

Abstract

CRISPR-based technologies are paramount in genome engineering and synthetic biology. Prime editing (PE) is a technology capable of installing genomic edits without double-stranded DNA breaks (DSBs) or donor DNA. Prime editing guide RNAs (pegRNAs) simultaneously encode both guide and edit template sequences. They are more design intensive than CRISPR single guide RNAs (sgRNAs). As such, application of PE technology is hindered by the limited throughput of manual pegRNA design. To that end, we designed a software tool, Prime Induced Nucleotide Engineering Creator of New Edits (PINE-CONE), that enables high-throughput automated design of pegRNAs and prime editing strategies. PINE-CONE translates edit coordinates and sequences into pegRNA designs, accessory guides, and oligonucleotides for facile cloning workflows. To demonstrate PINE-CONE’s utility in studying disease-relevant genotypes, we rapidly design a library of pegRNAs targeting Alzheimer’s Disease single nucleotide polymorphisms (SNPs). Overall, PINE-CONE will accelerate the application of PEs in synthetic biology and biomedical research.
Keywords: CRISPR, prime editing, automation, genome engineering
Automation accelerates our ability to engineer living systems. As a result, synthetic biology has adopted design software and standardization to improve forward engineering.1,2 Experimental automation increases throughput, enables expanded assembly of genetic circuits and interrogation of genetics on a scale not attainable by manual efforts.3,4 CRISPR-based technologies are highly amenable to design automation and are functional in a broad range of organisms.5 This has made CRISPR-systems indispensable for the fields of synthetic biology and genome engineering.6 Canonical CRISPR technologies target DNA via inducing double stranded DNA breaks (DSBs) and are often subsequently repaired via nonhomologous end joining (NHEJ), or by homology directed repair (HDR) with exogenous DNA templates. However, DSBs can induce off-target mutations, apoptosis, and a destabilized karyotype.7−9 To address these shortcomings, new technologies have fused the programmability of CRISPR associated (Cas) proteins to enzymes capable of mediating DNA manipulations without DSBs including Cas9-fused recombinases, transposases, and deaminases.10−13 Deaminase fused-Cas9 base editing (BE) technologies have enabled single base pair chromosomal editing without the introduction of deleterious DSBs. Base editing consists of cytosine base editors (CBE), which mediate the change of C-to-T (or G-to-A), and adenine base editors (ABE), which facilitate the conversion of A-to-G (or T-to-C).14,15 To date, BEs have been used to interrogate genotype-to-phenotype relationships, engineer animal model of disease, and develop cell therapies.16 However, base editors can only facilitate the four transition mutations, are restricted to single nucleotide modifications within the editing window, and cannot facilitate insertion or removal of nucleotides.
CRISPR-Cas9 systems have been used with reverse transcriptases to facilitate highly efficient user programmed editing. For instance, CRISPey (Retron precISe Parallel Editing via homology) enables highly efficient editing in yeast.17 Alternatively, prime editors (PE) are a recently developed gene editing technology that is capable of introducing all 12 possible single nucleotide changes as well as small insertions and deletions without the need for DSBs or donor DNA templates.18 PEs are a fusion protein composed of a nicking Cas9 mutant fused to reverse transcriptase domain (Moloney Murine Leukemia Virus Reverse Transcriptase; MMLV-RT; Figure 1a). The PE protein is targeted to the editing site by a prime editing guide (pegRNA) which encodes three components: (i) a guide sequence, (ii) a primer binding sequence (PBS), and (iii) a reverse transcription template (RTT), which encodes the intended edit. The pegRNA directs the PE to the target locus, where Cas9 mediates a single-stranded DNA break (SSB) on the PAM-strand. The PBS of the pegRNA then hybridizes with the 3′ end of the nicked DNA strand resulting in a double-stranded DNA-RNA heteroduplex, with the edit on one strand and the wildtype (WT) sequence on the opposite strand. The nick on the 3′ end of the target DNA serves as the initiation point of polymerization by MMLV-RT, with the RTT sequence used as the template (Figure 1a).18 Endogenous DNA mismatch repair is then capable of incorporating the edit into the opposing strand resulting in the final editing product.
Figure 1.
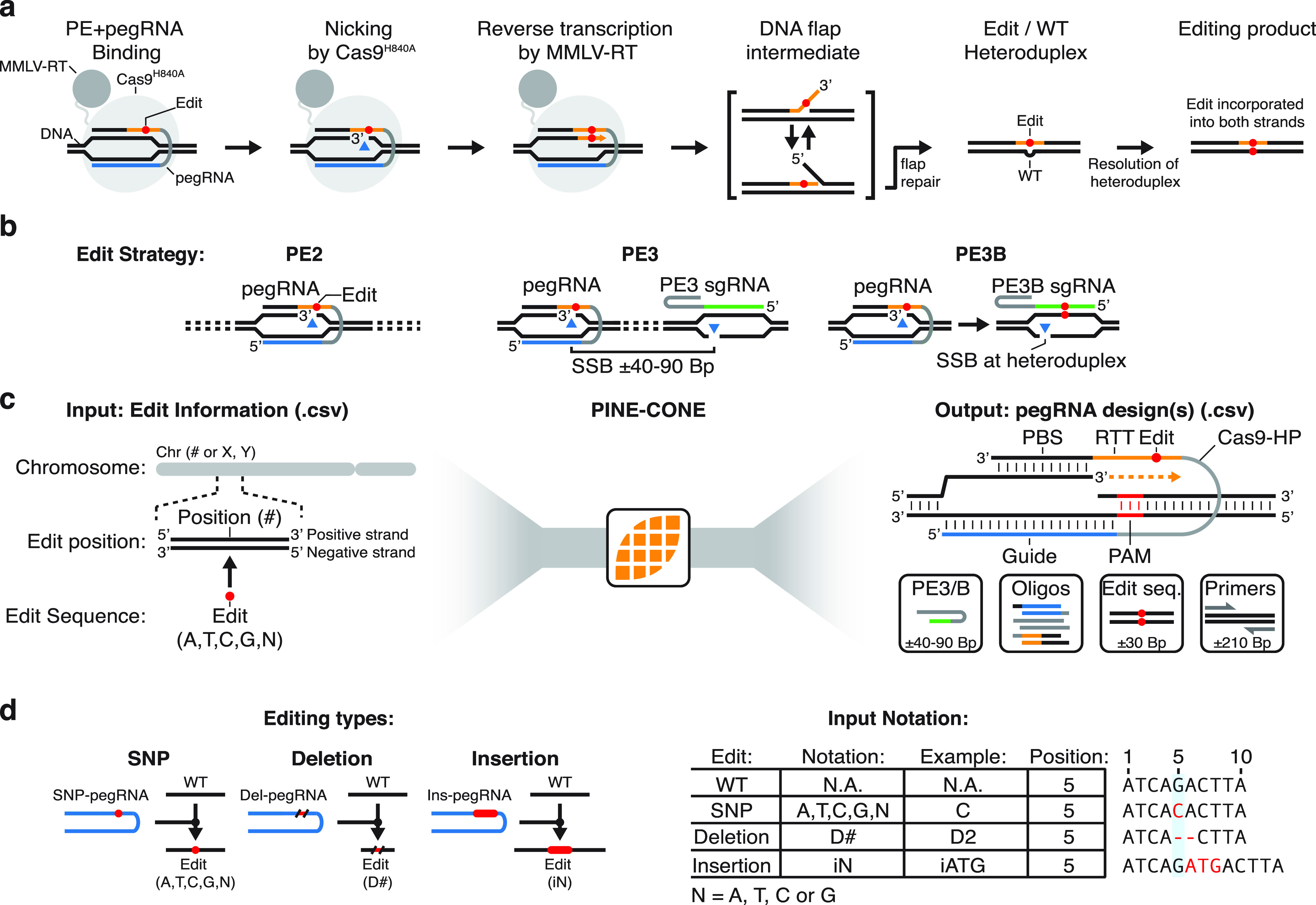
PINE-CONE automated design of Prime Editing Guide RNAs (pegRNAs). (a) Prime Editor (PE) utilizes a nicking Cas9 (Cas9H840A) fused to a Moloney Murine Leukemia Virus Reverse Transcriptase (MMLV-RT). The PE fusion is targeted to a specific locus via a prime editing Guide RNA (blue line), where the locus is subsequently nicked (blue triangle), exposing a 3′ OH. The MMLV-RT initiates reverse transcription from the free 3′ OH group using the pegRNA as the template for the edit (red circle). The flap intermediate and Edit/WT DNA heteroduplex is resolved via endogenous DNA repair. This results in the intended editing product incorporated into both DNA strands. (b) Prime editing strategies include PE2, PE3, and PE3B. PE2 utilizes a single pegRNA. PE3 utilizes a pegRNA matching the target locus and a separate sgRNA that targets upstream or downstream of the edit site. PE3b employs a sgRNA that is designed to nick the complement (WT) strand of the Edit/WT heteroduplex. (c) PINE-CONE takes edit information including chromosome, nucleotide position and intended editing product as input. pegRNAs are designed using DNA sequence data and include a guide directing Cas9 (blue), a Cas9-Hairpin (Cas9-HP, gray), reverse transcription template (RTT, orange), edit sequence (red circle), and primer binding sequence (PBS). The output file include PE3 or PE3B guides, oligonucleotides for cloning, intended edit DNA sequences, and PCR primers. (d) PINE-CONE is capable of designing multiple types of edits including single point mutations (e.g., SNPs), deletions, and insertions. Mutations are encoded in the input file in the format shown in the “input notation” panel.
Various PE-based strategies have been developed, including PE1, PE2, PE3, and PE3b18 (Figure 1b). Compared to PE1, PE2 utilizes an engineered MMLV-RT that significantly increases editing efficiency. PE3-based strategies utilize a pegRNA in combination with an accessory sgRNA targeting a SSB 40–90 base pairs (bp) upstream or downstream of the edit locus. Although PE3 results in higher targeting efficiencies it has been shown to result in increased indel formation. Finally, PE3b utilizes an accessory sgRNA that induces a nick on the complementary (WT) strand in the edit/WT heteroduplex. This favors mismatch repair to incorporate the edit into both strands of the target locus, which avoids transient DSBs and significantly reduces indel formation.
Although PE addresses many of the limitations of other CRISPR-based methods, the critical determinant in PE is the facile design of pegRNAs. Compared to the straightforward design of sgRNAs, pegRNA design requires proper placement of guide, PBS, RTT, and edit sequences. As such, the multifactorial design of pegRNAs results in higher complexity and limits manual design. To that end, we developed a freely accessible software tool, called Prime Induced Nucleotide Engineering Creator of New Edits (PINE-CONE) (https://github.com/xiaowanglab/PINE-CONE), that allows for the high-throughput design of pegRNAs. Overall, this tool will enable scientists from diverse fields to easily navigate their PE-based experiments by automating design of pegRNAs.
Results and Discussion
PINE-CONE is a software capable of turning basic edit information into pegRNA designs and accessory primers for PE workflows. The interface allows users to select from various organisms, such as human (hg38) and yeast (S288C) (Supplemental Table S1). PINE-CONE uses the organism selection to retrieve DNA sequence data from online reference genome’s web-based API (Supplementary Figure 1a). Because many laboratory strains and cell lines differ from their canonical reference genomes, PINE-CONE is also capable of running on locally stored sequence information (via the “Manual.txt” selection). Consequently, information obtained in the lab, such as by DNA sequencing, can be used to inform pegRNA design.
Edit input information is provided by a simple comma separated variable (.CSV) file (Supplemental Figure S1b). The Input file includes edit chromosome, coordinates, sequence and optional basic pegRNA parameters (RTT and PBS length). The input information is used by PINE-CONE to design pegRNAs encoding the intended edit along with PE3/B, cloning oligos intended edit sequences, and PCR primers (Figure 1c). PINE-CONE retrieves the wildtype (WT) target DNA sequences. In turn, edit information is used to design the intended editing products. Broadly speaking, PINE-CONE designs guides based off proximity to edit or by specificity. Specifically, to account for potential off-target effects, specificity scoring has been integrated into PINE-CONE’s pegRNA design. When designing pegRNAs against a reference genome, PINE-CONE retrieves “MIT Specificity scores”. In turn, PINE-CONE ranks these scores and uses the highest specificity guides available for pegRNA design. RTT sequence lengths are (i) defined by the user via the input file (“RT (Bp)”, Supplemental Figure S1b), or (ii) if the RTT input is blank, determined by PINE-CONE with a viable size (10–33 Bp). Similarly, PBS design are (i) defined by the user using a preferred PBS length, or (ii) if the PBS section is left blank, optimized by PINE-CONE using GC-content as the deterministic criteria as previous studies have shown that high-GC contents favor short PBSs while low GC-content favors longer PBSs.19 Because pegRNAs often require design of multiple guides, RTT and PBS lengths for experimental optimization,18 for most target loci PINE-CONE designs at least 2 pegRNAs. In addition, the user can enter multiple rows with systematic changes to RTT and/or PBS lengths to the same edit generating multiple pegRNA variants. Critically, PINE-CONE is capable of designing pegRNAs for a range of edits from single nucleotide edits, such as single nucleotide polymorphisms (SNPs/replacements), or deletions (Del, D) and insertions (Ins, i) (Figure 1d). Finally, PINE-CONE can design pegRNAs and accessory sgRNAs for various PE-based strategies including PE2, PE3, and PE3B18 (Figure 1b).
PINE-CONE’s outputs design results in a “.CSV” format and encodes edit information, pegRNA PE3 or 3B sgRNAs, cloning oligonucleotides and PCR primers. Edit information is encoded in “WT-to-Edit” format along with WT and Edit DNA sequences. PE3 or 3B sgRNA protospacer and target cleavage distance(s) are provided. The output file also includes oligonucleotides necessary for pegRNA and sgRNA cloning workflows (Supplemental Figure S2, Supplemental Table S2). Cloning of peg and sgRNAs uses straightforward restriction enzyme cloning and is compatible with an available CRISPR RNA expression vector. Since PCR and sequencing are often necessary in genome editing workflows, PINE-CONE also designs PCR and sequencing primers flanking the edit locus. Importantly, PINE-CONE designs primers with annealing temperatures that correlate with a commercially available TM calculator (Supplemental Figure S3). PINE-CONE is capable of plotting valid pegRNA loci in the form of a Circos plot for Human (hg38) and yeast (S288C) reference genomes.
For our initial validation of PINE-CONE’s functionality, we used PINE-CONE to design pegRNAs for targets in which pegRNAs had been previously experimentally validated by Anzalone et al., Kim et al., and Schene et al.(18−20) These targets included a broad spectrum of single nucleotide substitutions as well as small deletions. Overall, this analysis revealed that PINE-CONE generated pegRNA sequences with matching guides, PBS, and RTT sequences to previously published designs (Supplemental Figure S4).
Next, we employed PINE-CONE for the design of de novo pegRNA constructs that would be useful for disease modeling applications (Figure 2a). To test PINE-CONE’s ability to improve design automation, we assessed its ability to generate pegRNAs to target 24 diverse single nucleotide polymorphisms (SNPs) that have been previously been identified to be associated with increased risk of Alzheimer’s Disease (AD) (Figure 2b).21,22 Initially, we systematically assessed the effect of RTT length on pegRNA targeting by analyzing a cumulative 625 pegRNA designs at these 24 loci. We found longer RTT lengths expanded the editing window of PEs with 30 bp RTT sequences targeting up to 87% of loci (21/24). Concurrently, we analyzed the prevalence of valid PE3 and PE3B accessory sgRNA targets. PINE-CONE successfully designed PE3 sgRNAs for 79% of loci (19/24) and PE3B sgRNAs for 58% of loci (14/24) (Figure 2c). Circos style plots generated by PINE-CONE indicate valid pegRNA loci across numerous chromosomal contexts (Figure 2d).23 Finally, we analyzed the type of base conversions within our in silico experiment and found pegRNAs target a series of transition mutations accomplishable by BEs and PEs (38%). However, the majority (62%) of mutations consist of base transversions accomplishable solely through use of PEs (Figure 2e). This highlights the expanded editing scope of PEs and the ability of PINE-CONE to allow for pegRNA design automation.
Figure 2.
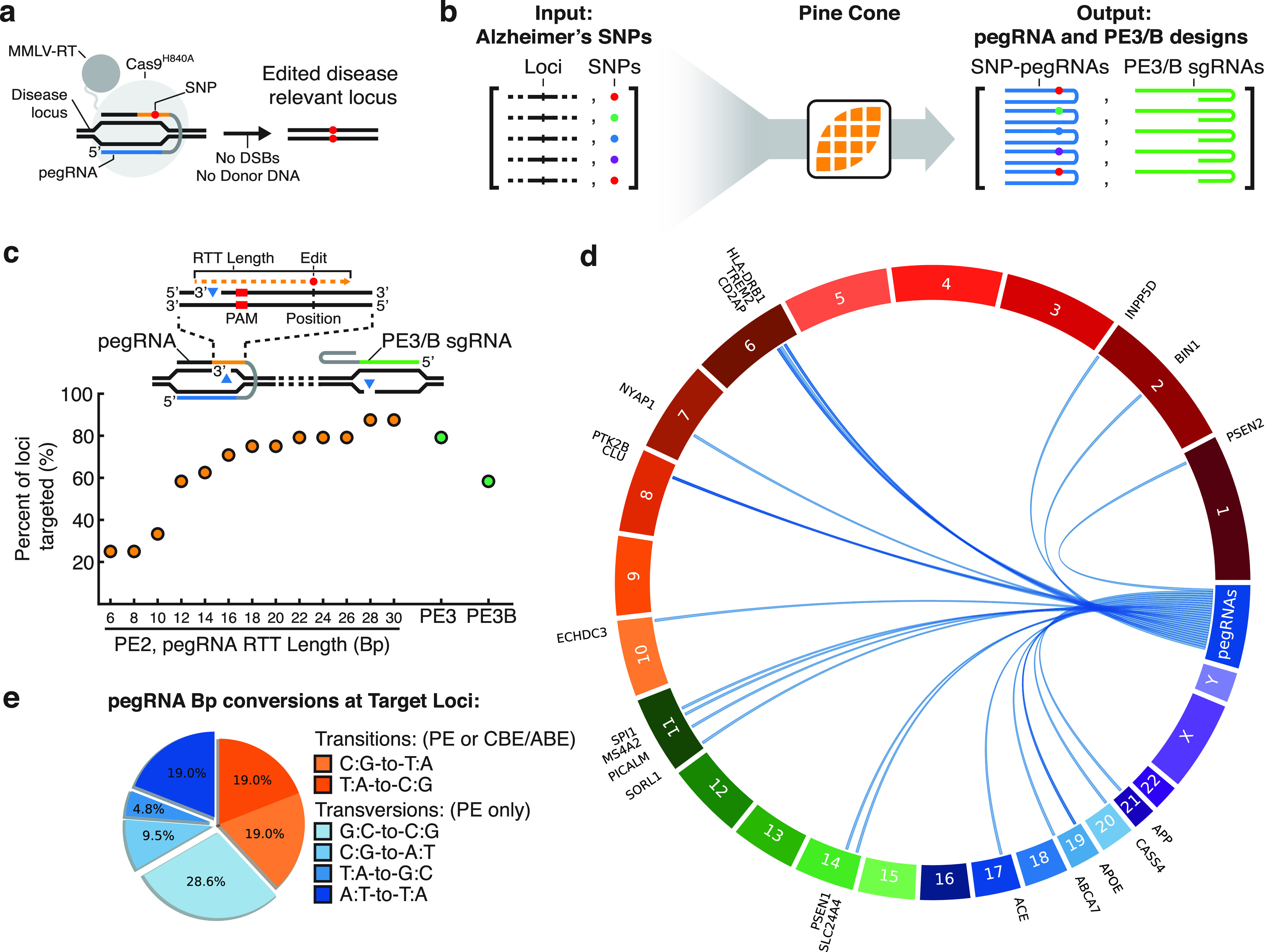
PINE-CONE design of pegRNAs of Alzheimer’s disease (AD)-related single nucleotide polymorphisms (SNPs). (a) Prime Editing mediated introduction of SNPs. A pegRNA targeting a disease locus encodes an edit, which is then incorporated into the target locus without the need for double-stranded DNA breaks (DSBs) or introduction of linearized donor DNA. (b) PINE-CONE rapidly analyzed and designed a library of pegRNAs and PE3 or PE3B sgRNAs for 24 AD-related loci. (c) The percent of loci targeted by pegRNAs was systematically analyzed for various RTT lengths. Inset schematic indicates valid edits that fall with the reverse transcription range. Longer RTT lengths expand the Prime Editing window and thus increase the number of targets up to 87% of loci with RTTs of 30 Bp (21/24). In parallel, PINE-CONE generated designs of PE3 sgRNAs for 79% of loci (19/24) and PE3B sgRNAs for 58% of loci (14/24). (d) PINE-CONE generated Circos mapping of pegRNAs to target loci indicates PINE-CONE successfully designs pegRNAs across numerous chromosome contexts. (e) A pie chart of PINE-CONE designed edits at the 24 AD-relevant loci. Transition mutations are accomplishable by cytosine base editors (CBEs), adenosine base editors (ABEs) and Prime Editors (PEs) (38%, orange). The majority of mutations consist of base transversion mutations (62%, blue).
Finally, to validate design of pegRNAs in an alternative organism in silico, we tested PINE-CONE on Saccharomyces cerevisiae S288C, an important host for biotechnology and synthetic biology. We utilized PINE-CONE to rapidly design pegRNAs for a series of loci including a series of auxotrophic marker genes. PINE-CONE was able to design pegRNAs that would induce a range of genetic modifications including introducing premature stop codons, short deletions resulting in frameshift knockouts, and insertions of LoxP site flanking target loci (Supplemental Figure S5). Collectively, this demonstrates PINE-CONE is capable of automating pegRNAs design in multiple organism contexts for a variety of applications.
Tools for rapid design and implementation of genome engineering techniques are important for their broad adoption. As such, multiple pegRNA design tools have recently become available. For instance, pegFinder designs pegRNAs viain silico alignment of WT and intended edit products.24 Multicrispr is a R package for a wide range of CRISPR-based strategies including pegRNA designs.25 PrimeDesign is capable of designing pegRNAs for genome-wide and saturation mutagenesis.26 These tools are effective and each offers unique functionality; however, they require prior generation of the intended editing product with flanking DNA sequences. Consequently, this may reduce throughput and increase the likelihood of user imparted errors. We sought to develop a tool that enables direct integration of nucleotide coordinates and straightforward editing nomenclature. To that end, PINE-CONE automates pegRNA design for multiple species, offers flexible RTT and PBS specification, and requires only numerical DNA positional information and simple editing notation.
In summary, PINE-CONE is capable of designing a range of edits and systematically analyzing pegRNA designs. Specifically, we demonstrated design of a series of pegRNA libraries in multiple contexts for both disease study and synthetic biology. Altogether, PINE-CONE increases ease of pegRNA design and significantly accelerates PE-based workflows.
Methods
PINE-CONE was written in Python with the user interface (UI) constructed using Tkinter. PINE-CONE source code, executables, and example files are provided for download at the Xiao Lab GitHub (https://github.com/xiaowanglab/PINE-CONE). A callable python script version for integration into genome-wide design pipelines is also available.
DNA sequences of each reference genome are accessed via API hosted by UCSC genome browser (Supplemental Table S1). For API based retrieval of genomic DNA sequence, PINE-CONE limits searches to 1 search per 0.5 s to avoid high frequency requests. After DNA retrieval, PINE-CONE conducts a bidirectional PAM search based off of user preferred RTT length. If RTT length is undefined, PINE-CONE will identify a viable RTT length given the availability of PAMs. Guide sequences are defined from available PAM motifs and will retrieve MIT specificity scores from UCSC browser web api if selected (via the “high specificity” preference). PINE-CONE utilizes the highest specificity guides available. The PBS is either (i) of a user defined length or (ii) PINE-CONE will design the PBS based off of GC-content. Guide, Cas9 hairpin, RTT, and PBS are combined to create pegRNA sequences. PINE-CONE attempts to design at least 2 guides per target locus.
PINE-CONE’s ability to design pegRNAs was first tested by comparing sequence output of loci tested in Anzalone et al., Kim et al., and Schene et al.(18−20) Coordinates edited were determined by Basic Local Alignment Tool (BLAT) analysis of edit locations. The subsequent coordinates and edit nucleotides were provided to PINE-CONE in an input .csv matching the format (Supplemental Figure S1). Output pegRNA sequences were aligned back to experimentally validated pegRNAs assuming use of the same scaffold sequence. This input file is available for download at the Xiao Lab GitHub.
For design of pegRNAs to target AD-related loci, PINE-CONE curated list of alleles identified by genome wide association study (ref (21)) and via physiological importance (ref (22)). For systematic analysis of RTT length. Loci were downloaded from UCSC Genome Browser hg38 and were analyzed locally by PINE-CONE. The list of valid RTT lengths was then used to query pegRNA designs against the human genome (hg38).
To test in silico pegRNA design on Saccharomyces cerevisiae (S288C), the coordinates for marker loci were downloaded from SGD and used to either introduce stop codons at points in the ORF, deletions 4–5 Bp in length or via insertion loxP sites by 2 pegRNAs flanking the coding sequence.
Acknowledgments
We thank the members of the X.W. and D.A.B. laboratories for constructive comments in the preparation of this manuscript. X.W. is supported by NIH grant (R01-GM106081, GM131405). D.A.B. is supported by (R21-AG056706 and R01-GM121698).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.0c00445.
Supplementary Figures S1–S5; Supplementary Tables S1 and S2 (PDF)
Author Contributions
K.S.B. designed and authored code for PINE-CONE. S.J.T. and K.S.B. designed in silico disease allele experiments. K.S.B., S.J.T., D.A.B., and X.W. analyzed experiments and edited the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Bartley B. A.; Choi K.; Samineni M.; Zundel Z.; Nguyen T.; Myers C. J.; Sauro H. M. (2019) PySBOL: A Python Package for Genetic Design Automation and Standardization. ACS Synth. Biol. 8 (7), 1515–1518. 10.1021/acssynbio.8b00336. [DOI] [PubMed] [Google Scholar]
- Nielsen A. A. K.; Der B. S.; Shin J.; Vaidyanathan P.; Paralanov V.; Strychalski E. A.; Ross D.; Densmore D.; Voigt C. A. (2016) Genetic Circuit Design Automation. Science 352 (6281), aac7341. 10.1126/science.aac7341. [DOI] [PubMed] [Google Scholar]
- HamediRad M.; Chao R.; Weisberg S.; Lian J.; Sinha S.; Zhao H. (2019) Towards a Fully Automated Algorithm Driven Platform for Biosystems Design. Nat. Commun. 10 (1), 5150. 10.1038/s41467-019-13189-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iverson S. V.; Haddock T. L.; Beal J.; Densmore D. M. (2016) CIDAR MoClo: Improved MoClo Assembly Standard and New E. Coli Part Library Enable Rapid Combinatorial Design for Synthetic and Traditional Biology. ACS Synth. Biol. 5 (1), 99–103. 10.1021/acssynbio.5b00124. [DOI] [PubMed] [Google Scholar]
- Stemmer M.; Thumberger T.; del Sol Keyer M.; Wittbrodt J.; Mateo J. L. (2015) CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS One 10 (4), e0124633 10.1371/journal.pone.0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standage-Beier K.; Wang X. (2017) Genome Reprogramming for Synthetic Biology. Front. Chem. Sci. Eng. 11 (1), 37–45. 10.1007/s11705-017-1618-2. [DOI] [Google Scholar]
- Fu Y.; Foden J. A.; Khayter C.; Maeder M. L.; Reyon D.; Joung J. K.; Sander J. D. (2013) High-Frequency off-Target Mutagenesis Induced by CRISPR-Cas Nucleases in Human Cells. Nat. Biotechnol. 31 (9), 822–826. 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihry R. J.; Worringer K. A.; Salick M. R.; Frias E.; Ho D.; Theriault K.; Kommineni S.; Chen J.; Sondey M.; Ye C.; Randhawa R.; Kulkarni T.; Yang Z.; McAllister G.; Russ C.; Reece-Hoyes J.; Forrester W.; Hoffman G. R.; Dolmetsch R.; Kaykas A. (2018) P53 Inhibits CRISPR–Cas9 Engineering in Human Pluripotent Stem Cells. Nat. Med. 24 (7), 939–946. 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- Kosicki M.; Tomberg K.; Bradley A. (2018) Repair of Double-Strand Breaks Induced by CRISPR–Cas9 Leads to Large Deletions and Complex Rearrangements. Nat. Biotechnol. 36 (8), 765–771. 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standage-Beier K.; Brookhouser N.; Balachandran P.; Zhang Q.; Brafman D. A.; Wang X. (2019) RNA-Guided Recombinase-Cas9 Fusion Targets Genomic DNA Deletion and Integration. CRISPR J. 2 (4), 209–222. 10.1089/crispr.2019.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikind B.; Bessen J. L.; Thompson D. B.; Hu J. H.; Liu D. R. (2016) A Programmable Cas9-Serine Recombinase Fusion Protein That Operates on DNA Sequences in Mammalian Cells. Nucleic Acids Res. 44 (20), 9758–9770. 10.1093/nar/gkw707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. P.; Wang H. H. (2019) An Engineered Cas-Transposon System for Programmable and Site-Directed DNA Transpositions. CRISPR J. 2 (6), 376–394. 10.1089/crispr.2019.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees H. A.; Liu D. R. (2018) Base Editing: Precision Chemistry on the Genome and Transcriptome of Living Cells. Nat. Rev. Genet. 19 (12), 770–788. 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor A. C.; Kim Y. B.; Packer M. S.; Zuris J. A.; Liu D. R. (2016) Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 533 (7603), 420–424. 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli N. M.; Komor A. C.; Rees H. A.; Packer M. S.; Badran A. H.; Bryson D. I.; Liu D. R. (2017) Programmable Base Editing of A•T to G•C in Genomic DNA without DNA Cleavage. Nature 551 (7681), 464–471. 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone A. V.; Koblan L. W.; Liu D. R. (2020) Genome Editing with CRISPR–Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat. Biotechnol. 38 (7), 824–844. 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- Sharon E.; Chen S.-A. A.; Khosla N. M.; Smith J. D.; Pritchard J. K.; Fraser H. B. (2018) Functional Genetic Variants Revealed by Massively Parallel Precise Genome Editing. Cell 175 (2), 544–557. 10.1016/j.cell.2018.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone A. V.; Randolph P. B.; Davis J. R.; Sousa A. A.; Koblan L. W.; Levy J. M.; Chen P. J.; Wilson C.; Newby G. A.; Raguram A.; Liu D. R. (2019) Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 576 (7785), 149–157. 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. K.; Yu G.; Park J.; Min S.; Lee S.; Yoon S.; Kim H. H. (2020) Predicting the Efficiency of Prime Editing Guide RNAs in Human Cells. Nat. Biotechnol. 1–9. 10.1038/s41587-020-0677-y. [DOI] [PubMed] [Google Scholar]
- Schene I. F.; Joore I. P.; Oka R.; Mokry M.; van Vugt A. H. M.; van Boxtel R.; van der Doef H. P. J.; van der Laan L. J. W.; Verstegen M. M. A.; van Hasselt P. M.; Nieuwenhuis E. E. S.; Fuchs S. A. (2020) Prime Editing for Functional Repair in Patient-Derived Disease Models. Nat. Commun. 11 (1), 5352. 10.1038/s41467-020-19136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle B. W.; Grenier-Boley B.; Sims R.; et al. (2019) Genetic Meta-Analysis of Diagnosed Alzheimer’s Disease Identifies New Risk Loci and Implicates Aβ, Tau, Immunity and Lipid Processing. Nat. Genet. 51 (3), 414–430. 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman S.; Brookhouser N.; Brafman D. A. (2020) Using Human Induced Pluripotent Stem Cells (HiPSCs) to Investigate the Mechanisms by Which Apolipoprotein E (APOE) Contributes to Alzheimer’s Disease (AD) Risk. Neurobiol. Dis. 138, 104788. 10.1016/j.nbd.2020.104788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M. I.; Schein J. E.; Birol I.; Connors J.; Gascoyne R.; Horsman D.; Jones S. J.; Marra M. A. (2009) Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 19, 1639. 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow R. D.; Chen J. S.; Shen J.; Chen S. (2020) A Web Tool for the Design of Prime-Editing Guide RNAs. Nat. Biomed. Eng. 1–5. 10.1038/s41551-020-00622-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagwat A. M.; Graumann J.; Wiegandt R.; Bentsen M.; Welker J.; Kuenne C.; Preussner J.; Braun T.; Looso M. (2020) Multicrispr: GRNA Design for Prime Editing and Parallel Targeting of Thousands of Targets. Life Sci. Alliance 3 (11), e202000757. 10.26508/lsa.202000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J. Y.; Anzalone A. V.; Grünewald J.; Lam K. C.; Shen M. W.; Liu D. R.; Joung J. K.; Pinello L. (2020) PrimeDesign Software for Rapid and Simplified Design of Prime Editing Guide RNAs. bioRxiv 10.1101/2020.05.04.077750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.