

Abstract
Interleukin (IL)-10 is elevated in the autoimmune disease systemic lupus erythematosus (SLE). Here we show that conventional dendritic cells (cDCs) from pre-disease lupus-prone B6.NZM Sle1/Sle2/Sle3 triple congenic (TCSle) mice produce more IL-10 than wild type congenic cDCs upon TLR stimulation, and this overproduction is prevented by blocking the type I IFN receptor (IFNAR) with specific antibodies. Priming wild type cDCs with type I IFN mimics the IL-10 overproduction of TCSle cDCs. The MAP kinase ERK is more phosphorylated in lupus cDCs, partially contributing to IL-10 overproduction. Moreover, we found that TCSle cDCs express higher levels of IL-27 upon TLR7/TLR9 stimulation, and IFNAR blockade reduced IL-27 levels in TCSle cDCs. These results suggest that dysregulated type I IFNs in cDCs contribute to the increased IL-10 and IL-27 in SLE. Since IL-27 neutralization did not inhibit TLR-induced IL-10 production, we propose that type I IFNs enhanced IL-10 in TCSle cDCs independently from IL-27. Moreover, RNA sequencing analysis of a cohort of SLE patients reveals higher gene expression of these cytokines in SLE patients expressing a high IFN signature. Since IL-27 and IL-10 have both pro- and anti-inflammatory effects, our results also suggest that these cytokines can be modulated by the therapeutic IFN blockade in trials in SLE patients and have complex effects on the autoimmune response.
Keywords: innate immunity, cytokines, MAPK, Lupus, Type I Interferons
Graphical Abstract
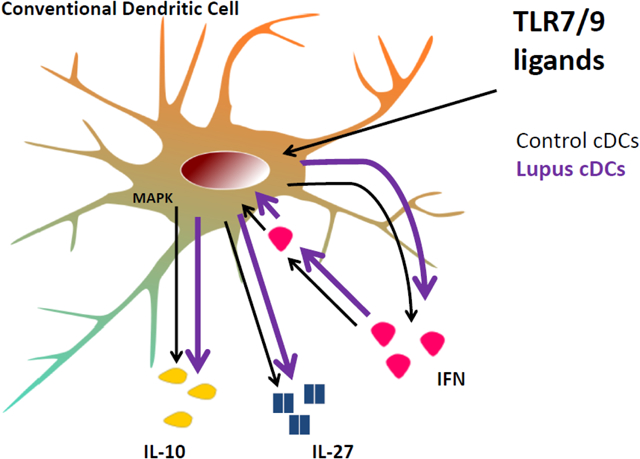
Model of a type I IFN - IL-27 / IL-10 molecular network augmented in murine lupus cDCs. Murine lupus cDCs overproduce IL-10 and IL-27 due to heightened autocrine I-IFN Signature. Furthermore, elevated IL-10 and IL-27 correlate with high IFN Signature in SLE patients.
Introduction
Systemic lupus erythematous (SLE) is a complex multisystem disorder, in which multiple immune abnormalities and autoantibodies result in tissue damage [1]. Lupus pathogenesis is multifactorial - including many genetic and environmental factors [2, 3]. However, the molecular mechanisms that mediate disease remain to be fully elucidated.
Overexpression of type I interferons (I-IFN) and IFN-stimulated genes (ISGs) are considered pathogenic in SLE [4]. SLE patients express an I-IFN signature [5–7] and clinical trials are aiming to block I-IFN or its receptor [8, 9]. We have previously found that conventional dendritic cells (cDCs) from lupus-prone B6.NZMSle1/Sle2/Sle3 triple congenic (TCSle) mice also express an I-IFN signature before disease development [10]; moreover, it has been recently shown that RNA sensing cDCs are also pivotal for lupus pathogenesis [11]. These results suggest that the I-IFN signature in lupus can be the result of abnormalities in DC function, particularly in response to environmental triggers and cell death [12, 13]. I-IFNs are a family of cytokines with multiple effects on the immune response and it is important to understand the downstream consequences of the I-IFN signature expressed by lupus DCs. DCs from single congenic lupus-prone B6.Sle3 mice produce high levels of pro-inflammatory cytokines IL-12, IL-6, and TNFα in response to LPS [14], which can drive T cell hyperactivity and disease progression [14]. Here, we used TCSle cDCs to investigate the regulation of IL-10, an anti-inflammatory cytokine less studied in this lupus model.
IL-10 has broad anti-inflammatory properties by suppressing production of pro-inflammatory cytokines and despite being a B cell growth factor (reviewed in [15]), it was shown to restrain autoantibody production in lupus-prone mice [16]. In apparent contradiction, serum levels of IL-10 are often elevated in SLE patients with active disease [17] and in relatives of SLE patients [18, 19]. It is therefore important to understand the causes of IL-10 dysregulation in lupus.
Polymorphisms in the Il10 promoter are found in SLE and suggest a genetic cause for the elevated levels of IL-10 present in some SLE patients [20]. However, these polymorphisms are not ubiquitous in SLE patients and the Il10 locus resides outside the susceptibility regions of lupus-prone mouse strains, suggesting a different cause for IL-10 dysregulation in most SLE patients and in murine lupus.
Autocrine I-IFNs were shown to contribute to TLR-induced IL-10 production in wild type cells via mechanisms that include activation of MAP kinase ERK in B cells [21] and autocrine production of IL-27 in macrophages in response to LPS [22]. However, others have shown IL-27-independent production of IL-10 in macrophages infected with Mycobacterium tuberculosis [23] and we have previously shown that the IFN-beta enhancement of IL-10 production in B6 DCs was not dependent on IL-27 [24]. IL-27 is a member of the IL-12 family that is produced by antigen-presenting cells [25] and can have either pro- or anti-inflammatory effects on T and B cells and in autoimmunity [26]. It is still controversial whether IL-27 production is abnormal in SLE patients [27, 28]. Serum levels of IL-27 have been found increased in SLE and correlate with disease activity and levels of autoantibodies [27]. These findings are in contrast with further work demonstrating that disease treatment increased IL-27 levels [29]. Since I-IFNs are elevated in PBMCs from SLE patients [5–7] and in murine TCSle DCs [10], we hypothesized that increased I-IFN may augment IL-10 production in TCSle DCs. Here, we show that TCSle cDCs overproduce IL-10 and IL-27 upon TLR stimulation because of the increased autocrine triggering of the I-IFN receptor (IFNAR) as part of the I-IFN signature. The increased production of IL-10, induced by autocrine I-IFN, is in part mediated through increased phosphorylation of ERK. These results in murine cDCs mirror the higher gene expression of IL27 and IL10 that we found in immune cells from SLE patients with a high I-IFN signature. Our results highlight an unexpected ramification of the over-activated I-IFN pathway in SLE and provide further relevance for IL-10 and IL-27 in lupus autoimmunity.
Methods
Mice
6–12 weeks old female C57BL/6 (B6) and B6.NZMSle1/Sle2/Sle3 triple congenic (TCSle) mice were purchased from Jackson Laboratory, bred and maintained in our colony, and used following institutional guidelines approved by the Institutional Animal Care and Use Committee of Temple University, which is an AAALAC-accredited facility.
Bone Marrow-Derived Dendritic Cells
Mouse bone marrow-derived DCs were generated as previously described [10]. Briefly, bone marrow precursors were cultured in complete IMDM (10% FBS, antibiotics and 2-mercaptoethanol) containing 3.3 ng/ml GM-CSF (BD Biosciences, San Jose, USA) or 1% conditioned media from GM-CSF-secreting B7H1 cells. At day 6–8, cDCs were stimulated with LPS (100 ng/ml), R848 (1 μg/ml), or CpG 1826 (10 μg/ml) and harvested after 1.5 hours for Western Blot and 6 hours for RT-PCR, while supernatants and cells were collected after 24 hours for ELISA and Flow analysis. Anti-IFNAR (10 μg/ml, clone MAR1–5A3, Leinco Technologies I-400 or Gentex GTX14637) or isotype control antibody (10 μg/ml, IgG1, Leinco Technologies I-102 or Gentex GTX35014) were added 24 hours and again 30 minutes before or 1 hour before TLR stimulation. ERK inhibitor (10 μM, PD98059, Cell Signaling #9900) was used 30 minutes before TLR stimulation. 1μg/ml anti-IL-27 (Polyclonal goat IgG, R&D AF1834) or 1μg/ml IgG control antibodies (R&D AB-108-C) were added one hour prior to TLR stimulation.
Quantitative RT-PCR
Gene expression in cDCs was analyzed by quantitative real-time RT-PCR (qPCR) using TaqMan probes. Briefly, RNA was extracted using ZymoResearch (Irvine, CA, USA) Quick-RNA MiniPrep (R1055). Complementary DNA (cDNA) was synthesized using a cDNA archive kit (Applied Biosystems, Foster City, CA, USA). Pre-synthesized TaqMan primers and probes for Ifnb and Irf7 [Mm00516788], Isg15 [Mm01705338], Il10 [Mm00439614_m1] were from Applied Biosystems. Relative quantification of gene expression was calculated to housekeeping gene Cyclophilin [10].
ELISA
IL-10, CXCL10 and IL-27 production were measured using an IL-10 ELISA kit from BD, a CXCL10 ELISA kit from R&D, or anti-mouse IL-27p28 and biotin-conjugated anti-mouse IL-27p28 antibodies from R&D (Minneapolis, MN, USA) as described in [25]. Alternatively, IL-27 was also measured by mouse IL-27 ELISA Ready-Set-Go! Kit from eBioscience, following manufacturer’s instructions.
Flow Cytometry
cDCs were harvested 24h post-stimulation then stained with FVDef780 (Invitrogen), rat anti-mouse CD16/CD32 (clone 2.4G2, BioLegend), APC-conjugated hamster anti-mouse CD11c (N418, eBioscience), PE-Cy7-conjugated rat anti-mouse CD11b (M1/70) and FITC-conjugated hamster anti-mouse CD40 (HM40–3, BD) then acquired on a BD FACSCanto. FlowJo (FlowJo LLC, Ashland, Oregon) software was used for data analysis.
Western Blots
Western blots were performed as previously described [10]. Anti-ERK1/2 (9107) and Anti-Phospho-ERK1/2 (4370) from Cell Signaling Technology (Beverly, MA, USA) were used to probe for ERK activation. Anti-Mouse anti-GAPDH (Santa Cruz Biotechnology, Dallas, TX, USA) was used as a loading control. IR Dye 800-goat anti-rabbit and IR Dye 680 goat anti-mouse (LI-COR Biosciences, Lincoln, NE, USA) were used to probe primary antibodies. Blots were visualized using Odyssey Infrared Imaging System (LI-COR Biosciences).
RNA Sequencing
A publicly available dataset of RNA Sequencing of whole blood samples from a cohort of 99 SLE patients and 18 healthy controls available in Gene Expression Omnibus accession number GSE72509 was analyzed. RPKM values were converted to transcripts per million (TPMs), which were used for downstream analyses. To measure the I-IFN signature, the expression of 27 ISGs was analyzed using mEV software and compared to IL-10 and IL-27.
Statistical analysis
Data were analyzed with Prism software (GraphPad, La Jolla, CA, USA) using two-tailed unpaired t-test and Mann-Whitney test for comparison between two groups and ANOVA with post-hoc Tukey’s multiple comparison for multiple groups. Significance was determined as p-values < 0.05. The principal component analysis of the I-IFN Signature was calculated using JMP software.
Results and Discussion
cDCs from TCSle mice overexpress Il10 RNA constitutively and overproduce IL-10 in response to TLR ligands
cDCs generated from TCSle mice constitutively express an I-IFN signature that precedes disease onset [10]. In addition to classic I-IFN signature genes (i.e. Ifnb, Isg15, and Irf7), we found that unstimulated TCSle cDCs also express basal levels of Il10 RNA higher than wild type congenic C56BL/6 (B6) cDCs (Figure 1a), while IL-10 protein was undetectable in the supernatants of unstimulated cDCs (results by ELISA not shown). These results suggest that IL-10 is primed for translation in TCSle cDCs.
Figure 1.
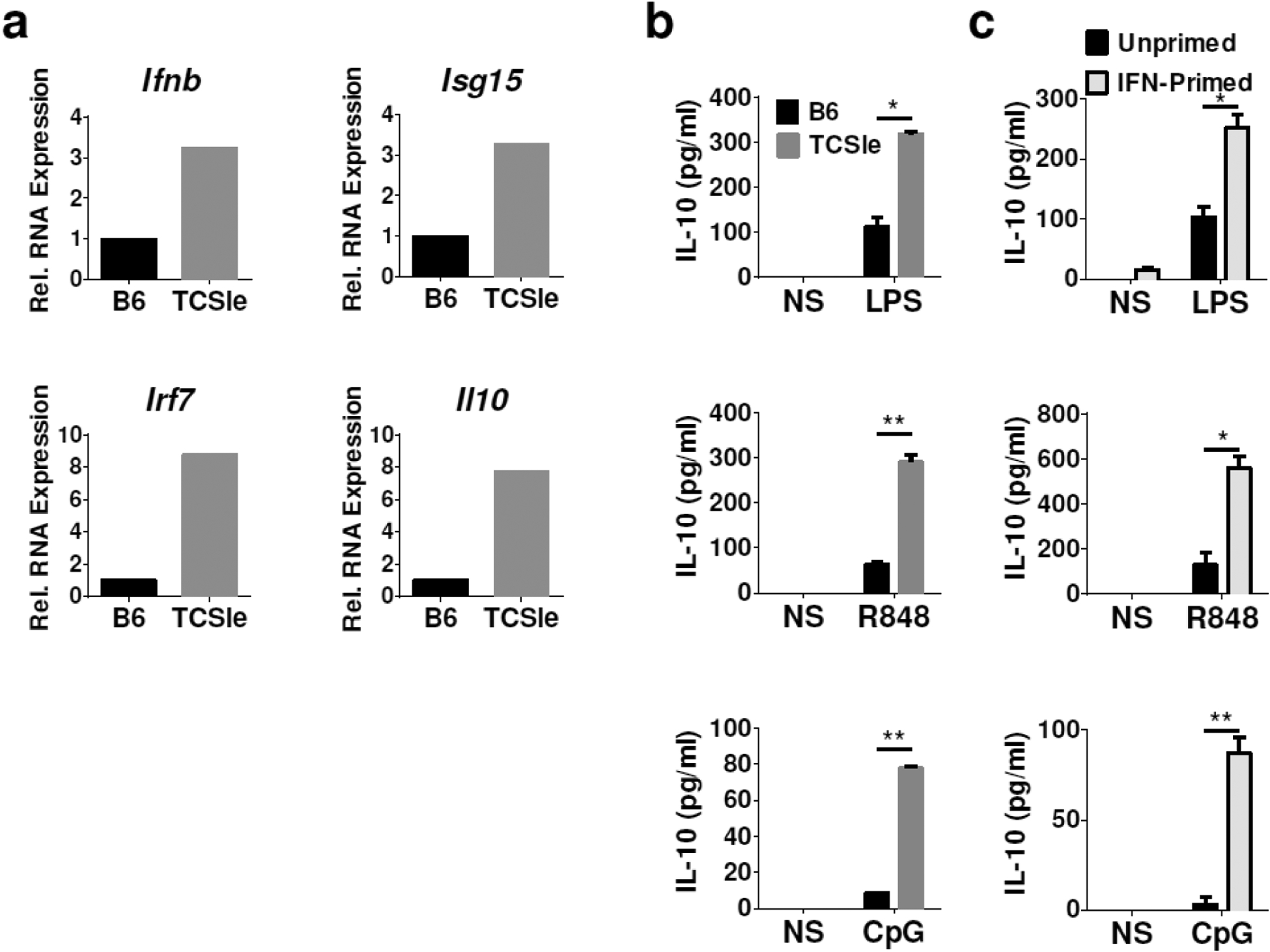
The I-IFN signature and IL-10 in TCSle cDCs. (a) Basal gene expression was measured by qRT-PCR in TCSle cDCs and normalized to unstimulated B6 cDCs. (b) B6 and TCSle cDCs were stimulated with TLR4 ligand LPS (100 ng/ml), TLR7 ligand R848 (1 μg/ml) and TLR9 ligand CpG (10 μg/ml) for 24 hours. (c) B6 cDCs were stimulated with TLR ligands after 48 hours priming with 1500 U/ml of IFN-alpha. IL-10 was measured by ELISA. Figures are representative of at least 3 independent experiments, conducted with 3 cDCs cultures from 3 individual mice per strain.
Furthermore, we found that upon stimulation with major inducers of IL-10, namely the TLR4 ligand LPS, TLR7 ligand R848 (an imiquimod-like compound) and TLR9 ligand CpG, TCSle cDCs secrete 3–4 folds more IL-10 than B6 cDCs in response to all three TLR ligands (Figure 1b). These results suggest that signals leading to IL-10 production are overactive in TCSle cDCs.
Priming B6 cDCs with I-IFN mimics the IL-10 overproduction of TCSle cDCs
We next asked whether I-IFN could induce IL-10 overproduction. To mimic the chronic exposure to I-IFNs that the TCSle cDCs are subject to [10], we primed B6 cDCs with IFN-alpha for 48 hours before TLR stimulation. We found that priming B6 cDCs with I-IFN enhances IL-10 production upon stimulation with TLR ligands LPS, R848, and CpG (Figure 1c). IFN-priming alone induced some IL-10 production, however at the lower limit of detection - indicating that IFN-priming requires a second stimulus (e.g. TLR triggering) to promote IL-10 production. Priming with I-IFN prior to TLR stimulation increased IL-10 production in B6 cDCs to levels similar to those of the TCSle cDCs, suggesting that the increased IL-10 production in TCSle cDCs is due to overexpressed autocrine I-IFN.
IFNAR blockade reduces IL-10 and IL-27 production of TCSle cDCs in response to LPS
To test whether IL-10 overproduction by TCSle cDCs is indeed due to increased autocrine I-IFNs, we used an antibody blocking the I-IFN receptor (anti-IFNAR), which we have previously found to be able to inhibit IFN responses upon IFNAR stimulation [30]. We pretreated cDCs with anti-IFNAR or isotype control antibodies 24 hours before stimulating with LPS, as an example of TLR ligand. IFNAR blockade significantly decreased IL-10 production by TCSle cDCs (Figure 2a), while we observed lesser inhibition in B6 cDCs.
Figure 2.
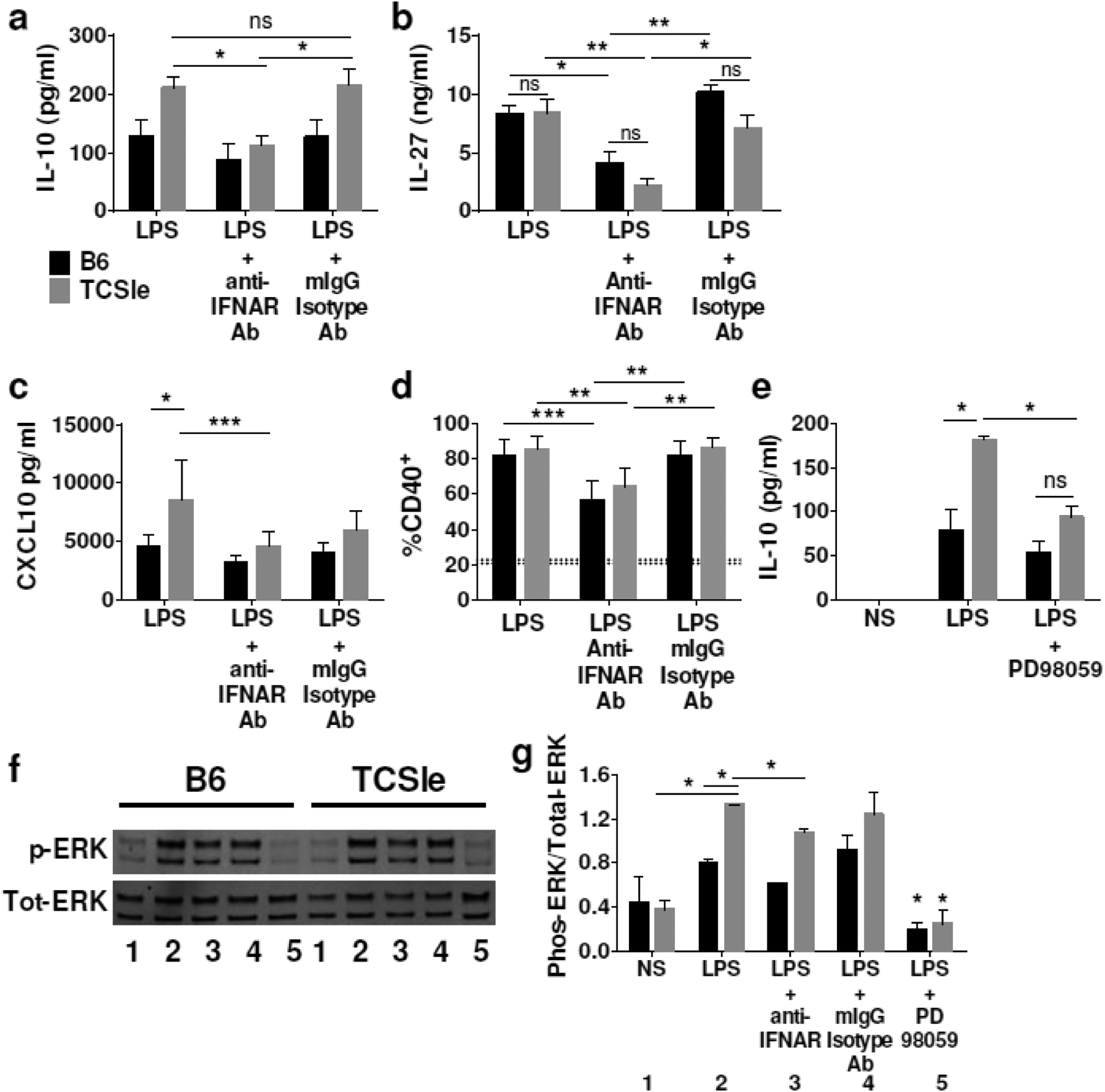
Effects of IFNAR blockade on IL-10, IL-27 and ERK in TCSle cDCs in response to LPS. cDCs were treated with anti-IFNAR antibody or isotype control antibody for (a, b) 24 hours and then again for 30 minutes prior to stimulation with LPS or (c, d) one hour prior to stimulation with LPS. (a-b) Supernatants were collected 24 hours after LPS stimulation and (a) IL-10, (b) IL-27 and (c) CXCL10 were measured by ELISA. (d) cDCs were collected at 24 hours after stimulation and stained. Samples were gated on singlets, scatter gate, live cells, CD11c+ CD11b+, CD40+. Dotted lines represent CD40 expression in PBS treated B6 (20.6%) or TCSle (22.7%) (e) cDCs were treated with ERK inhibitor PD98059 for 30 minutes prior to LPS stimulation. Supernatants were collected 24 hours after LPS stimulation and IL-10 was measured by ELISA. (f) Representative Western blot and (g) averages and SD of the densitometries of Western Blots for phosphorylated ERK and total ERK. cDCs were treated with anti-IFNAR antibodies, or with ERK inhibitor PD98059 and LPS stimulation as above. Cells were harvested in ice cold western lysis buffer 1.5 hours after LPS stimulation. Western blots were probed for total and phosphorylated ERK. Number code: 1) Not stimulated, 2) LPS, 3) LPS + anti-IFNAR, 4) LPS + isotype control, 5) LPS + PD98059. (a-g) Averages and SD of 4–6 replicates from 3–4 experiments. (a-g) Statistical significance was calculated by ANOVA and post-hoc Tukey’s multiple comparison test. *p < 0.05, **p < 0.01, *** p < 0.001 ns means not significant.
IL-27 is induced by TLR stimulation in wild type DCs [25] and participates in I-IFN-induced IL-10 production in T cells and macrophages [22, 31]. Therefore, we asked whether blocking autocrine I-IFNs could affect IL-27 production in lupus cDCs. We found that TCSle and B6 cDCs produced similar amounts of IL-27 upon LPS stimulation, and IFNAR blockade significantly reduced IL-27 production in TCSle cDCs and in B6 cDCs (Figure 2b), supporting the role of I-IFNs in the regulation of IL-27 in lupus cDCs.
We further tested if IFNAR neutralization could reduce CXCL10 production and costimulatory molecule CD40 up-regulation in response to LPS stimulation, chosen as two examples of outcomes downstream of TLR and dependent on IFNAR. While IFNAR neutralization reduced TCSle cDC CXCL10 production in response to LPS, CXCL10 production in B6 cDC was not significantly reduced (Figure 2c). In contrast, IFNAR neutralization significantly decreased CD40 expression on TCSle and B6 cDCs in response to LPS (Figure 2d).
These results suggest that the overexpression of I-IFN, as part of the I-IFN signature in TCSle cDCs, leads to intrinsic I-IFN priming and overproduction of IL-10 and CXCL10 in response to LPS, which can be normalized with IFNAR neutralization. Moreover, IL-27 production and CD40 expression, although they were not augmented in TCSle cDCs, were anyway reduced by anti-IFNAR Ab in both B6 and TCSle cDCs, supporting a role for autocrine I-FNs in the induction of IL-27 as well.
ERK activation contributes to IFN-induced IL-10 production in TCSle cDCs in response to LPS
A number of mechanisms have been described for the I-IFN-induced IL-10 production [21]. The MAP kinase ERK participates in IL-10 production upon many stimuli [32, 33] and it was reported to be abnormally active in B cells of lupus-prone mice [34]. We asked whether ERK activation contributes to IL-10 induced by autocrine I-IFN. First, we used the ERK inhibitor PD98095 to determine if IL-10 overexpression in TCSle cDCs is ERK-dependent. We found that ERK inhibition significantly decreases IL-10 production in LPS-stimulated TCSle cDCs (Figure 2e). ERK inhibition was verified by western blot for phospho-ERK (Figure 2f–g). This result suggests that ERK is required for maximal IL-10 production in response to TLR stimulation in TCSle cDCs.
To determine whether autocrine I-IFN contributes to ERK activation, we stimulated cDCs in the presence of an IFNAR blocking antibody, as above. We found that TCSle cDCs have 30% higher levels of phosphorylated ERK after stimulation with LPS than B6 cDCs. IFNAR blockade slightly reduced LPS-induced ERK phosphorylation, suggesting that autocrine I-IFN partially contributes to the increased ERK activation in TCSle cDCs (Figure 2f–g). Together our results indicate that IL-10 overexpression in TCSle cDCs is the result of autocrine I-IFN and is mediated in part by ERK activation. Previous studies have found ERK inhibition to be protective in Sle models [35] although IL-10 was not assessed. ERK inhibitor may have been beneficial in this model by suppressing IL-10 over-production in TCSle cDCs.
IFNAR Blockade reduces IL-10, CD40, and CXCL10 production of TCSle cDCs in response to R848 and CpG stimulation
TLR7 and TLR9 are important receptors in the recognition of nucleic acids, the main autoantigens and adjuvants in lupus pathogenesis [11]. To test whether IL-10 overproduction by TCSle cDCs in response to R848 and CpG is also due to increased autocrine I-IFNs, we again utilized an anti-IFNAR neutralizing antibody. We pretreated cDCs with anti-IFNAR Ab or isotype control antibodies 1 hour before stimulating with CpG or R848. Compared to the isotype control, IFNAR blockade significantly decreased IL-10 production by TCSle cDCs in response to R848. Furthermore, the significant difference between IL-10 production of TCSle and B6 cDC in response to R848 was lost in IFNAR-neutralizing conditions (Figure 3a). Similarly, the significant difference between IL-10 production of TCSle and B6 cDC in response to CpG was lost in IFNAR-neutralizing conditions, with the reduction of IL-10 production by TCSle cDCs (Figure 3b).
Figure 3.
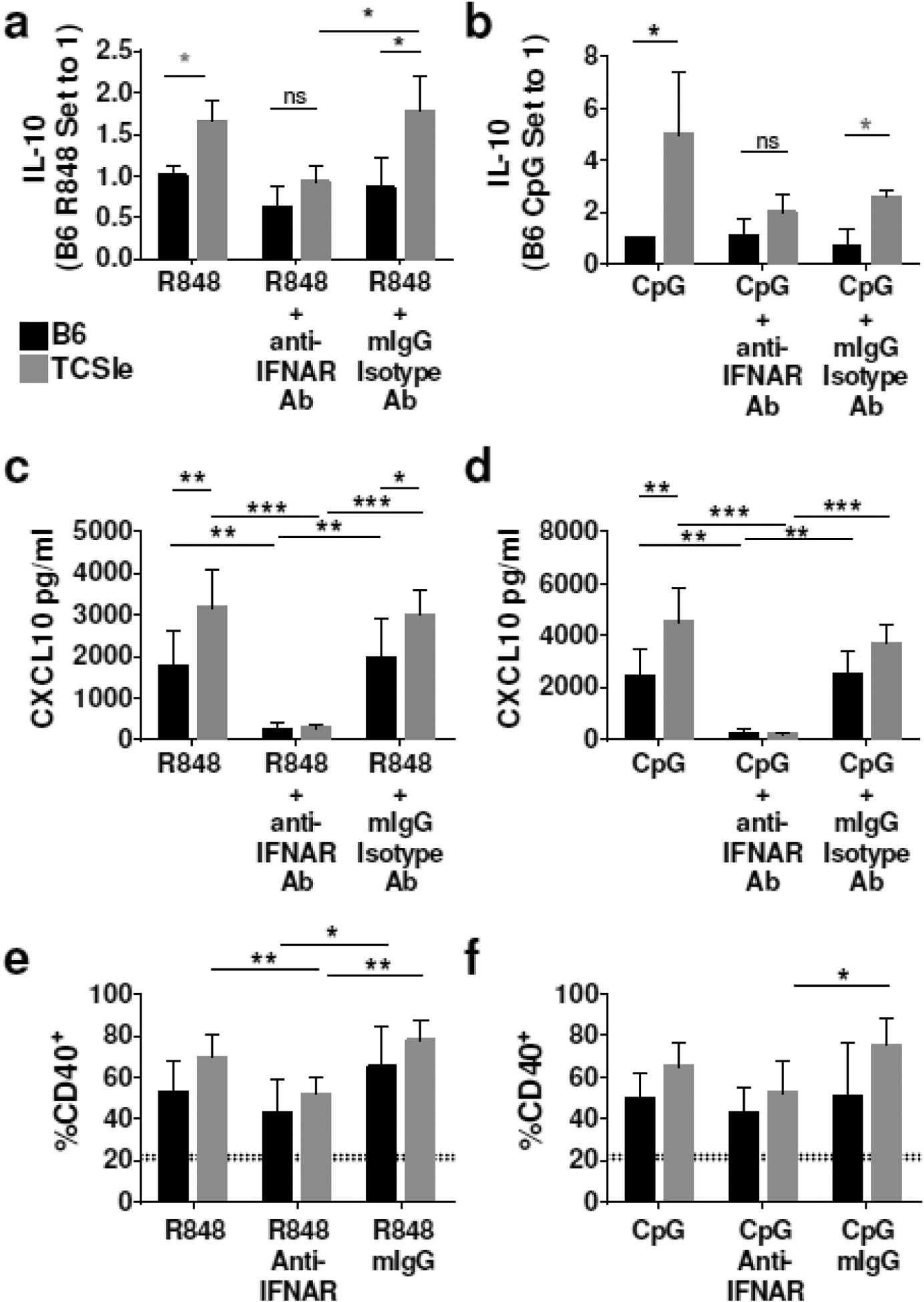
IFNAR blockade reduces IL-10 and CD40, and CXCL10 in TCSle cDCs response to CpG and R848 stimulation. (a-f) cDCs were treated with anti-IFNAR antibody or mIgG isotype control for 1 hour prior to stimulation with R848 (1 μg/ml) and CpG (10 μg/ml) for 24 hours. (a-b) Supernatants were collected 24 hours after stimulation and IL-10 was measured by ELISA. (c-d) Supernatants were collected 24 hours after stimulation and CXCL10 was measured by ELISA. (e-f) cDCs were collected at 24 hours after stimulation and stained. Samples were gated on singlets, scatter gate, live cells, CD11c+ CD11b+, CD40+. Dotted lines represent PBS treated B6 (20.6%) or TCSle (22.7%) CD40 expression (a-f) Averages and SE of 4–6 replicates from 3 experiments. (a-b) Due to inter-experimental variation, B6 stimulation condition is set to 1. Statistical significance was calculated by ANOVA and post-hoc Tukey’s multiple comparison test with the exception of (a-b) gray stars representing statistical significance calculated by unpaired t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
As previously observed by our lab [10], TCSle cDCs produced significantly more CXCL10 in response to R848 or CpG than B6 cDCs. We found that IFNAR neutralization ablated the CXCL10 production in response to R848 and CpG of both TCSle cDCs and B6 cDCs (Figure 3c, d). We further tested if IFNAR neutralization could reduce costimulatory molecule CD40 expression in response to R848 and CpG stimulation. Compared to the isotype control, IFNAR blockade significantly decreased expression of CD40 by TCSle cDCs in response to R848 (Figure 3e) and CpG (Figure 3f), suggesting that the neutralization of IFNAR led to inhibition of IL-10 and CD40 as much as of the ISG CXCL10.
We have previously found that CXCL10 production in response to CpG was IFNAR and Stat2 - dependent in wild-type cDCs, while CXCL10 production in response to LPS was only partially IFNAR/Stat2 - dependent in wild-type cDCs [30]. A different dependency from autocrine I-IFNs of different TLRs can explain the modest reduction in CXCL10 in response to LPS in TCSle cDCs (Figure 2c) and the ablation of CXCL10 in response to R848 and CpG in both B6 and TCSle cDCs (Figure 3c, d). The same ablation of CXCL10 by TCSle cDCs in response to R848 and CpG also suggests that a sufficient concentration of anti-IFNAR Ab was utilized. As IFNAR neutralization brought IL-10 production of TCSle cDCs down to similar levels seen in B6 cDCs, we conclude that a basal amount of IL-10 production in response to R848 or CpG is IFN-independent but dysregulated I-IFN can enhance IL-10 production.
cDCs from TCSle mice overproduce IL-27 in response to TLR7 and TLR9 ligands
To further analyze the production of IL-27 in lupus cDCs, we measured IL-27 upon stimulation with R848 and CpG and found that, in contrast to LPS, TCSle cDCs produced significantly higher levels of IL-27 than B6 cDCs upon TLR7 and TLR9 stimulation. Furthermore, IFNAR neutralization significantly reduced IL-27 production of both TCSle and B6 cDCs in response to R848 and CpG stimulation (Figure 4a, b), indicating a role for autocrine I-IFNs in the production of IL-27 in cDCs. These results are in agreement with our previous findings that TCSle cDCs show stronger differences of I-IFN and ISG expression with B6 cDCs upon stimulation with CpG or R848 rather than with LPS [10], and confirm that TCSle cDCs hyper-respond to the receptors recognizing nucleic acids.
Figure 4.
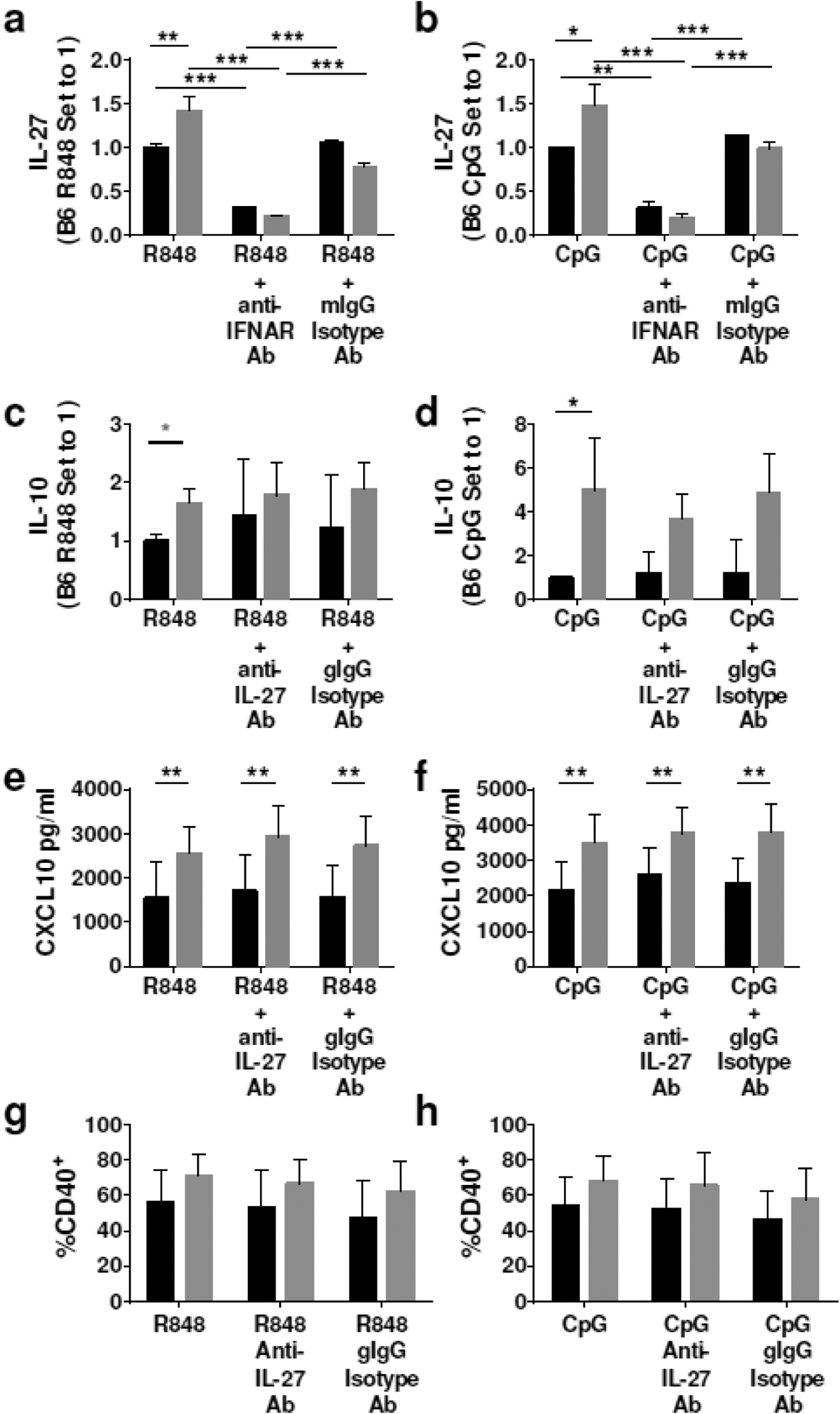
IFNAR blockade prevents IL-27 production in cDCs. (a-b) cDCs were treated with anti-IFNAR antibody (10 μg/ml) or mouse IgG isotype control (10 μg/ml) for 1 hour prior to stimulation with R848 (1 μg/ml) and CpG (10 μg/ml) for 24 hours. (c-h) cDCs were treated with anti-IL-27 antibody (1 μg/ml) or goat IgG isotype control (1 μg/ml) for 1 hour prior to stimulation with R848 (1 μg/ml) and CpG (10 μg/ml) for 24 hours. (a-b) IL-27 was measured by IL-27 ELISA Ready-Set-Go!™ Kit in the supernatants collected 24 hours after TLR stimulation. (c-d) IL-10 was measured by ELISA in the supernatants collected 24 hours after TLR stimulation. (e-f) Supernatants were collected 24 hours after stimulation and CXCL10 was measured by ELISA. (g-h) cDCs were collected at 24 hours after stimulation and stained. Samples were gated on singlets, scatter gate, live cells, CD11c+ CD11b+, CD40+. Dotted lines represent PBS treated B6 (20.6%) or TCSle (22.7%) CD40 expression (a-e) Results show an average and SE of 4–6 replicates from 3 experiments. (a-d) Due to inter-experimental variation, B6 stimulation condition is set to 1. Statistical significance was calculated by ANOVA with post-hoc Tukey’s multiple comparison test with the exception of (c) gray star representing statistical significance calculated by unpaired t-test. *p < 0.05, **p < 0.01, ns means not significant.
A molecular axis involving LPS-IFN-IL-27-IL10 was previously reported in wild type murine macrophages and T cells, in which IL-27 participates in I-IFN-induced IL-10 production in response to LPS [22, 31]. However, others have reported an IL-27-independent IL-10 production in infected macrophages [23] and we have previously found that IL-27 was not necessary for the IL-10 enhancement induced by IFN-beta in B6 cDCs [24], suggesting that IL-27 may not be required to augment IL-10 in cDCs. To test whether this is the case for TCSle cDCs, we used an anti-IL-27 neutralizing Ab in TCSle cDCs and B6 cDCs stimulated by TLR7 and TLR9 ligands, and we found that IL-27 neutralization did not reduce IL-10 production of TCSle cDCs in response to R848 (Figure 4c) nor CpG (Figure 4d) stimulation. Furthermore, IL-27 neutralization did not reduce CXCL10 production (Figure 4e–f) nor CD40 expression (Figure 4g–h) in response to R848 and CpG, indicating lack of effects on IFN-dependent outcomes. Since IFNAR neutralization was able to reduce production of both IL-10 and IL-27, and this neutralization was more pronounced in TCSle cDCs than B6 cDCs, our results indicate a clear role for type I IFN in IL-10 and IL-27 expression in both B6 and TCSle cDCs.
Our work shows a distinct regulation of cytokines in cDCs by I-IFN, which is very active in TCSle DCs, while the production of IL-27/IL-10 is less affected by IFNAR neutralizing antibodies in B6 DCs due to lesser contribution of I-IFNs. Notably, we found that IL-10 is not downstream of IL-27, as it was reported in macrophages [22], but it is parallel in cDCs, suggesting a novel regulation in these cells. We have previously reported an I-IFN-dependent, IL-27-independent production of IL-10 in B6 cDCs [24] and now we show the same regulation in lupus cDCs; others have reported it in macrophages in vivo during chronic Mycobacterium Tuberculosis infection [23], suggesting a generalized role for this regulation of cytokines.
I-IFN-IL-27-IL-10 are highly expressed in immune cells of SLE patients
To determine whether the same I-IFN-IL-27-IL-10 molecular network that we have found in murine lupus is also present in human SLE, we analyzed a publicly available dataset of RNA sequencing performed on whole blood samples from a Michigan patient cohort, consisting of 99 SLE patients and 18 healthy controls (Gene Expression Omnibus accession number GSE72509)[36]. Further details on this cohort are described in [36]. We first analyzed the I-IFN signature expressed in these samples, choosing 27 ISGs (Supplemental Figure 1), and confirmed that this cohort of SLE patients expressed higher levels of ISGs than healthy controls. Then we analyzed the gene expression of IL27 and IL10 and found that both cytokines were significantly higher in SLE patients than in age- and sex-matched healthy controls (Figure 5a–b). To determine a link between type I IFNs and these higher levels of IL27 and IL10, we divided the SLE patients into two groups: those with a high I-IFN signature and those with an I-IFN signature similar to the healthy controls. We found that both IL27 and IL10 levels were significantly higher only in the SLE patients with a high I-IFN signature, while no significant difference in IL27 and IL10 levels were found between SLE patients and healthy controls who had similar levels of ISGs (Figure 5c–d). These results strongly suggest that in human SLE the I-IFN signature is associated with higher levels of IL-27 and IL-10. This association could be linked to multiple cell subsets present in the whole blood, extending the results beyond DCs. A causative link between I-IFNs, IL-27 and IL-10 has been previously proposed in normal immune cells [22, 31]. Our results in murine cDCs suggest that an excess of I-IFNs leads to overproduction of IL-27 and IL-10 in parallel. Since many immune cells can produce these cytokines, including T cells and B cells, we propose that while the I-IFN-IL-27-IL-10 network is a physiological mechanism that regulates the expression of these cytokines, the abnormally high levels of I-IFNs in SLE patients lead to intrinsic IFN priming of IL-27 and IL-10 production in many immune subsets, including cDCs.
Figure 5.
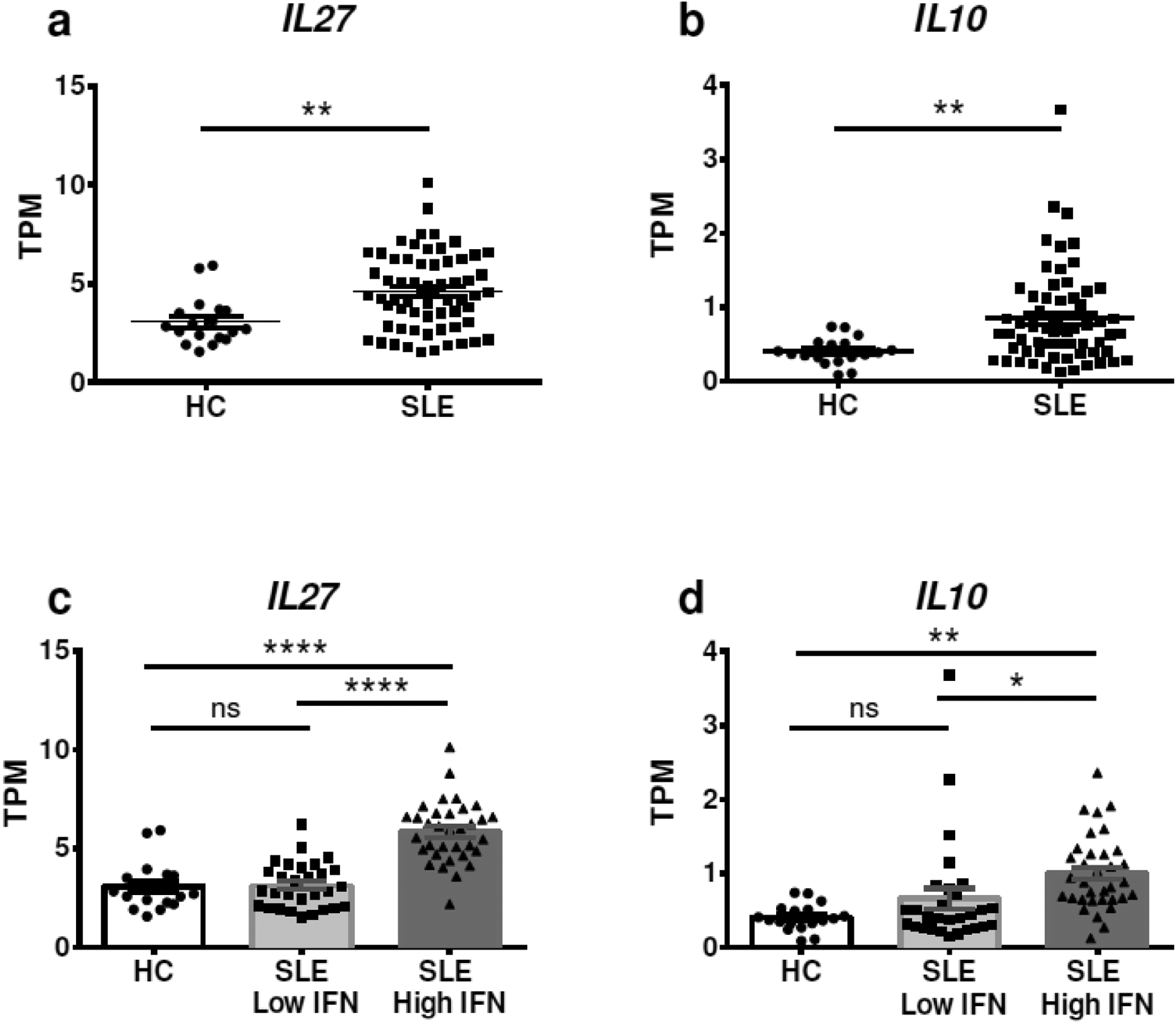
Increased expression of IL10 and IL27 in a publicly available RNAseq data-set. RNAseq data from whole blood of 18 healthy controls and 99 SLE patients was obtained from NCBI Gene Expression Omnibus (GEO), accession number GSE72509. Data were analyzed using GraphPad Prism software, and (a) IL27 and (b) IL10 gene expression (presented as Transcripts Per Million [TPM]) was analyzed and compared between SLE patients and age- and sex-matched healthy controls. SLE patients were then divided in those expressing a low or high I-IFN signature, determined by analyzing the principal component analysis of 27 ISGs, and the expression of (c) IL27 and (d) IL10 gene expression was compared to age- and sex-matched healthy controls (HC). Statistical significance was calculated by Mann-Whitney test in (a) and (b), and by ANOVA with post-hoc Tukey’s multiple comparison test in (c) and (d). Individual samples, averages and SE are shown. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns means not significant.
In conclusion, we propose that murine lupus cDCs overproduce IL-10 and IL-27 as the result of an enhancement by the increased I-IFNs that is highly active in lupus. Importantly, we show that IL-10 and IL-27 levels correlate with the IFN Signature in SLE patients. Clinical trials focused on neutralizing type I IFN-IFNAR (IFNα (Sifalimumab, Rontalizumab, IFNκ and Anifrolumab) have not consistently been able to reduce disease severity in SLE patients [37–39]. Our results suggest that I-IFN blockade, by suppressing IL-27 and IL-10, may have opposite effects on lupus disease: it may block the promotion of autoantibodies but also suppress anti-inflammatory mechanisms. We speculate that future analyses of results from clinical trials utilizing anti-IFNAR antibodies in SLE patients would benefit from examining the effects on IL-27 and IL-10 production, and the opposite effects that these cytokines have on the immune response in SLE.
Supplementary Material
Acknowledgements
We would like to thank the Temple Infections and Autoimmunity Interest Group for stimulating discussions.
Funding
This work was supported by the NIH, NIAID grant R01-AI076423, the FCCC-Temple University Nodal grant, and the Lupus Research Institute, now Lupus Research Alliance, Innovative Grant (SG); NIH NIAMS grant R01-AR061569 (RC). PMG was partially supported by the Lupus Foundation’s Goldie Simon Preceptorship Award.
Footnotes
Publisher's Disclaimer: This is the author manuscript accepted for publication and has undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record.
Conflict of interest
The authors have no financial conflicts of interest.
References
- 1.Rose Noel R., I. R. M. (2014) The Autoimmune Diseases. Academic Press, Elsevier. [Google Scholar]
- 2.Deng Y and Tsao BP (2017) Updates in Lupus Genetics. Curr Rheumatol Rep 19, 68. [DOI] [PubMed] [Google Scholar]
- 3.Liu Z and Davidson A (2012) Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med 18, 871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elkon KB and Stone VV (2011) Type I interferon and systemic lupus erythematosus. J Interferon Cytokine Res 31, 803–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V (2003) Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197, 711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW (2003) Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 100, 2610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK (2007) High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun 8, 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petri M, Wallace DJ, Spindler A, Chindalore V, Kalunian K, Mysler E, Neuwelt CM, Robbie G, White WI, Higgs BW, Yao Y, Wang L, Ethgen D, Greth W (2013) Sifalimumab, a human anti-interferon-alpha monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis Rheum 65, 1011–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morand EF, Trasieva T, Berglind A, Illei GG, Tummala R (2018) Lupus Low Disease Activity State (LLDAS) attainment discriminates responders in a systemic lupus erythematosus trial: post-hoc analysis of the Phase IIb MUSE trial of anifrolumab. Ann Rheum Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sriram U, Varghese L, Bennett HL, Jog NR, Shivers DK, Ning Y, Behrens EM, Caricchio R, Gallucci S (2012) Myeloid Dendritic Cells from B6.NZM Sle1/Sle2/Sle3 Lupus-Prone Mice Express an IFN Signature That Precedes Disease Onset. The Journal of Immunology 189, 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Celhar T, Hopkins R, Thornhill SI, De Magalhaes R, Hwang SH, Lee HY, Yasuga H, Jones LA, Casco J, Lee B, Thamboo TP, Zhou XJ, Poidinger M, Connolly JE, Wakeland EK, Fairhurst AM (2015) RNA sensing by conventional dendritic cells is central to the development of lupus nephritis. Proc Natl Acad Sci U S A 112, E6195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, Buttaro B, Caricchio R, Gallucci S, Tukel C (2015) Amyloid-DNA Composites of Bacterial Biofilms Stimulate Autoimmunity. Immunity 42, 1171–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallo PM and Gallucci S (2013) The dendritic cell response to classic, emerging, and homeostatic danger signals. Implications for autoimmunity. Front Immunol 4, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu J, Liu X, Xie C, Yan M, Yu Y, Sobel ES, Wakeland EK, Mohan C (2005) T cell hyperactivity in lupus as a consequence of hyperstimulatory antigen-presenting cells. J Clin Invest 115, 1869–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mosser DM and Zhang X (2008) Interleukin-10: new perspectives on an old cytokine. Immunol Rev 226, 205–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baglaenko Y, Manion KP, Chang N-H, Gracey E, Loh C, Wither JE (2016) IL-10 Production Is Critical for Sustaining the Expansion of CD5+ B and NKT Cells and Restraining Autoantibody Production in Congenic Lupus-Prone Mice. PLOS ONE 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novikov A, Aleksandrova E, Verizhnikova Z, Panafidina T, Popkova T, Karateev D, Luchihina E, Nasonov E (2016) Cytokine Profiles in Systemic Lupus Erythematosus and Rheumatoid Arthritis. Annals of the Rheumatic Diseases 75, 907. [Google Scholar]
- 18.Llorente L, Richaud-Patin Y, Couderc J, Alarcon-Segovia D, Ruiz-Soto R, Alcocer-Castillejos N, Alcocer-Varela J, Granados J, Bahena S, Galanaud P, Emilie D (1997) Dysregulation of interleukin-10 production in relatives of patients with systemic lupus erythematosus. Arthritis Rheum 40, 1429–35. [DOI] [PubMed] [Google Scholar]
- 19.Llorente L, Richaud-Patin Y, Wijdenes J, Alcocer-Varela J, Maillot MC, Durand-Gasselin I, Fourrier BM, Galanaud P, Emilie D (1993) Spontaneous production of interleukin-10 by B lymphocytes and monocytes in systemic lupus erythematosus. Eur Cytokine Netw 4, 421–7. [PubMed] [Google Scholar]
- 20.Chong WP, Ip WK, Wong WH, Lau CS, Chan TM, Lau YL (2004) Association of interleukin-10 promoter polymorphisms with systemic lupus erythematosus. Genes Immun 5, 484–92. [DOI] [PubMed] [Google Scholar]
- 21.Liu BS, Cao Y, Huizinga TW, Hafler DA, Toes RE (2014) TLR-mediated STAT3 and ERK activation controls IL-10 secretion by human B cells. Eur J Immunol 44, 2121–9. [DOI] [PubMed] [Google Scholar]
- 22.Iyer SS, Ghaffari AA, Cheng G (2010) Lipopolysaccharide-mediated IL-10 transcriptional regulation requires sequential induction of type I IFNs and IL-27 in macrophages. J Immunol 185, 6599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNab FW, Ewbank J, Howes A, Moreira-Teixeira L, Martirosyan A, Ghilardi N, Saraiva M, O’Garra A (2014) Type I IFN Induces IL-10 Production in an IL-27-Independent Manner and Blocks Responsiveness to IFN-γ for Production of IL-12 and Bacterial Killing in Mycobacterium tuberculosis-Infected Macrophages. The Journal of Immunology 193, 3600–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yen JH, Kong W, Hooper KM, Emig F, Rahbari KM, Kuo PC, Scofield BA, Ganea D (2015) Differential effects of IFN-β on IL-12, IL-23, and IL-10 expression in TLR-stimulated dendritic cells. Journal of Leukocyte Biology 98, 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hooper KM, Yen JH, Kong W, Rahbari KM, Kuo PC, Gamero AM, Ganea D (2017) Prostaglandin E2 Inhibition of IL-27 Production in Murine Dendritic Cells: A Novel Mechanism That Involves IRF1. J Immunol 198, 1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meka RR, Venkatesha SH, Dudics S, Acharya B, Moudgil KD (2015) IL-27-induced modulation of autoimmunity and its therapeutic potential. Autoimmun Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu F, Song L, Yang N, Li X (2013) Glucocorticoid downregulates expression of IL-12 family cytokines in systemic lupus erythematosus patients. Lupus 22, 1011–6. [DOI] [PubMed] [Google Scholar]
- 28.Li TT, Zhang T, Chen GM, Zhu QQ, Tao JH, Pan HF, Ye DQ (2010) Low level of serum interleukin 27 in patients with systemic lupus erythematosus. J Investig Med 58, 737–9. [DOI] [PubMed] [Google Scholar]
- 29.Xia LP, Li BF, Shen H, Lu J (2015) Interleukin-27 and interleukin-23 in patients with systemic lupus erythematosus: possible role in lupus nephritis. Scand J Rheumatol 44, 200–5. [DOI] [PubMed] [Google Scholar]
- 30.Xu J, Lee MH, Chakhtoura M, Green BL, Kotredes KP, Chain RW, Sriram U, Gamero AM, Gallucci S (2016) STAT2 Is Required for TLR-Induced Murine Dendritic Cell Activation and Cross-Presentation. Journal of immunology (Baltimore, Md. : 1950) 197, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, Ernst M, Saris CJ, O’Shea JJ, Hunter CA (2007) Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol 8, 1363–71. [DOI] [PubMed] [Google Scholar]
- 32.Lucas M, Zhang X, Prasanna V, Mosser DM (2005) ERK activation following macrophage FcgammaR ligation leads to chromatin modifications at the IL-10 locus. J Immunol 175, 469–77. [DOI] [PubMed] [Google Scholar]
- 33.Gallo P, Goncalves R, Mosser DM (2010) The influence of IgG density and macrophage Fc (gamma) receptor cross-linking on phagocytosis and IL-10 production. Immunol Lett 133, 70–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu K, Liang C, Liang Z, Tus K, Wakeland EK (2005) Sle1ab mediates the aberrant activation of STAT3 and Ras-ERK signaling pathways in B lymphocytes. J Immunol 174, 1630–7. [DOI] [PubMed] [Google Scholar]
- 35.Wu T, Qin X, Kurepa Z, Kumar K, Liu K, Kanta H, Zhou XJ, Satterthwaite AB, Davis LS, Mohan C (2007) Shared signaling networks active in B cells isolated from genetically distinct mouse models of lupus. Journal of Clinical Investigation 117, 2186–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hung T, Pratt GA, Sundararaman B, Townsend MJ, Chaivorapol C, Bhangale T, Graham RR, Ortmann W, Criswell LA, Yeo GW, Behrens TW (2015) The Ro60 autoantigen binds endogenous retroelements and regulates inflammatory gene expression. Science 350, 455–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riggs JM, Hanna RN, Rajan B, Zerrouki K, Karnell JL, Sagar D, Vainshtein I, Farmer E, Rosenthal K, Morehouse C, de los Reyes M, Schifferli K, Liang M, Sanjuan MA, Sims GP, Kolbeck R (2018) Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Science & Medicine 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, Illei GG, Drappa J, Wang L, Yoo S, for the Investigators CD (2017) Anifrolumab, an Anti-Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis & Rheumatology 69, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lauwerys BR, Ducreux J, Houssiau FAA (2014) Type I interferon blockade in systemic lupus erythematosus: where do we stand? Rheumatology (Oxford, England) 53, 1369–1376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.