

Abstract

Herein, we report the stereoselective and convergent synthesis of resolvin E4, a newly identified specialized pro-resolving mediator. This synthesis proves the absolute configuration and exact olefin geometry. Key elements of the successful strategy include a highly stereoselective MacMillan organocatalytic oxyamination, a Midland Alpine borane reduction, and the use of a 1,4-pentadiyne unit as a linchpin building block. The application of reaction telescoping in several of the synthetic transformations enabled the preparation of the resolvin E4 methyl ester in 10% yield over 10 steps (longest linear sequence). The physical property (UV–Vis and LC–MS/MS) data of synthetic resolvin E4 matched those obtained from biologically produced material.
Introduction
Inflammation is a consequence of the immune system responding to injurious stimuli and constitutes an essential, protective strategy with the aim of restoring cellular homeostasis. Recent efforts concerning the mechanisms involved in the resolution of acute inflammation have provided evidence for a new superfamily of endogenous lipid mediators named specialized pro-resolving mediators (SPMs).1 These oxygenated polyunsaturated fatty acids are biosynthesized in the presence of lipoxygenase and cyclooxygenase enzymes.2 SPMs are chemically labile molecules formed in nano- to picogram amounts in vivo(3) and exhibit anti-inflammatory and pro-resolving bioactions, often in the low nano- to picomolar range.2,3 Additionally, SPMs are important in the process of clearing bacterial infections and participate in host defense, organ protection, pain reduction and also play a role in tissue remodeling.3 The E-series resolvins, derived from eicosapentaenoic acid (EPA), were among the first SPMs to be reported (Figure 1).4
Figure 1.

Reported E-series resolvins biosynthesized from EPA.
RvE1 and RvE2 have been subjected to clinical trial development programs5 as well as drug discovery efforts with the aim of establishing new pro-resolution agonists.6 The active resolution processes governed by SPMs are considered a biomedical paradigm shift.7
In 2019, Serhan and co-workers reported a new SPM and named it resolvin E4 (RvE4) based on its potent physiologic actions.8 This SPM is produced by human macrophages and neutrophils during physiologic hypoxic conditions (1-5% O2). In contrast to the three earlier reported E-series resolvins, this SPM is formed after two consecutive lipoxygenation reactions (Scheme 1).8 Earlier, 18S-configured epimers of RvE1, RvE2, and RvE3 have been identified.9 In the first step of the biosynthesis of RvE4, 15S-HpEPE is formed by 15-LOX, while the second lipoxygenation step is catalyzed by 5-LOX. Reductions of the hydroperoxide intermediates 15S-HpEPE and 5S-HpEPE are facilitated by peroxidase activity (Scheme 1).
Scheme 1. Proposed Biosynthetic Pathway for RvE4.

The need for absolute configuration assignments and materials of high chemical purity for further biological investigations and targeted lipidomic analyses has spurred great interest in the synthesis of the E-series resolvins.9a,10,11 Herein, we report the total synthesis of RvE4 (1) together with results from LC–MS/MS matching experiments that established its structure as (5S,6E,8Z,11Z,13E,15S,17Z)-5,15-dihydroxyicosa-6,8,11,13,17-pentaenoic acid.
Results and Discussion
An overview of the retrosynthetic analysis applied to the tentatively assigned structure of RvE4 (1) is shown in Scheme 2. The observation of a central (Z,Z)-1,4-pentadiene structural motif contained within the C2-symmetric C4–C16-domain of the molecule resulted in the first two disconnections being based on the Sonogashira cross-coupling reaction12 followed by Z-selective hydrogenation. This analysis identified three key fragments 3, 5, and 1-trimethylsilyl-1,4-pentadiyne (4), the latter serving the role of a linchpin, to be convergently assembled in the synthesis.
Scheme 2. Overview of the Key Retrosynthetic Disconnections Made for RvE4 (1).

Fragment 3 was disconnected back to cis-4-heptenal (6) with an enantioselective, organocatalytic oxyamination13 as well as a Takai olefination14 planned as the two pivotal steps in the forward direction. The Carreira alkynylation was chosen as the key transformation for furnishing fragment 5, with the intent of later transforming the acetylene moiety into the corresponding E-vinyl halide functionality needed for the planned palladium cross-coupling chemistry.
The project commenced with the construction of ω-3 fragment 3, starting from commercially available and affordable cis-4-heptenal (6). To this end, different α-oxidation protocols were first examined based on literature protocols (Table 1).
Table 1. Different Protocols Examined for the Organocatalytic Oxyamination of cis-4-Heptenal (6).

In light of these results, we settled on an enantioselective, organocatalytic α-oxyamination using 10 mol % d-proline and nitrosobenzene in CHCl3 based on the procedure developed by the MacMillan group.13d A solvent switch to ethanol preceded the NaBH4-based reduction of the in situ masked aldehyde functionality, and then the comparatively weak O–N bond was cleaved using zinc and acetic acid. After this sequence, a chromatographic purification step was introduced. The overall yield obtained for the described synthetic sequence was 80%, and chiral HPLC analysis of the α-aminoxylated alcohol intermediate 21 before zinc reduction to 7 showed an enantiomeric excess of 98% (Supporting Information).
The next objective was the regioselective TBS-protection of the secondary alcohol present in the 1,2-diol system in 7, and this was achieved by first masking the primary alcohol as the corresponding bulky pivaloyl ester and then adding a catalytic amount of 4-dimethylaminopyridine (DMAP) together with an excess of TBS triflate to the reaction mixture, yielding bis-protected 8 in 81% after column chromatography. A DIBAL-H reduction then cleanly did away with the pivaloyl moiety, and the primary alcohol was obtained in a crude form after work up and removal of volatiles under high-vacuum. This material was directly subjected to a Dess–Martin oxidation15 to give the corresponding aldehyde. Passing the crude material through a short plug of silica gel to remove periodinane-related residues was found beneficial before the next reaction. Finally, the vinyl iodide portion in 3 was installed by anE-selective Takai olefination (>97:3, 1H NMR analysis) with a combined yield of 78% over three steps.
The synthetic sequence depicted in Scheme 3 is shorter than our previously reported preparation16 of 3 when the step count for one-pot reactions is taken into account. Furthermore, this approach comes with other benefits: for example, (i) the catalytic, highly enantioselective oxyamination replaces the rather expensive use of chiral pool starting materials of unreliable supply, (ii) the thoughtful use of reaction telescoping17 allows for the conduction of several transformations without the need to isolate, purify, and handle sensitive intermediates, and (iii) cryogenic conditions combined with an array of hazardous reagents and additives have been avoided.
Scheme 3. Organocatalytic Approach to the Construction of ω-3 Fragment 3.

Turning our attention to the preparation of α-fragment 5, the first step was the straight-forward esterification of lactone 9 in basic methanol and a subsequent copper-catalyzed Stahl aerobic oxidation18 of the resulting primary alcohol 10, affording 11 in good yield. The Carreira alkynylation19 between aldehyde 11 and 2-methylbut-3-yn-2-ol was studied next, and we found that a yield of 50% could be achieved if a solution of the aldehyde in toluene was added dropwise with the aid of a syringe pump, over a 24 h period, to two equivalents of the corresponding alkynylzinc species of said alkyne (Scheme 4). Slow addition is often needed for α-unbranched aliphatic aldehydes in order to minimize the competing aldol self-condensation pathway.20
Scheme 4. Initial Approach toward 5 Utilizing the Carreira Alkynylation.

Surprisingly, however, chiral HPLC analysis of the 2-naphthoate derivative of 12 revealed that the obtained enantiomeric excess was only 34% in this case (Supporting Information), which is significantly lower than what we have previously obtained for other structurally similar substrates in hitherto unpublished work. Hence, in light of this outcome, the alkynylation sequence was put to the side in favor of an alternative approach (Scheme 5).
Scheme 5. Application of the Midland (S)-Alpine Borane Reduction in the Preparation of Fragment 16.

Capitalizing on the β-silicon effect, an aliphatic Friedel–Crafts acylation between acid chloride 13 and bis(trimethylsilyl)acetylene in the presence of Lewis acidic AlCl3, gave ketone 14 in 72% yield.21 Gram-scale asymmetric reduction of the alkynyl ketone was achieved by the addition of the Midland (S)-Alpine borane reagent22 in tetrahydrofuran (THF) at 0 °C, followed by swift removal of the solvent to give essentially neat conditions, ultimately furnishing the desired propargylic alcohol 15 in 96% enantiomeric excess and 89% yield after workup and purification (Supporting Information). The secondary alcohol in 15 was then protected using TBS chloride and imidazole in dichloromethane, followed by a solvent switch to methanol and addition of K2CO3, effectively removing the TMS-group attached to alkyne 5 in 91% overall yield.
At this stage, it was necessary to convert the terminal acetylene into the corresponding E-vinyl iodide, and this was achieved by a two-step process: first, free radical hydrostannation was initiated using a catalytic amount of azobisisobutyronitrile (AIBN), with excess tributyltin hydride added to ensure complete equilibration to the desired geometrical isomer, and then, iododestannylation was performed, yielding 16 in 74% over two steps.
The first of the two planned Sonogashira cross-coupling reactions was performed using catalytic amounts of Pd(PPh3)2Cl2/CuI, which cleanly effected the union between vinyl iodide 16 and linchpin 4 in 98% yield. Given the inherent lability of the resulting diyne system in 17, especially to basic reaction conditions, protiodesilylation was performed in a mild manner by the employment of AgNO3 and KCN,23 affording the terminal alkyne 18 in 65% yield.
The same catalyst system was then used again for the final Sonogashira carbon–carbon bond-forming reaction between alkyne 18 and vinyl iodide 3, giving the complete carbon skeleton 19 in 78%. The two internal, conjugated triple bonds were reduced in 70% yield using the tried-and-tested Lindlar hydrogenation protocol which involves the utilization of a mixed solvent system consisting of EtOAc/pyridine/1-octene.24 The inclusion of pyridine helps to modulate and control the activity of the heterogeneous catalyst, and 1-octene serves as a sacrificial olefin, the presence of which aids in minimizing competing over-reduction as the reaction nears completion. Removal of the two TBS-groups in 20 was first attempted using tetra-n-butylammonium fluoride (TBAF) in THF; however, significant byproduct formation was observed, leading to a diminished yield and difficulties during the purification process. A different deprotection approach was thus sought and found. Subjecting 20 instead to a catalytic amount of acetic chloride in methanol25 afforded RvE4 methyl ester (2) in 66% yield (Scheme 6) and chemical purity >97% (Supporting Information). The NMR- (1H, 13C, and COSY), MS-, and UV-data were all in accordance with the structure of 2 (Supporting Information).
Scheme 6. Sonogashira Cross-Coupling Reactions and Z-Selective Hydrogenation to Complete the Synthesis of RvE4 Methyl Ester (2).

MRM LC–MS/MS Matching Experiments
Since SPMs are formed in the nano- to picogram range in vivo, direct NMR analyses for structural verification are not viable. In order to ascertain that our synthetically prepared material was identical to that of authentic RvE4 (1) produced in vitro, matching experiments were conducted. Due to the chemically sensitive nature of this and other SPMs,26 hydrolysis was performed just prior to the LC–MS/MS experiments, as earlier reported.8 In Figure 2, top panel, the targeted MRM chromatogram from biogenic RvE4 (1) is shown together with an MS/MS spectrum displaying the molecular ion at m/z 333 (M – H) as well as the accompanying daughter ions (m/z 315 (M – H – H2O), m/z 271 (M – H – H2O – CO2), m/z 253 (M – H – 2H2O – CO2), m/z 235, m/z 217 (235 – H2O), m/z 199 (217 – H2O), m/z 191 (235 – CO2), and m/z 173 (235 – H2O – CO2) and m/z 115). In the middle panel, the chromatographic behavior of synthetically produced RvE4 (1), with an identical observed retention time (12.9 min) to that of the authentic material, is shown. Next, the result from coinjection of the biologically produced material and synthetically produced RvE4 (1) appears in the bottom panel, resulting in both coelution as well as an overall matching MS/MS fragmentation fingerprint. Additionally, the UV–Vis spectrum was in agreement with the original isolation of RvE4 (1).8 Overall, these results confirm that the synthetic material matched the biogenic material.
Figure 2.

MRM chromatograms and MS/MS spectra obtained from the matching experiments.
Conclusions
A total synthesis providing multi-milligram quantities of the methyl ester 2 of the SPM RvE4 (1) has been reported in 10% yield over 10 steps (longest linear sequence). Several of the reactions were performed using telescoping techniques, establishing the basis for an efficient total synthesis. Moreover, the successful use of the organocatalytic MacMillian oxyamination reaction is presented. The application of stereoselective organocatalytic protocols offers many advantages in the total synthesis of natural products.27 The integrity of the synthetically prepared material was demonstrated through matching experiments with authentic material obtained from human macrophages and neutrophils during hypoxic conditions. These results showed that synthetic and biologically produced RvE4 (1) matched, thus establishing both the absolute configurations of the carbinol atoms as well as an overall alkene geometry. Collectively, this provided evidence for the complete stereochemical assignment as (5S,6E,8Z,11Z,13E,15S,17Z)-5,15-dihydroxyicosa-6,8,11,13,17-pentaenoic acid.
Experimental Section
General Information
Unless otherwise stated, all commercially available reagents and solvents were used in the form they were supplied without any further purification. The stated yields are based on the isolated material. All sensitive reactions were performed under an argon or nitrogen atmosphere using Schlenk techniques. Reaction flasks were covered with aluminum foil during sensitive reactions and storage to minimize exposure to light. Thin layer chromatography was performed on silica gel 60 F254 aluminum-backed plates fabricated by Merck. Flash column chromatography was performed on silica gel 60 (40–63 μm) produced by Merck. NMR spectra were recorded on a Bruker AVII400 or Bruker DPX300 spectrometer at 400 or 300 MHz, respectively for 1H NMR and at 101 or 75 MHz, respectively for 13C NMR. Coupling constants (J) are reported in hertz, and chemical shifts are reported in parts per million (δ) relative to the central residual protium solvent resonance in 1H NMR (CDCl3 = δ 7.26, DMSO-d6 = δ 2.50 and MeOD = δ 3.31) and the central carbon solvent resonance in 13C NMR (CDCl3 = δ 77.00 ppm, DMSO-d6 = δ 39.43 and MeOD = δ 49.00). Optical rotations were measured using a PerkinElmer 341 polarimeter. Mass spectra were recorded at 70 eV on a Micromass Prospec Q or Micromass QTOF 2 W spectrometer using ESI as the method of ionization. High-resolution mass spectra were recorded at 70 eV on a Micromass Prospec Q or Micromass QTOF 2W spectrometer using ESI as the method of ionization. HPLC-analyses were performed using an AD-H stationary phase (CHIRALPAK, 4.6 × 250 mm, particle size 5 μm, from Diacel Corporation) or a C18 stationary phase (Eclipse XDBC18, 4.6 × 250 mm, particle size 5 μm, from Agilent Technologies), applying the conditions stated. The UV–Vis spectrum was recorded using an Agilent Technologies Cary 8485 UV–Vis spectrophotometer using quartz cuvettes.
(+)-(S,Z)-Hept-4-ene-1,2-Diol (7)
Diol 7 was prepared according to the literature with minor adjustments.13d,13e Nitrosobenzene (536 mg, 5.00 mmol, 1.00 equiv) and d-proline (58.0 mg, 0.500 mmol, 10.0 mol %) were dissolved in CHCl3 (2.5 mL) and cooled to 0 °C. cis-4-Heptenal (6, 1.98 mL, 1.68 g, 15.0 mmol, 3.00 equiv) was added dropwise, and the reaction was stirred at 0 °C for 2 h. The reaction mixture was then added dropwise to a solution of NaBH4 (567 mg, 15.0 mmol, 3.00 equiv) in EtOH (30 mL) at 0 °C and stirred at this temperature for an additional 2 h. The solvent was removed in vacuo and to the product was added sat. aq. NaHCO3 (10 mL) followed by extraction with EtOAc (3 × 10 mL). The combined organic phase was dried (Na2SO4) and concentrated in vacuo. The product was dissolved in EtOH/AcOH (3:1, 28.0 mL), and zinc powder (3.27 g, 50.0 mmol, 10.0 equiv) was added. The reaction mixture was stirred at room temperature overnight, filtrated through Celite, and concentrated in vacuo. The material thus obtained was purified by flash column chromatography (SiO2, gradient elution, 50–70% EtOAc in hexane) to give the desired diol 7 (522 mg, 4.01 mmol, 80%) as a clear oil. Rf (50% EtOAc in hexane, visualized by KMnO4-stain) = 0.32; [α]D25 = +9.5 (c 1.0, CHCl3) [Lit.28 [α]D = +9.0 (c 1.0, CHCl3)]; 1H NMR (400 MHz, MeOD): δ 5.55–5.35 (m, 2H), 3.67–3.55 (m, 1H), 3.50 (dd, J = 11.1, 4.3 Hz, 1H), 3.42 (dd, J = 11.1, 6.5 Hz, 1H), 2.35–2.23 (m, 1H), 2.22–2.12 (m, 1H), 2.08 (p, J = 7.4 Hz, 2H), 0.97 (t, J = 7.5 Hz, 3H); 13C{1H} NMR (101 MHz, MeOD): δ 134.6, 125.8, 73.4, 66.8, 32.3, 21.6, 14.6; HRESIMS m/z: 153.0885 [M + Na]+ (calcd for C7H14O2Na, 153.0886).
A
small amount of the α-aminoxylated alcohol intermediate was
kept for HPLC analysis. The enantiomeric excess (98%) was determined
by HPLC analysis using a chiral column (AD-H, i-PrOH/hexane,
5:95, 1.0 mL/min): tr(major) = 19.54 min, tr(minor) = 26.13 min.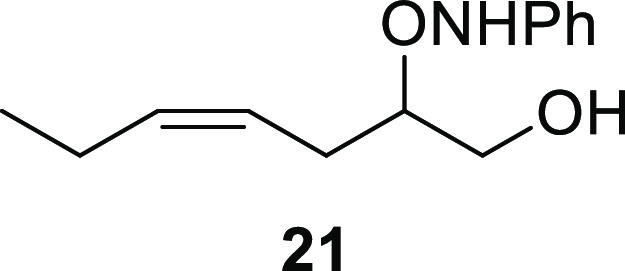
(Z)-2-((Phenylamino)oxy)hept-4-en-1-ol (21)
Nitrosobenzene (96.0 mg, 0.884 mmol, 1.00 equiv) and dl-proline (10.4 mg, 88.4 μmol, 10.0 mol %) were dissolved in CHCl3 (0.45 mL) and cooled to 0 °C. cis-4-Heptenal (6, 0.354 mL, 300 mg, 2.65 mmol, 3.00 equiv) was added dropwise, and the reaction was stirred at 0 °C for 2 h. The reaction mixture was added dropwise to a solution of NaBH4 (102 mg, 2.65 mmol, 3.00 equiv) in EtOH (5.4 mL) at 0 °C and stirred at the same temperature for an additional 2 h. The solvent was removed in vacuo, and to the crude product was added sat. aq. NaHCO3 (1.8 mL) followed by extraction with EtOAc (3 × 2 mL) and the combined organic phase was dried (Na2SO4), filtrated and concentrated in vacuo. The crude product thus obtained was filtrated through a short plug of silica gel (50% EtOAc in hexane) to yield the desired α-aminoxylated alcohol intermediate 21 (176 mg, 0.796 mmol, 90%) as a yellow oil. Rf (20% EtOAc in hexane, visualized by UV and KMnO4 stain) = 0.28; 1H NMR (400 MHz, CDCl3): δ 7.38–7.26 (m, 3H), 7.05–7.01 (m, 2H), 5.63–5.51 (m, 1H), 5.48–5.38 (m, 1H), 4.10–3.96 (m, 1H), 3.88 (dd, J = 12.1, 2.8 Hz, 1H), 3.79 (dd, J = 12.1, 6.5 Hz, 1H), 2.50 (app dt, J = 13.7, 6.6 Hz, 1H), 2.37 (app dt, J = 14.7, 7.5 Hz, 1H), 2.10 (p, J = 7.3 Hz, 1H), 1.00 (t, J = 7.5 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 148.5, 134.7, 129.2 (2C), 123.8, 122.7, 115.0 (2C), 84.0, 64.9, 28.1, 20.8, 14.3; HRESIMS m/z: 244.1306 [M + Na]+ (calcd for C13H19NO2Na, 244.1308).
(S,Z)-2-((tert-Butyldimethylsilyl)oxy)hept-4-en-1-yl Pivalate (8)
Diol 7 (200 mg, 1.54
mmol, 1.00 equiv) was dissolved
in a 1:1 mixture of CH2Cl2/pyridine (4.6 mL)
and cooled to 0 °C. Then, trimethylacetyl chloride (0.227 mL,
1.85 mmol, 1.20 equiv) was added dropwise. The reaction mixture was
stirred at 0 °C until deemed complete by TLC (30% EtOAc in hexane,
∼2 h). TBSOTf (0.88 mL, 3.83 mmol, 2.50 equiv) was then added
dropwise followed by addition of one crystal of DMAP. Stirring was
continued at 0 °C until deemed complete by TLC (30% EtOAc in
hexane, ∼2 h). The reaction mixture was quenched with sat.
aq. NaHCO3 (10 mL), extracted with EtOAc (3 × 5 mL),
dried (Na2SO4), filtrated, and concentrated in vacuo. The crude product thus obtained was purified by
flash chromatography (SiO2, 2% EtOAc in hexane) to yield 8 (409 mg, 1.25 mmol, 81%) as a clear oil. Rf (3.5% EtOAc in hexane, visualized by KMnO4-stain) = 0.29; [α]D25 = +7.0 (c 0.1, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ
5.53–5.43 (m, 1H), 5.40–5.33 (m,1H), 3.99 (dd, J = 11.0, 5.2 Hz, 1H), 3.93 (dd, J = 11.0,
5.3 Hz, 1H), 3.87 (p, J = 5.6 Hz, 1H), 2.36–2.17
(m, 2H), 2.04 (p, J = 7.5 Hz, 2H), 1.21 (s, 9H),
0.96 (t, J = 7.5 Hz, 3H), 0.88 (s, 9H), 0.08–0.07
(2 × s, 6H); 13C{1H} NMR (101 MHz, CDCl3): δ 178.6, 134.2, 124.1, 70.3, 67.8, 38.9, 32.6, 27.4
(3C), 25.9 (3C), 20.8, 18.2, 14.3, −4.5, −4.5; HRESIMS m/z: 351.2325 [M + Na]+ (calcd
for C18H36O3SiNa, 351.2326).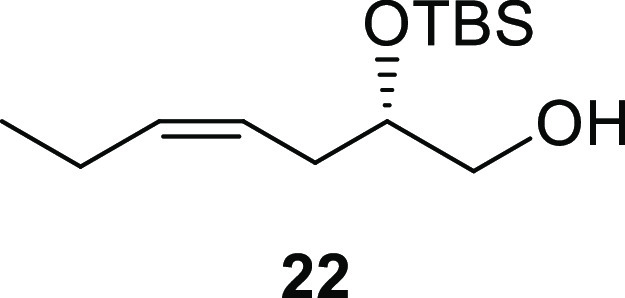
(S,Z)-2-((tert-Butyldimethylsilyl)oxy)hept-4-en-1-ol (22)
The pivalate 8 (674 mg, 2.05 mmol, 1.00 equiv) was dissolved in hexane (4.8 mL) and cooled to 0 °C. DIBAL-H (1.0 M in hexane, 5.13 mL, 5.13 mmol, 2.50 equiv) was added dropwise, and the reaction mixture was stirred until deemed complete by TLC (20% EtOAc in hexane, ∼2 h). MeOH (2.7 mL) was added to quench the reaction followed by addition of sat. aq. potassium sodium tartrate (27 mL). The aqueous phase was extracted with Et2O (3 × 7 mL). The combined organic phase was dried (Na2SO4), filtrated, concentrated in vacuo, and then kept under high vacuum for 2 h. The resulting alcohol intermediate was used without further purification in the next step. Rf (20% EtOAc in hexane, visualized by KMnO4-stain) = 0.48; [α]D25 = +19.8 (c 0.1, CH2Cl2); 1H NMR (400 MHz, CDCl3): δ 5.53–5.40 (m, 1H), 5.39–5.25 (m, 1H), 3.79–3.71 (m, 1H), 3.56 (dd, J = 11.0, 3.6 Hz, 1H), 3.44 (dd, J = 11.0, 5.5 Hz, 1H), 2.35–2.17 (m, 2H), 2.05 (p, J = 7.4 Hz, 2H), 1.86–1.73 (br s, 1H), 0.96 (t, J = 7.5 Hz, 3H), 0.91 (s, 9H), 0.10 (s, 3H), 0.09 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 134.3, 124.2, 72.9, 66.1, 32.1, 26.0 (3C), 20.8, 18.2, 14.3, −4.3, −4.5; HRESIMS m/z: 267.1751 [M + Na]+ (calcd for C13H28O2SiNa, 267.1751).
tert-Butyl(((S,1E,5Z)-1-iodoocta-1,5-dien-3-yl)oxy)dimethylsilane (3)
Alcohol 22 was dissolved in CH2Cl2 (60 mL) and cooled to 0 °C. The Dess–Martin periodinane reagent (1.04 g, 2.45 mmol, 1.20 equiv) was then added in one portion. The reaction mixture was removed from the cooling bath, and stirring was continued for 4 h. The reaction was quenched by addition of sat. aq. Na2S2O3 (13 mL), and the phases were separated. The aqueous phase was extracted with CH2Cl2 (2 × 15 mL). The combined organic phase was dried (Na2SO4), filtrated, and concentrated in vacuo. The crude product thus obtained was filtrated through a short plug of silica gel (10% EtOAc in hexane, Rf (10% EtOAc in hexane) = 0.52) to remove the leftover and spent DMP-reagent and then concentrated in vacuo.
A 50 mL flask was charged with CrCl2 (2.02 g, 16.4 mmol, 8.00 equiv), and the salt was dried under high-vacuum using a heat gun, cooled to ambient temperature, and covered with aluminum foil. Dry THF (2.8 mL) and dry dioxane (16 mL) were added, and the resulting suspension was cooled to 0 °C before CHI3 (2.03 g, 5.13 mmol, 2.50 equiv) was added. The reaction mixture was stirred at room temperature for 2 h, at which point the mixture turned from dark-green to red-brown. The reaction mixture was cooled to 0 °C, and the aldehyde dissolved in dry dioxane (2.0 mL) was added in a dropwise manner, and the reaction mixture was stirred for 2 h at room temperature. The reaction was quenched by the addition of sat. aq. NH4Cl (10 mL). The aqueous phase was extracted with Et2O (3 × 7 mL). The combined organic phase was washed successively with sat. aq. Na2S2O3 (∼2 mL) and brine, dried (Na2SO4), filtrated, and concentrated in vacuo. The crude product thus obtained was purified by flash chromatography (SiO2, gradient elution, 0–1% EtOAc in hexane) to yield vinyl iodide 3 (586 mg, 1.60 mmol, 78% from 8, E/Z = 97:3) as a pale yellow oil. Rf (1% EtOAc in hexane, visualized by UV and KMnO4 stain) = 0.50; [α]D20 = −4.1 (c 0.3, benzene); 1H NMR (400 MHz, CDCl3): δ 6.54 (dd, J = 14.3, 5.7 Hz, 1H), 6.21 (dd, J = 14.4, 1.3 Hz, 1H), 5.53–5.43 (m, 1H), 5.38–5.24 (m, 1H), 4.16–3.99 (m, 1H), 2.29–2.17 (m, 2H), 2.08–1.97 (m, 2H), 0.96 (t, J = 7.5 Hz, 3H), 0.89 (s, 9H), 0.05 (s, 3H), 0.04 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 148.9, 134.4, 123.8, 75.9, 75.2, 35.7, 26.0 (3C), 20.9, 18.4, 14.4, −4.5, −4.7; HRESIMS m/z: 389.0768 [M + Na]+ (calcd for C14H27IOSiNa, 389.0768).
Methyl 5-Hydroxypentanoate (10)
To a solution of δ-valerolactone (9, 5.0 g, 50 mmol, 1.0 equiv) in MeOH (50 mL) was added triethylamine (2.4 mL, 17 mmol, 34 mol %). The reaction was stirred for 18 h at room temperature. The reaction mixture was then filtrated through a short plug of silica gel, and the plug was washed with additional MeOH and concentrated in vacuo to yield the desired methyl ester 10 (6.6 g, 50 mmol, quant.) as a clear oil. The product was used as is in the next reaction. The spectroscopic data was in agreement with previously reported data.291H NMR (400 MHz, DMSO-d6): δ 4.38 (t, J = 5.2 Hz, 1H), 3.58 (s, 3H), 3.38 (app td, J = 6.4, 5.2 Hz, 2H), 2.30 (t, J = 7.4 Hz, 2H), 1.70–1.47 (m, 2H), 1.46–1.32 (m, 2H); 13C{1H} NMR (101 MHz, DMSO): δ 173.4, 60.2, 51.2, 33.1, 31.8, 21.2.
Methyl 5-Oxopentanoate (11)
Alcohol 10 (200 mg, 1.51 mmol, 1.00 equiv) was dissolved in MeCN (15 mL). [Cu(MeCN)4]OTf (28.3 mg, 75.1 μmol, 5.00 mol %) and commercial Stahl aerobic oxidation TEMPO solution (0.47 mL) were added, and the reaction was stirred open to air until complete as deemed by TLC. The reaction mixture was diluted with Et2O (15 mL) and filtrated through a short plug of silica gel. The plug was washed with additional Et2O. The solvent was removed in vacuo, and the crude product thus obtained was purified by flash column chromatography (SiO2, 16% EtOAc in hexane) to yield the desired aldehyde 11 (158 mg, 1.21 mmol, 80%) as a clear oil. The spectroscopic data was in agreement with previously reported data.30Rf (14% EtOAc in hexane, visualized by KMnO4-stain) = 0.15; 1H NMR (400 MHz, CDCl3): δ 9.77 (t, J = 1.3 Hz, 1H), 3.67 (s, 3H), 2.53 (td, J = 7.2, 1.3 Hz, 2H), 2.37 (t, J = 7.2 Hz, 2H), 1.95 (p, J = 7.2 Hz, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 201.6, 173.5, 51.8, 43.0, 33.1, 17.5.
Methyl (S)-5-((tert-Butyldimethylsilyl)oxy)-8-hydroxy-8-methylnon-6-ynoate (12)
Zn(OTf)2 (559 mg, 1.54 mmol, 2.00 equiv) was added to a flame-dried flask under argon and dried under high vacuum at 120 °C overnight. The flask was cooled and vented with argon, before (1R, 2S)-(−)-N-methylephedrine (289 mg, 1.61 mmol, 2.10 equiv) was added. The flask was purged with argon (∼15 min), and then, toluene (1.3 mL) and triethylamine (225 μL, 1.61 mmol, 2.10 equiv) were added. The reaction mixture was stirred for 2 h followed by the addition of 2-methyl-3-butyn-2-ol (149 μL, 1.54 mmol, 2.00 equiv). The content of the flask was stirred for 20 min before starting the addition of aldehyde 11 (100 mg, 0.768 mmol, 1.00 equiv), dissolved in toluene (1.8 mL) using a syringe pump over 24 h. The reaction was quenched by the addition of sat. aq. NH4Cl (∼5 mL) and extracted with ether (5 × 5 mL). The organic phase was dried (Na2SO4), filtrated, and concentrated in vacuo. The material thus obtained was purified by flash chromatography (SiO2, 50% EtOAc in hexane), yielding diol 12 (82.3 mg, 0.384 mmol, 50%) as a clear oil. Rf (50% EtOAc in hexane, visualized by KMnO4-stain) = 0.13; [α]D25 = −0.9 (c 1.0, MeOH); 1H NMR (400 MHz, MeOD): δ 4.33 (t, J = 6.4 Hz, 1H), 3.66 (s, 3H), 2.37 (t, J = 7.3 Hz, 2H), 1.81–1.72 (m, 2H) 1.71–1.61 (m, 2H), 1.46 (s, 6H); 13C{1H} NMR (101 MHz, MeOD): δ 175.7, 90.3, 83.6, 65.4, 62.4, 52.0, 49.4, 38.2, 34.4, 31.7, 21.9; HRESIMS m/z: 237.1098 [M + Na]+ (calcd for C11H18O4Na, 237.1097).
Methyl 5,8-Dihydroxy-8-methylnon-6-ynoate (rac-12)
2-Methyl-3-butyn-2-ol (74.5 μL,
0.786 mmol, 1.00
equiv) in THF (34 mL) was cooled to −78 °C. nBuLi (1.6 M in hexane, 0.983 mL, 1.57 mmol, 2.00 equiv) was added
dropwise over 15 min. The reaction mixture was stirred for 30 min
before aldehyde 11 (100 mg, 0.768 mmol, 1.00 equiv) in
THF (1.74 mL) was added dropwise. After 2 h at −78 °C,
the reaction was quenched by addition of sat. aq. NH4Cl
(7 mL) and extracted with EtOAc (3 × 5 mL). The combined organic
phase was dried (Na2SO4), filtrated, and concentrated in vacuo. The material thus obtained was purified by flash
chromatography (SiO2, 50% EtOAc in hexane), yielding diol rac-12 (62.0 mg, 0.289 mmol, 37%) as a clear oil. The obtained
experimental data matched that given for compound 12.
(S)-7-Methoxy-7-oxo-1-(trimethylsilyl)hept-1-yn-3-yl 2-Naphthoate (23)
Propargylic alcohol 12 (20.0 mg, 93.3 μmol, 1.00 equiv) was dissolved in CH2Cl2 (0.43 mL) and cooled to 0 °C. Triethylamine (39.0 μL, 0.280 mmol, 3.00 equiv) was added followed by DMAP (1.10 mg, 9.00 μmol, 10.0 mol %). Next, 2-naphthoyl chloride (21.4 mg, 0.112 mmol, 1.20 equiv) was added in one portion. The reaction mixture was allowed to warm up to room temperature and stirred overnight. The solvent was removed under a gentle stream of argon, and the material thus obtained was purified by flash column chromatography (SiO2, gradient elution, 10–30% EtOAc in hexane) to give the desired naphthalate 23 (31.5 mg, 85.6 μmol, 92%) as a white solid. The enantiomeric excess (34%) was determined by HPLC analysis using a chiral column (AD-H, i-PrOH/hexane, 15:85, 1.0 mL/min): tr(major) = 17.44 min, tr(minor) = 21.99 min. Rf (30% EtOAc in hexane, visualized by UV and KMnO4 stain) = 0.18; [α]D25 = +10.5 (c 0.6, CHCl3); 1H NMR (300 MHz, MeOD): δ 8.61 (s, 1H), 8.13–7.80 (m, 4H), 7.61 (p, J = 7.1 Hz, 2H), 5.72 (t, J = 5.8 Hz, 1H), 3.66 (s, 3H), 2.46 (t, J = 6.6 Hz, 2H), 1.93 (m, 4H), 1.47 (s, 6H); 13C{1H} NMR (101 MHz, MeOD): δ 175.4, 166.9, 137.1, 133.9, 132.2, 130.4, 129.7, 129.4, 128.9, 128.3, 128.0, 126.0, 92.2, 79.7, 65.6, 65.4, 52.1, 35.3, 34.1, 31.6, 21.7 (2C); HRESIMS m/z: 391.1516 [M + Na]+ (calcd for C22H24O5Na, 391.1516).
The exact same procedure was followed for the preparation of racemic naphthalate. Yield: 30.8 mg, 0.0837 mmol, 90%. The obtained experimental data matched that given for compound 23.
Methyl 5-Oxo-7-(trimethylsilyl)hept-6-ynoate (14)
A flame-dried flask under argon was charged with AlCl3 (10.5 g, 78.8 mmol, 1.30 equiv) and CH2Cl2 (75 mL) at 0 °C. A solution of bis(trimethylsilyl)acetylene (10.4 g, 60.8 mmol, 1.00 equiv) and methyl 4-(chloroformyl)butyrate (13, 10.0 g, 60.8 mmol, 1.00 equiv) in CH2Cl2 (75 mL) was then added in a dropwise manner over 15 min. The reaction mixture was stirred at 0 °C for 30 min, warmed up to room temperature over a period of 45 min, and then cooled back down to 0 °C. The reaction was quenched by the addition of 1 M HCl (80 mL) and stirred for 10 min. The resulting thick suspension was vacuum filtrated through a short plug of silica gel directly into a separatory funnel, and the plug was washed with additional fresh CH2Cl2 (50 mL). The phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 50 mL). The combined organic phase was dried (Na2SO4), filtrated, and concentrated in vacuo. The crude product thus obtained was purified by flash column chromatography (SiO2, 10% EtOAc in hexane) to yield the desired product 14 (9.90 g, 43.7 mmol 72%) as a yellow oil. The spectroscopic data was in agreement with previously reported data.31Rf (10% EtOAc in hexane, visualized by KMnO4-stain) = 0.27; 1H NMR (400 MHz, CDCl3): δ 3.68 (s, 3H), 2.65 (t, J = 7.2 Hz, 2H), 2.37 (t, J = 7.3 Hz, 2H), 1.97 (p, J = 7.3 Hz, 2H), 0.24 (s, 9H); 13C{1H} NMR (101 MHz, CDCl3): δ 186.8, 173.5, 101.9, 98.3, 51.8, 44.3, 32.9, 19.1, −0.6 (3C); HRESIMS m/z: 249.0917 [M + Na]+ (calcd for C11H18O3SiNa, 249.0917).
Methyl (S)-5-Hydroxy-7-(trimethylsilyl)hept-6-ynoate (15)
Ketone 14 (5.66 g, 25.0 mmol, 1.00 equiv) was azeotropically dried with 2-MeTHF (2 × 15 mL) and then placed under high vacuum for 30 min. The flask was vented with argon and cooled to −10 °C, and (S)-Alpine-borane solution (0.5 M in THF, 100 mL, 50.0 mmol, 2.00 equiv) was added over a period of 15 min. Most of the THF solvent was immediately removed under vacuum with efficient stirring while warming up to 0 °C. The resulting, highly viscous reaction mixture was then allowed to warm to room temperature and stirred overnight. Next, the reaction mixture was cooled to 0 °C, and acetaldehyde (1.40 mL, 1.10 g, 25.0 mmol, 1.00 equiv) was added in a dropwise manner. After 15 min, diethyl ether (100 mL) was added, followed by the dropwise addition of ethanolamine (3.00 mL, 3.00 g, 50.0 mmol, 2.00 equiv). The reaction mixture was stirred for 30 min at 0 °C, warmed to room temperature, and then stirred an additional hour. The white, solid 9-BBN-ethanolamine complex was removed by filtration, and the filtrate was washed with water (2 × 30 mL). The organic phase was dried (Na2SO4), filtrated, and concentrated in vacuo. The crude product thus obtained was purified by flash column chromatography (SiO2, gradient elution, 10–20% EtOAc in hexane) to give the desired product 15 (5.10 g, 22.3 mmol, 89%) as a clear oil. Rf (20% EtOAc in hexane, visualized by KMnO4-stain) = 0.21; [α]D20 = −1.0 (c 1.0, CHCl3) [Lit.32 [α]D = −0.7 (c 1.1, CHCl3)]; 1H NMR (400 MHz, CDCl3): δ 4.37 (t, J = 6.2 Hz, 1H), 3.67 (s, 3H), 2.38 (t, J = 7.1 Hz, 2H), 1.91 (s, 1H), 1.85–1.68 (m, 4H), 0.16 (s, 9H); 13C{1H} NMR (101 MHz, CDCl3): δ 174.0, 106.5, 89.8, 62.5, 51.7, 37.0, 33.7, 20.7, 0.0 (3C); HRESIMS m/z: 251.1073 [M + Na]+ (calcd for C11H20O3SiNa, 251.1074).
Methyl 5-Hydroxy-7-(trimethylsilyl)hept-6-ynoate (rac-15)
Ketone 14 (200 mg, 0.884
mmol, 1.00 equiv)
was azeotropically dried with 2-MeTHF (2 × 1 mL) and then placed
under high vacuum for 30 min. The flask was cooled to 0 °C, and
9-BBN-H (0.5 M in THF, 3.53 mL, 1.77 mmol, 2.00 equiv) was added,
and approximately half the solvent volume was removed under vacuum
at room temperature. The reaction mixture was stirred for 72 h before
acetaldehyde (0.05 mL, 0.884 mmol, 1.00 equiv) was added dropwise,
and the reaction mixture was stirred for an additional hour. The reaction
mixture was diluted with Et2O (5 mL), and ethanolamine
(53.0 μL, 0.884 mmol, 1.00 equiv) was added in a dropwise manner.
After 30 min, the reaction mixture was concentrated in vacuo to give a yellow oil together with some solid material. Water (5
mL) was added, and the aqueous phase was extracted with Et2O (3 × 3 mL). The organic phase was dried (Na2SO4), filtrated, and concentrated in vacuo.
The crude material thus obtained was purified by flash column chromatography
(SiO2, gradient elution, 10–20% EtOAc in hexane)
to give the desired racemic product rac-15 (109 mg, 0.477
mmol, 54%) as a clear oil. The obtained experimental data matched
that given for compound 15.
(S)-7-Methoxy-7-oxo-1-(trimethylsilyl)hept-1-yn-3-yl 2-Naphthoate (24)
Propargylic alcohol 15 (25.0 mg, 0.109 mmol, 1.00 equiv) was dissolved in CH2Cl2 (0.5 mL) and cooled to 0 °C. Triethylamine (46.0 μL, 0.327 mmol, 3.00 equiv) was added followed by DMAP (1.30 mg, 10.9 μmol, 10.0 mol %). Next, 2-naphthoyl chloride (25 mg, 0.131 mmol, 1.20 equiv) was added in one portion. The reaction mixture was allowed to warm up to room temperature and stirred overnight. The solvent was removed under a gentle stream of argon, and then, hexane (2 mL) and sat. aq. NaH2PO4 (2 mL) were added. After 5 min of vigorous stirring, the organic phase was separated and the aqueous phase was extracted with hexane (2 × 2 mL). The combined organic phase was dried (Na2SO4), filtrated, and concentrated in vacuo. The material thus obtained was purified by flash column chromatography (SiO2, gradient elution, 0–10% EtOAc in hexane) to give the desired naphthalate 24 (39.0 mg, 0.102 mmol, 93%) as a white solid. The enantiomeric excess (96%) was determined by HPLC analysis using a chiral column (AD-H, i-PrOH/hexane, 2:98, 1.0 mL/min): tr(major) = 13.63 min, tr(minor) = 12.54 min. Rf (10% EtOAc in hexane, visualized by UV and KMnO4 stain) = 0.22; [α]D25 = +6.2 (c 0.2, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.64 (s, 1H), 8.08 (dd, J = 8.6, 1.7 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.89 (dd, J = 8.4, 2.1 Hz, 2H), 7.64–7.52 (m, 2H), 5.72 (t, J = 6.2 Hz, 1H), 3.68 (s, 3H), 2.44 (t, J = 7.3 Hz, 2H), 2.07–1.96 (m, 2H), 1.95–1.87 (m, 2H), 0.18 (s, 9H); 13C{1H} NMR (101 MHz, CDCl3): δ 173.7, 165.7, 135.8, 132.6, 131.5, 129.6, 128.5, 128.3, 127.9, 127.3, 126.8, 125.5, 102.3, 91.2, 64.6, 51.8, 34.5, 33.6, 20.7, 0.0 (3C); HRESIMS m/z: 405.1492 [M + Na]+ (calcd for C22H26O4SiNa, 405.1493).
The exact same procedure was followed for the preparation of racemic naphthalate. Yield: 30.0 mg, 78.5 μmol, 72%. The obtained experimental data matched that given for compound 24.
Methyl (S)-5-((tert-Butyldimethylsilyl)oxy)hept-6-ynoate (5)
Propargylic alcohol 15 (3.90 g, 17.1 mmol, 1.00 equiv) was dissolved in CH2Cl2 (45 mL). Imidazole (2.33 g, 34.2 mmol, 2.00 equiv) and tert-butyldimethylsilyl chloride (3.86 g, 25.6 mmol, 1.50 equiv) were added in a successive manner at room temperature. The reaction mixture was stirred overnight and then the solvent was removed in vacuo. The material was dissolved in methanol (172 mL) and then cooled to 0 °C. Next, K2CO3 (4.74 g, 34.2 mmol, 2.00 equiv) was added in one portion, and the reaction mixture was allowed to warm to room temperature. The reaction was followed by TLC analysis (product is observed just below the starting material with 5% EtOAc in hexane as the eluent), and when deemed complete by TLC analysis (∼1 h), the flask was cooled back down to 0 °C. The reaction was quenched by the addition of phosphate buffer (132 mL, pH = 7), and the reaction mixture was stirred for 5 min. NaCl (∼10 g) was added, and the aqueous phase was extracted with hexane (5 × 50 mL). The combined organic phase was dried (Na2SO4), filtrated, and concentrated in vacuo. The crude material thus obtained was purified by flash column chromatography (SiO2, 5% EtOAc in hexane) to give the desired product 5 (4.21 g, 15.6 mmol, 91%) as a clear oil. The spectroscopic data was in agreement with previously reported data.33Rf (5% EtOAc in hexane, visualized by KMnO4-stain) = 0.24; [α]D20 = −36.2 (c 0.2, MeOH) [Lit.34 [α]D = −36 (c 0.2, MeOH)]; 1H NMR (400 MHz, CDCl3): δ 4.35 (td, J = 6.0, 2.1 Hz, 1H), 3.65 (s, 3H), 2.37 (d, J = 2.1 Hz, 1H), 2.34 (t, J = 7.2 Hz, 2H), 1.85–1.65 (m, 4H), 0.88 (s, 9H), 0.12 (s, 3H), 0.09 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 173.9, 85.3, 72.5, 62.4, 51.6, 37.9, 33.8, 25.9, 20.7, 18.3, −5.0, −4.5; HRESIMS m/z: 293.1543 [M + Na]+ (calcd for C14H26O3SiNa, 293.1543).
Methyl (S,E)-5-((tert-Butyldimethylsilyl)oxy)-7-iodohept-6-enoate (16)
Vinyl iodide 16 was prepared following the procedure by Sulikowski et al.35 Alkyne 5 (100 mg, 0.370 mmol, 1.00 equiv) was dissolved in benzene (7.5 mL), and then, nBu3SnH (0.30 mL, 1.11 mmol, 3.00 equiv) and AIBN (10.0 mg, 60.9 μmol, 16.5 mol %) were added. The reaction was heated to 80 °C (oil bath) for 2 h. The reaction mixture was then cooled to room temperature, and the solvent was removed in vacuo. The crude product thus obtained was purified by flash column chromatography (SiO2, 1% Et2O in hexane) to give the desired product as a clear oil which was used directly in the next reaction. Rf (5% Et2O in hexane, visualized with KMnO4 stain) = 0.35.
To a solution of the stannane intermediate (170 mg, 0.303 mmol, 1.00 equiv) in dry CH2Cl2 (1.2 mL) was added dropwise a solution of I2 (115 mg, 0.454 mmol, 1.50 equiv) in dry CH2Cl2 (1.7 mL) until the resulting solution maintained a slight pink color. The reaction was stirred for an additional 20 min, followed by the addition of sat. aq. Na2S2O3 (3 mL), H2O (2 mL), and sat. aq. NaHCO3 (3 mL). The mixture was stirred for an additional 5 min, the phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was dried (MgSO4), filtrated, and concentrated in vacuo. The crude product thus obtained was purified by flash column chromatography (SiO2, 5% Et2O in hexane) to obtain the vinyl iodide 16 (109 mg, 0.274 mmol, 74% over two steps) as a clear oil. The spectroscopic data was in agreement with previously reported data.35Rf (5% Et2O in hexane, visualized by UV and KMnO4 stain) = 0.23; [α]D20 = −25.8 (c 1.7, CHCl3) [Lit.35 [α]D = −25.8 (c 1.7, CHCl3)]; 1H NMR (400 MHz, CDCl3): δ 6.50 (dd, J = 14.4, 6.0 Hz, 1H), 6.22 (dd, J = 14.4, 1.3 Hz, 1H), 4.10 (app qd, J = 5.9, 1.3 Hz, 1H), 3.67 (s, 3H), 2.31 (t, J = 7.3 Hz, 2H), 1.86–1.56 (m, 2H), 1.56–1.46 (m, 2H), 0.89 (s, 9H), 0.04 (s, 3H), 0.03 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 174.0, 148.9, 76.2, 74.9, 52.2, 36.9, 34.0, 25.9 (3C), 20.4, 18.3, −4.4, −4.8; HRESIMS m/z: 421.0666 [M + Na]+ (calcd for C14H27IO3SiNa, 421.0666).
Methyl (S,E)-5-((tert-Butyldimethylsilyl)oxy)-12-(trimethylsilyl)dodeca-6-en-8,11-diynoate (17)
Vinyl iodide 16 (818 mg, 2.05 mmol, 1.00 equiv) was dissolved in THF (11 mL). The solution was cooled to 0 °C before Pd(PPh3)2Cl2 (72.1 mg, 0.103 mmol, 5.0 mol %), CuI (46.9 mg, 0.246 mmol, 12.0 mol %), and Et3N (416 mg, 0.57 mL, 4.11 mmol, 2.00 equiv) were added. The alkyne 4 (702 mg, 0.89 mL, 5.15 mmol, 2.50 equiv) was dissolved in THF (0.63 mL) and added dropwise. The reaction mixture was allowed to slowly warm to ambient temperature and stirred in the dark for an additional 16 h. After completion, the reaction mixture was filtrated through a short plug of silica gel (15% EtOAc in hexane) and concentrated in vacuo. The crude product thus obtained was purified by flash column chromatography (SiO2, 5% EtOAc in hexane) to obtain the coupled product 17 (820 mg, 2.02 mmol, 98%) as an orange/brown oil. Rf (5% EtOAc in hexane, visualized by UV and KMnO4 stain) = 0.25; [α]D25 = −0.7 (c 0.7, benzene); 1H NMR (400 MHz, CDCl3): δ 6.07 (dd, J = 15.9, 5.6 Hz, 1H), 5.65–6.60 (m, 1H), 4.19–4.15 (m, 1H), 3.66 (s, 3H), 3.33 (app d, J = 2.2 Hz, 2H), 2.29 (t, J = 7.4 Hz, 2H), 1.69–1.61 (m, 2H), 1.54–1.48 (m, 2H), 0.89 (s, 9H), 0.16 (s, 9H), 0.04 (s, 3H), 0.03 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 174.0, 146.0, 109.1, 99.8, 85.4, 83.5, 79.1, 72.3, 51.6, 37.3, 34.1, 26.0 (3C), 20.5, 18.3, 11.7, 0.1 (3C), −4.3, −4.7; HRESIMS m/z: 429.2251 [M + Na]+ (calcd for C22H38O3Si2Na, 429.2252).
Methyl (S,E)-5-((tert-Butyldimethylsilyl)oxy)dodeca-6-en-8,11-diynoate (18)
The TMS-protected acetylene 17 (66.0 mg, 0.162 mmol, 1.00 equiv) was dissolved in THF (2.55 mL) and EtOH (1.54 mL). A solution of AgNO3 (190 mg, 0.642 mmol, 3.90 equiv) in a mixture of EtOH and H2O (1:1, 1.80 mL) was added dropwise and stirred for 40 min. The reaction mixture went from dark yellow to black after the addition of the AgNO3 solution. KCN (73.8 mg, 1.13 mmol, 7.00 equiv) was dissolved in H2O (1.3 mL) and added dropwise at room temperature (precipitation was observed during this stage). The reaction was stirred for 2 h and quenched by the addition of H2O (20 mL) and diluted by EtOAc (30 mL). The phases were separated, and the aqueous phase was extracted with EtOAc (3 × 30 mL). The combined organic phase was dried (Na2SO4), filtrated, and concentrated in vacuo. The crude product thus obtained was purified by flash chromatography (SiO2, 5% EtOAc in hexane) to obtain product 18 (35.3 mg, 0.105 mmol, 65%) as a yellow oil. Rf (10% EtOAc in hexane, visualized by UV and KMnO4 stain) = 0.21; [α]D25 = −1.8 (c 0.6, benzene); 1H NMR (400 MHz, CDCl3): δ 6.08 (dd, J = 15.8, 5.4 Hz, 1H), 5.63 (dq, J = 15.9, 2.0 Hz, 1H), 4.17 (qd, J = 5.7, 1.6 Hz, 1H), 3.66 (s, 3H), 3.30 (t, J = 2.4 Hz, 2H), 2.30 (t, J = 7.4 Hz, 2H), 2.08 (t, J = 2.7 Hz, 1H), 1.69–1.60 (m, 2H), 1.53–1.47 (m, 2H), 0.90 (s, 9H), −0.04 (s, 3H), −0.02 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 174.1, 146.3, 108.8, 83.1, 79.3, 78.2, 72.2, 69.0, 51.6, 37.2, 34.1, 26.0 (3C), 20.5, 18.3, 10.4, −4.4, −4.8; HRESIMS m/z: 357.1855 [M + Na]+ (calcd for C19H30O3SiNa, 357.1856).
Methyl (5S,6E,13E,15S,17Z)-5,15-Bis((tert-butyldimethylsilyl)oxy)icosa-6,13,17-trien-8,11-diynoate (19)
Vinyl iodide 3 (51 mg, 0.14 mmol, 1.0 equiv) was dissolved in THF (1.0 mL), and the solution was cooled to 0 °C before Pd(PPh3)2Cl2 (5.0 mg, 7.1 μmol, 5.0 mol %), CuI (3.0 mg, 15 μmol, 11 mol %), and Et3N (52 mg, 40 μL, 0.28 mmol, 2.0 equiv) were added. Alkyne 18 (56 mg, 0.17 mmol, 1.2 equiv) was dissolved in THF (1.0 mL) and added dropwise. The reaction mixture was allowed to slowly warm up to rt and stirred in the dark for 16 h. After completion, the reaction mixture was filtrated through a plug of silica gel (15% EtOAc in hexane) and concentrated in vacuo. The crude product thus obtained was purified by flash chromatography (SiO2, gradient elution, 2.5–5% EtOAc in hexane) to obtain product 19 (62 mg, 0.11 mmol, 78%) as a clear oil. Rf (10% EtOAc in hexane, visualized by UV and KMnO4 stain) = 0.40; [α]D20 = +1.9 (c 0.30, benzene); 1H NMR (400 MHz, CDCl3): δ 6.12 (dd, J = 15.9, 5.2 Hz, 1H), 6.07 (dd, J = 15.9, 5.5 Hz, 1H), 5.69–5.61 (m, 2H), 5.50–5.42 (m, 1H), 5.35–5.27 (m, 1H), 4.19–4.13 (m, 2H), 3.66 (s, 3H), 3.42 (t, J = 2.2 Hz, 2H), 2.30 (t, J = 7.4 Hz, 2H), 2.27–2.18 (m, 2H), 2.02 (pd, J = 7.2, 1.5 Hz, 2H), 1.69–1.61 (m, 2H), 1.52–1.47 (m, 2H), 0.95 (t, J = 7.5 Hz, 3H), 0.89 (s, 18H), 0.05 (s, 3H), 0.04 (s, 3H), 0.03 (s, 3H), 0.02 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 174.1, 146.2, 146.1, 134.1, 124.2, 109.0, 108.6, 83.8, 83.6, 79.2, 79.0, 72.6, 72.2, 51.6, 37.2, 36.0, 34.1, 26.0 (6C), 20.9, 20.5, 18.4, 18.3, 14.3, 11.3, −4.4, −4.5, −4.7, −4.8; HRESIMS m/z: 595.3608 [M + Na]+ (calcd for C33H56O4Si2Na, 595.3609).
Methyl (5S,6E,8Z,11Z,13E,15S,17Z)-5,15-Bis((tert-butyldimethylsilyl)oxy)icosa-6,8,11,13,17-pentaenoate (20)
Diyne 19 (10 mg, 18 μmol, 1.0 equiv) was dissolved in a mixture of EtOAc/pyridine/1-octene (10:1:1, 0.3 mL) under argon. Lindlar’s catalyst (7.0 mg) was added, and the flask was evacuated and refilled with hydrogen gas twice. After 1 h, additional Lindlar’s catalyst (7.0 mg) was added. The reaction was stirred for another 4 h and filtrated through a short plug of silica gel (15% EtOAc in hexane) and concentrated in vacuo. The crude product thus obtained was purified by flash chromatography (SiO2, 1% EtOAc in hexane) to obtain product 20 (7.0 mg, 12 μmol, 70%) as a pale yellow oil. Rf (10% EtOAc in hexane, visualized by UV and KMnO4 stain) = 0.38; [α]D20 = +36.8 (c 0.25, benzene); 1H NMR (400 MHz, CDCl3): δ 6.49–6.40 (m, 2H), 5.99 (app td, J = 10.9, 2.0 Hz, 2H), 5.68 (dd, J = 15.2, 5.9 Hz, 1H), 5.64 (dd, J = 15.2, 6.2 Hz, 1H), 5.49–5.32 (m, 4H), 4.21–4.16 (m, 2H), 3.66 (s, 3H), 3.05 (app t, J = 7.4 Hz, 2H), 2.32 (t, J = 7.3 Hz), 2.28–2.22 (m, 2H), 2.03 (app p, J = 7.4 Hz, 2H), 1.72–1.64 (m, 2H), 1.53–1.49 (m, 2H), 0.95 (t, J = 7.5 Hz, 3H), 0.91 (s, 9H), 0.90 (s, 9H), 0.06 (2 × s, 6H), 0.04 (s, 3H), 0.03 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 174.2, 137.3, 137.2, 133.7, 129.3, 129.1, 128.7, 128.6, 124.8, 124.6, 124.3, 73.2, 72.9, 51.6, 37.8, 36.5, 34.2, 26.6, 26.4 (6C), 20.9 (2C), 18.4 (2C), 14.4, −4.1, −4.3, −4.6, −4.6; HRESIMS m/z: 599.3922 [M + Na]+ (calcd for C33H60O4Si2Na, 599.3922).
Methyl (5S,6E,8Z,11Z,13E,15S,17Z)-5,15-Dihydroxyicosa-6,8,11,13,17-pentaenoate (2)
The bis-TBS-protected intermediate 20 (10 mg, 17 μmol, 1.0 equiv) was azeotroped with 2-MeTHF (2 × 1 mL) and then cooled to 0 °C before a solution of AcCl in dry MeOH (0.13 mL, 2.6 mmol, 15 mol %) was added (the solution was prepared just prior to use by adding freshly distilled AcCl (3.0 μL) to dry MeOH (2.0 mL) under argon). The reaction mixture was stirred for 4 h at 0 °C. The reaction mixture was diluted with CH2Cl2 (0.3 mL) prior to neutralization with a 10% aq. solution of NaHCO3 (20 μL) and washed with H2O (0.2 mL). The organic phase was dried (Na2SO4), filtrated, and concentrated in vacuo. The crude product thus obtained was purified by flash chromatography (SiO2, 40% EtOAc in hexane) to afford the RvE4 methyl ester (2, 4.0 mg, 12 μmol, 66%) as a clear oil. The chemical purity (>97%) was determined by HPLC analysis (Eclipse XDB-C18, MeOH/H2O 76:24, 1.0 mL/min): tr(minor) = 7.88, 10.89 and 11.69 min, and tr(major) = 9.37 min. Rf (40% EtOAc in hexane, visualized by UV and KMnO4 stain) = 0.17; [α]D20 = +8.4 (c 0.5, MeOH); UV–Vis (MeOH) λmax 242 nm (log ε = 4.64); 1H NMR (400 MHz, MeOD): δ 6.61–6.54 (m, 2H), 6.01 (td, J = 11.0, 4.9 Hz, 2H), 5.69 (app ddd, J = 15.5, 9.2, 6.5 Hz, 2H) 5.51–5.34 (m, 4H), 4.13 (p, J = 6.8 Hz, 2H), 3.66 (s, 3H), 3.10 (tt, J = 7.5, 1.7 Hz, 2H), 2.36 (t, J = 7.2 Hz, 2H), 2.35–2.27 (m, 2H), 2.06 (app dp, J = 7.4, 0.8 Hz, 2H), 1.73–1.63 (m, 2H), 1.57–1.51 (m, 2H), 0.96 (t, J = 7.5 Hz, 3H); 13C{1H} NMR (101 MHz, MeOD): δ 175.8, 137.8, 137.5, 134.6, 130.3, 130.2, 129.6 (2C), 126.3 (2C), 125.5, 73.2, 72.8, 52.0, 37.6, 36.3, 34.6, 27.4, 22.1, 21.7, 14.5; HRESIMS m/z: 371.2192 [M + Na]+ (calcd for C21H32O4Na, 371.2193).
LC–MS/MS MRM Matching Experiments
Following a literature procedure,36 a solution of RvE4 methyl ester (2, 5.0 μg, 14 nmol) in MeOH was concentrated under a gentle stream of nitrogen gas, dissolved in THF (500 μL), and cooled to −78 °C. To the resulting solution was added 1 M LiOH (50 μL, 50 μmol) and distilled water (one drop, ∼20 μL) and the reaction mixture was stirred in a 4 °C cold room for 24 h. The reaction mixture was then concentrated under a gentle stream of nitrogen gas and reconstituted with MeOH (500 μL). The identity of the compound was verified by UV–Vis and LC–MS/MS. The chemical yield of the RvE4 free acid 1 was 69% (3.3 μg, 9.9 nmol) post saponification (based on UV–Vis) and was determined to be >95% pure by targeted MRM LC–MS/MS. The physical properties of synthetic RvE4 (1) and biogenic RvE4 (1) were analyzed on a QTRAP 5500 mass spectrometer (Sciex, Framingham, MA, USA) equipped with a LC20AD UFLC (Shimadzu, Tokyo, Japan) with a Poroshell EC-C18 column (100 mm × 4.6 mm × 2.7 μm; Agilent Technologies, Santa Clara, CA, USA) kept at 50 °C. RvE4 (1) was monitored by targeted multiple reaction monitoring (m/z 333 > 115) and enhanced product ion mode in negative polarity. RvE4 (1) was eluted at a flow rate of 0.5 mL/min with a gradient of LC–MS grade methanol/water/acetic acid from 50/50/0.01 v/v/v to 98/2/0.01 v/v/v. Data were acquired and analyzed with Analyst version 1.6.2 (Sciex).8
Acknowledgments
The scholarship to A.F.R. by the Department of Pharmacy is gratefully acknowledged. M.A. gratefully acknowledges the Research Council of Norway (RCN) for a grant (FRINATEK, 262901). This work was partly supported by the Research Council of Norway through the Norwegian NMR Package in 1994 and partly supported by the Research Council of Norway through the Norwegian NMR Platform, NNP (226244/F50). C.N.S. contributions are supported by NIH grant R01GM038765 (USA) and NIH Program Project P01GM095467 (USA).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c02913.
1H-, 13C NMR, and UV–Vis data of RvE4 methyl ester (2) and all synthetic intermediates (PDF)
Author Contributions
§ A.F.R., K.G.P., and M.A. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- a Serhan C. N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tabas I.; Glass C. K. Anti-Inflammatory Therapy in Chronic Disease: Challenges and Opportunities. Science 2013, 339, 166–172. 10.1126/science.1230720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan C. N.; Petasis N. A. Resolvins and Protectins in Inflammation-Resolution. Chem. Rev. 2011, 111, 5922–5943. and references cited therein 10.1021/cr100396c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan C. N.; Levy B. D. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J. Clin. Invest. 2018, 128, 2657–2669. 10.1172/jci97943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Serhan C. N.; Clish C. B.; Brannon J.; Colgan S. P.; Chiang N.; Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000, 192, 1197–1204. 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Serhan C. N.; Hong S.; Gronert K.; Colgan S. P.; Devchand P. R.; Mirick G.; Moussignac R.-L. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J.; Serhan C. N. Identification and structure elucidation of the pro-resolving mediators provides novel leads for resolution pharmacology. Br. J. Pharmacol. 2019, 176, 1024–1037. 10.1111/bph.14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shimamura K.; Matsuda M.; Miyamoto Y.; Yoshimoto R.; Seo T.; Tokita S. Identification of a stable chemerin analog with potent activity toward ChemR23. Peptides 2009, 30, 1529–1538. 10.1016/j.peptides.2009.05.030. [DOI] [PubMed] [Google Scholar]; b Arita M.; Oh S. F.; Chonan T.; Hong S.; Elangovan S.; Sun Y.-P.; Uddin J.; Petasis N. A.; Serhan C. N. Metabolic Inactivation of Resolvin E1 and Stabilization of Its Anti-inflammatory Actions. J. Biol. Chem. 2006, 281, 22847–22854. 10.1074/jbc.m603766200. [DOI] [PubMed] [Google Scholar]; c Hesselink J. M. K.; Chiosi F.; Costagliola C. Resolvins and aliamides: lipid autacoids in ophthalmology - what promise do they hold?. Drug Des., Dev. Ther. 2016, 10, 3133–3141. 10.2147/DDDT.S112389. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Murakami Y.; Fukuda H.; Muromoto R.; Hirashima K.; Ishimura K.; Fujiwara K.; Ishihara J.; Matsuda T.; Watanabe M.; Shuto S. Design and Synthesis of Benzene Congeners of Resolvin E2, a Proresolving Lipid Mediator, as Its Stable Equivalents. ACS Med. Chem. Lett. 2020, 11, 479–484. 10.1021/acsmedchemlett.9b00596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fullerton J. N.; Gilroy D. W. Resolution of inflammation: a new therapeutic frontier. Nat. Rev. Drug Discovery 2016, 15, 551–567. 10.1038/nrd.2016.39. [DOI] [PubMed] [Google Scholar]; b Dalli J. Does promoting resolution instead of inhibiting inflammation represent the new paradigm in treating infections?. Mol. Aspects Med. 2017, 58, 12–20. 10.1016/j.mam.2017.03.007. [DOI] [PubMed] [Google Scholar]
- Norris P. C.; Libreros S.; Serhan C. N. Resolution metabolomes activated by hypoxic environment. Sci. Adv. 2019, 5, eaax4895 10.1126/sciadv.aax4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Isobe Y.; Arita M.; Iwamoto R.; Urabe D.; Todoroki H.; Masuda K.; Inoue M.; Arai H. Stereochemical assignment and anti-inflammatory properties of the omega-3 lipid mediator resolvin E3. J. Biochem. 2013, 153, 355–360. 10.1093/jb/mvs151. [DOI] [PubMed] [Google Scholar]; b Oh S. F.; Pillai P. S.; Recchiuti A.; Yang R.; Serhan C. N. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Invest. 2011, 121, 569–581. 10.1172/JCI42545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recent total synthesis of E-series resolvins:; a Fukuda H.; Ikeda H.; Muromoto R.; Hirashima K.; Ishimura K.; Fujiwara K.; Aoki-Saito H.; Hisada T.; Watanabe M.; Ishihara J.; Matsuda T.; Shuto S. Synthesis of Resolvin E3, a Proresolving Lipid Mediator, and Its Deoxy Derivatives: Identification of 18-Deoxy-resolvin E3 as a Potent Anti-Inflammatory Agent. J. Org. Chem. 2020, 85, 14190–14200. 10.1021/acs.joc.0c01701. [DOI] [PubMed] [Google Scholar]; b Urbitsch F.; Elbert B. L.; Llaveria J.; Streatfeild P. E.; Anderson E. A. A Modular, Enantioselective Synthesis of Resolvins D3, E1, and Hybrids. Org. Lett. 2020, 22, 1510–1515. 10.1021/acs.orglett.0c00089. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Saito S.; Nanba Y.; Morita M.; Kobayashi Y. The Hudrlik-Peterson Reaction of Secondary cis-TMS-Epoxy Alcohols and its Application to the Synthesis of the Fatty Acid Intermediates. Synlett 2019, 30, 1085–1089. 10.1055/s-0037-1611809. [DOI] [Google Scholar]; d Nesman J. I.; Tungen J. E.; Vik A.; Hansen T. V. Stereoselective synthesis of the specialized pro-resolving and anti-inflammatory mediator resolvin E1. Tetrahedron 2020, 76, 130821. 10.1016/j.tet.2019.130821. [DOI] [Google Scholar]
- For a recent review on total synthesis and biological evaluations of E-series resolvins, see; Vik A.; Hansen T. V.. Stereoselective syntheses and biological activities of E-series resolvins. Org. Biomol. Chem. 2021 10.1039/D0OB02218G. [DOI] [PubMed] [Google Scholar]
- Sonogashira K.; Tohda Y.; Hagihara N.; Sonogashira K.; Tohda Y.; Hagihara N. A convenient synthesis of acetylenes: catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. 10.1016/s0040-4039(00)91094-3. [DOI] [Google Scholar]
- a Gotoh H.; Hayashi Y. Diphenylprolinol silyl ether as a catalyst in an asymmetric, catalytic and direct α-benzoyloxylation of aldehydes. Chem. Commun. 2009, 3083–3085. 10.1039/b902287b. [DOI] [PubMed] [Google Scholar]; b Hayashi Y.; Umekubo M.; Hirama T. Prolinate Salts as Catalysts for α-Aminoxylation of Aldehyde and Associated Mechanistic Insights. Org. Lett 2017, 19, 4155–4158. 10.1021/acs.orglett.7b01433. [DOI] [PubMed] [Google Scholar]; c Carpenter J.; Northrup A. B.; Chung d.; Wiener J. J. M.; Kim S.-G.; MacMillan D. W. C. Total Synthesis and Structural Revision of Callipeltoside C. Angew. Chem., Int. Ed. 2008, 47, 3568–3572. 10.1002/anie.200800086. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Brown S. P.; Brochu M. P.; Sinz C. J.; MacMillan D. W. C. The Direct and Enantioselective Organocatalytic α-Oxidation of Aldehydes. J. Am. Chem. Soc. 2003, 125, 10808–10809. 10.1021/ja037096s. [DOI] [PubMed] [Google Scholar]; e Donohoe T. J.; Lindsay-Scott P. J.; Parker J. S.; Callens C. K. A. New modes for the Osmium-Catalysed Oxidative Cyclization. Org. Lett. 2010, 12, 1060–1063. 10.1021/ol100046a. [DOI] [PubMed] [Google Scholar]
- Takai K.; Nitta K.; Utimoto K. Simple and selective method for RCHO → (E)-RCH=CHX conversion by means of a CHX3–CrCl2 system. J. Am. Chem. Soc. 1986, 108, 7408–7410. [Google Scholar]
- Dess D. B.; Martin J. C. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 1983, 48, 4155–4156. 10.1021/jo00170a070. [DOI] [Google Scholar]
- Tungen J. E.; Gerstmann L.; Vik A.; de Mateis R.; Colas R. A.; Dalli J.; Chiang N.; Serhan C. N.; Kalesse M.; Hansen T. V. Resolving Inflammation: Synthesis, Configurational Assignment, and Biological Evaluation of RvD1n-3 DPA. Chem. Eur. J. 2019, 25, 1476–1480. 10.1002/chem.201806029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y. Pot economy and one-pot synthesis. Chem. Sci. 2016, 7, 866–880. 10.1039/C5SC02913A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover J. M.; Stahl S. S. Highly Practical Copper(I)/TEMPO Catalyst System for Chemoselective Aerobic Oxidation of Primary Alcohols. J. Am. Chem. Soc. 2011, 133, 16901–16910. 10.1021/ja206230h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz D. E.; Fässler R.; Carreira E. M. Facile Enantioselective Synthesis of Propargylic Alcohols by Direct Addition of Terminal Alkynes to Aldehydes. J. Am. Chem. Soc. 2000, 122, 1806–1807. 10.1021/ja993838z. [DOI] [Google Scholar]
- Boyall D.; Lopez F.; Sasaki H.; Frantz D.; Carreira E. M. Enantioselective Addition of 2-Methyl-3-butyn-2-ol to Aldehydes: Preparation of 3-Hydroxy-1-butynes. Org. Lett. 2000, 26, 4233–4236. 10.1021/ol006791r. [DOI] [PubMed] [Google Scholar]
- Birkofer L.; Ritter A.; Uhlenbrauck H. Siliconorganic compounds. XXI. Substitution and addition reactions with silylated acetylenes. Chem. Ber. 1963, 96, 3280–3288. 10.1002/cber.19630961226. [DOI] [Google Scholar]
- Midland M. M.; McDowell D. C.; Hatch R. L.; Tramontano A. Reduction of α,β-acetylenic ketones with B-3-pinanyl-9-borabicyclo[3.3.1]nonane. High asymmetric induction in aliphatic systems. J. Am. Chem. Soc. 1980, 102, 867–869. 10.1021/ja00522a083. [DOI] [Google Scholar]
- Schmidt H. M.; Arens J. F. Cleavage of the carbon–silicon bond in 1-alkynylsilanes by silver nitrate: Protection of a terminal triple bond in hydrogenation reactions with the trimethylsilyl group. Rec. Trav. Chim. Pays-Bas 1967, 86, 1138–1142. 10.1002/recl.19670861013. [DOI] [Google Scholar]
- a Aursnes M.; Tungen J. E.; Vik A.; Dalli J.; Hansen T. V. Stereoselective synthesis of protectin D1: a potent anti-inflammatory and proresolving lipid mediator. Org. Biomol. Chem. 2014, 12, 432–437. 10.1039/c3ob41902a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Aursnes M.; Tungen J. E.; Vik A.; Colas R.; Cheng C. C.; Dalli J.; Serhan C. N.; Hansen T. V. Total Synthesis of the Lipid Mediator PD1n-3 DPA: Configurational Assignments and Anti-inflammatory and Pro-resolving Actions. J. Nat. Prod. 2014, 77, 910–916. 10.1021/np4009865. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tungen J. E.; Aursnes M.; Hansen T. V. Stereoselective synthesis of maresin 1. Tetrahedron Lett. 2015, 56, 1843–1846. 10.1016/j.tetlet.2015.02.080. [DOI] [Google Scholar]; d Tungen J. E.; Aursnes M.; Dalli J.; Arnardottir H.; Serhan C. N.; Hansen T. V. Total Synthesis of the Anti-inflammatory and Pro–resolving Lipid Mediator MaR1n-3 DPA Utilizing an sp3–sp3 Negishi Cross–Coupling Reaction. Chem. Eur. J. 2014, 20, 14575–14578. 10.1002/chem.201404721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A. T.; Mondal E. A Highly Efficient and Useful Synthetic Protocol for the Cleavage of tert-Butyldimethylsilyl (TBS) Ethers Using a Catalytic Amount of Acetyl Chloride in Dry Methanol. Synlett 2003, 5, 694–698. 10.1055/s-2003-38360. [DOI] [Google Scholar]
- Hansen T. V.; Dalli J.; Serhan C. N. The novel lipid mediator PD1n-3 DPA: An overview of the structural elucidation, synthesis, biosynthesis and bioactions. Prostaglandins Other Lipid Mediat. 2017, 133, 103–110. 10.1016/j.prostaglandins.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a MacMillan D. W. C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. 10.1038/nature07367. [DOI] [PubMed] [Google Scholar]; b Waser M.Asymmetric Organocatalysis in Natural Product Syntheses; Springer: Wien, 2012. [PubMed] [Google Scholar]; c Xiang S.; Tan B. Advances in asymmetric organocatalysis over the last 10 years. Nat. Commun. 2020, 11, 3786. 10.1038/s41467-020-17580-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey E. J.; Shirahama H.; Yamamoto H.; Terashima S.; Venkateswarlu A.; Schaaf T. K. Stereospecific Total Synthesis of Prostaglandins E3 and F3. J. Am. Chem. Soc. 1971, 93, 1490–1491. 10.1021/ja00735a032. [DOI] [PubMed] [Google Scholar]
- Reid B. T.; Mailyan A. K.; Zakarian A. Total Synthesis of (+)- Guadinomic Acid via Hydroxyl-Directed Guanidylation. J. Org. Chem. 2018, 83, 9492–9496. 10.1021/acs.joc.8b01214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannett P. M.; Nagel D. L.; Reilly P. J.; Lawson T.; Sharpe J.; Toth B. The Capsaicinoids: Their Separation, Synthesis, and Mutagenicity. J. Org. Chem. 1988, 53, 1064–1071. 10.1021/jo00240a024. [DOI] [Google Scholar]
- Götz K.; Liermann J. C.; Thines E.; Anke H.; Opatz T. Structure elucidation of hypocreolide A by enantioselective total synthesis. Org. Biomol. Chem. 2010, 8, 2123–2130. 10.1039/c001794a. [DOI] [PubMed] [Google Scholar]
- Yang P.; Zhong J.; Ji K.; Yin J.; Li S.; Wei S.; Zhou Y.; Wang L.; Wang M.; Bian Q. Catalytic asymmetric synthesis of Leukotriene B4. Tetrahedron: Asymmetry 2017, 28, 1596–1601. 10.1016/j.tetasy.2017.09.013. [DOI] [Google Scholar]
- Treilhou M.; Fauve A.; Pougny J.-R.; Prom J.-C.; Veshambre H. Use of Biological Catalysts for the Preparation of Chiral Molecules. 8. Preparation of Propargylic Alcohols. Application in the Total Synthesis of Leukotriene B4. J. Org. Chem. 1992, 57, 3203–3208. 10.1021/jo00037a044. [DOI] [Google Scholar]
- Primdahl K. G.; Tungen J. E.; Aursnes M.; Hansen T. V.; Vik A. An efficient total synthesis of leukotriene B4. Org. Biomol. Chem. 2015, 13, 5412–5417. 10.1039/c5ob00473j. [DOI] [PubMed] [Google Scholar]
- Boer R. E.; Gimnez-Bastida J. A.; Boutaud O.; Jana S.; Schneider C.; Sulikowski G. A. Total Synthesis and Biological Activity of the Arachidonic Acid Metabolite Hemiketal E2. Org. Lett. 2018, 20, 4020–4022. 10.1021/acs.orglett.8b01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M.; Rao M. K.; Reddanna P.; Li C. H.; Tu C.-P. D.; Corey E. J.; Reddy C. C. I. Specificity of the glutathione S-transferases in the conversion of leukotriene A4 to leukotriene C4. Arch. Biochem. Biophys. 1987, 259, 536–547. 10.1016/0003-9861(87)90520-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.