

Abstract
mRNA-based therapies and vaccines constitute a disruptive technology with the potential to revolutionize modern medicine. Chemically modified 5′ cap structures have provided access to mRNAs with superior translational properties that could benefit the currently flourishing mRNA field. Prime examples of compounds that enhance mRNA properties are antireverse cap analog diastereomers that contain an O-to-S substitution within the β-phosphate (β-S-ARCA D1 and D2), where D1 is used in clinically investigated mRNA vaccines. The compounds were previously found to have high affinity for eukaryotic translation initiation factor 4E (eIF4E) and augment translation in vitro and in vivo. However, the molecular basis for the beneficial “thio-effect” remains unclear. Here, we employed multiple biophysical techniques and captured 11 cap analog-eIF4E crystallographic structures to investigate the consequences of the β-O-to-S or -Se substitution on the interaction with eIF4E. We determined the SP/RP configurations of β-S-ARCA and related compounds and obtained structural insights into the binding. Unexpectedly, in both stereoisomers, the β-S/Se atom occupies the same binding cavity between Lys162 and Arg157, indicating that the key driving force for complex stabilization is the interaction of negatively charged S/Se with positively charged amino acids. This was observed for all structural variants of the cap and required significantly different conformations of the triphosphate for each diastereomer. This finding explains why both β-S-ARCA diastereomers have higher affinity for eIF4E than unmodified caps. Binding affinities determined for di-, tri-, and oligonucleotide cap analogs suggested that the “thio-effect” was preserved in longer RNAs. Our observations broaden the understanding of thiophosphate biochemistry and enable the rational design of translationally active mRNAs and eIF4E-targeting drugs.
Introduction
Phosphorothioate (PS) modification of (oligo)nucleotides is one of the most frequently used synthetic approaches for investigating nucleic acid metabolism or for improving the pharmacokinetics of therapeutic or diagnostic molecules.1−6 Many applications of thio-modified oligonucleotides, such as the elucidation of ion-binding sites on RNA7 and identification of phosphate sites important for ligand or protein binding or for catalysis,8−12 assume that this modification has little or no effect on the secondary structure of nucleic acids. However, it has been shown that incorporation of the PS moiety alters the negative charge distribution13 and can significantly alter the conformation of nucleic acids, thus changing their biological activity.14,15 Furthermore, substitution of a nonbridging oxygen in the phosphate residue with sulfur usually generates an additional stereogenic center at the phosphorus atom yielding a pair of diastereomers,16,17 which are often differentially recognized by specific proteins or enzymes.18−24 Interestingly, a natural PS backbone modification was shown to be an epigenetic mark in bacterial DNA,25−27 and recently it was also found in both prokaryotic and eukaryotic rRNAs.28
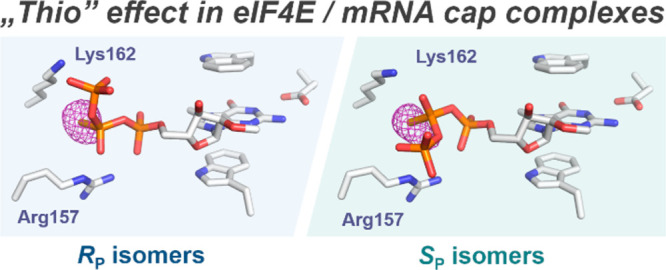
The 7-methylguanosine (m7G) cap structure is present at the 5′ end of all eukaryotic mRNAs (Figure 1A), protecting them from rapid degradation by 5′-exonucleases and regulating many important gene expression steps, including mRNA splicing, nuclear export, initiation of translation, and mRNA turnover.29,30 Chemical modification of the phosphate moieties in the 5′ cap structure has emerged as a particularly appealing strategy to stabilize transcripts and improve their translational activity,31,32 providing a new way to improve mRNA based therapeutics.33−36 Recently, several clinical trials on the development of mRNA vaccines against SARS-CoV-2 gave very promising results.37,38 We have previously reported a series of mRNA cap analogs carrying a PS moiety as reagents for mRNA preparation.39 We have shown that the two analogs termed β-S-ARCA (ARCA stands for Anti-Reverse Cap Analog, meaning that the ribose of 7-methylguanosine was blocked by a methyl group to prevent priming of in vitro transcription at m7G, which produces inactive mRNA), carrying an O-to-S substitution within the β-phosphate of the triphosphate chain, have particularly beneficial biological properties. The analogs, differing in the absolute configurations at the β-phosphate (Figure 1B), were labeled D1 and D2 according to their elution order from the RP HPLC column. β-S-ARCA D1 and D2 had, to different extents, decreased susceptibility to cleavage by Dcp2 decapping enzyme, and increased affinity for eukaryotic translation initiation factor 4E (eIF4E). Interestingly, isomer D1 had a 2-fold higher affinity for eIF4E than D2 and 4-fold higher affinity than the unmodified parent compound,39 which resulted in higher translational efficiency in vitro and in cultured cells for mRNAs incorporating either of these analogs.31 This indicates that the “thio-effect” resulting from a single-atom substitution is sufficient to evoke biologically relevant effects (Figure 1C). mRNAs incorporating β-S-ARCAs also had elongated cellular half-lives and showed augmented protein expression. Because of these favorable properties, β-S-ARCA D1 has been applied in experimental mRNA-based anticancer vaccines40,41 and is currently used as a capping reagent for mRNAs under clinical investigation in multiple trials. However, the RP/SP-absolute configurations of the β-modified PS cap analogs and the electronic and structural consequences of this modification have not been elucidated so far.
Figure 1.

Structure and biological properties of mRNA 5′ cap analogs studied in this work. (A) Schematic structure of mRNA including its 5′ end. (B) Structure of β-S-ARCA (m27,2′-OGppSpG) and its asymmetric phosphorothioate residue. (C) Equilibrium association constants of cap/eIF4E complexes39 and relative translation efficiency of capped mRNAs in HC11 cells31 for ARCA and both diastereomers of β-S-ARCA. (D) Structures of di- and trinucleotide cap analogs studied in this work along with β-S-ARCA (PDB codes for the reported cap-eIF4E complex are provided under each cap formula). Asterisks (*) indicate stereogenic P atoms that generate D1 and D2 isomers.
Here, we performed X-ray crystallography structural studies on a series of therapeutically relevant β-phosphorothioate and β-phosphoroselenoate mRNA 5′ cap analogs (Figure 1D),39,42,43 including β-S-ARCA isomers, to elucidate the molecular mechanism of their tight interaction with eIF4E as well as the differences in the recognition of diastereomers.31,39 To this end, we determined 11 original X-ray crystal structures of eIF4E in complexes with ligands carrying the O to S or O to Se substitutions at the β-position of the triphosphate chain, which enabled us to define both the differences and the common features of their recognition. We also determined binding affinities of model compounds and RNAs incorporating them for eIF4E, using two independent methods to verify whether the observed “thio-effect” is an artifact observed only for dinucleotide cap analogs or is preserved in longer RNAs.
Results and Discusion
Chemical Modification of the Cap Has No Effect on eIF4E Protein Conformation
To explain the “thio-effect” on the molecular level, we determined the structures of β-S-ARCA (m27,2′-OGppSpG) D1 and D2 (Figure 2) and related compounds (Figure 3) in complex with murine eIF4E (meIF4E; residues 28–217), which is 99% identical and 100% similar to the human protein. All the structures were solved at 1.7–2.2 Å resolution, most of them in the P1 space group (Table S1) with four copies of the protein-cap complex in a unit cell, except for complexes with m27,2′-OGppSpA D1 and D2, which were solved in the C2 space group with two copies of the complex (the overall crystal packing was very similar in both space groups). We were able to find noncrystallographic symmetry operators between those copies and verified that their conformation and interaction patterns are consistent (except for m7GppSpApG D2, which is discussed later). Protein conformations in the structures of β-S-ARCA diastereomer complexes are very similar to that of murine eIF4E in complex with an unmodified cap (m7GpppG) reported earlier (Figure 2A; RMSD 1.41 and 1.29 Å for D1 and D2, respectively, RMSD 0.61 Å between D1 and D2; all the atoms were included).44
Figure 2.

X-ray structures of β-phosphorothio(/seleno)ate mRNA 5′ cap analogs in complexes with eukaryotic translation initiation factor 4E. (A) Structures of β-S-ARCAs and m7GpppG (PDB ID: 1L8B) in complexes with eIF4E—simulated annealing omit maps (green mesh) are contoured at 3.0 σ; the electron density corresponding to the first transcribed nucleotide (TSS) is not visible in any of these structures. (B) Structures of β-selenophosphate cap analogs in complexes with eIF4E—anomalous maps (purple mesh) are contoured at 5.0 σ. (C) Resonance structures of disubstituted phosphate and phosphorothioate residues.
Figure 3.
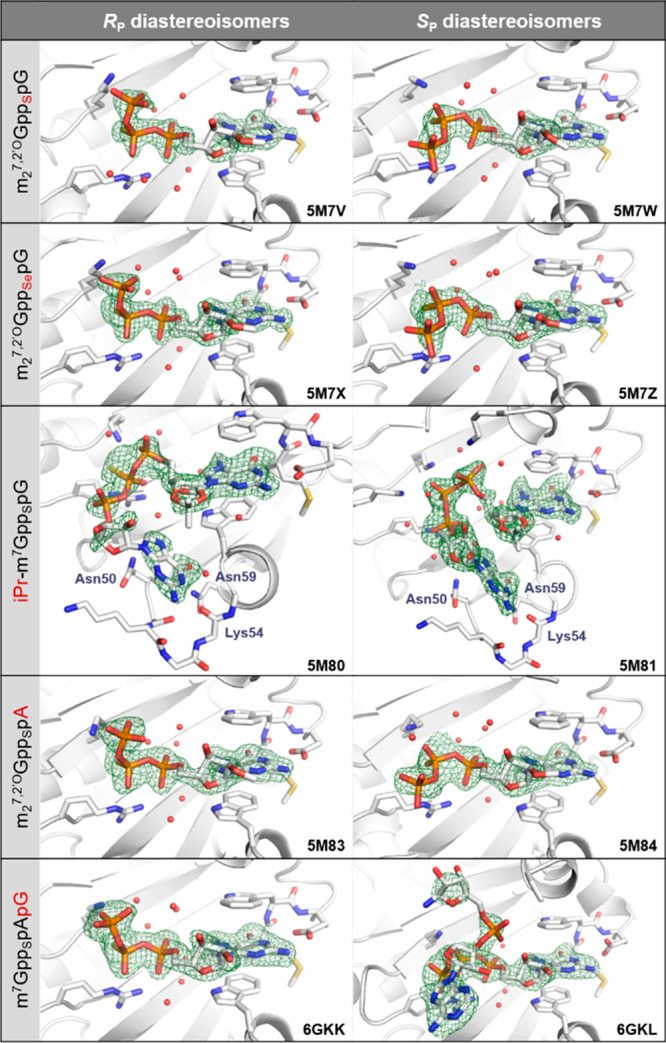
Close-up view of the binding pocket in X-ray structures of eIF4E in complex with cap analogs modified within the β-phosphate moiety. Simulated annealing omit maps (Fo–Fc) contoured at 3σ are represented as a green mesh.
The Absolute Configuration of β-Phosphorus Affects the Cap-Binding Mode by eIF4E
Surprisingly, the ligand conformations adopted by β-S-ARCA D1 and D2 are quite different. In both diastereomers, 7-methylguanosine and γ-phosphate moieties have similar binding modes, similar to the previously observed structures of mono- and dinucleotide caps in complex with either murine or human eIF4E.44,45 However, the position and conformation of β- and α-phosphates in β-S-ARCA D1 and D2 are markedly distinct from each other (Figure 2A). β-S-ARCA D1 adopts a conformation similar to that determined previously for m7GpppG,44 with the sulfur atom of the β-thiophosphate placed in a small cavity between a positively charged Lys162 and Arg157 (Figure 2A). β-S-ARCA D2 is bound in a different conformation, in which the β-phosphate appeared to rotate around the Pβ–Oβ,γ bond by ∼120° compared to D1, and α-phosphate points outside of the binding pocket (Movie 1). Interestingly, for both diastereomers, sulfur atoms appear to be localized in the same position of the cap binding pocket, which suggests that the stereochemistry of β-phosphorus determines the arrangement of the triphosphate bridge. Unfortunately, the initial data did not enable us to clearly distinguish sulfur from oxygen based on the electron density; hence, an unambiguous assignment of the absolute configuration of the β-phosphorus atom required additional experiments. To this end, we cocrystallized eIF4E in complex with the corresponding phosphoroselenoate cap analogues (β-Se-ARCA = m27,2′-OGppSepG) and collected diffraction data at the wavelength corresponding to the maximum absorption of selenium (0.97957 Å – 12.657 keV).46 This enabled unambiguous determination of the position of selenium atoms in the complex.
The phosphoroselenoate cap analogs have biological properties similar to those of phosphorothioate analogs (Table 1, Table S2; m27,2′-OGppSepG D1 should be compared to m27,2′-OGppSpG D1 and m27,2′-OGppSepG D2 to m27,2′-OGppSpG D2),42 so we expected that they should be closely related in structure and the molecular mechanism of binding to eIF4E. Indeed, the shapes of Fo–Fc electron densities for β-Se-ARCAs in the cap binding pocket are very similar to those observed for the corresponding β-S-ARCAs. The anomalous difference electron density maps revealed strong signals in both β-Se-ARCAs complexes (Figure 2B), confirming the absolute configurations around the β-phosphorus center for D1 and D2 as RP and SP, respectively, and clearly positioning the selenium between the side chains of Lys162 and Arg157 regardless of the absolute configuration. The occupancy of this positively charged cavity by the sulfur/selenium atom in β-S-ARCA D2 and β-Se-ARCA D2 is enabled by a significant conformational rearrangement of the 5′,5′-triphosphate chain, resulting in different positioning of the α-phosphate (and likely also a guanosine moiety, for which the electron density is unfortunately not visible in any of the analyzed structures) compared to m7GpppG or corresponding isomers D1.
Table 1. Relative Affinities of Di- and Trinucleotide mRNA cap Analogs and Capped RNAs for Murine eIF4E Determined Using Microscale Thermophoresis (MST) and Fluorescence Quenching Titration (FQT)a.
| compound | Pβ config. | KD FQT/nM | Kd,app MST/μM [C.I.] |
|---|---|---|---|
| m7GpppG | 106 ± 5b | n.d | |
| m27,2′-OGpppG | 92.6 ± 2.6b | n.d | |
| m27,2′-OGppSpG D1 | RP | 23.2 ± 0.8b | 3.2 [2.7, 3.8] |
| m27,2′-OGppSpG D2 | SP | 51.8 ± 5.9b | 5.2 [4.5, 6.1] |
| m27,2′-OGppSepG D1 | RP | 26.0 ± 0.5c | n.d. |
| m27,2′-OGppSepG D2 | SP | 52.6 ± 0.5c | n.d. |
| iPr-m7GpppG | 345 ± 12d | n.d | |
| iPr-m7GppSpG D1 | RP | 179 ± 3d | n.d. |
| iPr-m7GppSpG D2 | SP | 108 ± 2d | n.d. |
| m7GpppA | 178 ± 3 | 22 [18, 28] | |
| 213 ± 24e | |||
| m27,2′-OGppSpA D1 | RP | 44.0 ± 1.4 | 4.9 [4.1, 5.9] |
| m27,2′-OGppSpA D2 | SP | 71.9 ± 2.1 | 8.1 [6.7, 9.9] |
| m7GpppApG | 26.6 ± 0.9 | 5.4 [4.6, 6.3] | |
| m7GppSpApG D1 | RP | 7.3 ± 0.4 | 1.7 [1.3, 2.0] |
| m7GppSpApG D2 | SP | 9.5 ± 0.4 | 1.9 [1.7, 2.2] |
| m27,2′-OGpppG-RNA35 | n.d. | 0.14 [0.11, 0.17] | |
| m27,2′-OGppSpG(D1)-RNA35 | RP | n.d. | 0.065 [0.055, 0.085] |
| pppG-RNA35 | n.d. | no binding |
n.d., not determined; C.I., 68.3% confidence interval.
Kowalska et al. 2008.
Kowalska et al. 2009.
Warminski et al. 2013.
Niedzwiecka et al. 2002.
The Nature of the “Thio-Effect” Is Mainly Electrostatic
Sulfur and selenium atoms form hydrogen bonds of similar strength to those with oxygen atoms.47 Therefore, their strong tendency to occupy space between the two positively charged groups is most likely caused by the electrostatic attraction between opposite charges. This, in turn, suggests that the negative charge in β-phosphorothioate and β-phosphoroselenoate moieties is localized at the heteroatom, instead of being equally distributed between the heteroatom and nonbridging β-oxygen. This hypothesis is consistent with an extensive analysis based on NMR, X-ray, and vibrational spectroscopy evidence,13 as well as ab initio calculations.48 These calculations led to the conclusion that the most accurate representation of charge localization in phosphorothioate esters and anhydrides in solution is a resonance structure with a single P–S bond and the negative charge localized at the sulfur atom (Figure 2C). We envisage that in the case of the studied cap analogues, the negatively charged S/Se atoms from the modified β-phosphate residue form a very strong contact with positively charged Arg157 and Lys162, which can be classified as a salt bridge. The presence of these strong stabilizing interactions involving S/Se atoms arguably explains why both the D1 and D2 isomers display increased affinity for eIF4E compared to m7GpppG. On the other hand, the conformational change observed for the triphosphate in D2 isomers entails different positioning of the α-phosphate in those complexes, which disrupts its interaction with Lys162 and is most likely responsible for the lower affinity to eIF4E observed for the D2 diastereomer as compared to D1. Comparison of the “z-score normalized”49 B-factors of D1 and D2 ligands in complexes with eIF4E show that the D2 isomer is generally more flexible than the D1 isomer, particularly at the β-phosphate, which is consistent with the number of observed contacts with the protein.
To further examine the significance of charge distribution within the phosphorothioate residue in arranging the conformation of the 5′,5′-triphosphate bridge, we determined crystal structures of a diastereomeric pair of another β-phosphorothioate cap analog, in which the m7G ribose ring is constrained by a 2′,3′-O,O-isopropylidene substituent (iPr-m7GppSpG).43 We have previously obtained iPr-m7GppSpG D1 and D2 as synthetically simpler alternatives to β-S-ARCA but found that iPr modification slightly destabilizes eIF4E/cap complexes, likely by affecting the conformational equilibrium of m7G pucker, which results in relatively lower translational efficiencies for capped mRNAs.43 Interestingly, we determined that the D2 isomer of iPr-m7GppSpG has higher affinity for eIF4E than the D1 isomer, in contrast to what was observed for β-S-ARCA, suggesting that the conformation of the triphosphate bridge may also be affected by the bulky substituent.43 Therefore, we were interested to obtain structural insights into iPr-m7GppSpG binding by eIF4E at the molecular level, to see whether it can provide a rationale for the observed biophysical and biological properties. As expected, the crystal structures of eIF4E/iPr-m7GppSpG complexes showed unnatural sugar puckering in m7G, resulting in the displacement of the γ-phosphate toward the C-terminal loop (residues 206–212) by ca. 2 and 3 Å for diastereomers D1 (RP) and D2 (SP), respectively (Figure 3). Consequently, for the D1 (RP) isomer, the positioning of the β-phosphate moiety is also altered, resulting in suboptimal contact with Arg157 and Lys162, which is probably the main reason for its lower affinity for eIF4E as compared to the D2 (SP) isomer. Despite the significant differences in the conformation of the triphosphate bridge between iPr-m7GppSpG diastereomers, the sulfur atoms are clearly attracted toward Lys162 and Arg157 side chains. This observation again confirms that the electrostatic interaction between basic amino acids and heteroatoms at the β-position of the triphosphate chain plays a dominant role in determining the arrangement of the cap analogues in complexes with eIF4E.
Chemically Modified Cap Analogs Reveal a New Potentially Druggable Binding Site on the Surface of eIF4E
Notably, in the case of the iPr-m7GppSpG D2 (SP) complex, the difference electron density map is very well-defined for the entire ligand molecule, revealing the binding mode of the second nucleoside (Figure 3). This unprecedented conformation of the cap in complex with eIF4E is stabilized by several hydrogen bonds to Lys54 and Asn59, engaging the Watson–Crick edge of guanine (N1, N2, and O6) as well as an edge-to-face contact between the nucleobase and the −CONH2 group of Asn50 (Figure S1A). Binding of the second base is accommodated by rotation of the Asn50 side chain, which exposes a small cavity formed by the N-terminal loop (residues 50–55). After a careful inspection of the refined structures of (SP)-m27,2′-OGppSpG and (SP)-m27,2′-OGppSepG complexes (D2 isomers), we observed some residual electron density peaks in a pocket formed by the N-terminal loop, which could not be satisfactorily modeled by water molecules (in contrast to the corresponding structures of RP isomers). The superposition of this map with the structure of the iPr-m7GppSpG D2 (SP) complex shows a very good colocalization of an unmodeled electron density with the guanine part of isopropylidene cap analogs (Figure S1B), suggesting that a similar binding mode may be relevant to m27,2′-OGppSpG and m27,2′-OGppSepG D2 (SP) complexes, but these conformers are apparently not fully occupied.
To verify whether the observed conformational differences between D1 and D2 isomers are more general phenomena related to the presence of sulfur (or selenium) atoms (“thio-effect”) or are observed due to the particular affinity of guanine to interact with the N-terminal loop pocket, we cocrystallized eIF4E in complexes with another pair of β-PS cap analogs, m27,2′-OGppSpA D1 and D2, which contained adenosine instead of guanosine at the transcription start site (TSS or +1 nucleotide; Figure 1A,D). Adenine differs from guanine in the hydrogen bonding pattern at the Watson–Crick edge; therefore, it should not form similar contacts with Lys54 and Asn59. Indeed, in these structures, we did not observe any unmodeled electron density peaks in the N-terminal loop region nor the rotation of the Asn50 side chain, suggesting the absence of nucleobase in the pocket. However, the simulated annealing Fo–Fc omit map showed some residual electron density peaks in the C-terminal loop region of eIF4E-m27,2′-OGppSpA D1 (RP) complex. Importantly, the density does not colocalize with the adenosine portion of an unmodified m7GpppA cap in complex with the human eIF4E reported earlier (Figure S2).45 Rather, it suggests that the α-phosphate is rotated around Pα-Oα,β by ∼120° compared to m7GpppA, placing the second nucleoside in a different region of the loop. The 3′-OH group in the TSS (which is the site of attachment of the RNA chain) for both m7GpppA and m27,2′-OGppSpA D1 is buried deep in the cap binding pocket, which suggests that these conformations cannot be accommodated by full-length mRNA. Nonetheless, the structures reveal new structural features in eIF4E that may be targeted for the design of small molecule translational inhibitors with anticancer activity.50−52 Importantly, for both diastereomers of m27,2′-OGppSpA, the sulfur atom is localized between Arg157 and Lys162 side chains and the conformations of the triphosphate bridge were essentially identical to the conformations of corresponding β-S-ARCAs (Figure 3). The absolute configurations of β-phosphorus atoms also match those of m27,2′-OGppSpG (namely, RP for the D1 isomer and SP for the D2 isomer).
Trinucleotide mRNA 5′ Cap Analogs Provide a More Detailed View of the Structure of the Translation Initiation Complex
We next aimed to verify whether the two distinct conformations observed for β-PS caps are also relevant to longer RNAs. To provide better model molecules to study the impact of the RNA chain on the cap-binding mode, we synthesized trinucleotide cap analogs (the third nucleotide was attached through a 3′-5′-phosphodiester linkage, identical to the natural mRNA 5′ end structure; Figure 1D, compounds m7GpppApG, m7GppSpApG D1 and D2). We obtained high-resolution structures of complexes with m7GpppApG and both diastereomers of m7GppSpApG. Notably, these are the first experimentally determined models of eIF4E in complex with a portion of mRNA longer than two nucleotides. The overall conformations of the triphosphate bridges of trinucleotide analogs are virtually identical to those of their dinucleotide counterparts (m7GpppA and m27,2′-OGppSpA; Figure 3), including the position of the sulfur atom, which means that the “thio-effect” is also preserved for trinucleotides. The structures also allowed us to assign the absolute configurations of the β-phosphorus atom for both PS diastereoisomers as RP for the D1 isomer and SP for the D2 isomer, which are analogous to the configurations determined for dinucleotides. The electron density maps for m7GpppApG and m7GppSpApG D1 (RP) are interpretable only for the 7-methylguanosine and triphosphate bridge portions, while the rest of the ligand is disordered with no apparent electron density for adenosine near the C-terminal loop. This suggests that the first transcribed nucleotide (TSS) and downstream nucleotides in mRNA are generally not involved in any specific contacts with eIF4E or that they form multiple weaker contacts that do not stabilize any particular conformation. This is in contrast to the eIF4E-m7GpppA X-ray structure, which showed an interaction between adenine and the C-terminal loop of the protein.45 However, as mentioned above, the conformation of adenosine observed in dinucleotide complexes could not be adopted by trinucleotide or longer RNAs, since its 3′-OH group is buried in the C-terminal loop, which makes no room for phosphodiester bonds and downstream nucleotides. Therefore, we hypothesize that the presence of the mRNA chain attached to the cap might influence the conformation of the flexible loop by steric hindrance. To verify this, we recorded 1H–15N HSQC spectra of eIF4E/m7GTP and eIF4E/m7GpppApG complexes and compared the chemical shifts of amide NH groups between those complexes. Indeed, large chemical shift perturbations (CSPs) were observed for resonances corresponding to residues 200–204, which form a hinge region of the C-terminal loop, indicating some conformational differences between the complexes (Figure 4, Figure S4). A similar effect was observed for trinucleotide cap analogs differing in the methylation status of adenosine at the TSS (m7GpppApG vs m7Gpppm6AmpG).53 Although the identity and methylation status of the TSS nucleotide have minor effects on the affinity of the cap for eIF4E, it might have some biological implications, since the C-terminal loop of eIF4E is phosphorylated at Ser209 in response to mitogenic stimuli and cytokines.54 Comparison of the HSQC spectra of mono- and trinucleotide complexes also showed significant shift differences for some amides (residues G160, L117, and N118) located on the surface of eIF4E. These CSPs indicate the position of the TSS (+1) and +2 nucleotides in the eIF4E/m7GpppApG complex, and hence imply a possible path taken by the RNA chain in complex with the full-length transcript (Figure 4).
Figure 4.

Chemical shift differences between m7GpppApG and m7GTP bound eIF4E color coded on the protein surface (PDB id: 1L8B) and fragments of 15N HSQC spectra of eIF4E/m7GpppApG (pink contours) and eIF4E/m7GTP (blue contours) presenting selected chemical shift perturbations (CSPs calculated as sqrt[ΔδH2 + (0.15·ΔδN)2]. A fragment of C-terminal loop that was not assigned in the spectra is colored red. Possible RNA path deduced from CSPs is indicated by a yellow dotted line.
Surprisingly, in the cap binding pocket of one of the eIF4E/-m7GppSpApG D2 (SP) complex subunits (chain A), we observed a very well-defined electron density, which allowed us to model almost the entire ligand, excluding only the nucleobase of the third nucleotide (Figures 3 and 5). This particular conformation of the cap is stabilized by an intramolecular hydrogen bond between m7G ribose (3′-OH) and phosphodiester linkage and probably by the contact between ribose of the third nucleotide and aspartate of the symmetry-related protein molecule (the one that belongs to the adjacent crystallographic unit cell −1 + x, y, z), and as such may be only an artifact of crystal packing. Nonetheless, it provides the idea of a possible alignment of the mRNA 5′ end related to eIF4E in biologically relevant complexes. This is consistent with the NMR chemical shift perturbation experiment (Figures 5 and S3A), showing that an unnatural alignment of the triphosphate bridge does not have to affect the path taken by the RNA chain along the eIF4E surface. Such a well-defined electron density corresponding to +1 and +2 nucleotides of the ligand is not observed in the other copy of the complex present in a crystallographic unit cell—a noncrystallographic symmetry (NCS)-related subunit (chain B), which differs only slightly (by ∼3.3 Å) in the position of the aspartate from analogous symmetry-related molecule (x, −1 + y, z). Instead, chain B revealed a new and interesting binding pattern. At a hydrogen bond distance from the ε-amine group of Lys162, we observed a large electron density (4.6 RMSD peak) in the Fo–Fc simulated annealing omit map, which can be attributed to the phosphorus atom of the phosphodiester linkage (Figure S3B). Such an interaction might compensate for the lack of a hydrogen bond between Lys162 and α-phosphate. It also allows the RNA chain to take the path along the β-strand core of eIF4E, similar to that observed for m7GpppApG by NMR CSP mapping (Figure 4).
Figure 5.

Structure of trinucleotide cap analog [m7GppSpApG D2 (SP), chain A] in complex with eIF4E. The sulfur atom is represented as a yellow sphere, and the missing part of the C-terminal loop (the electron density was not well-defined in the X-ray structure) is marked by a white dashed line. The surface of the protein was colored according to Coulombic potential (panel A) calculated using the UCSF Chimera tool55 or color coded by 15N HSQC chemical shift differences between m7GpppApG and m7GTP bound eIF4E (panel B).
The Conformation of the 5′,5′-Triphosphate Bridge Observed for SP (D2) Isomers May Be Biologically Relevant
To obtain a more comprehensive image of β-PS modification in the context of cap-eIF4E interactions, we superimposed all the structures determined in this work (excluding isopropylidene analogs). This superposition revealed the existence of two distinct alignments of the triphosphate bridge, dependent solely on the β-phosphorus stereochemistry (Figure 6). Comparing our structures with other X-ray structures of cap-bound translation initiation factor 4E deposited in the Protein Data Bank, it was found that the conformation characteristic of RP β-phosphorothioates is virtually identical to that of unmodified caps (m7GTP, m7GpppG, and m7GpppA) in complex with mammalian eIF4E proteins. Interestingly, the conformation characteristic of SP isomers closely resembles that of m7GpppG in complex with the parasite (Schistosoma mansoni, Sch) eIF4E protein (32% sequence identity and 51% similarity to human eIF4E).56 Similar to the SP β-phosphorothioate cap analogs in mammalian eIF4E, the α-phosphate of m7GpppG in the Sch-eIF4E complex points away from the lysine amine group, directing the second nucleoside to the N-terminal loop (Figure S5). It has been suggested that such conformation of the cap allows for the movement of the C-terminal loop of Sch-eIF4E toward the cap-binding pocket to clasp the ligand and consequently impacts the pathway taken by the mRNA chain in the translation initiation complex.56 However, mammalian eIF4Es have considerably shorter C-terminal loops, which cannot fully clasp the ligand. The structure of the eIF4E/m7GppSpApG D2 (SP) complex suggests that the RNA phosphodiester backbone is flexible enough to bypass the changes in alignment of the triphosphate bridge caused by PS modification.
Figure 6.
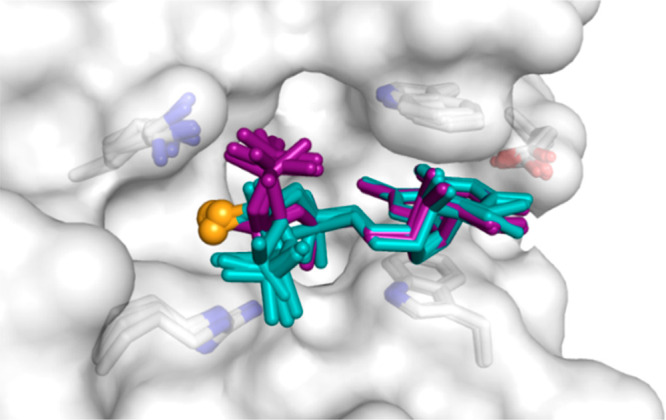
Superposition of eight β-S/β-Se cap-eIF4E structures analyzed in this work. The RP diastereoisomers are colored purple. The SP diastereoisomers are colored cyan, and the sulfur or selenium atoms are represented as orange spheres in all of the analogs. Only m7G and triphosphate moieties are shown for clarity. A single protein model is shown as a gray surface representation. The superposition reveals that S or Se atoms always occupy the same space, regardless of the absolute configuration around the phosphorus atom.
The “Thio-Effect” Is Preserved in Longer RNAs
Unfortunately, we did not succeed in crystallizing longer capped RNA fragments in complex with eIF4E. Therefore, to verify whether the “thio-effect” is preserved in longer RNAs, we focused on analyzing the thermodynamic properties of various cap-eIF4E complexes. First, to corroborate the structural information with thermodynamic data, we determined the dissociation constants (KD) for eIF4E-m27,2′-OGppSpA complexes by fluorescence quenching titration (FQT) and compared them to values previously determined for β-S-ARCA and β-Se-ARCA (Tables 1 and S2). Similar to guanine analogs, RP-β-PS modification in m7GppSpA D1 stabilizes the complex, yielding 4.0-fold lower KD compared to m7GpppA. Interestingly, the stabilization effect for SP modification in m7GppSpA D2 was slightly larger than that for the corresponding guanine analog (2.5-fold increase in binding compared to 1.8-fold increase for guanine analog), although no additional contacts were observed. Using the FQT method, we also determined the binding affinities of trinucleotide cap analogs, finding that the stabilizing “thio-effect” is preserved for those compounds, although the differences between stereoisomers are less pronounced. For trinucleotides, β-PS D1 increases binding affinity to eIF4E by ∼3.5-fold, which is slightly less than for dinucleotides, while D2 increases binding affinity by 2.8-fold, which is slightly higher than that for dinucleotides. Finally, we prepared 35-nt fragments of RNA capped with ARCA or β-S-ARCA D1 by in vitro transcription to determine their KD values for eIF4E. The FQT method was not suitable for this purpose because of the large sample consumption and high inner filter effect occurring in the case of RNA. To overcome this issue, we developed a microscale thermophoresis (MST) competition assay for eIF4E (Figure S6), based on a fluorescently labeled tightly binding probe [m7Gp5OC3(5)FAM, Supporting Information]. The assay enabled indirect comparison of eIF4E-cap dissociation constants for different ligands, based on the apparent binding constant values Kd,app derived from fluorescent probe replacement experiments (Figure S6C). To validate the MST assay, we first determined the Kd,app for ligands that had already been characterized by FQT. These two data sets (Table S2) show very good correlation with a Pearson’s r value of 0.994 (KD[FQT] vs Kd,app[MST], Figure S6B). The MST assay independently confirmed that the stabilization effect of β-PS was observed for both di- and trinucleotides. Finally, we determined Kd,app for β-S-ARCA D1- and ARCA-capped RNA. In this case, the β-PS modification also provided a statistically significant stabilizing effect, albeit less pronounced than in dinucleotides. Namely, RNA capped with β-S-ARCA D1 binds to eIF4E with an affinity 2.2-fold higher than ARCA-capped RNA. The thio-effect is then generally conserved from di- through tri- to oligonucleotide cap analogs, but its contribution to overall complex stability seems to be slightly diminished in the presence of the RNA chain.
Conclusions
In conclusion, a series of therapeutically relevant β-phosphorothioate-modified analogs of mRNA 5′ ending in complex with eukaryotic translation initiation factor 4E was structurally characterized. On the basis of the crystal structures of phosphoroselenoate analogs and their anomalous diffraction, we were able to unambiguously determine the position of selenium atoms and thus assign the absolute configuration of the asymmetric phosphorus atom in each diastereomer as well as indirectly confirm assignments for phosphorothioate analogs. The local environment of sulfur or selenium formed by positively charged amino acids clearly suggests that the negative charge in the modified phosphate residues is mainly localized at the S/Se atoms. The ionic contact between S/Se and basic amino acids was identified as a major factor in complex stabilization and the driving force for the alignment of ligands in the cap binding pocket. As a consequence, phosphorothioate cap analog diastereomers adopted two different sets of conformations, one of which has never been observed before in any mammalian eIF4E complex. In this conformation, adopted preferentially by SP β-phosphorothioate cap analogs, the triphosphate bridge is bound by the protein only through β and γ phosphates, lacking interaction between α-phosphate and Lys162. This is the likely reason for the lower affinity of SP isomers in comparison to RP isomers, but the electrostatic interactions between a negatively charged sulfur atom and two basic amino acids (Arg157 and Lys162) provide higher stabilization energy than the similar interactions involving nonmodified α and β phosphate residues with a delocalized charge in m7GpppG. The missing interaction involving α-phosphate is compensated to some extent in RNAs capped with β-S-ARCA D2 (SP), most likely by a contact between Lys162 and phosphodiester linkage, which is not present in dinucleotide analogs. On the other hand, the lower number of contacts involving the triphosphate bridge of SP diastereomers might facilitate dissociation of the transcript from eIF4E, which was suggested to be necessary for the loading of mRNA onto the 43S preinitiation complex and subsequent start codon scanning.57,58 This conclusion is consistent with the polysomal distribution experiment reported earlier, which showed that mRNAs capped with m27,2′-OGppSpG D2 (SP) are more efficiently recruited to polysomes in HC11 cells and are shifted to heavier polysomes than the analogous mRNAs capped with m27,2′-OGpppG.31 The structural insight into stabilization of cap/eIF4E complexes by phosphorothioate modification presented here provides an explanation of the observed biological properties of clinically relevant β-S-ARCA-capped RNAs and may guide the design of biologically superior mRNA caps that are chemically modified within the phosphodiester linkage between the TSS (+1) and +2 nucleotides. It also reveals two new potentially druggable sites on the eIF4E surface, which might be attractive targets for the inhibition of eukaryotic translation: (i) the N-terminal loop (residues 50–55), occupied by guanine in some of the structures, and (ii) the groove in the vicinity of residues 117–118, which is probably the path taken by the RNA chain in eIF4E/mRNA complexes.
Acknowledgments
This work was supported by the Foundation for Polish Science (TEAM/2016-2/13 and START 91.2018), the Ministry of Science and Higher Education (Poland, DI2012 024842), and the National Science Centre (ETIUDA UMO-2017/24/T/NZ1/00345 and UMO-2016/21/B/ST5/02556). Diffraction data were collected on BL14.1 at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin.59 The research was partially performed in the Biopolymers Laboratory, Division of Biophysics, Institute of Experimental Physics, Faculty of Physics, University of Warsaw, supported by the ERDF within the Innovation Economy Operational Programme POIG.02.01.00-14-122/09.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00864.
Accession Codes
The atomic coordinates and structural factors have been deposited in the Protein Data Bank under accession codes 5M7V (eIF4E-m27,2′OGppSpG D1), 5M7W (eIF4E-m27,2′OGppSpG D2), 5M7X (eIF4E-m27,2′OGppSepG D1), 5M7Z (eIF4E-m27,2′OGppSepG D2), 5M80 (eIF4E-iPr-m7GppSpG D1), 5M81 (eIF4E-iPr-m7GppSpG D2), 5M83 (eIF4E-m27,2′OGppSpA D1), 5M84 (eIF4E-m27,2′OGppSpA D2), 6GKJ (eIF4E-m7GpppApG), 6GKK (eIF4E-m7GppSpApG D1), and 6GKL (eIF4E-m7GppSpApG D2).
Author Contributions
J.J., J.K, J.D.G., and M.N. designed and directed the research. M.W. and E.N. performed the crystallographic experiments. M.W. and R.T. performed NMR experiments. M.W., J.K., and R.K. performed chemical syntheses. D.K. performed ITC and FQT experiments. M.W., J.K., and R.K. performed MST experiments. M.W., J.K., and J.J. wrote the first draft of the paper. All authors discussed, edited, and approved the final version of the manuscript.
The authors declare the following competing financial interest(s): J.J. and J.K. are co-inventors of patented beta-S-ARCA technology.
Supplementary Material
References
- Eckstein F. (1983) Phosphorothioate Analogues of Nucleotides—Tools for the Investigation of Biochemical Processes. Angew. Chem., Int. Ed. Engl. 22 (6), 423–439. 10.1002/anie.198304233. [DOI] [Google Scholar]
- Eckstein F. (2014) Phosphorothioates, Essential Components of Therapeutic Oligonucleotides. Nucleic Acid Ther. 24 (6), 374–387. 10.1089/nat.2014.0506. [DOI] [PubMed] [Google Scholar]
- Kawaguchi D.; Kodama A.; Abe N.; Takebuchi K.; Hashiya F.; Tomoike F.; Nakamoto K.; Kimura Y.; Shimizu Y.; Abe H. (2020) Phosphorothioate Modification of mRNA Accelerates the Rate of Translation Initiation to Provide More Efficient Protein Synthesis. Angew. Chem., Int. Ed. 59 (40), 17403–17407. 10.1002/anie.202007111. [DOI] [PubMed] [Google Scholar]
- Crooke S. T.; Vickers T. A.; Liang X.-h. (2020) , Phosphorothioate modified oligonucleotide–protein interactions. Nucleic Acids Res. 48 (10), 5235–5253. 10.1093/nar/gkaa299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Zhou W.-J.; Liu L.; Fan Z.; Tang H.; Yu R.-Q.; Jiang J.-H. (2020) In vivo mRNA imaging based on tripartite DNA probe mediated catalyzed hairpin assembly. Chem. Commun. 56, 8782–8785. 10.1039/D0CC03596C. [DOI] [PubMed] [Google Scholar]
- Stein C. A.; Castanotto D. (2017) FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 25 (5), 1069–1075. 10.1016/j.ymthe.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forconi M., and Herschlag D.. Chapter 15 - Use of Phosphorothioates to Identify Sites of Metal-Ion Binding in RNA. In Methods in Enzymology; Academic Press, 2009; Vol. 468, pp 311–333. [DOI] [PubMed] [Google Scholar]
- Ruffner D. E.; Uhlenbeck O. C. (1990) Thiophosphate interference experiments locate phosphates important for the hammerhead RNA self-cleavage reaction. Nucleic Acids Res. 18 (20), 6025–6029. 10.1093/nar/18.20.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suydam I. T., and Strobel S. A.. Chapter 1 - Nucleotide Analog Interference Mapping. In Methods in Enzymology; Academic Press, 2009; Vol. 468, pp 3–30. [DOI] [PubMed] [Google Scholar]
- Milligan J. F.; Uhlenbeck O. C. (1989) Determination of RNA-protein contacts using thiophosphate substitutions. Biochemistry 28 (7), 2849–2855. 10.1021/bi00433a016. [DOI] [PubMed] [Google Scholar]
- Vortler L. C.; Eckstein F. (2000) Phosphorothioate modification of RNA for stereochemical and interference analyses. Methods Enzymol. 317, 74–91. 10.1016/S0076-6879(00)17007-7. [DOI] [PubMed] [Google Scholar]
- Dertinger D.; Behlen L. S.; Uhlenbeck O. C. (2000) Using Phosphorothioate-Substituted RNA To Investigate the Thermodynamic Role of Phosphates in a Sequence Specific RNA–Protein Complex. Biochemistry 39 (1), 55–63. 10.1021/bi991769v. [DOI] [PubMed] [Google Scholar]
- Frey P. A.; Sammons R. D. (1985) Bond order and charge localization in nucleoside phosphorothioates. Science 228 (4699), 541. 10.1126/science.2984773. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Nikonowicz E. P. (2000) Phosphorothioate Substitution Can Substantially Alter RNA Conformation. Biochemistry 39 (19), 5642–5652. 10.1021/bi992712b. [DOI] [PubMed] [Google Scholar]
- Hyjek-Składanowska M.; Vickers T. A.; Napiórkowska A.; Anderson B. A.; Tanowitz M.; Crooke S. T.; Liang X.-h.; Seth P. P.; Nowotny M. (2020) Origins of the Increased Affinity of Phosphorothioate-Modified Therapeutic Nucleic Acids for Proteins. J. Am. Chem. Soc. 142 (16), 7456–7468. 10.1021/jacs.9b13524. [DOI] [PubMed] [Google Scholar]
- Eckstein F. (1985) Nucleoside Phosphorothioates. Annu. Rev. Biochem. 54 (1), 367–402. 10.1146/annurev.bi.54.070185.002055. [DOI] [PubMed] [Google Scholar]
- Knouse K. W.; deGruyter J. N.; Schmidt M. A.; Zheng B.; Vantourout J. C.; Kingston C.; Mercer S. E.; McDonald I. M.; Olson R. E.; Zhu Y.; Hang C.; Zhu J.; Yuan C.; Wang Q.; Park P.; Eastgate M. D.; Baran P. S. (2018) Unlocking P(V): Reagents for chiral phosphorothioate synthesis. Science 361 (6408), 1234. 10.1126/science.aau3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L. T.; Cohn M. (1982) Reactions of thio analogs of adenosine 5′-triphosphate catalyzed by methionyl-tRNA synthetase from Escherichia coli and metal dependence of stereospecificity. Biochemistry 21 (7), 1530–1534. 10.1021/bi00536a010. [DOI] [PubMed] [Google Scholar]
- O’Brian C. A.; Roczniak S. O.; Bramson H. N.; Baraniak J.; Stec W. J.; Kaiser E. T. (1982) A kinetic study of interactions of (RP)- and (SP)-adenosine cyclic 3′,5′-phosphorothioates with type II bovine cardiac muscle adenosine cyclic 3′,5′-phosphate dependent protein kinase. Biochemistry 21 (18), 4371–4376. 10.1021/bi00261a028. [DOI] [PubMed] [Google Scholar]
- Moore M. J.; Sharp P. A. (1993) Evidence for two active sites in the spliceosome provided by stereochemistry of pre-mRNA splicing. Nature 365, 364. 10.1038/365364a0. [DOI] [PubMed] [Google Scholar]
- Das R.; Melacini G. (2007) A Model for Agonism and Antagonism in an Ancient and Ubiquitous cAMP-binding Domain. J. Biol. Chem. 282 (1), 581–593. 10.1074/jbc.M607706200. [DOI] [PubMed] [Google Scholar]
- Lim J.; Kim H.-Y. (2020) Novel Applications of Biocatalysis to Stereochemistry Determination of 2′3′-cGAMP Bisphosphorothioate (2′3′-cGSASMP). ACS Omega 5 (23), 14173–14179. 10.1021/acsomega.0c01942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funder E. D.; Albæk N.; Moisan A.; Sewing S.; Koch T. (2020) Refining LNA safety profile by controlling phosphorothioate stereochemistry. PLoS One 15 (6), e0232603. 10.1371/journal.pone.0232603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stivers J. T.; Nawrot B.; Jagadeesh G. J.; Stec W. J.; Shuman S. (2000) Stereochemical Outcome and Kinetic Effects of Rp- and Sp-Phosphorothioate Substitutions at the Cleavage Site of Vaccinia Type I DNA Topoisomerase. Biochemistry 39 (18), 5561–5572. 10.1021/bi992429c. [DOI] [PubMed] [Google Scholar]
- Wang L.; Jiang S.; Deng Z.; Dedon P. C.; Chen S. (2019) DNA phosphorothioate modification—a new multi-functional epigenetic system in bacteria. FEMS Microbiology Reviews 43 (2), 109–122. 10.1093/femsre/fuy036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Cao B.; Aquino P.; Chiu T.-P.; Chen C.; Jiang S.; Deng Z.; Chen S.; Rohs R.; Wang L.; Galagan J. E.; Dedon P. C. (2020) Epigenetic competition reveals density-dependent regulation and target site plasticity of phosphorothioate epigenetics in bacteria. Proc. Natl. Acad. Sci. U. S. A. 117 (25), 202002933. 10.1073/pnas.2002933117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong T.; Chen S.; Wang L.; Tang Y.; Ryu J. Y.; Jiang S.; Wu X.; Chen C.; Luo J.; Deng Z.; Li Z.; Lee S. Y.; Chen S. (2018) Occurrence, evolution, and functions of DNA phosphorothioate epigenetics in bacteria. Proc. Natl. Acad. Sci. U. S. A. 115 (13), E2988. 10.1073/pnas.1721916115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Tang Y.; Dong X.; Zheng Y. Y.; Haruehanroengra P.; Mao S.; Lin Q.; Sheng J. (2020) RNA Phosphorothioate Modification in Prokaryotes and Eukaryotes. ACS Chem. Biol. 15 (6), 1301–1305. 10.1021/acschembio.0c00163. [DOI] [PubMed] [Google Scholar]
- Furuichi Y. (2015) Discovery of m7G-cap in eukaryotic mRNAs. Proc. Jpn. Acad., Ser. B 91 (8), 394–409. 10.2183/pjab.91.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M. J. (2005) From Birth to Death: The Complex Lives of Eukaryotic mRNAs. Science 309 (5740), 1514. 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- Grudzien-Nogalska E.; Jemielity J.; Kowalska J.; Darzynkiewicz E.; Rhoads R. E. (2007) Phosphorothioate cap analogs stabilize mRNA and increase translational efficiency in mammalian cells. RNA 13 (10), 1745–1755. 10.1261/rna.701307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H.; Kim M.; Seo Y.; Moon Y. S.; Lee H. J.; Lee K.; Lee H. (2018) Emergence of synthetic mRNA: In vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials 156, 172–193. 10.1016/j.biomaterials.2017.11.034. [DOI] [PubMed] [Google Scholar]
- van Dülmen M.; Rentmeister A. (2020) mRNA Therapies: New Hope in the Fight against Melanoma. Biochemistry 59 (17), 1650–1655. 10.1021/acs.biochem.0c00181. [DOI] [PubMed] [Google Scholar]
- Jackson N. A. C.; Kester K. E.; Casimiro D.; Gurunathan S.; DeRosa F. (2020) The promise of mRNA vaccines: a biotech and industrial perspective. npj Vaccines 5 (1), 11. 10.1038/s41541-020-0159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U.; Karikó K.; Türeci Ö. (2014) mRNA-based therapeutics — developing a new class of drugs. Nat. Rev. Drug Discovery 13 (10), 759–780. 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- Pastor F.; Berraondo P.; Etxeberria I.; Frederick J.; Sahin U.; Gilboa E.; Melero I. (2018) An RNA toolbox for cancer immunotherapy. Nat. Rev. Drug Discovery 17 (10), 751–767. 10.1038/nrd.2018.132. [DOI] [PubMed] [Google Scholar]
- Mulligan M. J.; Lyke K. E.; Kitchin N.; Absalon J.; Gurtman A.; Lockhart S.; Neuzil K.; Raabe V.; Bailey R.; Swanson K. A.; Li P.; Koury K.; Kalina W.; Cooper D.; Fontes-Garfias C.; Shi P.-Y.; Türeci Ö.; Tompkins K. R.; Walsh E. E.; Frenck R.; Falsey A. R.; Dormitzer P. R.; Gruber W. C.; Şahin U.; Jansen K. U. (2020) Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 586 (7830), 589–593. 10.1038/s41586-020-2639-4. [DOI] [PubMed] [Google Scholar]
- Jackson L. A.; Anderson E. J.; Rouphael N. G.; Roberts P. C.; Makhene M.; Coler R. N.; McCullough M. P.; Chappell J. D.; Denison M. R.; Stevens L. J.; Pruijssers A. J.; McDermott A.; Flach B.; Doria-Rose N. A.; Corbett K. S.; Morabito K. M.; O’Dell S.; Schmidt S. D.; Swanson P. A.; Padilla M.; Mascola J. R.; Neuzil K. M.; Bennett H.; Sun W.; Peters E.; Makowski M.; Albert J.; Cross K.; Buchanan W.; Pikaart-Tautges R.; Ledgerwood J. E.; Graham B. S.; Beigel J. H. (2020) An mRNA Vaccine against SARS-CoV-2 — Preliminary Report. N. Engl. J. Med. 383 (20), 1920–1931. 10.1056/NEJMoa2022483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska J.; Lewdorowicz M.; Zuberek J.; Grudzien-Nogalska E.; Bojarska E.; Stepinski J.; Rhoads R. E.; Darzynkiewicz E.; Davis R. E.; Jemielity J. (2008) Synthesis and characterization of mRNA cap analogs containing phosphorothioate substitutions that bind tightly to eIF4E and are resistant to the decapping pyrophosphatase DcpS. RNA 14 (6), 1119–1131. 10.1261/rna.990208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn A. N.; Diken M.; Kreiter S.; Selmi A.; Kowalska J.; Jemielity J.; Darzynkiewicz E.; Huber C.; Tureci O.; Sahin U. (2010) Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 17 (8), 961–971. 10.1038/gt.2010.52. [DOI] [PubMed] [Google Scholar]
- Beissert T.; Perkovic M.; Vogel A.; Erbar S.; Walzer K. C.; Hempel T.; Brill S.; Haefner E.; Becker R.; Türeci Ö.; Sahin U. (2020) A Trans-amplifying RNA Vaccine Strategy for Induction of Potent Protective Immunity. Mol. Ther. 28 (1), 119–128. 10.1016/j.ymthe.2019.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska J.; Lukaszewicz M.; Zuberek J.; Darzynkiewicz E.; Jemielity J. (2009) Phosphoroselenoate Dinucleotides for Modification of mRNA 5′ End. ChemBioChem 10 (15), 2469–2473. 10.1002/cbic.200900522. [DOI] [PubMed] [Google Scholar]
- Warminski M.; Kowalska J.; Buck J.; Zuberek J.; Lukaszewicz M.; Nicola C.; Kuhn A. N.; Sahin U.; Darzynkiewicz E.; Jemielity J. (2013) The synthesis of isopropylidene mRNA cap analogs modified with phosphorothioate moiety and their evaluation as promoters of mRNA translation. Bioorg. Med. Chem. Lett. 23 (13), 3753–3758. 10.1016/j.bmcl.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Niedzwiecka A.; Marcotrigiano J.; Stepinski J.; Jankowska-Anyszka M.; Wyslouch-Cieszynska A.; Dadlez M.; Gingras A.-C.; Mak P.; Darzynkiewicz E.; Sonenberg N.; Burley S. K.; Stolarski R. (2002) Biophysical Studies of eIF4E Cap-binding Protein: Recognition of mRNA 5′ Cap Structure and Synthetic Fragments of eIF4G and 4E-BP1 Proteins. J. Mol. Biol. 319 (3), 615–635. 10.1016/S0022-2836(02)00328-5. [DOI] [PubMed] [Google Scholar]
- Tomoo K.; Matsushita Y.; Fujisaki H.; Abiko F.; Shen X.; Taniguchi T.; Miyagawa H.; Kitamura K.; Miura K.-i.; Ishida T. (2005) Structural basis for mRNA Cap-Binding regulation of eukaryotic initiation factor 4E by 4E-binding protein, studied by spectroscopic, X-ray crystal structural, and molecular dynamics simulation methods. Biochim. Biophys. Acta, Proteins Proteomics 1753 (2), 191–208. 10.1016/j.bbapap.2005.07.023. [DOI] [PubMed] [Google Scholar]
- Wilds C. J.; Pattanayek R.; Pan C.; Wawrzak Z.; Egli M. (2002) Selenium-Assisted Nucleic Acid Crystallography: Use of Phosphoroselenoates for MAD Phasing of a DNA Structure. J. Am. Chem. Soc. 124 (50), 14910–14916. 10.1021/ja021058b. [DOI] [PubMed] [Google Scholar]
- Mishra K. K.; Singh S. K.; Ghosh P.; Ghosh D.; Das A. (2017) The nature of selenium hydrogen bonding: gas phase spectroscopy and quantum chemistry calculations. Phys. Chem. Chem. Phys. 19 (35), 24179–24187. 10.1039/C7CP05265K. [DOI] [PubMed] [Google Scholar]
- Liang C.; Allen L. C. (1987) Sulfur does not form double bonds in phosphorothioate anions. J. Am. Chem. Soc. 109 (21), 6449–6453. 10.1021/ja00255a034. [DOI] [Google Scholar]
- Schneider B.; Gelly J.-C.; de Brevern A. G.; Cerny J. (2014) Local dynamics of proteins and DNA evaluated from crystallographic B factors. Acta Crystallogr., Sect. D: Biol. Crystallogr. 70 (9), 2413–2419. 10.1107/S1399004714014631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerke N. J.; Aktas H.; Chen H.; Cantel S.; Reibarkh M. Y.; Fahmy A.; Gross J. D.; Degterev A.; Yuan J.; Chorev M.; Halperin J. A.; Wagner G. (2007) Small-Molecule Inhibition of the Interaction between the Translation Initiation Factors eIF4E and eIF4G. Cell 128 (2), 257–267. 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Chen X.; Kopecky D. J.; Mihalic J.; Jeffries S.; Min X.; Heath J.; Deignan J.; Lai S.; Fu Z.; Guimaraes C.; Shen S.; Li S.; Johnstone S.; Thibault S.; Xu H.; Cardozo M.; Shen W.; Walker N.; Kayser F.; Wang Z. (2012) Structure-Guided Design, Synthesis, and Evaluation of Guanine-Derived Inhibitors of the eIF4E mRNA–Cap Interaction. J. Med. Chem. 55 (8), 3837–3851. 10.1021/jm300037x. [DOI] [PubMed] [Google Scholar]
- Wan X.; Yang T.; Cuesta A.; Pang X.; Balius T. E.; Irwin J. J.; Shoichet B. K.; Taunton J. (2020) Discovery of Lysine-Targeted eIF4E Inhibitors through Covalent Docking. J. Am. Chem. Soc. 142 (11), 4960–4964. 10.1021/jacs.9b10377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski P. J.; Warminski M.; Kubacka D.; Ratajczak T.; Nowis D.; Kowalska J.; Jemielity J. (2020) The identity and methylation status of the first transcribed nucleotide in eukaryotic mRNA 5′ cap modulates protein expression in living cells. Nucleic Acids Res. 48 (4), 1607–1626. 10.1093/nar/gkaa032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuberek J.; Wyslouch-Cieszynska A.; Niedzwiecka A.; Dadlez M.; Stepinski J.; Augustyniak W.; Gingras A.-C.; Zhang Z.; Burley S. K.; Sonenberg N.; Stolarski R.; Darzynkiewicz E. (2003) Phosphorylation of eIF4E attenuates its interaction with mRNA 5′ cap analogs by electrostatic repulsion: Intein-mediated protein ligation strategy to obtain phosphorylated protein. RNA 9 (1), 52–61. 10.1261/rna.2133403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. (2004) UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25 (13), 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Liu W.; Zhao R.; McFarland C.; Kieft J.; Niedzwiecka A.; Jankowska-Anyszka M.; Stepinski J.; Darzynkiewicz E.; Jones D. N. M.; Davis R. E. (2009) Structural Insights into Parasite eIF4E Binding Specificity for m7G and m2,2,7G mRNA Caps. J. Biol. Chem. 284 (45), 31336–31349. 10.1074/jbc.M109.049858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Hellen C. U. T.; Pestova T. V. (2016) Toward the mechanism of eIF4F-mediated ribosomal attachment to mammalian capped mRNAs. Genes Dev. 30 (13), 1573–1588. 10.1101/gad.282418.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliseev B.; Yeramala L.; Leitner A.; Karuppasamy M.; Raimondeau E.; Huard K.; Alkalaeva E.; Aebersold R.; Schaffitzel C. (2018) Structure of a human cap-dependent 48S translation pre-initiation complex. Nucleic Acids Res. 46 (5), 2678–2689. 10.1093/nar/gky054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller U.; Förster R.; Hellmig M.; Huschmann F. U.; Kastner A.; Malecki P.; Pühringer S.; Röwer M.; Sparta K.; Steffien M.; Ühlein M.; Wilk P.; Weiss M. S. (2015) The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: Current status and perspectives. Eur. Phys. J. Plus 130 (7), 141. 10.1140/epjp/i2015-15141-2. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.