEPC‐treated water.
2.2.2. Rat production
sgRNAs (25 ng/μl each) and Cas9 mRNA (50 ng/μl) were combined in embryo injection buffer (10 mM Tris, pH 7.4, 0.1 mM EDTA), aliquoted, and stored at −80° C until use. Wistar (Charles River, Wilmington, MA) one‐cell embryos were collected from superovulated females and cultured in KSOM media (Sigma‐Aldrich, St. Louis, MO) at 37° C in 5% CO2/95% air. Embryos were briefly transferred to M2 medium and the nucleic acid mixture was injected into cytoplasm. Embryos that survived injection were transferred to the oviduct of day 0.5 postcoitum pseudopregnant Long Evans (Charles River, Wilmington, MA) recipient females.
2.2.3. Rat genotype analysis
Pups resulting from injected embryos were screened for DNA sequence changes in exon 3 of the Lrap gene by PCR/DNA sequence analysis. Briefly, a crude DNA extract was prepared from rat tail tips or ear punches using 150 μl QuickExtract (Epicenter, Madison, WI). An 846 bp amplicon (+/+) from exon 3 was PCR amplified with forward (Lrap F1: GCTGTCAGAACACAGACCCA) and reverse (Lrap R1: GGAATCTGGCTGGGGAAACA) primers. PCR products were sequenced directly or subcloned into pCR2.1‐TOPO (Invitrogen) and sequenced. For routine genotyping, PCR products were analyzed on 2% agarose in TAE buffer.
Because a naturally occurring polymorphism that creates a premature stop codon in the Grm2 gene is present in commercially available Wistar rats, and this polymorphism has been reported to influence the alcohol phenotype, 22 rats were also genotyped for this polymorphism. Those rats harboring the premature stop codon were eliminated from the Lrap pedigree.
2.2.4. Off target analysis
The sgRNA target sequences (sgRNA#1: GCCCAAAGATGTGGAGACAG; sgRNA#2: ATATGCTCACAAAAGGGCTC) were run through the Off –Targets tool of the Cas9 Online Designer site (http://cas9.wicp.net). The top 8 predicted off targets for each sgRNA were amplified from male Founder rat #5254 DNA and sequenced.
2.3. Quantitative RT‐PCR
Quantitative RT‐PCR (qRT‐PCR) was used to validate the disruption of Lrap in the genetically modified rats, and to assess levels of expression of module transcripts in the wild‐type (+/+), Lrap knockout (−/−) and Lrap heterozygous (+/−) rats.
2.3.1. RNA isolation, reverse transcription
Rat brain RNA was isolated using an RNAeasy Mini Kit with RNA MinElute for RNA cleanup (Qiagen, Germantown, MD). The cDNA first strand transcription was performed in duplicate using 1.5 ug of total RNA with the iScript RT mix for qRT‐PCR following the manufacturer's protocol (BioRad, Hercules, CA). cDNA was stored at −20°C until further use.
2.3.2. Primer design for SYBR green
Design for primers that assay various regions of the Lrap sequence were limited to specific regions of sequence unique to each of three exon sequences. Primers were selected using Primer3 (http://frodo.wi.mit.edu/) and are described in Reference 14. Primers for Ift81, P2rx4 and Txnip were obtained from Integrated DNA Technologies (Coralville IA), and targeted exon 12–13 of Ift81; exon 2–4 of P2rx4; and exon 3–4 of Txnip.
2.3.3. Measurement and quantification of RNA expression levels
Quantitative PCR was performed on a Roche LightCycler 480II Real Time PCR instrument, using Roche SYBR Green I Master Mix (Indianapolis, IN). Primers used to determine levels of the full length Lrap transcript spanned exons 1–3 (Exon 1 F2 and Exon 3 R1). 14 To assess levels of other exons, primers were used that spanned exons 1–2 (Exon 1 F2 and Exon 2 R1) or exons 2–3 (Exon 2 F1 and Exon 3 R1). 14 PCR was carried out in a 15 μl volume and a final concentration of 1X reaction buffer, 500 nM forward and reverse primers and 0.5 μl of cDNA reaction. All standard curve and validation reactions were performed in triplicate with NTC reactions for all primer sets, and all samples were done in duplicate. PCR cycling parameters were as follows: hot‐start at 95° C for 5 min, 50 cycles of 95°C for 10 s, 61°C for 20 s, 72°C for 45 s, followed by a dissociation curve measurement from 65 to 97°C. For relative quantification, Advanced Relative Quantification analysis with efficiency correction (standard curves) was performed using the LC480II data collection software release 1.5.0.39. Geometric averaging of three reference genes (Actb, CypA and Pgk1) was used to normalize target gene transcripts. Melt curve analysis for all assays was used to verify single product amplification and absence of primer dimers. NTC reactions for all primer sets were >5Cq from samples, except in the case of samples from Lrap−/− and/or Lrap+/− rats where Lrap was measured.
To calculate absolute amounts of the Lrap transcript, cDNA was amplified from Lrap+/+ rat brain RNA by PCR, using primers Exon1 F2 and Exon 3 R1, 14 and was purified on a D2500 column (Omega Biotek, Life Sciences Products, Frederick, CO). cDNA was quantified by absorbance and diluted to produce a standard curve (2.27 × 10−1 to 2.27 × 10−8 ng). qRT‐PCR was performed (primers Exon 1 F2 and Exon 3 R1) 14 using this cDNA and cDNA from brains of Lrap+/+, Lrap+/− and Lrap−/− rats, as described above. Amounts of transcripts were calculated based on regression analysis of the standard curve.
For the relative quantitation qRT‐PCR experiments, differences between genotypes were determined using the ΔΔCt method. Differences in ΔCt values were estimated using a one‐way ANOVA model with post hoc testing using estimated marginal means via the emmeans package (version 1.4.8) in R without multiple testing correction. Estimated ΔΔCt values were transformed into relative quantities (i.e., 2−ΔΔCt) compared with wild type. For the relative quantitation of Lrap, a repeated measures one‐way ANOVA was used to account for duplicate assays of individual samples. For absolute quantitation qRT‐PCR experiments, values were log base 10 transformed prior to statistical analysis and then subjected to differential expression analysis using a one‐way ANOVA model with post hoc testing using marginal means without multiple testing correction.
2.4. Measures of impact of Lrap on alcohol consumption
Male Lrap+/+, Lrap−/− and Lrap+/− rats, 90 days old, were given a choice of 10% alcohol or water, as previously described. 23 Data on alcohol consumption obtained daily during the second and third weeks of 2‐bottle choice alcohol consumption were used for statistical analyses (g/kg body weight of alcohol consumed/day). To measure daily “alcohol preference”, we divided the volume of a 10% alcohol solution consumed by the total volume of fluid consumed for each of the tested animals. The (geometric) average of daily alcohol consumption and alcohol preference measures for each week was used in analyses.
A linear mixed model was used to determine the effect of genotype (Lrap+/+, Lrap+/−, and Lrap−/−) on amount of alcohol consumed (log base 2 transformed), alcohol preference (log base 2 transformed), daily fluid consumption in milliliters, and weight of the rat in grams. This model included a random effect to account for multiple measures on the same rat and a heterogeneous covariance structure that allowed for differences in variance between genotypes. Post hoc testing was done using least squares estimates and no multiple testing correction. The Satterthwaite method for estimated degrees of freedom was used for fixed effects testing and post hoc testing. The linear mixed model was executed using the MIXED procedure in SAS Statistical Software (version 9.4; Cary NC).
2.5. Measures of impact of Lrap on the brain transcriptome
To assess global changes in gene expression in response to disruption of the Lrap gene, total RNA was extracted from whole brain of three alcohol‐naïve male rats from each of the three genotypes (Lrap+/+, Lrap+/−, and Lrap−/−) using the RNeasy Midi Kit with additional cleanup performed using the RNeasy Mini Kit (Qiagen, Valencia, CA). Sequencing libraries were constructed using the Illumina TruSeq Stranded RNA Library Prep Kit with Ribo‐Zero (Illumina, San Diego, CA), in accordance with the manufacturers protocol. This library prep kit includes the removal of ribosomal RNA. Library quality was assessed using the Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). Samples were sequenced (2 × 100 paired‐end reads) on the Illumina HiSeq2500 (Illumina, San Diego, CA) with four libraries multiplexed per lane.
The pipeline for the RNASeq analysis and differential expression analysis is described below. Reads were trimmed to remove adapters and low‐quality base calls using cutadapt. 24 Trimmed reads were aligned to the rn6 version of the rat genome using HISAT2. 25 A genome‐ and transcriptome–guided reconstruction was executed using the StringTie software for each genotype independently using the Ensembl rat transcriptome (rn6), and then genotype‐specific transcriptomes were merged into a single transcriptome 26 using the merge function within StringTie. Transcripts were annotated by comparing the merged transcriptome back to the Ensembl rat transcriptome (rn6) using 'gffcompare'. 27
For quantitating RNA expression levels, expression can be either estimated for individual isoforms (i.e., splice variants) or estimated as the sum of expression across all isoforms that map to a given gene, that is, sum of isoforms (SOI). In the expression analyses presented here, RNA expression estimates were derived separately, at both the isoform‐level and at the SOI‐level, and were analyzed separately. This was done to ensure the capture of all relevant information, particularly since lncRNA has been shown to affect transcript splicing, 4 , 28 and a change in the ratio of isoforms may be missed if only the total of all isoforms expressed from a gene was considered.
Individual isoforms and SOIs from the merged transcriptome were quantitated using RSEM. 29 Transcripts were tested for differential expression using the DESeq2 package in R. 30 A false discovery rate (FDR) was used to account for multiple testing. 31 Isoforms and SOIs were only included in these analyses if they could be annotated as a product of a protein‐coding gene (Ensembl annotated isoforms and novel isoforms associated with Ensembl annotated genes) and if the sum of the estimated read counts across all samples from the Lrap−/− and Lrap+/+ animals was greater than 50.
Genes were included in a functional enrichment analysis if their isoform or SOI expression levels were significantly different between Lrap+/+ and Lrap−/− rats (FDR < 0.10). Functional enrichment of KEGG pathways 32 and Gene Ontology (GO 33 ;) categories was determined using a Fisher's exact test where the background gene list only included genes with at least one isoform or SOI expressed in rat brain (total estimated read counts across six samples from the Lrap−/− and Lrap+/+ animals was greater than 50). KEGG annotation was retrieved using the KEGG API ([ 32 ; Release 88.0) and GO annotation was retrieved using the 'biomaRt' package in R ( 34 ; Ensembl Gene 94; version 2.36.1). Programs related to the RNA‐Seq analysis are available at http://github.com/SabaLab/LrapKO. The raw RNA‐Seq data are available through GEO (GSE1557079) and the processed data are available on PhenoGen (http://phenogen.org).
2.6. Identification of Lrap‐influenced transcripts associated with alcohol consumption
To further focus attention on the differentially expressed transcripts with a higher probability of being associated with the Lrap‐induced differences in alcohol consumption between the Lrap−/− and Lrap+/+ rats, we established three criteria for inclusion of the information from both the genetically manipulated rats and from the RI panel. 14 First, the SOI for a given gene had to be differentially expressed between Lrap+/+ and Lrap−/− rats (unadjusted p value <0.05). Second, the transcript levels of a gene (derived from microarray analysis) had to be significantly (Pearson correlation coefficient, unadjusted p value <0.05) correlated with alcohol consumption in the HXB/BXH RI panel. Finally, the transcript levels of a gene had to be correlated with Lrap expression (Pearson correlation coefficient, unadjusted p value <0.05) in the HXB/BXH RI panel.
2.7. Analysis of differential alternative splicing
RNASeq data provides an opportunity to detect differential alternative splicing events across conditions. Employing the replicates of RNASeq data across pairs of conditions (i.e., Lrap+/+ vs. Lrap−/− rats), we applied rMATS (replicate Multivariate Analysis of Transcript Splicing) 35 to identify differential alternative splicing (AS) events. rMATS provides a computational framework to identify all possible splicing events which are altered between two samples, by inspecting the status of exons/introns as they are included or excluded resulting from alternative splicing. We used sorted BAM (Binary Alignment/Map) files, obtained from aligning the raw RNASeq datasets against the rat reference genome using HISAT, 25 as input to rMATS. Lrap+/+ and Lrap−/− rats were examined for alterations in various splicing events. We have provided the reconstructed brain transcriptome as described above as input to rMATS and have used the default thresholds for remaining options. Briefly, rMATS enabled us to analyze the inclusion/exclusion of target exons/introns contributing to different types of alternative splicing events, namely skipped exon (SE), alternative 5′ splice site (A5SS), alternative 3′ splice site (A3SS), mutually exclusive exons (MXE) and retained intron (ReI), across any pair of conditions with replicates. An AS event is quantified based on the difference in the level of inclusion of an exon which is defined as the splice index or percentage spliced index (ψ score) between two samples or conditions and ranges between 0 and 1. Significant differences in the values of ψ for an exon, between a pair of conditions compared with a null distribution indicate its differential abundance. Alternative splicing events between Lrap+/+ and Lrap−/− rats for the isoforms that were differentially expressed were examined using reads that span splicing junctions and an FDR threshold of 0.10.
2.8. Informatic characterization of Lrap
2.8.1. Homology of Lrap across rat, mouse and human
To ascertain the possible presence of sequences similar to the DNA sequence in rat that gives rise to Lrap, the reference rat genome sequence for the region containing Lrap on the negative strand was obtained from the UCSC genome browser and variants including SNPs and short insertions and deletions were included from the SHR/WKY strains as reported by Hermsen et al. 36 The sequence was submitted to run BLASTN through the Ensembl website (v 98) against the human and mouse genomes, using distant homology search sensitivity with the default settings, except we required a minimum e‐value reporting of 1e‐3, word size of 7 and gap penalty of 2,2. Aligned blocks were converted to a bed file to include custom alignment tracks in the UCSC genome browser to generate a browser image for each species. 37
2.8.2. Overlap of RNA expression in a human cell line with homologous regions
Publicly available RNA sequencing reads from polyA‐ long RNA were downloaded from the UCSC Genome Browser [72]. These data were derived from the GM12878 cell line and are part of the Long RNASeq from ENCODE/Cold Spring Harbor Lab track (GEO Accession: GSM758572). 8 Reads were aligned to the human genome (hg38 version) using HISAT2. 25 The graphic depicting the RNASeq coverage was generated using the UCSC Genome Browser. 37
3. RESULTS
3.1. Identification of transcripts responsible for network cohesiveness
Examination of intramodular connectivity of all the transcripts contained in the module in which Lrap was identified as a hub gene, 14 indicated that Lrap was the most highly connected transcript using either correlation coefficients or topological overlap (connectivity) measures (Table 1). Furthermore, Lrap had the largest impact on module integrity/connectivity among all transcripts in the module (Table 1). In fact, more edges (connections) were disrupted when adjusting for Lrap than when adjusting for the module eigengene QTL. Theoretically, a connection between the products of two genes that was present (absolute value of the correlation coefficient > 0.3) in the original module, but was eliminated after adjusting for the effect of Lrap, represents an indirect association between the expression levels of those two gene products that is due to the influence of Lrap on both transcripts. Because of the evidence related to Lrap's central role in the cohesiveness of this module, and the desire to better understand the role of Lrap in determining the expression levels of the module transcripts, we chose to disrupt the Lrap gene and assess, not only the effects of the disruption on the expression of the transcripts in the module, and transcript levels throughout the brain, but also the effect of disruption on alcohol consumption.
TABLE 1.
Prioritization of transcripts within the alcohol consumption module for genetic manipulation
| Gene symbol | Genomic position | Correlation with alcohol consumption [correlation coefficient (p value)] | Association with alcohol consumption QTL on chromosome 12 [log base 2 difference (p value)] | Correlation with module eigengene [correlation coefficient (p value)] | Correlation‐based intramodular connectivity (rank within module) | Connectivity‐based intramodular connectivity (rank within module) | Number of connected nodes a in module (|correlation coefficient| > 0.3) | Percent of all edges in module that remain after adjustment for transcript |
|---|---|---|---|---|---|---|---|---|
| Lrap | chr12: 39.01 Mb | −0.55 (0.011) | −0.32 (0.0079) | 0.90 (4.1e‐08) | 10.66 (1) | 2.99 (1) | 16 | 41% |
| Ift81 | chr12: 39.42 Mb | −0.43 (0.051) | −0.17 (0.0015) | 0.88 (2.0e‐07) | 10.33 (2) | 2.66 (2) | 15 | 49% |
| Coq5 | chr12: 47.08 Mb | −0.50 (0.021) | −0.10 (0.0057) | 0.83 (2.6e‐06) | 9.94 (4) | 2.24 (3) | 15 | 65% |
| Txnip | chr2: 198.68 Mb | 0.61 (0.003) | 0.20 (0.0087) | −0.77 (4.6e‐05) | 9.09 (8) | 2.20 (4) | 13 | 74% |
| P2rx4 | chr12: 39.31 Mb | −0.63 (0.002) | −0.17 (0.0022) | 0.83 (3.6e‐06) | 9.86 (5) | 2.15 (5) | 16 | 62% |
| Tmem116 | chr12: 40.56 Mb | 0.34 (0.133) | 0.34 (0.0001) | −0.81 (7.5e‐06) | 9.95 (3) | 2.00 (6) | 16 | 49% |
| Maats1 | chr11: 64.98 Mb | −0.56 (0.008) | −0.05 (0.0772) | 0.65 (1.3e‐03) | 7.57 (14) | 1.95 (7) | 9 | 95% |
| GENE_27603 | chr6:42.79 Mb | −0.50 (0.021) | −0.12 (0.0298) | 0.70 (4.5e‐04) | 8.18 (11) | 1.74 (8) | 13 | 83% |
| Oas1f (formerly Oas1b) | chr12: 41.26 Mb | 0.34 (0.136) | 0.61 (0.0006) | −0.79 (2.2e‐05) | 9.61 (6) | 1.69 (9) | 16 | 53% |
| GENE_18197 | chr2:257.76 Mb | −0.40 (0.075) | −0.12 (0.0196) | 0.77 (3.9e‐05) | 9.25 (7) | 1.65 (10) | 15 | 68% |
| Parp3 | chr8: 115.17 Mb | −0.28 (0.221) | −0.05 (0.0159) | 0.67 (8.0e‐04) | 8.40 (10) | 1.47 (11) | 15 | 76% |
| GENE_03396 | chr1:259.43 Mb | 0.27 (0.240) | 0.22 (0.0032) | −0.65 (1.4e‐03) | 7.97 (12) | 1.44 (12) | 12 | 72% |
| Slc8b1 (formerly Slc24a6) | chr12: 41.57 Mb | 0.16 (0.496) | 0.11 (0.0004) | −0.62 (2.5e‐03) | 7.60 (13) | 1.42 (13) | 12 | 86% |
| Prkar1b | chr12: 17.61 Mb | 0.30 (0.192) | 0.07 (0.0721) | −0.68 (7.6e‐04) | 8.44 (9) | 1.41 (14) | 15 | 72% |
| GENE_04887.ISO_01 | chr10:21.98 Mb | 0.25 (0.266) | 0.08 (0.0113) | −0.49 (2.3e‐02) | 6.46 (16) | 1.28 (15) | 9 | 87% |
| Pcdhb5 | chr18: 30.40 Mb | −0.10 (0.668) | −0.06 (0.0497) | 0.54 (1.2e‐02) | 6.92 (15) | 1.25 (16) | 10 | 82% |
| Anxa11 | chr16: 3.87 Mb | 0.25 (0.271) | 0.02 (0.3611) | −0.50 (2.0e‐02) | 6.29 (17) | 1.05 (17) | 12 | 92% |
Note: Gene symbols that begin with 'GENE' are unannotated transcripts that were identified in the original transcriptome reconstruction. All correlation coefficients are based on a parametric Pearson correlation. None of the p values have been adjusted for multiple testing.
Nodes refer to transcripts in the module.
3.2. Lrap−/− rat production
Two sgRNA sequences were used with the CRISPR/Cas9 system to create a deletion in exon 3 of Lrap. The sgRNA sequences were designed based on the genomic sequence of the Brown Norway (BN) rat (Rn6) (Figure 1A). It should be noted that the parental strains of the HXB/BXH RI panel, BN‐Lx/Cub and SHR/OlaIpcv, used for our QTL mapping and correlation analysis, are derived from the BN and the Wistar strains, respectively. We ascertained that the chosen sequences were also a perfect match to genomic sequences in the same location in the Wistar rat genome. PCR/DNA sequence analysis of 9 founders derived from injected embryos of Wistar rats revealed that 2 were wild type (Lrap+/+), 3 had small deletions at one or both sgRNA binding sites, and 4 had deletions spanning the area between the sgRNA binding sites.
FIGURE 1.
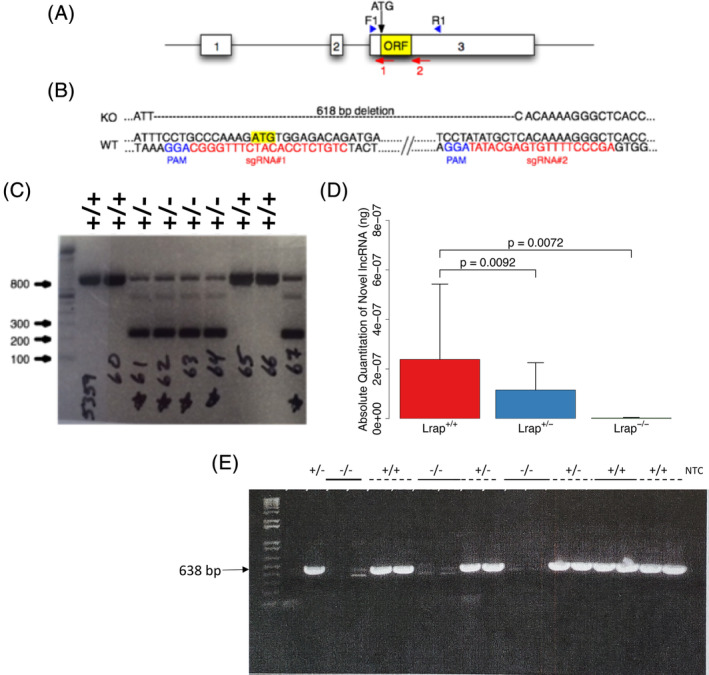
Lrap−/− rat strategy and characterization. (A) Diagram of Lrap locus illustrating exons (numbered white boxes), the potential open reading frame (yellow ORF box), the putative start codon (ATG), sgRNA target sites (numbered red arrows) and the location of the forward and reverse PCR genotyping primers (blue arrowheads). (B) Partial Lrap wild type (Lrap+/+) and knockout (Lrap−/−) rat genomic DNA sequence. In red are the sgRNA target sequences used for CRISPR/Cas9 gene targeting. The CRISPR protospacer adjacent motifs (PAM) are shown in blue. The yellow highlighted ATG is the putative start codon. Note that the Lrap−/− sequence harbors a 618 bp deletion. (C) Lrap PCR genotype analysis. Ethidium bromide stained agarose gel of Lrap PCR products from Lrap wild type (Lrap+/+) and Lrap+/− rats. Ladder is in bp. (D) Validation of differences in Lrap RNA expression using qRT‐PCR. RNA expression levels of exons 1–3 in rat brains are reported as absolute quantity (n = 3/genotype). This PCR product is undetectable in the Lrap−/− rats, since the exon 3 primer (Exon 3 R1) targets the excised region of exon 3. 14 All statistical analyses were done on the log transformed data and results are reported based on the back transformation of mean values and mean values plus one standard error. All p values were calculated using a one‐way ANOVA with post hoc pairwise comparisons. (E) qRT‐PCR products from Lrap+/+, Lrap+/− and Lrap−/− rats. Ethidium bromide stained agarose gel of products from qRT‐PCR of naïve Lrap+/+, Lrap+/− and Lrap−/− rats, using primers for exons 1 and 3: Exon 1 F2‐Exon 3 R1. 14 With the exception of the first lane (+/−), all other samples are run as duplicates
Two of the Wistar founders with the large deletions were mated to Wistar females to establish germline transmission, but only one of these produced germline transmission. Founder rat #5254 (aka 4B.3) appeared heterozygous for a 618 bp deletion (Figure 1B) within which was contained a 2 bp deletion/3 bp insertion (indels) near sgRNA#1 binding site when compared with the BN genome in this region (not shown). All offspring from this founder were genotyped for the 618 bp deletion using the PCR analysis illustrated in Figure 1C. From 27 offspring, 16 were heterozygous for the wild type (Lrap+/+) and 618 bp deletion alleles. Note that the Lrap+/+ allele produces an 846 bp fragment whereas the knockout allele (Lrap−/−) produces a 228 bp fragment. The PCR amplicons from all heterozygous F1 animals that were shipped for breeding to the University of Colorado were sequenced to verify the fidelity of the mutated locus.
Analysis of the predicted top 8 off‐target mutation sites for each sgRNA in Founder rat #5254 revealed no off‐target mutations.
3.3. Lrap−/− rat characterization
Rats were bred at the University of Colorado and genotyped by Transnetyx (Cordova, TN). As shown in Figure 1D,E, the absolute quantification of the Lrap transcript by qRT‐PCR, measured using primers that spanned the full length Lrap transcript (exons 1–3, with the reverse primer aligning to the exon 3 deleted region), indicated that there was a significant effect of genotype (F 2,5 = 12.49; p = 0.011). More specifically, Lrap was significantly decreased in male Lrap+/− rats (p = 0.0092) and not detectable in brains of male Lrap−/− rats. qRT‐PCR was also used to determine relative levels of the Lrap transcript in the wild type (Lrap+/+), Lrap+/− and Lrap−/− rats, using the primers spanning exons 1–3, and similar results were found. Relative transcript levels were also reduced for Lrap−/− rats (relative expression compared with Lrap+/+ = 0.002; p value = 2.8 × 10−5) when qRT‐PCR was performed using primers spanning exons 2–3 (using the reverse primer targeting the deleted region of exon 3). 14 The reduction in Lrap+/− rats from the area spanning exons 2–3 was suggestive (relative expression compared with Lrap+/+ = 0.31; p value = 0.061). When primers spanning exons 1–2 were used, 14 transcript levels were also reduced but to a lesser extent (Lrap−/− relative to Lrap+/+ = 0.16, p value = 0.041; Lrap+/− relative to Lrap+/+ = 0.56, p value = 0.48).
3.4. Effect of disruption of Lrap on alcohol drinking behavior
Because the original candidate coexpression module was identified from correlations/associations with alcohol consumption in a two‐bottle choice paradigm with 24‐h access to a 10% ethanol solution, 14 an identical phenotype was measured in the genetically manipulated rats. There was a suggestive effect of genotype on alcohol consumption (F 2,22.2 = 3.19; p = 0.061). Male Lrap+/+ (Wistar) rats drank an average of 1.1 g of alcohol/kg of body weight/day (Figure 2A). In the Lrap−/− rats, alcohol consumption was significantly increased to 2.1 g/kg/day (range 0.7–4.4 g/kg/day, p = 0.021). The increase in alcohol consumption is consistent with our finding that the expression level of Lrap in brains of the HXB/BXH RI rats was negatively correlated with amount of alcohol consumed (Table 1). In the Lrap+/− rats, alcohol consumption was increased to 1.5 g/kg/day, and this intermediate value was not significantly different from the level observed in the Lrap−/− rats (p = 0.26) or the level observed in the Lrap+/+ rats (p = 0.28). When we calculated “alcohol preference” for the Lrap+/+, Lrap+/− and Lrap−/− rats, the alcohol preference ratio for the Lrap+/+ rats averaged 0.10, a value indicating that the ethanol solution was aversive to these animals (Figure 2B). There was a significant effect of genotype on alcohol preference (F 2,21.3 = 3.75; p = 0.040). The alcohol preference value (0.18) for the Lrap−/− rats was significantly higher than that for the Lrap+/+ rats (p = 0.014) while the alcohol preference ratio for the Lrap+/− rats fell between those two values and was not significantly different from either (p = 0.16 compared with Lrap+/+ and p = 0.29 compared with Lrap−/−). Group average total fluid intake was not significantly different among the Lrap+/+, Lrap+/− and Lrap−/− animals (Figure 2C; F 2,19.6 = 0.21; p = 0.81). Body weight did not differ significantly among the three genotype groups (Figure 2D; F 2,17 = 0.06; p = 0.94).
FIGURE 2.
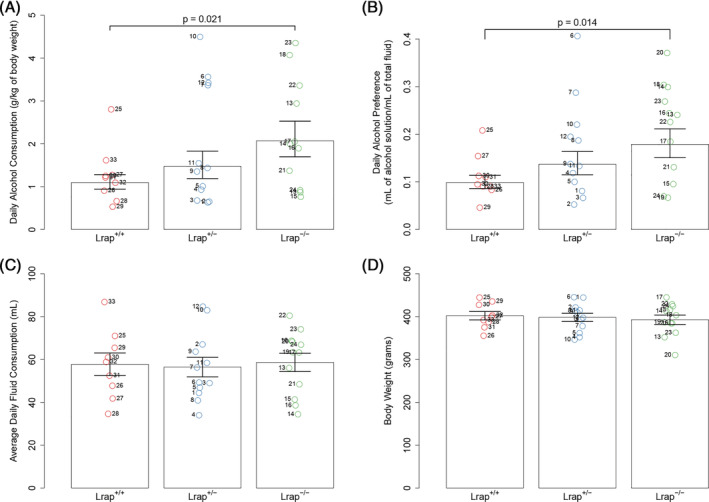
Effect of Lrap on alcohol consumption in genetically modified rats. Alcohol consumption by male rats (n = 9–12 rats/genotype) from the three genotypes (Lrap+/+, Lrap+/−, and Lrap−/−) was determined using the two‐bottle choice paradigm with 10% ethanol solution. Circles represent values for individual rats. Each point is labeled with a number to facilitate access to raw data if needed. For alcohol consumption and alcohol preference, means ±1 standard error from the linear mixed model on log transformed values were transformed back to the original scale and reported in the graphic. For all four outcomes, post hoc pairwise comparisons were made between all three groups and only comparisons with a p value less than 0.05 are reported. (A) Alcohol consumption in grams per kilogram of body weight. (B) Alcohol preference measured as the volume of alcohol solution divided by the total volume of fluids consumed. (C) Average daily fluid consumption in milliliters. No significant (p < 0.05) differences among genotypes were found. (D) Body weight in grams. There were no significant differences in body weight (p < 0.05) among genotypes
3.5. Effect of disruption of Lrap on brain gene expression (RNASeq)
3.5.1. Global brain gene expression levels in male Lrap+/+, Lrap+/− and Lrap−/− rats
On average, 54 million paired end reads were sequenced per single brain sample (range 36.6–61.4 million) with three individual brain samples per genotype. Of those, between 85% and 95% of reads per sample aligned to the Rn6 version of the rat genome. The merged reconstructed transcriptome included 145,807 isoforms (both polyadenylated and non‐polyadenylated) representing 114,700 gene products. Novel isoforms were annotated to an Ensembl protein‐coding gene if they shared at least one exon junction with an annotated isoform of an Ensembl protein‐coding gene. Of the 38,988 transcript isoforms annotated to a protein‐coding gene, 33,063 had expression levels above our pre‐determined background (more than 50 total reads from Lrap−/− and Lrap+/+ rats). The 33,063 isoforms that were expressed above the background, could be assigned to 14,114 protein‐coding Ensembl genes. All nine libraries passed quality control standards based on (1) number of reads sequenced (>10 million), (2) percent of reads eliminated during trimming (<10%), (3) percent of reads aligning to the genome (>80% of trimmed reads), and (4) percent of reads aligned to transcriptome (>60% of trimmed reads).
For protein‐coding genes which were the source of more than one isoform, the total number of reads that mapped to all the isoforms were summed (Sum Of Isoforms), and these values were subjected to a separate statistical analysis. The process of quantifying both individual isoforms and the composite of all isoforms (SOI) derived from a single gene was predicated on the known function of certain lncRNAs as modulators of alternative splicing. 38
At an FDR threshold of 0.10, 782 isoforms were differentially expressed (DE) between brains of Lrap−/− and Lrap+/+ rats. An independent analysis showed that 72 SOIs were differentially expressed when one summed all the reads that could be assigned to each gene and compared the means of these summed reads between the Lrap−/− and Lrap+/+ rats. The total of the differentially expressed isoforms and total gene products (SOIs) amounted to 750 products of annotated protein coding genes (i.e., some of differentially expressed isoforms were annotated to the same SOI and/or for some isoforms, the SOI was also significantly, differentially, expressed).
To explore differential alternative splicing between Lrap+/+ and Lrap−/− rats (i.e., isoforms present in one line and not present in the other), we used the rMATS algorithm [73] to specifically examine differences in the number of reads that covered individual alternate splice junctions among the 782 isoforms that were differentially expressed between the Lrap+/+ and Lrap−/− rats. Of the 782 differentially expressed isoforms, only 601 could be tested in this additional manner, because the remaining isoforms did not meet the minimum sequencing coverage needed for the rMATS algorithm to confidently associate them with one of the alternative splicing event types detected by this software. Overall, 133 of the 601 tested isoforms were associated with at least one novel alterative splicing event (FDR < 0.10, Lrap+/+ vs. Lrap−/−). A significant novel alternative splicing event indicates that the splicing event, e.g., a skipped exon, occurred in one group (Lrap+/+ or Lrap−/−) but not the other. Skipped Exon (SE), Retained Intron (ReI), and Mutually Exclusive Exon (MXE) were found to be the most prevalent types of significant alternative splicing events detected across the isoforms (103 [77%] out of 133 isoforms had at least one of these three types of splicing events). MXE refers to the situation in which two different exons within a gene coding region, when expressed, can determine the quantity of isoform expressed in Lrap+/+ versus Lrap−/− rats. This results in a transcript isoform exhibiting expression in one condition (e.g., Lrap+/+) when the first exon is expressed, while in the second condition (Lrap−/−), a different exon and its corresponding transcript isoform are expressed. As a result, splicing events, especially SEs and MXEs, would likely influence the protein coding sequence arising from the expressed isoforms. The remaining 468 differentially expressed isoforms in our study did not meet our statistical criteria for identifying novel alternative splicing events distinguishing Lrap+/+ from Lrap−/− rats and hence likely reflect differences in expression levels of the same isoforms between the Lrap+/+ and genetically manipulated, Lrap−/−, rats.
Of the transcripts that were part of the module originally associated with alcohol consumption, 14 only Lrap and P2rx4 transcripts were found to be differentially expressed in brains of Lrap+/+ compared with Lrap−/− rats in this analysis (Table 2). In the particular case of P2rx4, the SOI was significantly different between the Lrap+/+ and Lrap−/− rats, but the ratio of the two isoforms remained constant in each of the genotypes. This indicated that Lrap in this case may be affecting the overall transcription of P2rx4 rather than changing the levels of alternatively spliced isoforms. Of the 12 annotated genes in the module, one gene, Pcdhb5, was not detected in the RNASeq data set. Differential expression results for the remaining 11 annotated genes in the module indicated that they were not differentially expressed between Lrap+/+ and Lrap−/− rats when measured as the sum of expression of all isoforms or when examining each isoform separately (Table 2). We also performed qRT‐PCR analysis of transcripts of P2rx4, Ift81 and Txnip, which were three of the most highly connected genes in the co‐expression module associated with alcohol consumption (Figure 3). Similar to the RNASeq results, the levels of expression of Ift81 and Txnip were unchanged in the Lrap+/− and Lrap−/− rats (F 2,6 = 0.08 and p = 0.92; F 2,6 = 0.51 and p = 0.63, respectively), while decreases were indicated in the expression of P2rx4 in both Lrap+/− and Lrap−/− rats (F 2,6 = 3.93 and p = 0.08).
TABLE 2.
RNASeq expression levels of transcripts from the original alcohol consumption module in brains of genetically manipulated rats
| Gene symbol | Gene description | SOI‐level quantitation | ||||||
|---|---|---|---|---|---|---|---|---|
| Median of normalized read count—Lrap−/− | Median of normalized read count—Lrap+/− | Median of normalized read count—Lrap+/+ | Ratio of Lrap−/− to Lrap+/+ | Ratio of Lrap+/− to Lrap+/+ | Unadjusted p value | FDR | ||
| Anxa11 | annexin A11 | 1485 | 1423 | 1540 | 0.96 | 0.92 | 0.975 | 0.99 |
| Coq5 | coenzyme Q5, methyltransferase | 324 | 309 | 314 | 1.03 | 0.98 | 0.855 | 0.96 |
| Ift81 | intraflagellar transport 81 | 324 | 314 | 372 | 0.87 | 0.84 | 0.258 | 0.71 |
| Lrap a | long non‐coding RNA for alcohol preference | 14 | 40 | 86 | 0.16 | 0.47 | 1.5E‐14 | 1.80E‐11 |
| Maats1 | MYCBP‐associated, testis expressed 1 | 91 | 110 | 109 | 0.83 | 1.01 | 0.386 | 0.79 |
| P2rx4 | purinergic receptor P2X 4 | 197 | 187 | 280 | 0.70 | 0.67 | 0.006 | 0.26 |
| Parp3 | poly (ADP‐ribose) polymerase family, member 3 | 112 | 107 | 110 | 1.01 | 0.97 | 0.694 | 0.91 |
| Prkar1b | protein kinase cAMP‐dependent type 1 regulatory subunit beta | 6976 | 6766 | 6193 | 1.13 | 1.09 | 0.171 | 0.63 |
| Slc8b1(formerly Slc24a6) | solute carrier family 8 member B1 | 97 | 110 | 91 | 1.07 | 1.21 | 0.511 | 0.85 |
| Tmem116 | transmembrane protein 116 | 78 | 94 | 81 | 0.96 | 1.15 | 0.960 | 0.99 |
| Txnip | thioredoxin interacting protein | 1441 | 1575 | 1291 | 1.12 | 1.22 | 0.816 | 0.95 |
| Oas1f(formerly Oas1b) | 2 '‐5 'oligoadenylate synthetase 1F | 44 | 40 | 46 | 0.96 | 0.88 | 0.920 | 0.98 |
Only the isoform targeted for knockout was included for Lrap.
FIGURE 3.
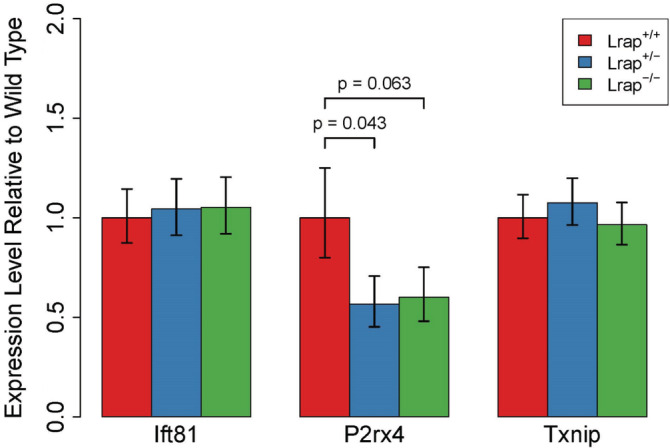
qRT‐PCR validation of difference in mRNA expression of genes from the alcohol consumption candidate module. The expression levels of three of the transcripts most highly connected to the hub transcript (Lrap) were assessed in brains of alcohol naive male Lrap wild type (+/+), Lrap heterozygous (+/−) and Lrap knockout (−/−) rats (n = 3) by qRT‐PCR. All statistical analyses were done on the delta Ct data and results are reported based on the transformation of mean values and mean values plus one standard error. All p values were calculated using a linear model (Ift81, P2rx4, Txnip) with post hoc pairwise comparisons
3.6. Informatics analysis of functional correlates of Lrap sensitive transcripts
To examine functional enrichment of the significantly differentially expressed transcripts of protein‐coding genes (Lrap+/+ vs. Lrap−/−, FDR < 0.10), we utilized KEGG pathways and Gene Ontology terms. One KEGG pathway (“Tight Junction”) was significantly enriched for differentially expressed gene products (Figure 4A; criteria: FDR for enrichment<0.10; fold enrichment >2 and 3 or more differentially expressed genes or isoforms had to be present in a category). This “Tight Junction” pathway (rno04530) contained 19 genes with either an isoform or a SOI that was differentially expressed between Lrap−/− and Lrap+/+ rat brains (Figure 4B). Likewise, 15 GO terms were significantly enriched for genes with either an isoform or a SOI that was differentially expressed (FDR for enrichment<0.10; fold enrichment >2, and 3 or more differentially expressed genes/isoforms had to be present in a category). One of the top 5 GO terms was “Response to Ethanol” (GO:0045471). That category contains 15 genes with differentially expressed isoforms or SOIs (Figure 4C). In both the Tight Junction and Response to Ethanol categories it was also noted that the primary differences in expression levels were at the isoform level rather than the SOI (Table S1).
FIGURE 4.
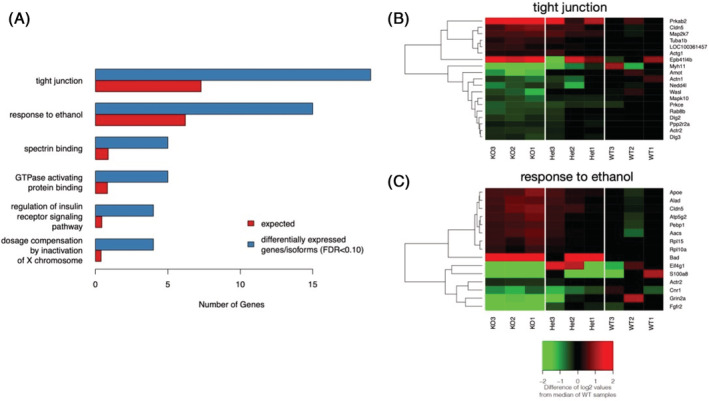
Functional enrichment of KEGG pathways and Gene Ontology (GO) terms in transcripts differentially expressed between Lrap knockout and wild type rat brains. Genes associated with isoforms and/or SOI that were differentially expressed (FDR < 0.10) were included. The background data set for enrichment include genes with at least one isoform and/or SOI that was expressed in brain and tested for differential expression. (A) Terms and pathways enriched for isoforms/SOI whose expression levels were altered by the genetic manipulation of Lrap. All pathways/terms included in the figure: (1) contained 3 or more differentially expressed isoform/SOI, (2) meet a significance threshold of an FDR <0.10, and (3) had a fold enrichment (observed number of differentially expressed genes divided by the number of differentially expressed genes expected by chance) of at least 2. Fifteen GO terms met all three criteria but only the top 5 (by p value) are included in the graphic for simplicity. Differentially expressed isoforms/SOI associated with the (B) Tight Junction KEGG pathway or associated with the (C) Response to Ethanol GO term. Expression values are represented in this heatmap as the difference in log2 transformed and library size adjusted read counts for a sample and the median of this transformed read count in the wild type rats
3.7. Identification of Lrap‐influenced transcripts associated with alcohol consumption
As noted above, there were many isoforms and some SOIs whose RNA expression levels were influenced by the genetic manipulation of Lrap, but not all of these are necessarily associated with, or determinants of, the level of alcohol consumption. To focus on Lrap‐influenced transcripts that are likely to contribute to the relationship between Lrap and alcohol consumption, we combined gene expression information from the Lrap−/− RNASeq experiment and our original data that included Affymetrix Exon Array brain expression data and alcohol consumption measures from the HXB/BXH RI rat strains. 14 Of the 14,114 protein coding Ensembl genes (representing 33,063 isoforms) that were expressed above background in the RNASeq experiment, 8770 were also represented as expressed transcripts in the microarray experiment. We used a multi‐level statistical strategy to arrive at association between gene product and phenotype. Of the 8770, the expression levels of 209 were significantly (unadjusted p < 0.05) correlated with alcohol consumption in the HXB/BXH RI panel. Furthermore, 59 of those transcripts were also significantly correlated with Lrap expression in the HXB/BXH RI panel (unadjusted p value<0.05). Finally, of the 59 transcripts correlated with both alcohol consumption and Lrap expression in the HXB/BXH RI panel, 6 had an SOI that was also differentially expressed within the RNASeq data (unadjusted p value < 0.05) between Lrap−/− and Lrap+/+ rats (Table 3). All six are differentially expressed in the direction predicted from the correlation with Lrap in the microarray experiment, i.e., a positive correlation between the transcript and Lrap in the microarray experiment results in an expression ratio less than 1 for the comparison of Lrap−/− to Lrap+/+. To arrive at the identity of the six genes considered to be associated with both Lrap function and alcohol consumption, these candidates had to traverse three levels of statistical scrutiny (i.e., correlated with alcohol consumption across HXB/BXH RI panel, expression correlated with Lrap expression across the HXB/BXH RI panel, and differentially expressed between Lrap+/+ and Lrap−/− rats).
TABLE 3.
Possible mediators of the effect of Lrap on alcohol consumption
| Gene symbol | Gene description | Physical location (RN6) | Correlation with alcohol consumption HXB/BXH RI Panel (microarrays) | Correlation with Lrap expression in HXB/BXH RI panel (microarrays) | eQTL in HXB/BXH RI panel (microarrays) | SOI‐level analysis of Lrap−/− vs Lrap+/+ (RNA‐Seq) | SOI‐level analysis of Lrap+/− vs Lrap−/− (RNA‐Seq) | Combination of correlation with Lrap and alcohol consumption in the HXB/BXH and differentiial expression in Lrap−/− and Lrap+/+ | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Correlation Coefficient (p value) | Correlation Coefficient (p value) | Position (95% Bayesian credible interval) | Genome‐wide p value | Ratio of KO to WT | Unadjusted p value | Ratio of HET to WT | Unadjusted p value | p Value | FDR | |||
| Cndp1 | carnosine dipeptidase 1 | Chr18: 81.5 Mb | 0.63 (0.002) | −0.51 (0.0193) | Chr6: 6.6 Mb [3.6 to 145.8] | 0.21 | 1.77 | 0.036 | 1.38 | 0.032 | 1.7E‐06 | 9.5E‐04 |
| Ninj2 | ninjurin 2 | Chr4: 152.6 Mb | 0.53 (0.012) | −0.63 (0.0024) | Chr4: 166.1 Mb [136.6 to 166.1] | 0.025 | 1.3 | 0.045 | 1.06 | 0.411 | 1.3E‐06 | 9.5E‐04 |
| P2rx4 | purinergic receptor P2X 4 | Chr12: 39.3 Mb | −0.63 (0.002) | 0.84 (<0.0001) | Chr12: 39.1 Mb [39.1 to 41.5] | 0.034 | 0.7 | 0.006 | 0.67 | 0.003 | 3.3E‐11 | 2.9E‐07 |
| Slc35c2 | solute carrier family 35 member C2 | Chr3: 161.8 Mb | −0.57 (0.008) | 0.53 (0.0142) | Chr17: 61.9 Mb [2.3 to 89.6] | 0.375 | 1.47 | 0.001 | 1.44 | 0.002 | 1.4E‐07 | 2.4E‐04 |
| Zfr2 | zinc finger RNA binding protein 2 | Chr7: 11.3 Mb | 0.46 (0.035) | −0.46 (0.0364) | Chr9: 117.7 Mb [15.3 to 120] | 0.232 | 1.38 | 0.028 | 1.14 | 0.229 | 3.6E‐05 | 4.2E‐03 |
| Znhit6 | zinc finger, HIT‐type containing 6 | Chr2: 251.5 Mb | −0.48 (0.028) | 0.51 (0.0174) | Chr2: 251 Mb [146.8 to 259.9] | 0.409 | 0.64 | 0.009 | 0.92 | 0.972 | 4.3E‐06 | 1.7E‐03 |
Note: Sums of isoforms (SOI) are included if they were correlated with alcohol consumption in the HXB/BXH recombinant inbred rat panel using the microarray data (unadjusted p value<0.05), correlated with Lrap in the HXB/BXH RI panel (unadjusted p value<0.05), and differentially expressed between the Lrap−/− and the wild type Lrap+/+ rat in the RNASeq study (unadjusted p value<0.05).
3.8. Lrap characteristics and similarities with mouse and human sequences
A cross‐species comparison for Lrap generated information that a similar transcript exists in the mouse, and the mouse DNA sequence producing this transcript (A930024E05Rik) resides in a region syntenic to the rat genome coding for Lrap. Certain regions of the DNA sequences for Lrap and A930024E05Rik have high homology. Particularly evident is the homology between the areas coding for what can be described as exon 1 in both species, as well as the region that was deleted in the Lrap−/− rats (Figure 5). It should be noted that in the rats, the deleted DNA region appears in an area characteristic of an exon. In the mouse, the homologous region starts within an exon sequence but a portion of the sequence of this homologous region is beyond a canonical splice signal sequence. The deletion would disrupt a splicing signal that would be necessary for linking Exon 2 and 3 in the mouse. It should also be noted that the sequences for Lrap in mouse and rat are on opposite strands (sense for mouse and antisense for rat). In human, an Lrap‐like sequence (AC145422.1) is evident in a DNA region that is syntenic with both mouse and rat Lrap‐like regions and lies on the sense strand in this region. The current annotation of transcripts produced from this region of the human DNA indicates the presence of a transcript generated from two exons, with these exons being separated by a long (12,169 bp) intronic region (Figure 5A). The region homologous to the region that we deleted in the rat genome maps to the intronic region in the human. On closer examination, however, and using long polyA‐ RNA‐Seq data from the ENCODE project on the GM12878 cell line, we noted that the region of the human DNA homologous to the area that we deleted in the rat produced RNA reads that could represent the presence of an unannotated exon of the human ortholog of Lrap being transcribed in the region identified as AC14522.1 (Figure 5E).
FIGURE 5.

Regions of the mouse and human genome homologous to the region of the rat genome that contains Lrap. The entire genomic sequence of Lrap, that is, including introns, from the rat was compared with the mouse genome and the human genome. (A) Alignment of the Lrap genomic sequence to the human genome. The green blocks in the first track denote areas of homology in the human DNA sequence. The numeric label on each box corresponds to a row in (F). The intensity of the green color is related to the percent identity of the two sequences. The area highlighted in gray designates the homologous region of the human genome that was deleted in the rat. The second track 'GENCODE v29 Comprehensive Transcript Set (+ only)' contains an annotated human transcript, AC145422.1, produced from the same strand that is homologous to the Lrap sequence in this region. (B) Alignment of the Lrap genomic sequence to the mouse genome. The green blocks in the first track denote areas of homology in the mouse DNA sequence. The numeric label on each box corresponds to a row in (G). The intensity of the green color is related to the percent identity of the two sequences. The area highlighted in gray designates the homologous region of the mouse genome that was deleted in the rat. The second track 'GENCODE VM20 Comprehensive Transcript Set (+ only)' contains an annotated mouse transcript, A930024E05Rik, produced from the same strand that is homologous to the Lrap sequence in this region. (C) Human and mouse homologous sequences mapped onto the rat genome. The first track, 'Human Alignment', highlights the regions of the rat genome that are homologous to the human genome using the same coloring and labeling as in (A). The second track, 'Mouse Alignment', highlights the regions of the rat genome that are homologous to the mouse genome using the same coloring and labeling as in (B). The third track, 'Lrap', contains the original structure of Lrap derived from the transcriptome reconstruction in the SHR and BN‐Lx brain RNASeq data in blue and the region of Lrap that was eliminated in the knockout rats in red. Please note that the homologous regions have been superimposed and intervening regions are not to the same scale. In addition, since Lrap was transcribed from the negative strand in rat but is homologous to regions on the positive strand in both human and mouse, the orientation of (C) has been flipped so that the first exon of Lrap is on the left and the last exon of Lrap is on the right. (D) Genetic variants within the genomic area of Lrap. This panel provides information on polymorphisms (SNPs and indels) distinguishing the Wistar Kyoto rat strain and the related SHR rat strain from the BN rat strain (reference strain). This illustration also indicates the indels and SNPs that disrupt the possible ORF in the BN sequence. (E) RNA expression in human of Lrap locus. The genomic area depicted in this panel covers chr12:121,579,800–121,593,949 bp of the human genome (hg38). The first track 'GENCODE v29 Comprehensive Transcript Set (+ only)' contains an annotated human transcript, AC145422.1, produced from the plus strand. The second track 'LRAP (WKY rn6) alignment to Human' includes regions of human genome that are homologous to the region of the rat genome that produces the Lrap transcript. The intensity of the green color is related to the percent identity of the two sequences. The area highlighted in gray designates the homologous region of the human genome that was deleted in the rat. The third and final track 'GM12878 ENCODE/CSHL PolyA‐ (+ strand)' indicates the number of RNASeq reads that align to the region. RNASeq reads generated by the ENCODE/Cold Spring Harbor Lab were derived from long polyA‐ sequence of the GM12878 cell line and are publicly available through the UCSC Genome Browser and through the Gene Omnibus Database (GEO Accession: GSM758572). (F) Summary of Lrap alignment to the human genome contains additional detail, including percent homology, about the alignment of Lrap to the human genome. The numeric labels on the rows correspond to the green blocks in (A) with the same label. (G) Summary of Lrap alignment to the mouse genome contains additional detail, including percent homology, about the alignment of Lrap to the mouse genome. The numeric labels on the rows correspond to the green blocks in (B) with the same label
4. DISCUSSION
It was by serendipity (and a false premise) that we chose the region to delete in Lrap to produce the Lrap−/− rats. The deleted region resembled an open reading frame (ORF) in the BN rat reference DNA, but the current comparison across species indicated that this is not an ORF in either mouse or human. Although the region resembling the ORF in rat, appears as an ORF in the reference BN genome, in other strains of rats, and particularly Wistar‐derived strains, the putative ORF is disrupted by an indel and SNPs (shown in Figure 5 for Wistar‐derived strains). The Wistar strain was used to generate our Lrap−/− and Lrap+/− rats. The maintenance of sequences homologous (but not identical) to the deleted region in species from mouse to human may indicate that the deleted region in the rat DNA, even though it is not apt to be translated, still has a functional significance. Since our genetic manipulation of Lrap produced a lesser reduction of the expression of other segments of Lrap (e.g., Exons 1 and 2), one might conclude that the excised sequence is an important component of the mechanism of action of Lrap. However, we do note the possibility that the decrease in levels of exons 1 and 2 in the Lrap−/− rat could also contribute to the observed results.
We had previously postulated that the disruption of Lrap would result in increased alcohol consumption, based on our prior finding of a negative correlation between alcohol consumption and brain Lrap levels across the rat HXB/BXH RI panel. 14 Our results with the Wistar rats in which Lrap was disrupted confirmed this hypothesis, showing that alcohol consumption was doubled in Lrap−/− rats, and increased by 50% in the Lrap+/− animals, when compared with the Lrap+/+ rats. When we calculated an alcohol preference ratio (volume of 10% alcohol solution/total volume of fluid consumed/day), we noted a similar order, with the Lrap−/− rats displaying the highest preference ratio. We also noted that the constitutive deletion of this portion of Lrap altered the expression of transcripts of 750 protein‐coding genes in brain when comparing adult Lrap+/+ and Lrap−/− rats. When considering a change in the levels of such a number of transcripts by deletion of a segment of a lncRNA in our studies, one is drawn to consider functions of lncRNA that are more global than discrete (i.e., acting on transcription or stability of a particular transcript or a small number of transcripts). 39 Two more global functions that have been described for lncRNA, are chromatin structure modification through interaction with histones 40 and modulation of alternative splicing. 41 By examining the effect of knockdown of 39 lncRNAs in three human cell lines, Wendt Porto et al. 41 identified 17,525 alternative splicing events. Many of these events were cell‐specific and were a result of lncRNA‐induced alternative splicing related to RNA binding protein pre‐mRNA interactions at splice junctions. 41 Another mechanism by which lncRNA can produce large scale changes in splicing is by modulating the phosphorylation of splicing factors or by direct interactions “hijacking” splicing factors. 4 The pattern (isoform level changes) and extent of changes we noted in protein‐coding transcripts (i.e., 708 of the 782 differentially expressed isoforms did not have corresponding changes at the SOI level) may well indicate that Lrap is a lncRNA that has as its major function, the modulation of alternative splicing. That is, the genetic deletion of a portion of Lrap primarily changed the quantitative relationship between the isoforms derived from a particular gene, rather than the total quantity of transcripts generated from a gene. We also examined more thoroughly the character of differences in splicing events (as opposed to quantitative differences in isoform expression levels) occurring between the Lrap+/+ and Lrap−/− rats. The rMATS algorithm distinguishes between five types of alternative splicing events and can indicate the differential presence of an isoform produced from a particular gene across experimental conditions compared. Within the 601 differentially expressed isoforms analyzed with the rMATS algorithm, 133 isoforms were predominantly unique to either Lrap+/+ or Lrap−/− rats. In other words, these isoforms were specifically expressed in either Lrap+/+ and Lrap−/− strains. This would indicate that Lrap can change both quantitative and qualitative characteristics of splicing.
On the other hand, the total quantity of transcript derived from the gene P2rx4 was significantly different between Lrap+/+ and Lrap−/− rats, but the ratio of the two isoforms of this gene remained constant. It was of interest that P2rx4 was the one gene product in the module in which Lrap was designated as the “hub” gene (“most connected”) that was significantly changed in its level of expression in brain between Lrap+/+ and Lrap−/− rats. In addition, its expression level was significantly correlated with levels of Lrap expression across the HxB/BxH panel of recombinant inbred rat strains. One needs to be cognizant that the coexpression relationships that determine segregation of transcripts into a module when using WGCNA, 16 do not necessarily indicate that the transcripts included within a module have an identical form of co‐regulation, 42 and coexpression may be a result of coordinated responses, through different mechanisms, to a particular cellular state or signal.
However, the significant correlation between Lrap expression and the total levels of P2rx4 expression, as well as P2rx4 expression and alcohol consumption, bears some additional attention. P2rx4 is a ligand‐gated calcium channel activated by ATP, 43 and its function has been linked by numerous studies to control of alcohol consumption in rats and mice. 44 , 45 , 46 , 47 Many lncRNAs are expressed in a tissue and/or cell‐specific manner, 8 , 48 and one could invoke a “guilt by association” approach 49 to hypothesize in which brain cells Lrap is directly influencing the expression of other transcripts. The high correlation of Lrap expression with expression of P2rx4 could indicate that these two transcripts may be localized to the same cell type. P2rx4 shows a broad distribution in the CNS and is present in both neurons and glial cells. 49 Data generated by Dong et al 50 indicate that the expression of the Lrap homolog (A930024E05Rik) in adolescent (P17) mice is highest in microglia compared with other cell types in brain, and thus a relationship between Lrap and P2rx4 expression may be most evident in microglia. Microglia are ubiquitous throughout the brain and if Lrap is localized to microglia, the distribution of the Lrap mouse homolog A930024E05Rik noted in the Allen Mouse Brain Atlas would be instructive. 51 The Allen Brain Atlas shows that Lrap appears at low levels throughout the adult mouse brain and resembles the expected distribution of microglia. It is of interest that our original interpretation of the function of the co‐expressed components of the module, for which Lrap was the hub gene, led us to conclude that the components of the module were related to “immune function, energy metabolism, calcium homeostasis and glial‐neuronal communication”. 14
Furthermore, we examined the expression values for Lrap in liver tissue from the BN‐Lx/Cub and SHR/OlaIpcv that we assayed using RNA‐Seq in a manner similar to the methods used here. 52 There was evidence for Lrap expression in both read coverage and coverage of specific exon junctions of Lrap (http://phenogen.org). There was also evidence of expression of the mouse homolog from the Expression Atlas 53 in the Ensembl database (http://ensembl.org) across several tissues including liver, lung, spleen, colon, heart, and kidney and across several brain regions with the highest expression in brain and testis. For the human homolog, we examined data from GTEx 54 available through the UCSC genome browser. 37 Similar to mouse, this transcript was expressed across many tissue tissues with the highest expression in testis and brain. Further research is needed to fully understand the relationship of expression of Lrap in different tissues with alcohol consumption and other phenotypes.
As stated above, our RNASeq studies of the adult brain transcriptome indicated extensive changes in gene expression between Lrap+/+ and Lrap−/− rats. When the genes with an isoform or a SOI that was differentially expressed in brains of adult Lrap+/+ and Lrap−/− rats were subjected to functional enrichment analysis using GO or KEGG, the most significant pathway using KEGG, was “Tight Junction”. In the CNS, tight junctions are evident between astrocytes and oligodendrocytes and capillary endothelial cells, as well as between glia and axons and in the myelin sheath. 55 , 56 , 57 The Tight Junction category encompasses MAGUK (zonula occludens, ZO) tight junction‐associated proteins that participate in signal transduction at specialized cell–cell junctions 58 and claudin 5, which is an essential functional component of endothelial tight junctions. 59 Current evidence shows that microglia participate in the integrity of the endothelial tight junction and modulation of microglial phenotype can alter brain endothelial permeability. 60 It is becoming evident that changes in endothelial permeability and increased entry of pattern recognition receptor ligands into brain, with generation of inflammatory responses, may contribute to increases in alcohol consumption by rodents 61 and possibly humans. 62
Among the top 5 GO terms that met our requirements for significance in enrichment (Figure 4A), transcripts in two of the five categories could be linked to receptor signaling (GTPase activating protein binding, regulation of insulin receptor signaling pathway) and one to neuronal membrane integrity (spectrin binding), while transcripts in one category were associated with X chromosome inactivation. X‐chromosome inactivation is a well‐studied epigenetic phenomenon associated with many lncRNA. 63 The last of these significantly enriched GO terms was “Response to Alcohol”. In this category were several isoforms of Actr2, actin related protein (ARP) homologs (with isoforms recognized through our RNASeq transcriptome reconstruction) and claudin 5, which was also part of the “Tight Junction” pathway in KEGG. The ARP is important in early synaptogenesis. 64 Two neurotransmitter receptor transcripts were also evident in the GO “Response to Alcohol” category: Cnr1 (cannabinoid receptor 1; CB1) and Grin2a (the GluN2A subunit of the NMDA receptor). The CB1 receptor has been linked to control of alcohol consumption in mice 65 and the NMDA receptor has been linked to alcohol tolerance and the hyperexcitability seen on withdrawal in alcohol‐dependent animals, including humans. 66
As attractive as it may seem to associate individual gene products that are differentially expressed between Lrap+/+ and Lrap−/− rats to differences in alcohol consumption, based on their function or inclusion in a particular GO category or KEGG pathway, one needs to, at least, consider that a candidate transcript should also be quantitatively related to the phenotype of interest. This assumption of quantitative genetics could be tested in our HXB/BXH RI panel of rats by measuring the correlation between levels of transcript expression and the phenotype of alcohol consumption (g/kg). Our analysis identified the products of six genes (Cndp1, Ninj2, P2rx4, Slc35c2, Zfr2 and Znhit6) that were differentially expressed between the Lrap−/− and Lrap+/+ rats, and correlated in their expression levels with both Lrap expression and alcohol consumption across the panel of HXB/BXH RI rat strains (Table 3). Based on the statistical significance of the various correlations and comparisons, P2rx4 stood out as the best candidate fulfilling all criteria, and its relationship to alcohol consumption was mentioned earlier. The functions of the other gene products can be related to neurological disorders, neurogenesis, inflammation/immune regulation and transcriptional activity. 67 , 68 , 69 , 70 , 71 , 72 , 73 , 74 , 75 These gene products may be considered as additional factors contributing to the phenotypes of alcohol consumption and/or alcohol preference in rats.
It should be noted that our experimental design is focused on ascertaining genetic components that generate a predisposition to particular phenotypes (i.e., predisposition to consumption of alcohol in the initial stages of exposure, not alcohol dependence). 14 Thus, our measures of gene expression and analysis of coexpression modules and networks takes place in animals not exposed to ethanol. We then measure alcohol consumption in another cohort from the same strains of rats, presuming that the gene expression patterns will remain constant in rats which are isogenic within a strain and are raised in an identical environment. The genetic differences between strains are considered to contribute to differences in gene expression and the subsequent differences in alcohol drinking and/or other phenotypes. Another caveat that needs to be noted, is that our studies were performed using male rats. The importance of lncRNAs in X chromosome inactivation and epigenetic programming would predicate that similar experiments be performed in female animals.
We have established the presence of a novel lncRNA in rat brain (Lrap) and produced evidence that homologous transcripts can be produced from syntenic regions of the genome of mice and humans. We have provided evidence of a significant negative correlation between Lrap expression and expression of P2rx4 in adult brain, and a significant correlation between both of these transcripts (Lrap and P2rx4) and levels of alcohol consumption across 21 strains of rats within a RI panel of rats (HXB/BXH). By disrupting a specific area of Lrap (an area also present in the genome of both mouse and human), we show that predicted changes (based on prior correlation analysis) take place in both the expression of P2rx4 and alcohol consumption when comparing the rats with the disrupted Lrap sequence and the Lrap+/+ controls. This information focuses attention on a region of the sequence of Lrap that may have functional implications. In addition, we show that constitutive disruption of Lrap produces broad based changes in the brain transcriptome, and show that Lrap effects are most evident in terms of alternative splicing of a large number of transcripts. Our results suggest that Lrap may be an important component controlling isoform expression, and that a subset of the genes whose expression is influenced by disruption of Lrap is predisposing the phenotype of elevated alcohol consumption. An important caveat is that our studies have been performed in male animals and extrapolation to females requires further work.
Supporting information
Table S1: Whole brain differential RNA expression results at both the isoform and the sum of isoform (SOI) level that include the statistical difference between Lrap+/+ and Lrap −/− rats. SOI and all associated isoforms were included in any of the isoforms for an SOI or the SOI were differentially expressed between Lrap+/+ and Lrap−/− rats (FDR < 0.10).
ACKNOWLEDGMENTS
The authors sincerely thank Carolyn Ferguson and Matthew McKay for help in the generation of the genetically modified rats, Seija Tillanen for assistance in the molecular biology experiments and Yinni Yu for help with the animal drinking studies. They would also like to thank the Integrative Neuroscience Initiative on Alcoholism‐Neuroimmune for their continued support and encouragement.
DATA AVAILABILITY
The data that support the findings of this study are openly available in Gene Expression Omnibus database at https://www.ncbi.nlm.nih.gov/geo/, reference number GSE1557079.
Saba LM, Hoffman PL, Homanics GE, et al. A long non‐coding RNA (Lrap) modulates brain gene expression and levels of alcohol consumption in rats. Genes, Brain and Behavior. 2021;20:e12698 10.1111/gbb.12698
[Correction added on 22 October 2020, after first online publication: The article category has been updated from “Review” to “Original Article” in this current version.]
Funding information Banbury Fund; Ethanol Mechanisms in GABAAR gene targeted mice, Grant/Award Number: R37AA010422; Mapping RNA Protein Interaction Networks in the Human Genome, Grant/Award Number: R01GM123314; Overall NIDA Core "Center of Excellence" in Transcriptomics, Systems Genetics and the Addictome, Grant/Award Number: P30DA044223; Role of noncoding RNA in Alcohol Action, Grant/Award Number: U01AA020889; The Heritable Transcriptome and Alcoholism, Grant/Award Number: R24AA013162
REFERENCES
- 1. Ernst C, Morton CC. Identification and function of long non‐coding RNA. Front Cell Neurosci. 2013;7:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sun Q, Hao Q, Prasanth KV. Nuclear Long noncoding RNAs: key regulators of gene expression. Trends Genet. 2018;34(2):142‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vance KW, Ponting CP. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 2014;30(8):348‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Romero‐Barrios N, Legascue MF, Benhamed M, Ariel F, Crespi M. Splicing regulation by long noncoding RNAs. Nucleic Acids Res. 2018;46(5):2169‐2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Long Y, Wang X, Youmans DT, Cech TR. How do IncRNAs regulate transcription? Sci Adv. 2017;3:eaao2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clark BS, Blackshaw S. Understanding the role of lncRNAs in nervous system development. Adv Exp Med Biol. 2017;1008:253‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pereira Fernandes D, Bitar M, Jacobs FMJ, Barry G. Long non‐coding RNAs in neuronal aging. Noncoding RNA. 2018;4(2):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775‐1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang A, Wang J, Liu Y, Zhou Y. Mechanisms of Long non‐coding RNAs in the assembly and plasticity of neural circuitry. Front Neural Circuits. 2017;11:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fochler S, Morozova TV, Davis MR, et al. Genetics of alcohol consumption in Drosophila melanogaster . Genes Brain Behav. 2017;16(7):675‐685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leggio L, Addolorato G, Cippitelli A, Jerlhag E, Kampov‐Polevoy AB, Swift RM. Role of feeding‐related pathways in alcohol dependence: a focus on sweet preference, NPY, and ghrelin. Alcohol Clin Exp Res. 2011;35(2):194‐202. [DOI] [PubMed] [Google Scholar]
- 12. Brasser SM, Castro N, Feretic B. Alcohol sensory processing and its relevance for ingestion. Physiol Behav. 2015;148:65‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clarke TK, Adams MJ, Davies G, et al. Genome‐wide association study of alcohol consumption and genetic overlap with other health‐related traits in UKbiobank (N=112 117). Mol Psychiatry. 2017;22(10):1376‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saba LM, Flink SC, Vanderlinden LA, et al. The sequenced rat brain transcriptome—its use in identifying networks predisposing alcohol consumption. FEBS J. 2015;282(18):3556‐3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doss S, Schadt EE, Drake TA, Lusis AJ. Cis‐acting expression quantitative trait loci in mice. Genome Res. 2005;15(5):681‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mulligan MK, Abreo T, Neuner SM, et al. Identification of a functional non‐coding variant in the GABA a receptor alpha2 subunit of the C57BL/6J mouse reference genome: major implications for neuroscience research. Front Genet. 2019;10:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blednov YA, Borghese CM, Ruiz CI, et al. Mutation of the inhibitory ethanol site in GABAA rho1 receptors promotes tolerance to ethanol‐induced motor incoordination. Neuropharmacology. 2017;123:201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA‐guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bassett AR, Tibbit C, Ponting CP, Liu JL. Highly efficient targeted mutagenesis of drosophila with the CRISPR/Cas9 system. Cell Rep. 2014;6(6):1178‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou Z, Karlsson C, Liang T, et al. Loss of metabotropic glutamate receptor 2 escalates alcohol consumption. Proc Natl Acad Sci U S A. 2013;110(42):16963‐16968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tabakoff B, Saba L, Printz M, et al. Genetical genomic determinants of alcohol consumption in rats and humans. BMC Biol. 2009;7:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin MW. Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet. journal. 2011;17:10‐12. [Google Scholar]
- 25. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nature Biotechnology. 2019;37:907‐915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat Biotechnol. 2015;33(3):290‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript‐level expression analysis of RNA‐seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 2016;11(9):1650‐1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parikshak NN, Swarup V, Belgard TG, et al. Genome‐wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature. 2016;540(7633):423‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinform. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289‐300. [Google Scholar]
- 32. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353‐D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Consortium TGO . The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47(D1):D330‐D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/bioconductor package biomaRt. Nat Protoc. 2009;4(8):1184‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shen S, Park JW, Lu Z, Lin L, Henry MD, Wu YN, Zhou Q, Xing Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA‐Seq data. PNAS. 2014;111:E5593‐E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hermsen R, de Ligt J, Spee W, et al. Genomic landscape of rat strain and substrain variation. BMC Genomics. 2015;16:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996‐1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gonzalez I, Munita R, Agirre E, et al. A lncRNA regulates alternative splicing via establishment of a splicing‐specific chromatin signature. Nat Struct Mol Biol. 2015;22(5):370‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fernandes JCR, Acuna SM, Aoki JI, Floeter‐Winter LM, Muxel SM. Long non‐coding RNAs in the regulation of gene expression: physiology and disease. Noncoding RNA. 2019;5(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Khalil AM, Guttman M, Huarte M, et al. Many human large intergenic noncoding RNAs associate with chromatin‐modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106(28):11667‐11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Porto FW, Daulatabad SV, Janga SC. Long non‐coding RNA expression levels modulate cell‐type‐specific splicing patterns by altering their interaction landscape with RNA‐binding proteins. Genes (Basel). 2019;10(8):593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Y, Zha H, Wang JZ, Chu CH. Gene Co‐regulation vs. Co‐expression. Poster Proceedings of the International Conference on Research in Computational Molecular Biology (RECOMB), San Diego, CA; 2004. pp. 232‐233.
- 43. Stokes L, Layhadi JA, Bibic L, Dhuna K, Fountain SJ. P2X4 receptor function in the nervous system and current breakthroughs in pharmacology. Front Pharmacol. 2017;8:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wyatt LR, Finn DA, Khoja S, et al. Contribution of P2X4 receptors to ethanol intake in male C57BL/6 mice. Neurochem Res. 2014;39(6):1127‐1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Franklin KM, Asatryan L, Jakowec MW, Trudell JR, Bell RL, Davies DL. P2X4 receptors (P2X4Rs) represent a novel target for the development of drugs to prevent and/or treat alcohol use disorders. Front Neurosci. 2014;8:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khoja S, Huynh N, Asatryan L, Jakowec MW, Davies DL. Reduced expression of purinergic P2X4 receptors increases voluntary ethanol intake in C57BL/6J mice. Alcohol. 2018;68:63‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Franklin KM, Hauser SR, Lasek AW, Bell RL, McBride WJ. Involvement of Purinergic P2X4 receptors in alcohol intake of high‐alcohol‐drinking (HAD) rats. Alcohol Clin Exp Res. 2015;39(10):2022‐2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Quinn JJ, Chang HY. Unique features of long non‐coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47‐62. [DOI] [PubMed] [Google Scholar]
- 49. Wolfe CJ, Kohane IS, Butte AJ. Systematic survey reveals general applicability of "guilt‐by‐association" within gene coexpression networks. BMC Bioinform. 2005;6:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dong X, Chen K, Cuevas‐Diaz Duran R, et al. Comprehensive identification of Long non‐coding RNAs in purified cell types from the brain reveals functional LncRNA in OPC fate determination. PLoS Genet. 2015;11(12):e1005669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. McCarthy M. Allen brain atlas maps 21,000 genes of the mouse brain. Lancet Neurol. 2006;5(11):907‐908. [DOI] [PubMed] [Google Scholar]
- 52. Harrall KK, Kechris KJ, Tabakoff B, et al. Uncovering the liver's role in immunity through RNA co‐expression networks. Mamm Genome. 2016;27(9–10):469‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Papatheodorou I, Moreno P, Manning J, et al. Expression atlas update: from tissues to single cells. Nucleic Acids Res. 2020;48(D1):D77‐D83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mele M, Ferreira PG, Reverter F, et al. Human genomics. The human transcriptome across tissues and individuals. Science. 2015;348(6235):660‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Abbott NJ, Ronnback L, Hansson E. Astrocyte‐endothelial interactions at the blood‐brain barrier. Nat Rev Neurosci. 2006;7(1):41‐53. [DOI] [PubMed] [Google Scholar]
- 56. Gonzalez‐Mariscal L, Betanzos A, Nava P, Jaramillo BE. Tight junction proteins. Prog Biophys Mol Biol. 2003;81(1):1‐44. [DOI] [PubMed] [Google Scholar]
- 57. Niu J, Tsai HH, Hoi KK, et al. Aberrant oligodendroglial‐vascular interactions disrupt the blood‐brain barrier, triggering CNS inflammation. Nat Neurosci. 2019;22(5):709‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gonzalez‐Mariscal L, Betanzos A, Avila‐Flores A. MAGUK proteins: structure and role in the tight junction. Semin Cell Dev Biol. 2000;11(4):315‐324. [DOI] [PubMed] [Google Scholar]
- 59. Van Itallie CM, Anderson JM. Claudin interactions in and out of the tight junction. Tissue Barriers. 2013;1(3):e25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mehrabadi AR, Korolainen MA, Odero G, Miller DW, Kauppinen TM. Poly(ADP‐ribose) polymerase‐1 regulates microglia mediated decrease of endothelial tight junction integrity. Neurochem Int. 2017;108:266‐271. [DOI] [PubMed] [Google Scholar]
- 61. Mayfield J, Harris RA. The Neuroimmune basis of excessive alcohol consumption. Neuropsychopharmacology. 2017;42(1):376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Warden A, Erickson E, Robinson G, Harris RA, Mayfield RD. The neuroimmune transcriptome and alcohol dependence: potential for targeted therapies. Pharmacogenomics. 2016;17(18):2081‐2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee JT. Lessons from X‐chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009;23(16):1831‐1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Spence EF, Kanak DJ, Carlson BR, Soderling SH. The Arp2/3 complex is essential for distinct stages of spine synapse maturation, including synapse Unsilencing. J Neurosci. 2016;36(37):9696‐9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Balla A, Dong B, Shilpa BM, et al. Cannabinoid‐1 receptor neutral antagonist reduces binge‐like alcohol consumption and alcohol‐induced accumbal dopaminergic signaling. Neuropharmacology. 2018;131:200‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Krystal JH, Petrakis IL, Mason G, Trevisan L, D'Souza DC. N‐methyl‐D‐aspartate glutamate receptors and alcoholism: reward, dependence, treatment, and vulnerability. Pharmacol Ther. 2003;99(1):79‐94. [DOI] [PubMed] [Google Scholar]
- 67. Babizhayev MA. Biochemical, biomedical and metabolic aspects of imidazole‐containing dipeptides with the inherent complexity to neurodegenerative diseases and various states of mental well‐being: a challenging correction and Neurotherapeutic pharmaceutical biotechnology for treating cognitive deficits, depression and intellectual disabilities. Curr Pharm Biotechnol. 2014;15(8):738‐778. [DOI] [PubMed] [Google Scholar]
- 68. Araki T, Milbrandt J. Ninjurin2, a novel homophilic adhesion molecule, is expressed in mature sensory and enteric neurons and promotes neurite outgrowth. J Neurosci. 2000;20(1):187‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang J, Fa J, Wang P, et al. NINJ2‐ a novel regulator of endothelial inflammation and activation. Cell Signal. 2017;35:231‐241. [DOI] [PubMed] [Google Scholar]
- 70. Lu L, Hou X, Shi S, Korner C, Stanley P. Slc35c2 promotes Notch1 fucosylation and is required for optimal notch signaling in mammalian cells. J Biol Chem. 2010;285(46):36245‐36254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Feng S, Shi T, Qiu J, et al. Notch1 deficiency in postnatal neural progenitor cells in the dentate gyrus leads to emotional and cognitive impairment. FASEB J. 2017;31(10):4347‐4358. [DOI] [PubMed] [Google Scholar]
- 72. He F, Umehara T, Tsuda K, et al. Solution structure of the zinc finger HIT domain in protein FON. Protein Sci. 2007;16(8):1577‐1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cloutier P, Poitras C, Durand M, et al. R2TP/Prefoldin‐like component RUVBL1/RUVBL2 directly interacts with ZNHIT2 to regulate assembly of U5 small nuclear ribonucleoprotein. Nat Commun. 2017;8:15615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Malinova A, Cvackova Z, Mateju D, et al. Assembly of the U5 snRNP component PRPF8 is controlled by the HSP90/R2TP chaperones. J Cell Biol. 2017;216(6):1579‐1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fu M, Blackshear PJ. RNA‐binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat Rev Immunol. 2017;17(2):130‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Whole brain differential RNA expression results at both the isoform and the sum of isoform (SOI) level that include the statistical difference between Lrap+/+ and Lrap −/− rats. SOI and all associated isoforms were included in any of the isoforms for an SOI or the SOI were differentially expressed between Lrap+/+ and Lrap−/− rats (FDR < 0.10).
Data Availability Statement
The data that support the findings of this study are openly available in Gene Expression Omnibus database at https://www.ncbi.nlm.nih.gov/geo/, reference number GSE1557079.