

Abstract
Background:
Cancer patients receiving anthracycline-based chemotherapy (Anth-bC) may experience early cardiac fibrosis, which could be an important contributing mechanism to the development of impaired left ventricular (LV) function. Substance P, a neuropeptide that predominantly acts via the neurokinin 1 receptor (NK-1R), contributes to adverse myocardial remodeling and fibrosis in other cardiomyopathies. We sought to determine if NK-1R blockade is effective against doxorubicin (Dox – a frequently used Anth-bC)-induced cardiac fibrosis and cardiomyocyte apoptosis. In addition, we explored the direct effects of Dox on cardiac fibroblasts.
Methods:
Male Sprague-Dawley rats were randomized to receive saline, six cycles of Dox (1.5 mg Dox/kg/cycle) or Dox with an NK-1R antagonist (L732138, 5 mg/kg/daily through Dox treatment). At 8 weeks after the initial dose of Dox, LV function and histopathological myocardial fibrosis and cell apoptosis were assessed. Collagen secretion was measured in vitro to test direct Dox activation of cardiac fibroblasts.
Results:
Rats undergoing Dox treatment (9 mg/kg cumulative dose) developed cardiac fibrosis and cardiomyocyte apoptosis. NK-1R blockade partially mitigated cardiac fibrosis while completely preventing cardiomyocyte apoptosis. This resulted in improved diastolic function. Furthermore, we found that Dox had direct effects on cardiac fibroblasts to cause increased collagen production and enhanced cell survival.
Conclusions:
This study demonstrates that cardiac fibrosis induced by Anth-bC can be reduced by NK-1R blockade. The residual fibrotic response is likely due to direct Dox effects on cardiac fibroblasts to produce collagen.
Keywords: Cardiotoxicity, Cardiac Fibrosis, Neurokinin-1 Receptor, Apoptosis
INTRODUCTION
Left ventricular (LV) dysfunction and heart failure (HF) in patients undergoing anthracycline-based chemotherapy (Anth-bC) limits the use of this otherwise effective class of anticancer drugs[1–3], which remain an essential curative component of treatment regimens for several tumor types including lymphoma and soft tissue sarcomas, and is a common adjuvant treatment for breast cancer[4]. Traditionally, the cardiotoxic effects of Anth-bC have been attributed to cardiomyocyte apoptosis due to DNA damage by topoisomerase 2β and formation of reactive oxygen species[5, 6]. However, cardiac extracellular matrix remodeling (cardiac fibrosis) represents an additional important mechanism contributing to impaired LV function and adverse outcomes after cancer treatment[7–9].
To this end, we have previously shown that substance P (SP) – an 11 amino acid neuropeptide found in sensory nerves, including the heart – exerts pro-fibrotic effects and induces adverse remodeling[10]. SP exerts its biological actions mainly via the neurokinin 1 receptor (NK-1R), which is found on cardiomyocytes[11], cardiac fibroblasts[10], and coronary endothelial cells[12]. We have reported that NK-1R blockade reduces cardiac remodeling including fibrosis, and improves cardiac function in animal models of pressure and volume overload[10, 13]. While NK-1R blockade reduces Anth-bC-induced cardiomyocyte apoptosis in vitro[14], it still remains unknown whether there is a role for the NK1-R in Anth-bC-induced cardiac fibrosis. Accordingly, in this study, we used a rat model of doxorubicine (Dox) induced cardiotoxicity that uses sequential cycles of chemotherapy, similar to the chronic repetitive dosing strategy used clinically in humans, to determine whether NK-1R blockade is effective against: 1) Dox-induced LV dysfunction and remodeling; 2) cardiac fibrosis; and 3) cardiomyocyte apoptosis in vivo. In addition, we explored the possibility that Dox acts directly on cardiac fibroblasts to contribute to cardiac fibrosis.
MATERIALS AND METHODS
Animals
All animal studies conformed to the principles of the National Institutes of Health, Guide for the Care and Use of Laboratory Animals and all protocols were approved by Wake Forest University Animal Care and Use Committee. Adult male 8 week old Sprague-Dawley rats were housed under standard environmental conditions and maintained on commercial rat chow and tap water ad libitum. Rats were anesthetized with 2 % inhaled isoflurane for intra-peritoneal (IP) Dox administration (Figure 1). At experimental endpoint, euthanasia was performed by removal of the heart following IP injection of sodium pentobarbital (50 mg/kg).
Figure 1. Study Design and Dox and NK-1 Receptor Antagonist Dosing scheme.
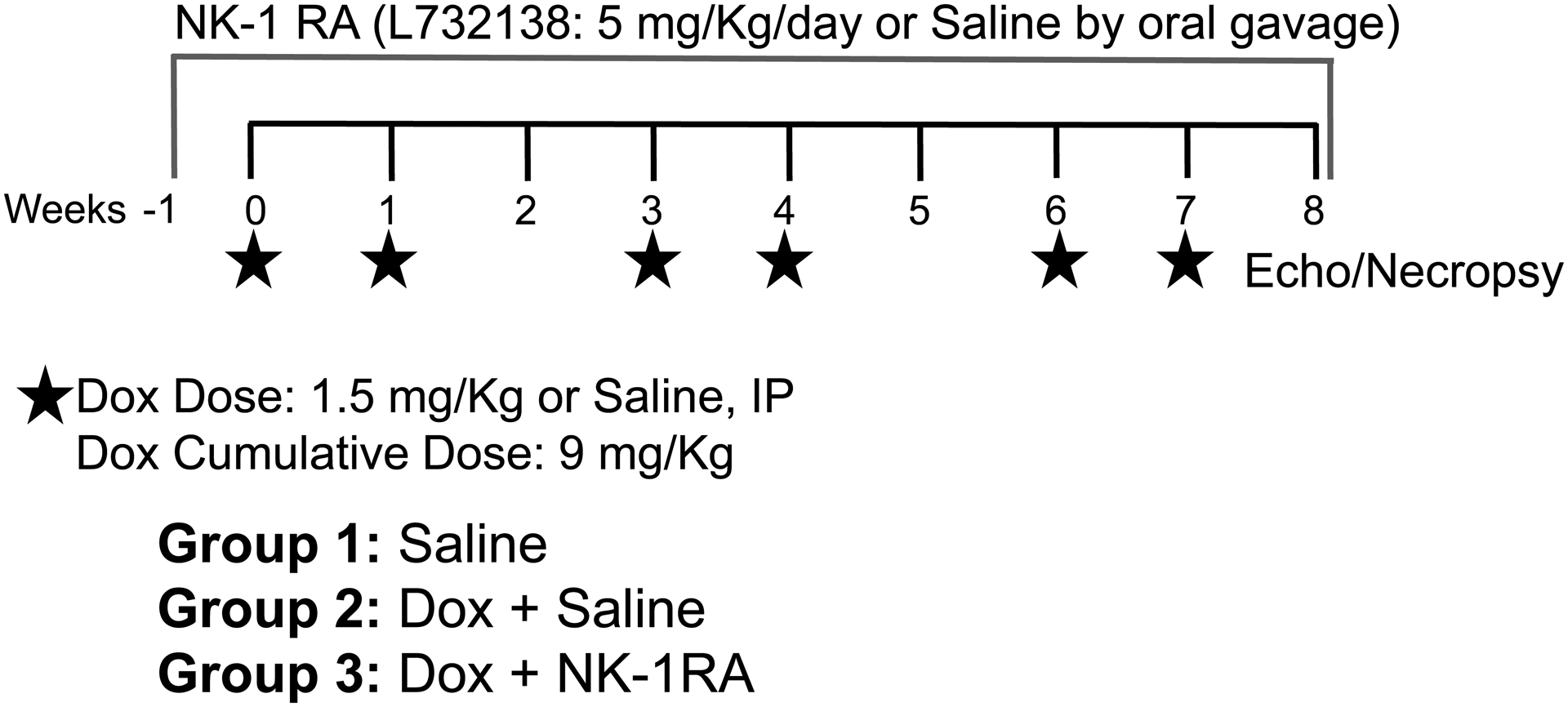
Rats were treated with 1.5 mg/kg/week IP with a week of rest between every two doses for 8 weeks (Group 2) or 0.5 ml of IP saline (Group 1). Group 3 started the NK-1R antagonist treatment (L732138, 5 mg/kg/day by daily oral gavage) 5 days prior to initiation of chemotherapy and continued throughout the 8 weeks of Dox treatment.
In vivo Studies
Rats were randomized into 3 groups: 1) Saline, 2) Dox + Saline and 3) Dox + L732138 (Figure 1). The Dox regimen consisted of 1.5 mg/kg/week IP by a single injection, with a week of rest between every two doses, for an overall treatment period of 8 weeks after the initial Dox dose. Thus, the total cumulative dose administered across the 8-week period was 9 mg/kg. The Saline group received a weekly 0.5 ml saline IP injection with a week of rest every two doses for 8 weeks and served as the age-matched untreated control group (Group1). Treatment with the NK-1R antagonist L732138 (5 mg/kg/d diluted in saline and administered by daily oral gavage) was initiated 1 week prior to initiation of chemotherapy and continued for 8 weeks after the initial Dox dose (Group 3). The dosage of L732138 was based on our previous studies[10]. Dox treated rats were gavaged with saline (vehicle) as the Dox untreated group (Group 2, Dox + Saline). At the experimental endpoint and following euthanasia, the heart was removed and the LV separated from the right ventricle (RV) and the atria with the individual weights recorded. A transverse mid-section of the LV was fixed in 4% paraformaldehyde. Lung weight was also obtained after the removal of the trachea, with the pleural surface blotted dry. Additional methods for echocardiographic evaluation, histopathological assessments of cardiac fibrosis, cardiomyocyte apoptosis and cross-sectional areas, as well as in vitro studies can be found in Supplemental Materials.
Statistical Analyses
Results are presented as mean ± SEM. Group differences were determined by one-way ANOVA with Tukey’s correction; p values of ≤0.05 were considered significant. All analyses were performed using Graph Pad Prism version 7 for Windows (San Diego, CA).
RESULTS
Biometric and echocardiographic parameters
Results for delta body weight (BW, pre-necropsy BW - initial BW), LV, RV and lung weights are displayed in Table 1. Rats receiving Dox exhibited less BW gain over the experimental period; this was accentuated by treatment with L732138. LV mass trended to be decreased in rats treated with Dox, however it did not reach statistical significance. On the other hand, rats treated with Dox and the NK-1R antagonist had significantly lower LV mass. In contrast, when LV mass was indexed to BW (LV/BW), both groups of Dox treated rats exhibited significantly higher LV mass. Lung weights did not change among groups. However, after lung weights were indexed to BW, lung weights were significantly higher in both Dox treated animal groups. RV weights were significantly lower in rats treated with Dox and Saline and this was further accentuated by treatment with the NK-1R antagonist. However, there were no statistical differences between groups when RV weights were normalized to BW. Echocardiographic parameters are displayed in Table 2 and Figure 2. No differences in LV mass, LVIDd or IVSd were observed between groups. However, LVPWd tended to be decreased in rats treated with Dox and was significantly lower in rats treated with Dox and the NK-1R antagonist. LV ejection fraction (EF) tended to be decreased in rats treated with Dox + Saline and Dox + NK-1R antagonist compared to rats treated with Saline alone. However, it did not reach statistical significance (Saline: 80.1 ± 1.9 % vs. Dox + Saline: 69.5 ± 3.3 %, p=0.08; Saline vs. Dox + L732138: 69.5 ± 3.3, p=0.06, Figure 2A). LV fractional shortening (FS) tended to be decreased in the Dox + Saline group compared to the control Saline group, however the effect was not statistically significant (Saline: 50.3 ± 1.9 % vs. Dox + Saline: 40.8 ± 2.8 %, p=0.06, Figure 2B). On the other hand, animals treated with the NK-1R antagonist exhibited significantly lower LVFS compared to those treated with Saline (Saline vs. Dox + L732138: 40.9 ± 2.6 %, p=0.04, Figure 2B). Doppler-derived LV filling pressure as determined by E/e’ was significantly increased by Dox treatment (Saline: 15.4 ± 0.9 vs. Dox + Saline: 26.3 ± 2, p=0.0005, Figure 2C) and normalized by administration of L732138 (Dox + Saline vs. Dox + L732138: 16.8 ± 1, p=0.009, Figure 2C).
Table 1.
Biometric parameters
| Groups | Saline (n=9) | Dox + Saline (n=15) | Dox + L 32138 (n=8) | Group Difference P Value |
|---|---|---|---|---|
| Delta BW (Pre-necropsy BW – initial BW, g) | 238 ± 13 | 134 ± 9* | 129 ± 8 * † | <0.0001 |
| LV/BW | 1.88 ± 0.30 | 2.1 ± 0.1 | 2.29 ± 0.09* | 0.02 |
| RV/BW | 0.50 ± 0.08 | 0.51 ± 0.02 | 0.48 ± 0.02 | 0.62 |
| Lung/BW | 2.97 ± 0.08 | 3.37 ± 0.12* | 4.03 ± 0.07*† | <0.0001 |
Values reported as mean ± SEM. BW indicates body weight; LV, left ventricle; RV, right ventricle;
p<0.05 vs. Saline and
p<0.05 vs. Dox + Saline
Table 2.
Echocardiographic Parameters
| Groups | Dox + Saline (n=15) | Dox + L 32138 (n=8) | P Value |
|---|---|---|---|
| Heart Rate (bpm) | 343 ± 13 | 346 ± 10 | 0.84 |
| LVPWd (mm) | 1.78 ± 0.09 | 1.69 ± 0.06 | 0.42 |
| LVIDd (mm) | 7.53 ± 0.41 | 7.61 ± 0.18 | 0.85 |
| IVSd (mm) | 1.79 ± 0.33 | 1.65 ± 0.32 | 0.45 |
| LV mass (mg) | 995 ± 94 | 915 ± 36 | 0.45 |
| EF (%) | 69.5 ± 3.4 | 69.5 ± 3.2 | 0.98 |
| FS (%) | 40.8 ± 2.8 | 40.0 ± 2.6 | 0.98 |
Values reported as mean ± SEM. LVPWd, left ventricle posterior wall in diastole;
LVIDd, left ventricular internal diameter in diastole; IVSd, interventricular septum in diastole; EF, ejection fraction; FS, fraction of shortening.
Figure 2. Echocardiographic Evaluation of the Left Ventricular Function.
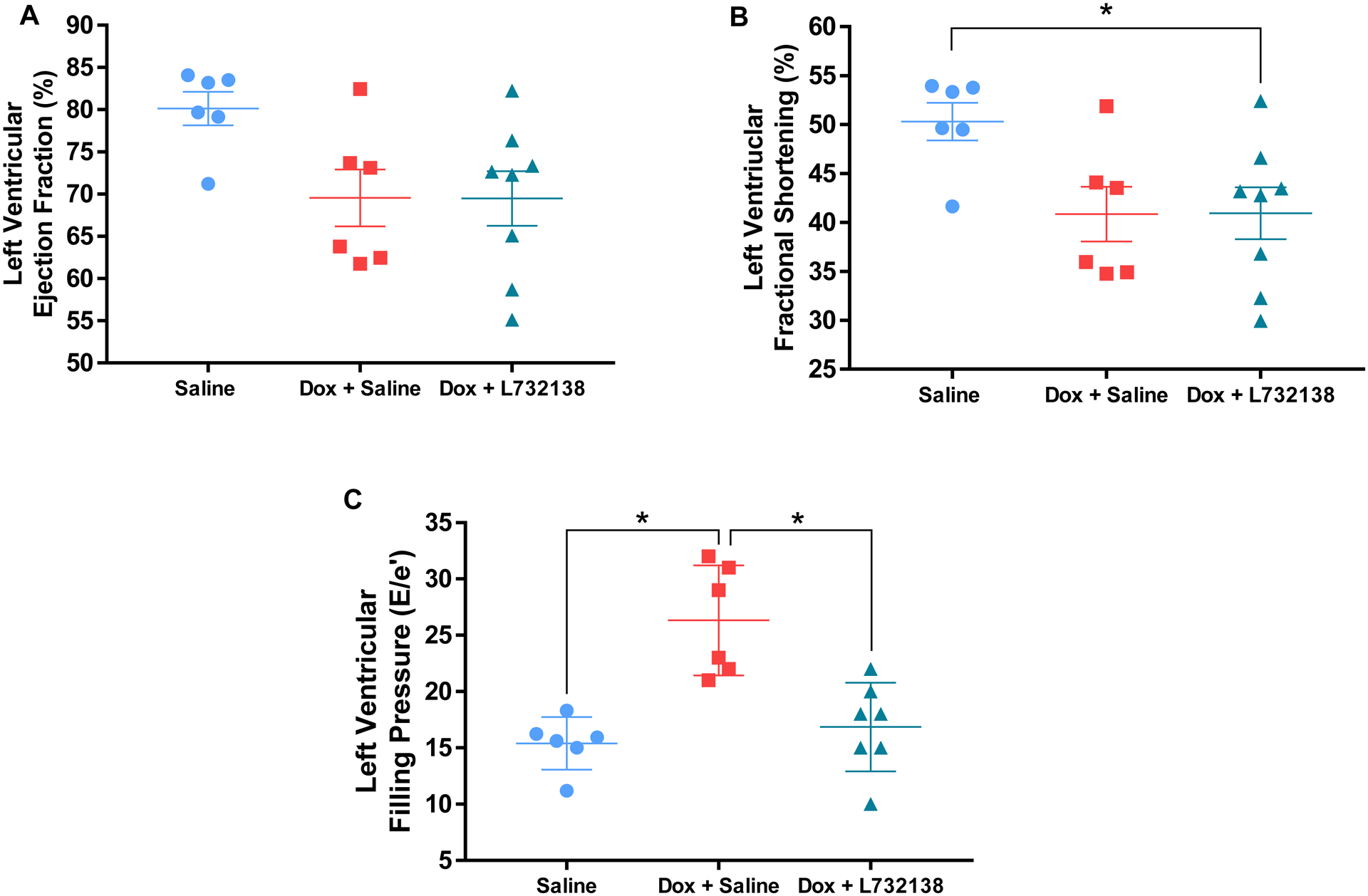
Left ventricular ejection fractlion (A), fractional shortening (B) and filling pressure (E/e’)(C) were measured in rats treated with Saline (blue dots, n=6), Dox + Saline (red squares, n=6) and Dox + L732138 (green triangles, n=8). All values are mean ± SEM. *p<0.05.
Collagen Volume Fraction
Dox treatment induced a significant increase in collagen deposition in the LV consistent with interstitial cardiac fibrosis (Figure 3A and B). Treatment with the NK-1R antagonist ameliorated, but did not completely prevent collagen deposition induced by Dox treatment (Figure 3A and B).
Figure 3. Assesment of LV Myocardial Fibrosis and Remodelling.
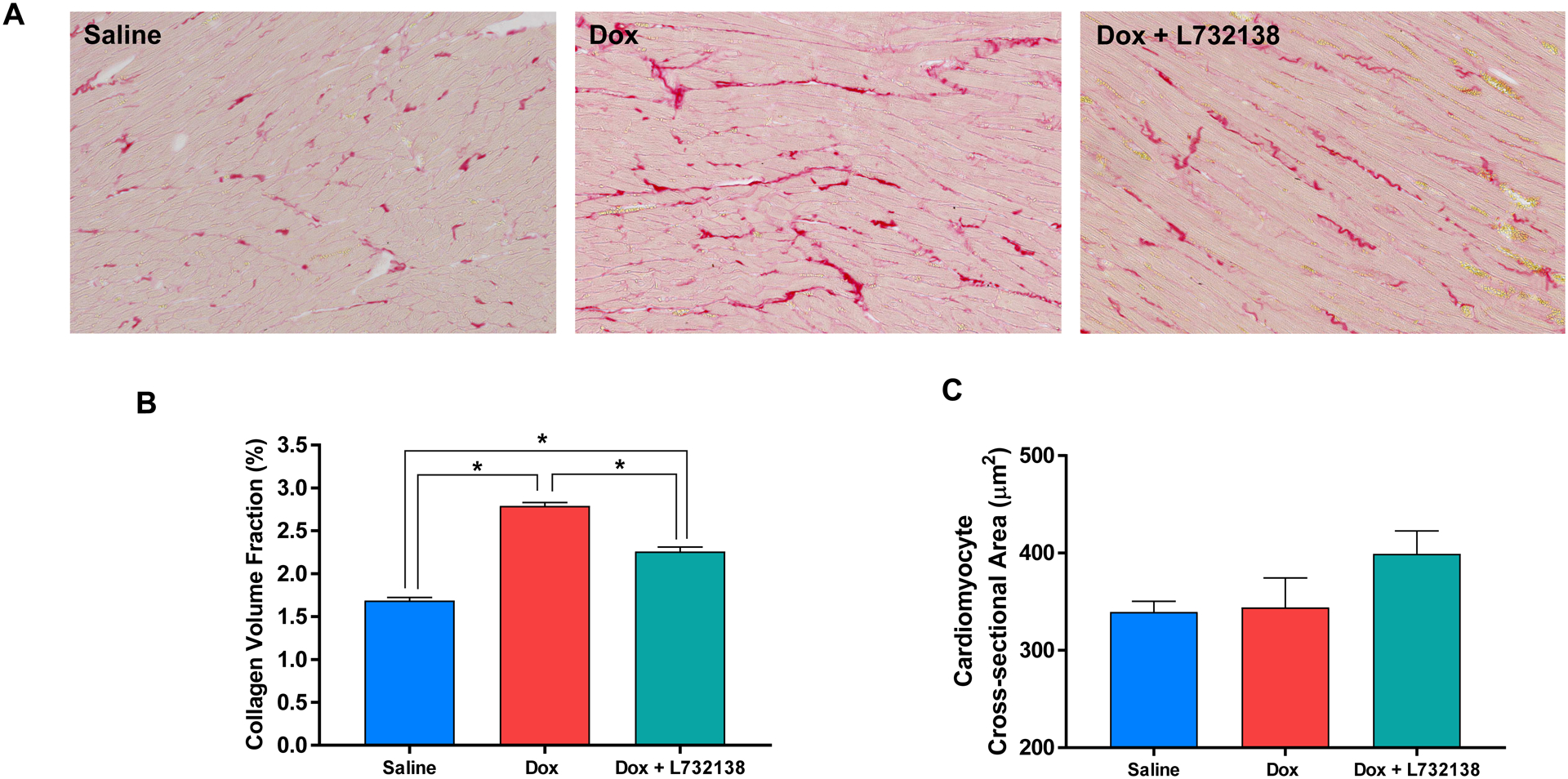
(A) Representative microphotographs, (B) graphical representation of collagen volume fraction and (C) cardiomyocyte cross-section area of left ventricles from rats treated with Saline (n=6, blue bar), Dox + Saline (n=6, red bar) and Dox + L732138 (n=8, green bar). All values are mean ± SEM. * p<0.0001.
Cardiomyocyte Cross-Sectional Area
We measured cardiomyocyte cross-sectional area in WGA stained LV sections to assess effects of Dox an cardiomyocyte size (Figure 3C). Treatment of rats with Dox did not affect cardiomyocyte cross-sectional area. Similarly, L732138 did not alter cross-sectional area.
Apoptosis
Dox treatment led to a 12-fold increase in the percent of TUNEL-positive cells in LV tissue (Figure 4A and B, Saline: 2.9 ± 0.9 % vs. Dox + Saline: 34.2 ± 4.14 %, p<0.001). More specifically, cardiomyocyte apoptosis was significantly increased by Dox (Figure 4C), however, apoptosis of interstitial cells was not (Figure 4D). NK-1R blockade reduced the percent of TUNEL-positive cells to 11.2 ± 1.9 % (Figures 4A and B), which did not differ from saline treated hearts (p=0.12).
Figure 4. Assesment of Cardiac Cell Apoptosis.
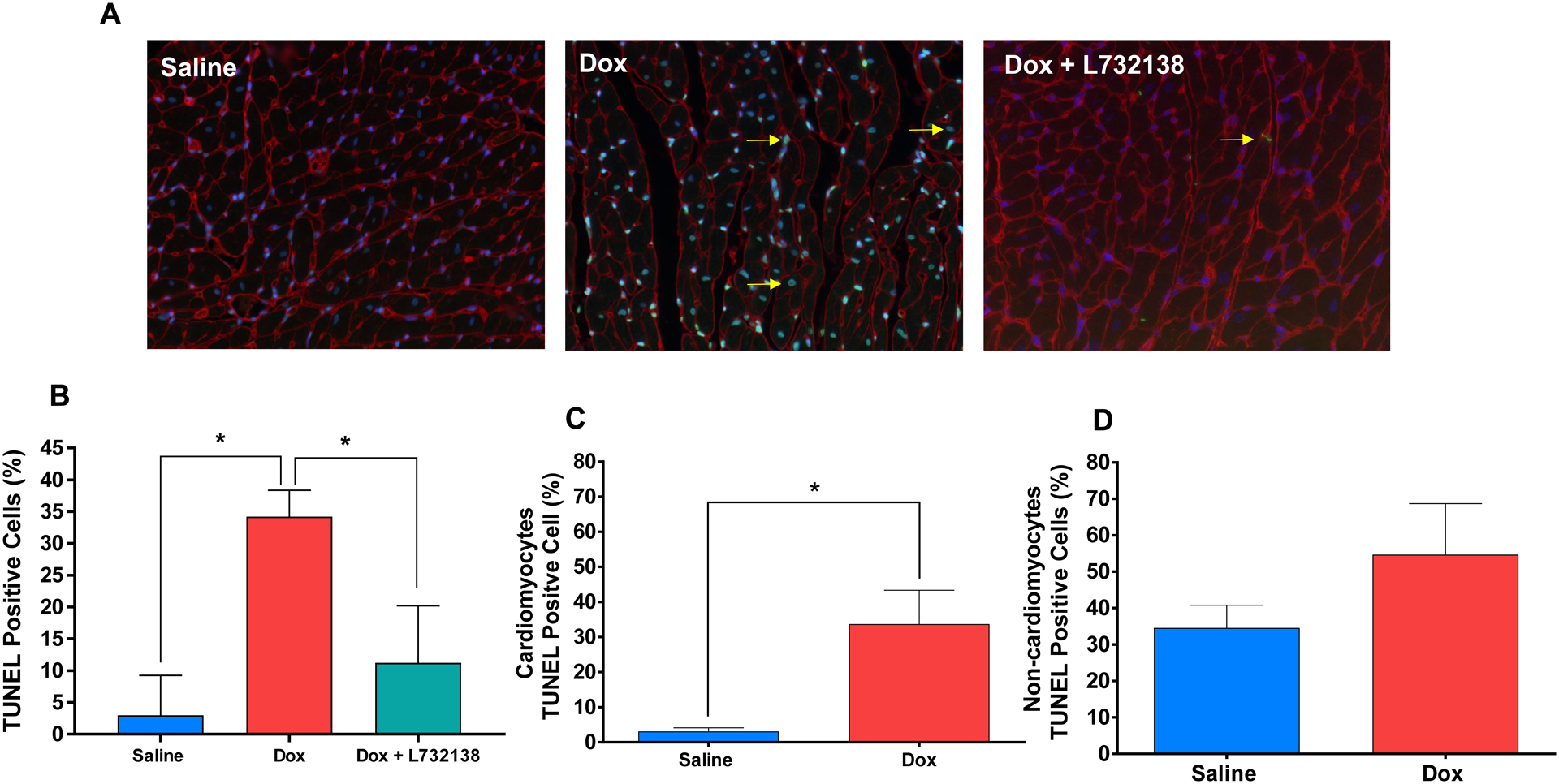
(A) Representative images (WGA = red, nuclei = blue, apoptotic nuclei = green and a few examples are denoted with a yellow arrow) and (B) quantification of TUNEL staining of the left ventricles of rats treated with Saline (n=6, blue bar), Dox + Saline (n=6, red bar) and Dox + L732138 (n=8, green bar). After co-staining with WGA, TUNEL positive cells were quantified among cardiomyocytes (C) and (D) non-cardiomyocytes in Dox vs. Saline groups. All values are mean ± SEM. * p<0.0001.
Isolated Cardiac Fibroblasts Up-regulate Collagen Secretion in Response to Dox
To determine whether Dox directly induces activation of cardiac fibroblasts, we exposed isolated adult LV fibroblasts to increasing concentrations of Dox in vitro. We found a dose-dependent increase in secretion of hydroxyproline (collagen) as shown in Figure 5A.
Figure 5. Isolated Cardiac Fibroblasts Exposed to Dox in-vitro.
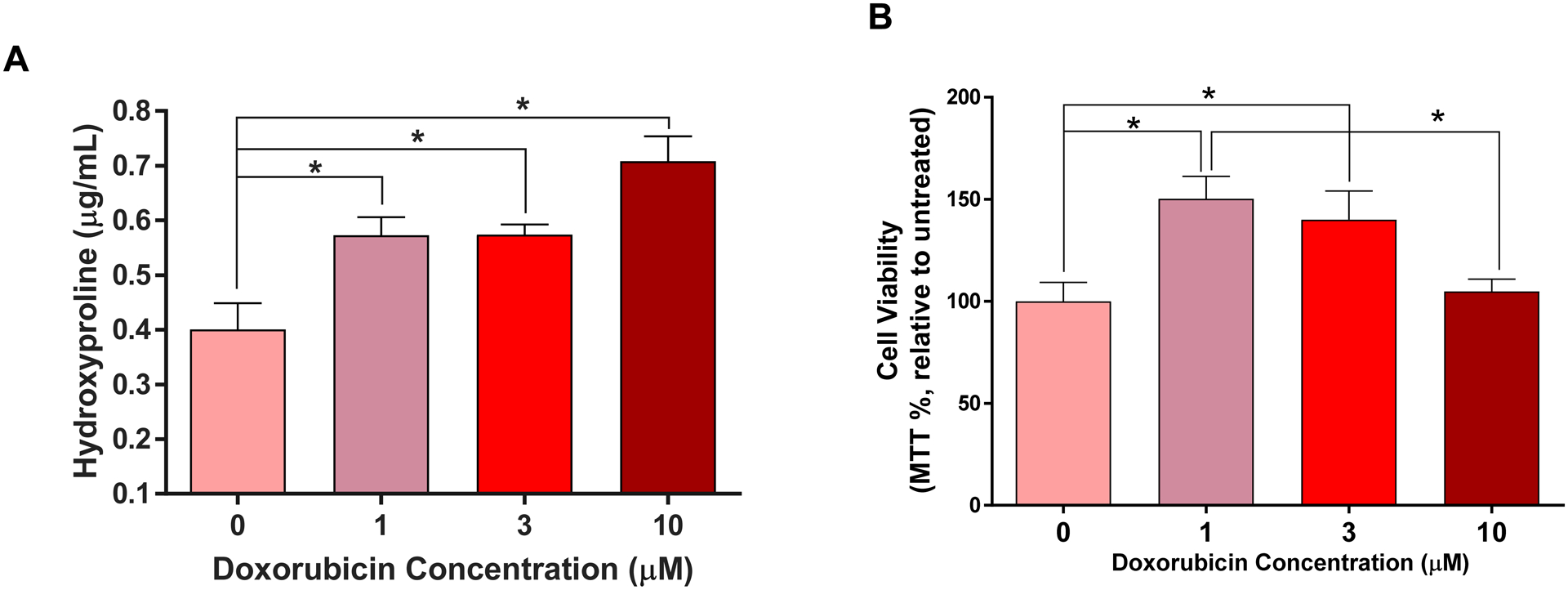
(A) Dose-response curve of hydroxyproline secreted from isolated cardiac fibroblasts exposed to increasing concentrations of Dox (0, 1, 3 and 10 μM). * p<0.0001 vs. 0 μM. (B) Cardiac fibroblasts viability determined by the MTT assay after exposure to increasing concentrations of Dox (0, 1, 3 and 10 μM). All values are mean ± SEM. * p<0.003 vs. 0 μM n=4.
Cardiac Fibroblast Viability
Isolated cardiac fibroblasts exposed to low and intermediate concentrations of Dox (1 and 3 μM) exhibited increased cell viability compared to vehicle treated cells (Figure 5B, p<0.05); those exposed to high Dox concentration (10 μM) exhibited a preserved cell viability compared to vehicle (p=0.31).
DISCUSSION
Our current study evaluates the effects of Dox treatment on the cardiac extracellular matrix (ECM) and its main regulating cell, the cardiac fibroblast and tests the effectiveness of NK-1R blockade in mitigating ECM and cardiac remodeling in response to Dox. Our results indicate: 1) interstitial cardiac fibrosis is established after the administration of a cumulative dose of 9 mg/kg of Dox; 2) treatment with an NK-1R antagonist partially mitigates cardiac fibrosis; 3) Dox also directly activates isolated cardiac fibroblasts to produce collagen; 4) Dox induces apoptosis in cardiomyocytes but not in cardiac fibroblasts, which preserve their viability both in vitro and in vivo; and 5) NK-1R blockade prevents cardiomyocyte apoptosis in vivo.
The molecular and cellular mechanisms of Dox-induced cardiotoxicity are multi-factorial and to date there are few effective strategies to prevent, predict and treat LV dysfunction in this otherwise effective cancer treatment. An important limitation of the current understanding of how Dox damages cardiac tissue is that the proposed mechanisms – DNA damage through topoisomerase 2β and oxidative stress – are focused exclusively on one cell type; the cardiomyocyte. Recently, cardiovascular magnetic resonance (CMR) measures of LV myocardial extracellular volume (ECV) fraction, which often represent cardiac fibrosis[15], are elevated in patients early after the initiation of Anth-bC[7]. ECV is also elevated in long term cancer survivors predominantly treated with Anth-bC, and is independent of other risk factors and cardiovascular comorbidities[8, 16]. Notably, increased ECV and histopathologic cardiac fibrosis, but not decreases in ejection fraction, predicted the late Anth-bC-induced mortality, in a mouse model of cardiotoxicity[17]. In other etiologies of heart failure, LV interstitial fibrosis contributes to LV dysfunction[7, 8]. Since fibrosis is one of the main contributors to impaired LV relaxation and filling[18], and is an important predictor of heart failure[19], understanding the underlying mechanisms by which Dox results in ECM remodeling, is important in uncovering novel and effective treatment strategies.
We have previously demonstrated that SP and the NK-1R modulate adverse myocardial and ECM remodeling in the hypertensive[10] and volume overloaded heart[13]. Similarly, others have shown the role of SP in the pathogenesis of myocarditis[20, 21] and magnesium-deficiency LV remodeling[22], including erlotinib-induced hypomagnesemia and cardiac dysfunction[23]. Interestingly, SP is known to mediate chemotherapy-induced nausea and vomiting (including Anth-bC) after binding to the NK-1R in the abdominal vagus and brainstem (nucleus tractus solitaries)[24]. Hence, we hypothesized that the SP/NK-1R axis may also be upregulated peripherally in cardiac sensory nerves and cardiac cells, mediating cardiac fibrosis after Anth-bC treatment. Therefore, an NK-1R antagonist could mitigate the development of cardiac fibrosis and diastolic dysfunction in this setting.
We tested our hypothesis by creating a clinically relevant rat model of Dox-induced cardiac fibrosis, designed to closely resemble a typical chemotherapy regimen (Figure 1). As shown in Figure 3, the establishment of cardiac fibrosis occurred after rats received a total cumulative dose of 9 mg/kg. These observations are consistent with our prior findings in a cohort of individuals undergoing cardiotoxic chemotherapy, in which expansion of the LV ECV is prominently observed early in individuals receiving Anth-bC with a cumulative dose of 375±0.56 mg/m2 (~8–10 mg/kg)[7]. Rats treated with the NK-1R antagonist (L732138) exhibited a reduction in cardiac fibrosis by approximately 50%, and had normal Doppler-derived transmitral early filling-to-mitral annular velocity ratio (E/e’), indicative of improved diastolic function (Figure 2C). This finding of a partial reduction in fibrosis by NK-1R blockade was somewhat intriguing because NK-1R blockade had completely prevented cardiac fibrosis in the hypertensive rat heart, while deletion of the SP gene completely prevented ECM remodeling in mice[13].
Since NK-1R blockade only partially mitigated cardiac fibrosis, it was essential to elucidate whether Dox treatment also exerted a direct effect on cardiac fibroblasts. We treated isolated adult cardiac fibroblasts with increasing concentrations of Dox and measured hydroxyproline levels in the media as a marker of collagen release. We found a dose-dependent secretion of collagen (Figure 5A), independent of cell proliferation (Dox 10 μM, Figure 5B). We also considered the possibility that Dox could cause SP secretion by isolated adult cardiac fibroblasts, which could in turn promote collagen production. However we could not detect SP in the media (data not shown), ruling out an autocrine SP effect in response to Dox. Our novel findings represent a new dimension in our understanding of Anth-bC-induced cardiotoxicity and underscores the fact that cardiac fibrosis in this setting is not only the result of cardiomyocyte death and replacement fibrosis. We conclude that cardiac fibrosis induced by Dox may be the result of multiple, non-mutually exclusive mechanisms that involve direct activation of cardiac fibroblasts and the NK-1R.
Amelioration of cardiac fibrosis by NK-1R blockade occurred in conjunction with prevention of cardiac cell apoptosis (Figure 4A and B), which was accounted for by cardiomyocyte apoptosis and not apoptosis of interstitial cells. This finding is consistent with the report by Robinson et al., showing that pretreatment of a cardiomyocyte cell line (H9C2) with the NK-1R antagonist aprepitant, increased cell viability after Dox treatment and decreased apoptotic cell death[14]. The lack of apoptosis observed in interstitial cells in vivo supports our in vitro findings with cultured cardiac fibroblasts where viability was preserved in the presence of Dox in these cells (Figure 5B). This is extremely interesting because it reveals a divergent cell-specific response where Dox induces cardiomyocyte apoptosis (death), while increasing fibroblast viability leading to increased ECM production and secretion. Interestingly, prevention of cardiomyocyte apoptosis by NK-1R blockade in vivo did not prevent the overall development of cancer therapeutics-related cardiac dysfunction which is defined by an LVEF drop of >10 percentage points[25]. These findings further support the notion that cardiomyocyte death may not be the main cause of Anth-bC LV systolic dysfunction and that those spared cardiomyocytes may still have a variety of toxicities (e.g. mitochondrial dysfunction, atrophy, loss of myofibers, calcium channel abnormalies) that ultimately impair their contractile capacity.
In cancer patients, LV mass index is inversely associated with the anthracycline dose and is a predictor of cardiovascular events[26]. In our study, LV weight (at necropsy) decreased with Dox treatment and was not affected by NK-1R blockade. On the other hand, after normalizing the LV weight to BW to account for BW loss, we found that a significantly increased the LV mass. In order to clarify whether this was the result of cardiomyocyte hypertrophy, cardiomyocyte cross-sectional area was evaluated. We found no overt changes between the groups (Figure 3C). These discrepancies between our rat study and patients could be due to the fact that our rats do not have cancer. Post-mortem studies of cancer patients and rodent models of cancer cachexia show characteristics of cardiac wasting or atrophy, as well as a decrease in LV weight and thinning of septal, interventricular and posterior walls by echocardiography[27, 28]. Rodent models of Dox-induced cardiotoxicity have shown conflicting data, with some demonstrating cardiomyocyte atrophy while other have shown hypertrophy[29, 30]. Moreover, our results and previous observations suggest that LV remodeling in response to Dox may vary depending on the model used, sex and underlying pathological co-morbidities. These are all issues for future studies to address.
The results of our study have important clinical implications. First, to date, the majority of studies have focused on cardiomyocyte injury as the main cause of cardiac dysfunction after Anth-bC. The results of our study strongly suggest that ECM remodeling is an additional mechanism contributing to cardiac dysfunction. Second, we demonstrate that the SP/NK-1R axis contributes to the establishment of cardiac fibrosis after Dox treatment. This finding is particularly significant since a commercially available NK-1R antagonist (aprepitant) is currently in clinical use to mitigate nausea and vomiting associated with highly-emetogenic chemotherapy such as Anthracyclines [31, 32]. However, aprepitant is only administed for up to 3 days each time a patient undergoes a chemotherapy session. Our study argues for continuous use of aprepitant throughout chemotherapy treatment. Third, our study shows for the first time that Dox directly activates cardiac fibroblasts, independent of cardiomyocyte injury, to produce excess collagen and increase viability. This finding represents a new dimension to the understanding of Anth-bC cardiotoxicity and underscores the need to better understand the intracellular pathways by which Dox activates fibroblasts.
Our study has the following limitations: first, we do not have evaluation of cardiac function to progression of frank heart failure. As such we are uncertain whether mitigating cardiac fibrosis could prevent further deterioration of LV systolic dysfunction or whether this decrease in LVEF may be transient. Second, the animals in this study do not have the co-morbidities that patients usually have such as hypertension, diabetes, obesity, coronary artery disease, and cancer itself. Therefore, the animals began the study with a relatively healthy myocardium. Third, the effects of chronic NK-1R antagonist administration were not evaluated in this study. While we have previously reported that mice lacking substance P (TAC1 −/−) did not exhibit cardiac abnormalities[13], further studies are required to assess the cardiovascular effects of NK-1R blockade in a cancer animal model. In conclusion, we demonstrated that interstitial cardiac fibrosis, independent from cardiomyocyte injury, occurs after treatment with Dox and can be partially ameliorated by NK-1R blockade. Further, Dox has direct effects on cardiac fibroblasts to induce a pro-fibrotic phenotype. Further studies are necessary to determine the short- and long-term LV functional benefits of myocardial fibrosis mitigation as well as the intracellular molecular mechanisms by which Dox promotes collagen secretion.
Supplementary Material
Acknowledgments:
We thank Ashley Davis for the technical help with animal studies and the Wake Forest University, School of Medicine Preclinical Photoacoustic and Ultrasound Imaging Core (S10RR033501).
Funding: This work was supported by a Research Award by the Foundation for Women’s Wellness (GCM), by the National Lung and Blood Institute at the National Institutes of Health (HL118740-01S1, WGH and GCM; HL093215, SPL), and by the Greater Milwaukee Foundation-Elsa Schoeneich Medical Research Fund (SPL).
Footnotes
DISCLOSURES
No conflicts of interest are declared by the author(s).
REFERENCES
- [1].Brenner H Long-term survival rates of cancer patients achieved by the end of the 20th century: a period analysis. Lancet (London, England). 2002;360:1131–5. [DOI] [PubMed] [Google Scholar]
- [2].Patnaik JL, Byers T, DiGuiseppi C, Dabelea D, Denberg TD. Cardiovascular disease competes with breast cancer as the leading cause of death for older females diagnosed with breast cancer: a retrospective cohort study. Breast cancer research : BCR. 2011;13:R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. 2015;131:1981–8. [DOI] [PubMed] [Google Scholar]
- [4].Mackey JR, Martin M, Pienkowski T, Rolski J, Guastalla JP, Sami A, et al. Adjuvant docetaxel, doxorubicin, and cyclophosphamide in node-positive breast cancer: 10-year follow-up of the phase 3 randomised BCIRG 001 trial. The Lancet Oncology. 2013;14:72–80. [DOI] [PubMed] [Google Scholar]
- [5].Moudgil R, Yeh ET. Mechanisms of Cardiotoxicity of Cancer Chemotherapeutic Agents: Cardiomyopathy and Beyond. Can J Cardiol. 2016;32:863–70 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vejpongsa P, Yeh ET. Topoisomerase 2beta: a promising molecular target for primary prevention of anthracycline-induced cardiotoxicity. Clin Pharmacol Ther. 2014;95:45–52. [DOI] [PubMed] [Google Scholar]
- [7].Meléndez GC, Jordan JH, D’Agostino RB Jr., Vasu S, Hamilton CA, Hundley WG. Progressive 3-Month Increase in LV Myocardial ECV After Anthracycline-Based Chemotherapy. JACC Cardiovascular imaging. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jordan JH, Vasu S, Morgan TM, D’Agostino RB Jr., Melendez GC, Hamilton CA, et al. Anthracycline-Associated T1 Mapping Characteristics Are Elevated Independent of the Presence of Cardiovascular Comorbidities in Cancer Survivors. Circulation Cardiovascular imaging. 2016;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nguyen KL, Hu P, Ennis DB, Shao J, Pham KA, Chen JJ. Cardiac MRI: a Translational Imaging Tool for Characterizing Anthracycline-Induced Myocardial Remodeling. Curr Oncol Rep. 2016;18:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dehlin HM, Manteufel EJ, Monroe AL, Reimer MH Jr., Levick SP. Substance P acting via the neurokinin-1 receptor regulates adverse myocardial remodeling in a rat model of hypertension. International journal of cardiology. 2013;168:4643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Walsh RJ, Weglicki WB, Correa-de-Araujo R. Distribution of specific substance P binding sites in the heart and adjacent great vessels of the Wistar white rat. Cell Tissue Res. 1996;284:495–500. [DOI] [PubMed] [Google Scholar]
- [12].Dehlin HM, Levick SP. Substance P in heart failure: the good and the bad. International journal of cardiology. 2014;170:270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Melendez GC, Li J, Law BA, Janicki JS, Supowit SC, Levick SP. Substance P induces adverse myocardial remodelling via a mechanism involving cardiac mast cells. Cardiovascular research. 2011;92:420–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Robinson P, Kasembeli M, Bharadwaj U, Engineer N, Eckols KT, Tweardy DJ. Substance P Receptor Signaling Mediates Doxorubicin-Induced Cardiomyocyte Apoptosis and Triple-Negative Breast Cancer Chemoresistance. Biomed Res Int. 2016;2016:1959270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ambale-Venkatesh B, Lima JA. Cardiac MRI: a central prognostic tool in myocardial fibrosis. Nat Rev Cardiol. 2015;12:18–29. [DOI] [PubMed] [Google Scholar]
- [16].Neilan TG, Coelho-Filho OR, Shah RV, Feng JH, Pena-Herrera D, Mandry D, et al. Myocardial extracellular volume by cardiac magnetic resonance imaging in patients treated with anthracycline-based chemotherapy. The American journal of cardiology. 2013;111:717–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Farhad H, Staziaki PV, Addison D, Coelho-Filho OR, Shah RV, Mitchell RN, et al. Characterization of the Changes in Cardiac Structure and Function in Mice Treated With Anthracyclines Using Serial Cardiac Magnetic Resonance Imaging. Circulation Cardiovascular imaging. 2016;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J. 2011;32:670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Assomull RG, Prasad SK, Lyne J, Smith G, Burman ED, Khan M, et al. Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J Am Coll Cardiol. 2006;48:1977–85. [DOI] [PubMed] [Google Scholar]
- [20].D’Souza M, Garza MA, Xie M, Weinstock J, Xiang Q, Robinson P. Substance P is associated with heart enlargement and apoptosis in murine dilated cardiomyopathy induced by Taenia crassiceps infection. The Journal of parasitology. 2007;93:1121–7. [DOI] [PubMed] [Google Scholar]
- [21].Robinson P, Garza A, Moore J, Eckols TK, Parti S, Balaji V, et al. Substance P is required for the pathogenesis of EMCV infection in mice. International journal of clinical and experimental medicine. 2009;2:76–86. [PMC free article] [PubMed] [Google Scholar]
- [22].Kramer JH, Phillips TM, Weglicki WB. Magnesium-deficiency-enhanced post-ischemic myocardial injury is reduced by substance P receptor blockade. Journal of molecular and cellular cardiology. 1997;29:97–110. [DOI] [PubMed] [Google Scholar]
- [23].Mak IT, Kramer JH, Chmielinska JJ, Spurney CF, Weglicki WB. EGFR-TKI, erlotinib, causes hypomagnesemia, oxidative stress, and cardiac dysfunction: attenuation by NK-1 receptor blockade. Journal of cardiovascular pharmacology. 2015;65:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Girish C, Manikandan S. Aprepitant: a substance P antagonist for chemotherapy induced nausea and vomiting. Indian journal of cancer. 2007;44:25–30. [DOI] [PubMed] [Google Scholar]
- [25].Plana JC, Galderisi M, Barac A, Ewer MS, Ky B, Scherrer-Crosbie M, et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: a report from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2014;15:1063–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Neilan TG, Coelho-Filho OR, Pena-Herrera D, Shah RV, Jerosch-Herold M, Francis SA, et al. Left ventricular mass in patients with a cardiomyopathy after treatment with anthracyclines. The American journal of cardiology. 2012;110:1679–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sweeney M, Yiu A, Lyon AR. Cardiac Atrophy and Heart Failure In Cancer. Cardiac failure review. 2017;3:62–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Springer J, Tschirner A, Haghikia A, von Haehling S, Lal H, Grzesiak A, et al. Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. Eur Heart J. 2014;35:932–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cove-Smith L, Woodhouse N, Hargreaves A, Kirk J, Smith S, Price SA, et al. An integrated characterization of serological, pathological, and functional events in doxorubicin-induced cardiotoxicity. Toxicol Sci. 2014;140:3–15. [DOI] [PubMed] [Google Scholar]
- [30].Moulin M, Piquereau J, Mateo P, Fortin D, Rucker-Martin C, Gressette M, et al. Sexual dimorphism of doxorubicin-mediated cardiotoxicity: potential role of energy metabolism remodeling. Circ Heart Fail. 2015;8:98–108. [DOI] [PubMed] [Google Scholar]
- [31].Basch E, Prestrud AA, Hesketh PJ, Kris MG, Feyer PC, Somerfield MR, et al. Antiemetics: American Society of Clinical Oncology clinical practice guideline update. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:4189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dupuis LL, Boodhan S, Holdsworth M, Robinson PD, Hain R, Portwine C, et al. Guideline for the prevention of acute nausea and vomiting due to antineoplastic medication in pediatric cancer patients. Pediatric blood & cancer. 2013;60:1073–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.