

Abstract
Increased glycolysis is the result of the sensing of glucose by hypothalamic neurons. The biochemical mechanisms underlying the control of hypothalamic glycolysis, however, remain to be elucidated. Here we showed that PFKFB3, the gene that encodes for inducible 6-phosphofructo-2-kinase (iPFK2), was expressed at high abundance in both mouse hypothalami and clonal hypothalamic neurons. In response to re-feeding, PFKFB3 mRNA levels were increased by 10-fold in mouse hypothalami. In the hypothalamus, re-feeding also decreased the phosphorylation of AMP-activated protein kinase (AMPK) (Thr172) and the mRNA levels of agouti-related protein (AgRP), and increased the mRNA levels of cocaine-amphetamine-related transcript (CART). Similar results were observed in N-43/5 clonal hypothalamic neurons upon treatment with glucose and/or insulin. In addition, knockdown of PFKFB3/iPFK2 in N-43/5 neurons caused a decrease in rates of glycolysis, which was accompanied by increased AMPK phosphorylation, increased AgRP mRNA levels and decreased CART mRNA levels. In contrast, overexpression of PFKFB3/iPFK2 in N-43/5 neurons caused an increase in glycolysis, which was accompanied by decreased AMPK phosphorylation and decreased AgRP mRNA levels and increased CART mRNA levels. Together, these results suggest that PFKFB3/iPFK2 responds to re-feeding, which in turn stimulates hypothalamic glycolysis and decreases hypothalamic AMPK phosphorylation and alters neuropeptide expression in a pattern that is associated with suppression of food intake.
Keywords: Inducible 6-phosphofructo-2-kinase, Hypothalamus, Glucose-sensing, Glycolysis, AMP-activated protein kinase, Neuropeptide
1. Introduction
The control of food intake is governed by the central nervous system, where the hypothalamus plays a crucial role. In the hypothalamus, certain neurons express orexigenic neuropeptide Y (NPY) and agouti-related protein (AgRP). In contrast, certain neurons express anorexigenic pro-opiomelanocortin (POMC) and cocaine-amphetamine-related transcript (CART) [1–5]. While investigating how nutrients are sensed by hypothalamic neurons, a number of studies have demonstrated that AMP-activated protein kinase (AMPK) is a key cellular energy sensor that responds to peripheral signals, for example, glucose and leptin, to control food intake [6–12]. For example, decreased hypothalamic AMPK signaling contributes to the anorectic effects caused by re-feeding and leptin [7,13,14]. On the other hand, the activation of hypothalamic AMPK signaling has been shown to abolish the anorectic effect induced by leptin [13]. Of interest, glycolysis, a major flux of glucose metabolism, is likely involved in the control of the energy status of hypothalamic neurons, thereby altering hypothalamic AMPK signaling and neuropeptide expression, in order to regulate food intake [15]. Despite these previous studies, the biochemical mechanisms underlying the linkage among glycolysis, AMPK signaling, and neuropeptide expression in the hypothalamus remain to be elucidated.
Physiologically, glycolysis is under the control of nutritional and hormonal signals that are associated with the transition between fasting and feeding [16–18]. At the cellular level, glycolysis is regulated at the level of glucose entry into cells via membrane glucose transporters, glucose phosphorylation to glucose-6-phosphate (G6P) by the activity of hexokinase (I–IV), and the conversion of fructose-6-phosphate (F6P) to fructose-1,6-bisphosphate (F1,6P2) [18]. For the past several decades, glycolysis in liver cells has been intensely studied. These studies have led to the demonstration of glucokinase (GK, also named as hexokinase IV) and 6-phosphofructo-1-kinase (6PFK1) as essential enzymes that control the rate-determining steps of glycolysis. Given the role for 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (6PFK2/FBP2) in determining the cellular levels of fructose-2,6-bisphosphate (F2,6P2), the most powerful allosteric activator of 6PFK1, 6PFK2/FBP2 is considered as a key regulatory enzyme that crucially controls glycolysis [18]. This concept, indeed, is supported by evidence obtained from various tissues and cells [19–25]. In the hypothalamus, much attention has paid to the role of GK in the control of neuronal glucose metabolism. In fact, in hypothalamic neurons, GK has been proposed as glucosensor, which is associated with the regulation of food intake [26–32]. As a key regulatory enzyme of glycolysis, 6PFK2/FBP2 is present in the brain [33]. The function of 6PFK2/FBP2, however, has not yet been fully characterized in hypothalamic neurons.
PFKFB1-PFKFB4 are four known PFKFB genes that have been shown to encode for different isoforms of 6PFK2/FBP2 in a wide variety of tissues [33–41]. Additionally, a given tissue may contain several isoforms of 6PFK2/FBP2. While there is diversity with respect to isoforms, the role of 6PFK2/FBP2 is simple: it determines F2,6P2 levels and controls glycolysis [41–47]. For instance, an increase in the kinase activity and/or a decrease in the bisphosphatase activity of 6PFK2/FBP2 lead to an increase in the concentrations of F2,6P2 [20]. The latter, in turn, activates 6PFK1 to increase glycolysis [18]. In the brain, six isoforms of 6PFK2/FBP2, encoded by four PFKFB genes, have been identified [33]. However, the PFKFB (6PFK2/FBP2) expression pattern in the hypothalamus has not been characterized. It is also not clear about the role of PFKFB (6PFK2/FBP2) in controlling glycolysis in relation to the regulation of food intake. The present study provides evidence to support a crucial role for PFKFB3/iPFK2, the most abundant PFKFB (6PFK2/FBP2) in hypothalamic neurons, in linking glycolysis and AMPK signaling and neuropeptide expression.
2. Methods and materials
2.1. Animal experiments
Wild-type C57BL/6J mice were fed ad libitum and maintained on a 12:12-h light-dark cycle (lights on at 06:00 h). A laboratory low-fat diet (Research Diets) contains 70% carbohydrate calories, 20% protein calories, and 10 fat calories. To examine the expression pattern of hypothalamic PFKFB genes, 4 male mice, at 12–14 weeks of age, were fasted for 4 h and subjected to collection of hypothalami as previously described [21]. Briefly, the optic chiasm was dissected away from the anterior portion of the hypothalamus, and the mammillary nuclei were removed from the posterior of the hypothalamus. Accordingly, the hypothalamus collected includes the arcuate, ventromedial, dorsomedial and paraventricular nuclei. To examine nutritional regulation of the expression of PFKFB genes, 12 male mice were fasted overnight (16 h) with or without re-feeding for an additional 4 h. Blood samples and hypothalami were collected and used for further analyses.
2.2. Measurement of plasma levels of glucose and insulin
The levels of plasma glucose were measured using metabolic kits (Sigma). The levels of plasma insulin were measured using ELISA kits (Crystal Chem, Downers Grove, IL, USA).
2.3. Cell culture and treatments
N-43/5 cells (Cellutions Biosystems, Ontario, Canada) are clonal mouse hypothalamic neurons that have been characterized previously by other investigators [15,48,49]. These cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 20 mM glucose, and 1% penicillin/streptomycin and maintained at 37 °C in an atmosphere of 5% CO2 as previously described [48]. Cells were harvested at 90% confluence. Total RNA was prepared as previously described [50,51] and used to examine the expression pattern of PFKFB genes. Some cells were seeded into 6-well plates and initially incubated with DMEM containing 20 mM glucose, a concentration used for maintaining N-43/5 cells, for 2 days. Thereafter, these cells were incubated with DMEM containing 0.5 mM glucose for an additional 24 h as described by Cheng et al. [15] and used for further analyses. To determine the effects of glucose and/or insulin, N-43/5 cells were incubated with DMEM supplemented with glucose (0.5 or 20 mM) in the presence or absence of insulin (100 nM) for 4 hours. The treated cells were then subjected to the assays to quantify PFKFB expression profile, rates of glycolysis, AMPK signaling, and neuropeptide expression as described below.
Based on the expression profile of PFKFB genes, PFKFB3/iPFK2 was chosen for further investigation. As such, PFKFB3/iPFK2 knockdown and overexpression experiments were performed in N-43/5 cells. To knock down PFKFB3/iPFK2, N-43/5 cells were transfected with siRNA against mouse PFKFB3/iPFK2 (Santa Cruz Biotech, Santa Cruz, CA, USA) or control (Ctrl) as previously described [24]. A similar procedure was used to over-express PFKFB3/iPFK2 in N-43/5 cells using a plasmid containing cDNA of PFKFB3. After transfection for 24 hours, the cells were harvested for further analyses.
2.4. Determination of hypothalamic gene expression
Total RNA prepared from frozen hypothalami and cells was used to determine the expression of PFKFB genes, as well as NPY, AgRP, POMC, and CART, as previously described [21]. The sequences of primers specific for mouse PFKFB1, PFKFB2, PFKFB3, and PFKFB4 were listed in Table 1.
Table 1.
Sequences of primers specific for PFKFB genes (cDNAs)
| Gene | Forward primer | Reverse primer |
|---|---|---|
| PFKFB1 | 5’-AAAGCCTCTAAGAGAGCAGCC TC | 5’-CACTTTAGTCGGTGTTCCTATCC |
| PFKFB2 | 5’-AAGGGCAGGGAAGACATGCTCAT | 5’-CTTTGGTGGGTACTCCAATCCAG |
| PFKFB3 | 5’-GATCTGGGTGCCCGTCGATCACCG | 5’-CAGTTGAGGTAGCGAGTCAGCTTC |
| PFKFB4 | 5’-CGTGGTGTCTGCATGACTAACTG | 5’-CGATACTGACCAACGTTGAATTC |
2.5. Measurement of rates of glycolysis
Each well (6-well plate) of cells were incubated with DMEM supplemented with 1 μCi [3-3H]-glucose for 3 h. After incubation, the medium was collected to determine the production of 3H2O (in disintegration per minute (DPM)) as described [52]. Rates of glycolysis are expressed as nanomoles of glucose metabolized per 3 h per milligram of protein.
2.6. Determination of hypothalamic AMPK signaling
Lysates of frozen hypothalami and N-43/5 cells were prepared and used for Western blot analyses as previously described [20,25,53]. Antibodies against total AMPK and/or phospho-AMPK (Thr172) are products of Cell .Signaling Technology, Inc (Danvers, MA).
2.7. Statistical methods
Numeric data are presented as means±S.E. Statistical significance was assessed by unpaired, 2-tailed ANOVA or Student t test. Differences were considered significant at the two-tailed P<.05.
3. Results
3.1. PFKFB3 is the PFKFB gene that is expressed at high abundance in mouse hypothalami and clonal hypothalamic neurons
Four PFKFB genes are known to encode six isoforms of 6PFK2/FBP2 in the brain. We addressed the expression pattern of hypothalamic PFKFB genes using primers specific for the four individual PFKFB genes. In mouse hypothalami, the expression of PFKFB3 is at high abundance, compared with PFKFB1, PFKFB2, and PFKFB4 (Fig. 1A). Similarly, in N-43/5 cells, PFKFB3 is expressed at high abundance, compared with the three other PFKFB genes (Fig. 1B). Of interest, two PFKFB4 products were detected in mouse hypothalami, but not in clonal hypothalamic neurons, suggesting that cells other than neurons express different PFKFB4 product(s). In combination, these results from the analysis of PFKFB mRNA levels served as the basis for the present study to address the role for PFKFB3/iPFK2 in regulating glycolysis in hypothalamic neurons, in the context of neuronal glucose sensing and neuropeptide expression.
Fig. 1.
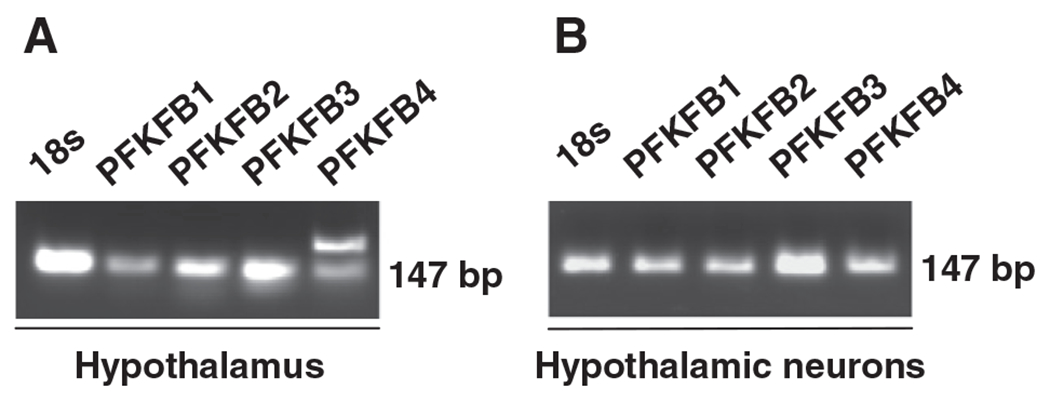
Identification of PFKFB3 as a PFKFB gene at high abundance in hypothalamic neurons. Total RNA was prepared from mouse hypothalami (A) and clonal hypothalamic neurons (B) and subjected to reverse transcription (RT)-PCR using primers designed specific for cDNA of each of the four PFKFB genes (PFKFB1 through PFKFB4). Representative PCR products were presented.
3.2. Re-feeding increases plasma levels of glucose and insulin and stimulates PFKFB3 expression
During re-feeding, glucose-sensing in hypothalamic neurons is involved in initiating signaling cascades to terminate feeding through feedback inhibitory mechanisms [54]. As a result of re-feeding, plasma levels of glucose and insulin, two essential factors that are involved in the control of food intake, were increased compared with their respective levels in fasted mice (Fig. 2, A and B). Next, the effects of fasting and re-feeding on PFKFB3 expression were examined. Compared with that in fasted mice, hypothalamic expression of PFKFB3 in re-fed mice was increased by 10-fold (Fig. 2C and D). Therefore, during the transition from fasting to re-feeding, increased hypothalamic PFKFB3 expression is associated with plasma signals (eg, glucose and insulin) in a pattern that contributes to suppression of food intake.
Fig. 2.
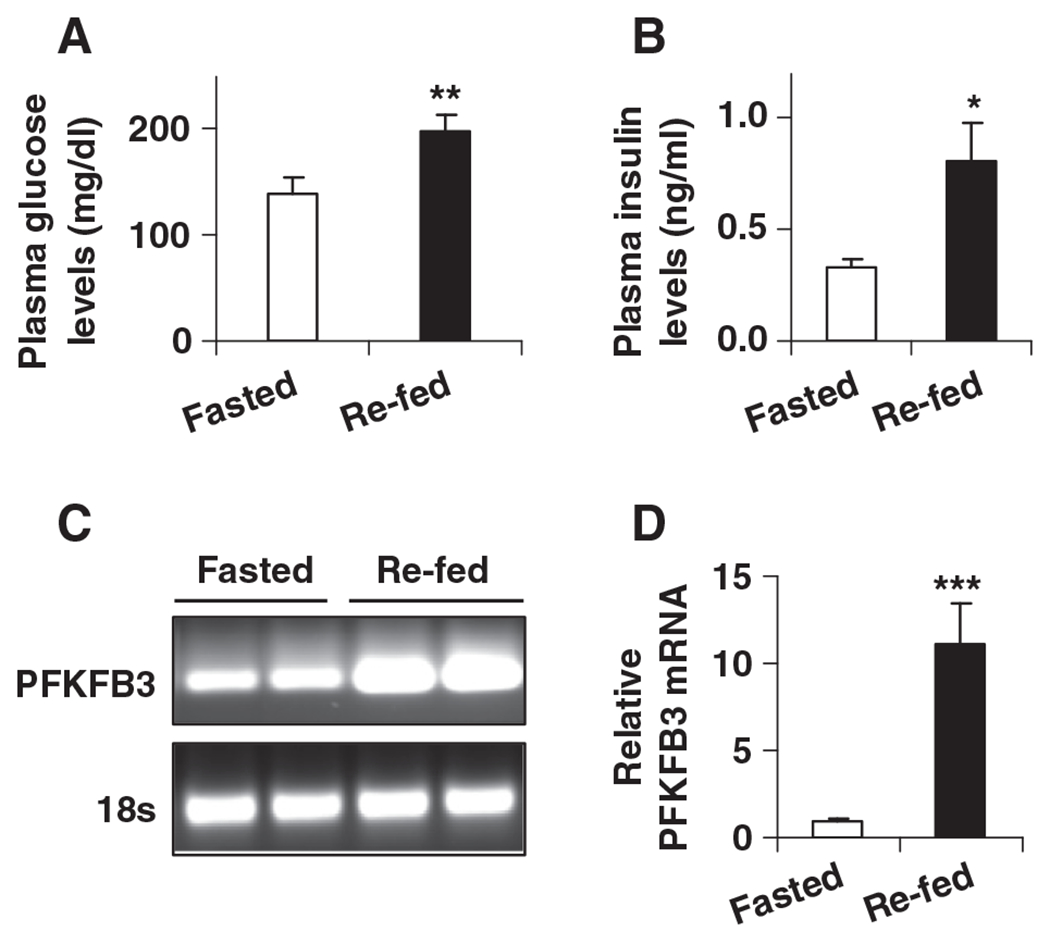
Changes in plasma levels of glucose and insulin and in PFKFB3 expression in mouse hypothalami. Male wild-type C57BL/6J mice, at the age of 12–14 weeks, were fed ad libitum. Before sample collection, mice were fasted for overnight (16 h) with or without re-feeding for an additional 4 h. For A and B, blood samples were collected and subjected to measurements of plasma levels of glucose (A) and insulin (B). (A) Plasma levels of glucose. (B) Plasma levels of insulin. For C and D, total RNA of hypothalami was prepared to examine the mRNA levels of PFKFB3 using real-time RT-PCR. (C) Representative PCR products. (D) Quantification of the mRNA levels of PFKFB3. For A–D, data are means±S.E., n=4–6. *P<.05; **P<.01; and ***P<.001 re-fed vs. fasted.
Hypothalamic AMPK and neuropeptides are of particular importance in the control of food intake. Therefore, the effects of fasting and re-feeding on hypothalamic AMPK phosphorylation and neuropeptide expression were examined. Compared with that in fasted mice, hypothalamic AMPK phosphorylation in re-fed mice was significantly decreased (Fig. 3A and B). In addition, hypothalamic AgRP expression was decreased and hypothalamic CART expression was increased in re-fed mice compared with controls (Fig. 3C). All of these changes contributed to suppression of food intake as documented previously by other investigators [7,55] and were accompanied by increased hypothalamic PFKFB3 expression (see above). Thus, a role for PFKFB3/iPFK2 in linking hypothalamic glycolysis and suppression of food intake is hypothesized.
Fig. 3.
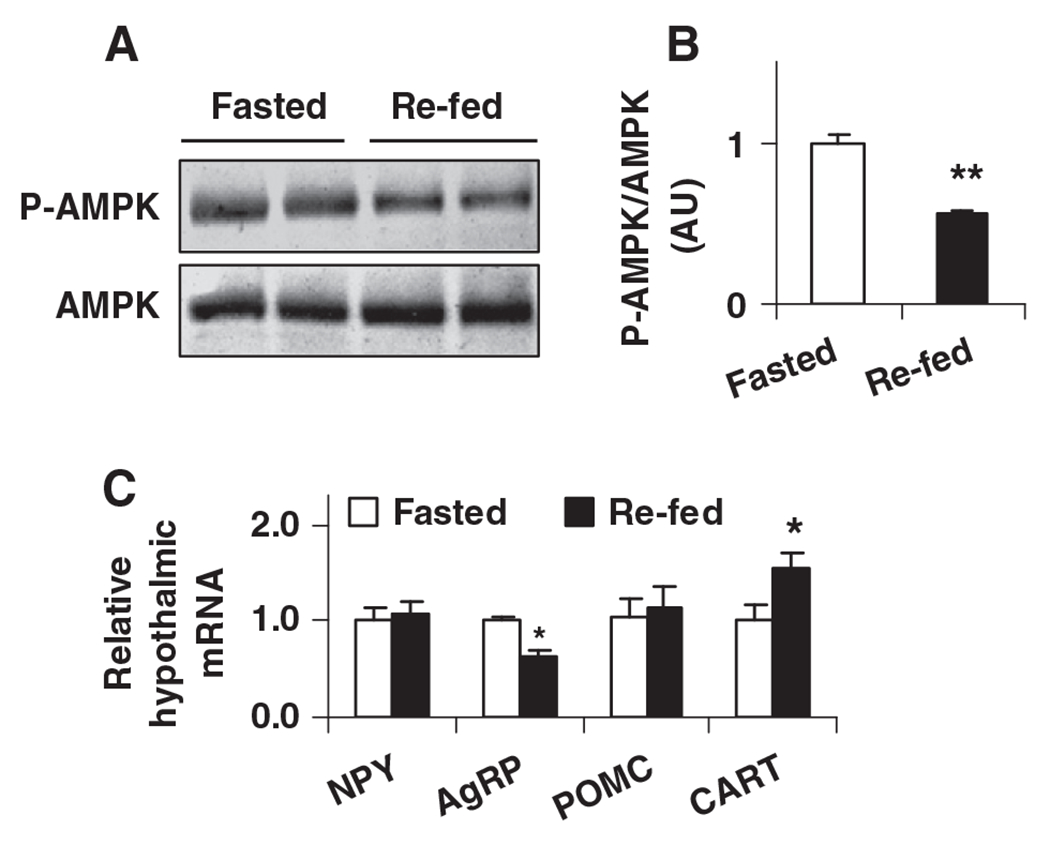
Physiological and nutritional regulation of AMPK phosphorylation and neuropeptide expression in mouse hypothalami. Male wild-type C57BL/6J mice, at the age of 12–14 weeks, were fed ad libitum. Before sample collection, mice were fasted for overnight (16 h) with or without re-feeding for an additional 4 h. Hypothalamus lysates were prepared to examine AMPPK signaling using Western blot analyses. (A) Amount of AMPK and phospho-AMPK (Thr172). (B) Ratios of phospho-AMPK/AMPK were calculated using densitometry. AU, arbitrary unit. (C) Total RNA was prepared from mouse hypothalami and subjected to real-time RT-PCR. NPY, neuropeptide Y; AgRP, agouti-related protein; POMC, pro-opiomelanocortin; and CART, cocaine-amphetamine-related transcript. For B and C, data are means±S.E., n=4–6. *P<.05 and **P<.01 re-fed vs. fasted (in B) for the same gene (in C).
3.3. Glucose and insulin stimulate PFKFB3 expression, increase glycolysis, and decrease AMPK phosphorylation in clonal hypothalamic neurons
Glucose and insulin, at increased levels, are 2 key signals associated with re-feeding. Therefore, effects of glucose and insulin on hypothalamic PFKFB3 expression and glycolysis were determined. In N-43/5 cells, glucose, supplemented at a concentration of 20 mM, significantly increased PFKFB3 expression compared with glucose at a concentration of 0.5 (the lowest concentration used to examine neuronal responses [15,49]) (Fig. 4A and 4B). Similarly, a stimulatory effect on PFKFB3 expression was observed in cells treated with insulin in the presence of either low (0.5 mM) or high (20 mM) glucose, indicating that insulin potentates the stimulatory effect of glucose on PFKFB3 expression in hypothalamic neurons. Consistent with increased PFKFB3 expression, rates of glycolysis were increased in neurons when treated with glucose (20 mM), insulin, or in combination (Fig. 4C). Collectively, these results suggest that PFKFB3/iPFK2 is a crucial regulator that stimulates neuronal glycolysis. Because the effects observed in clonal neurons recapitulated changes in hypothalami of re-fed mice, glucose and insulin are postulated as two essential signals that alter the hypothalamus during re-feeding.
Fig. 4.
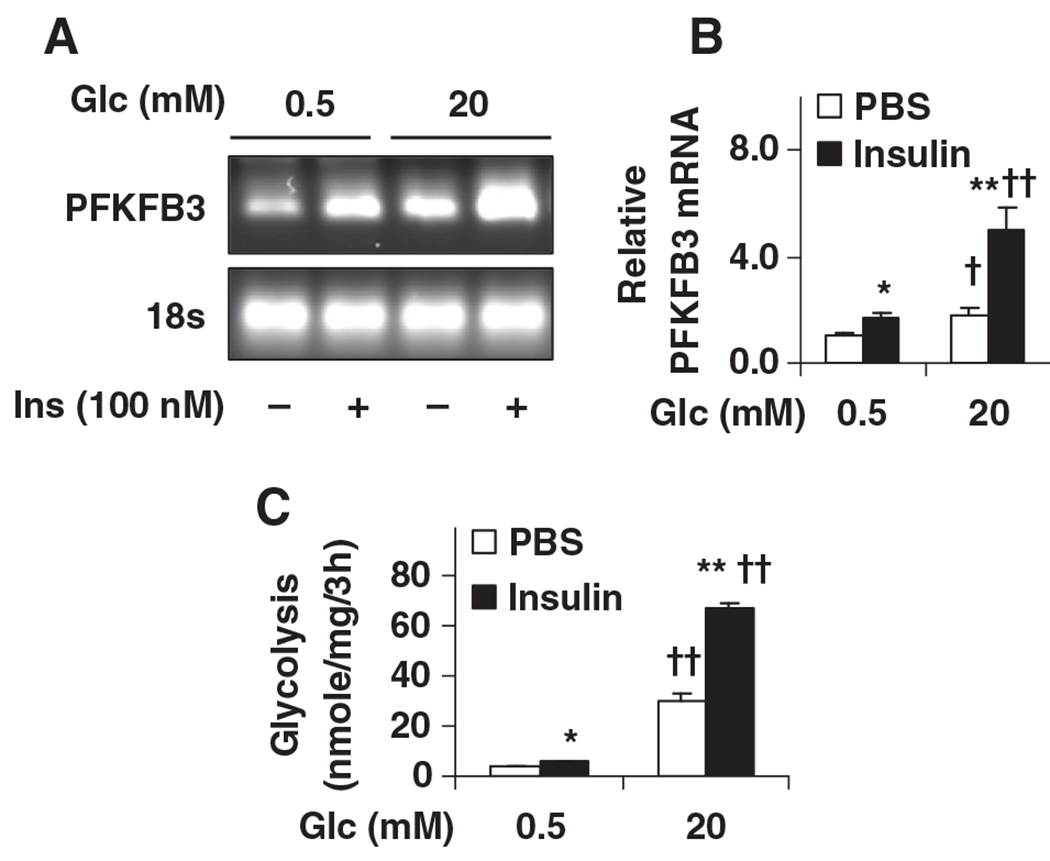
Stimulation of PFKFB3 expression and glycolysis in clonal hypothalamic neurons by glucose and insulin. N-43/5 cells, clonal hypothalamic neurons, were incubated with DMEM containing glucose (0.5 mM) for 24 h, and then treated with glucose (0.5 or 20 mM) in the presence or absence of insulin (100 nM) for 4 h. (A) and (B), the mRNA levels of PFKFB3 were quantified using real-time RT-PCR. (A) Representative PCR products. (B) Quantification of the mRNA levels of PFKFB3. Glc, glucose. (C) To quantify rates of glycolysis, 3H-glucose was added to the treated cells for 3 h prior to cell harvest. For B and C, data are means±S.E., n=4–6. *P<.05 and **P<.01 Insulin vs. PBS for the same condition (low or high glucose); †P<.05 and ††P<.01 high glc (20 mM) vs. low glc (0.5 mM) for the same treatment (PBS or Insulin).
The effects of glucose and insulin on neuronal AMPK phosphorylation were also examined. Compared with controls, AMPK phosphorylation was decreased in neurons treated with high levels of glucose, and in neurons treated with insulin in the presence of either low or high levels of glucose (Fig. 5A and 5B). These changes were inversely associated with neuronal PFKFB3 expression and rates of glycolysis, indicating a potential role for PFKFB3/iPFK2 in regulating glycolysis in relation to AMPK phosphorylation in hypothalamic neurons (see below).
Fig. 5.
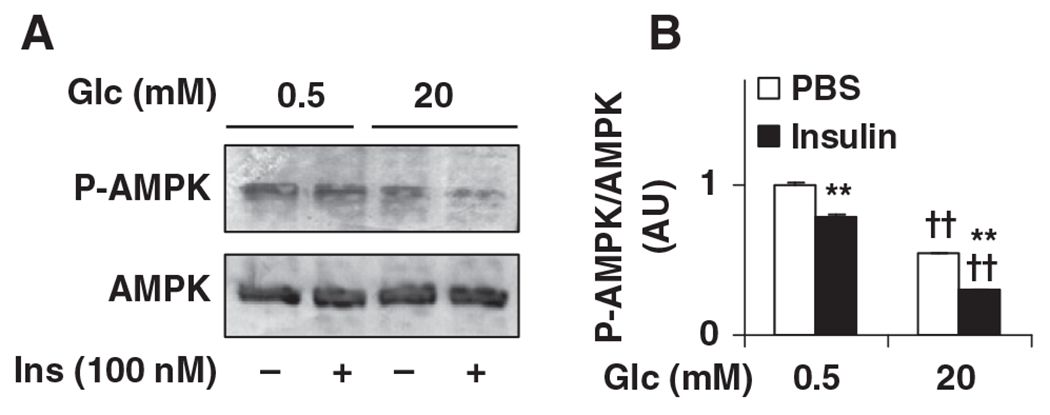
Reduction of AMPK phosphorylation in clonal hypothalamic neurons by glucose and insulin. N-43/5 cells were incubated with DMEM containing glucose (0.5 mM) for 24 h, and then treated with glucose (0.5 mM or 20 mM) in the presence or absence of insulin (100 nM) for 4 h. Cell lysates were prepared to examine AMPPK signaling using Western blot analyses. (A) Amount of AMPK and phospho-AMPK (Thr172). (B) Ratios of phospho-AMPK/AMPK were calculated using densitometry. AU, arbitrary unit; Glc, glucose. Data are means±S.E., n=4. **P<.01 Insulin vs. PBS for the same condition (low or high glc); ††P<.01 high glc (20 mM) vs. low glc (0.5 mM) for the same treatment (PBS or Insulin).
3.4. PFKFB3/iPFK2 knockdown or overexpression oppositely alters glycolysis, AMPK phosphorylation, and neuropeptide expression in clonal hypothalamic neurons
To address a direct role for PFKFB3/iPFK2 in regulating glycolysis and AMPK phosphorylation and neuropeptide expression in neurons, PFKFB3/iPFK2 knockdown or overexpression experiments were performed in N-43/5 cells. Compared with controls, PFKFB3/iPFK2 knockdown decreased, whereas PFKFB3/iPFK2 overexpression markedly increased, the mRNA levels of PFKFB3 (Fig. 6 A and B). Next, rates of glycolysis were examined. Compared with controls, rates of glycolysis in PFKFB3/iPFK2-knockdown cells were decreased by 25% (Fig. 6B). In contrast, rates of glycolysis in PFKFB3/iPFK2-overexpessing cells were increased by 2.4-fold compared with controls (Fig. 6B). Together, these results demonstrate a crucial role for PFKFB3/iPFK2 in stimulating neuronal glycolysis.
Fig. 6.
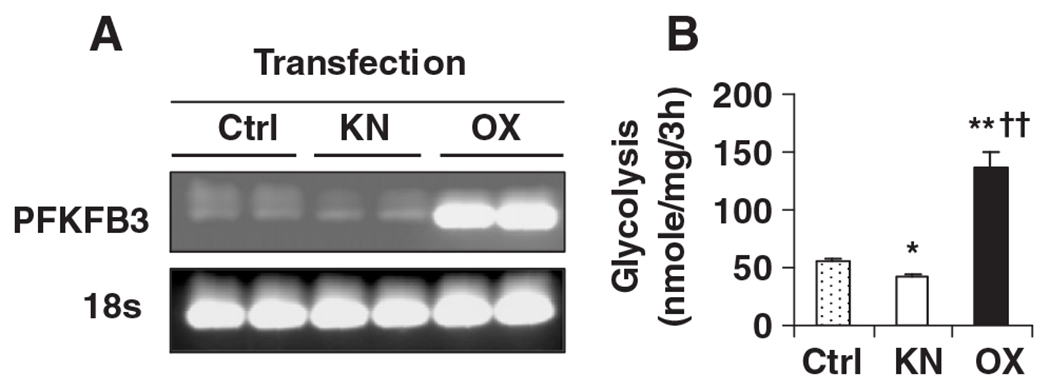
Modulation of neuronal glycolysis by altering PFKFB3 expression. For knockdown (KN) study, N-43/5 cells were transfected with siRNA against mouse PFKFB3/iPFK2 or control (Ctrl) for 24 h. For overexpression (OX) study, N-43/5 cells were transfected with a plasmid containing the cDNA of PFKFB3 or control (Ctrl) for 24 h. After transfection, the cells were harvested to verify PFKFB3 expression at the mRNA level (A) and/or subjected to the assay to quantify rates of glycolysis (B). For B, data are means±S.E., n=4–6. *P<.05 and **P<.01 vs. Ctrl; ††P<.01 vs. KN.
While neuronal glycolysis was altered, AMPK phosphorylation was increased in PFKFB3/iPFK2-knockdown cells and was deceased in PFKFB3/iPFK2-overexpressing cells compared with that in control cells (Fig. 7A and B). When neuropeptide expression was examined, the mRNA levels of AgRP were increased in PFKFB3/iPFK2-knockdown cells and deceased in PFKFB3/iPFK2-overexpressing cells, compared with those in control cells (Fig. 7C). In contrast, the mRNA levels of CART were decreased in PFKFB3/iPFK2-knockdown cells and increased in PFKFB3/iPFK2-overexpressing cells, compared with those in controls (Fig. 7C). Therefore, PFKFB3/iPFK2 also critically controls neuronal AMPK phosphorylation and neuropeptide expression. These results, in combination with changes in PFKFB3 expression and neuronal glycolysis, demonstrate a potential role for PFKFB3/iPFK2 in linking neuronal glycolysis and AMPK phosphorylation and neuropeptide expression.
Fig. 7.
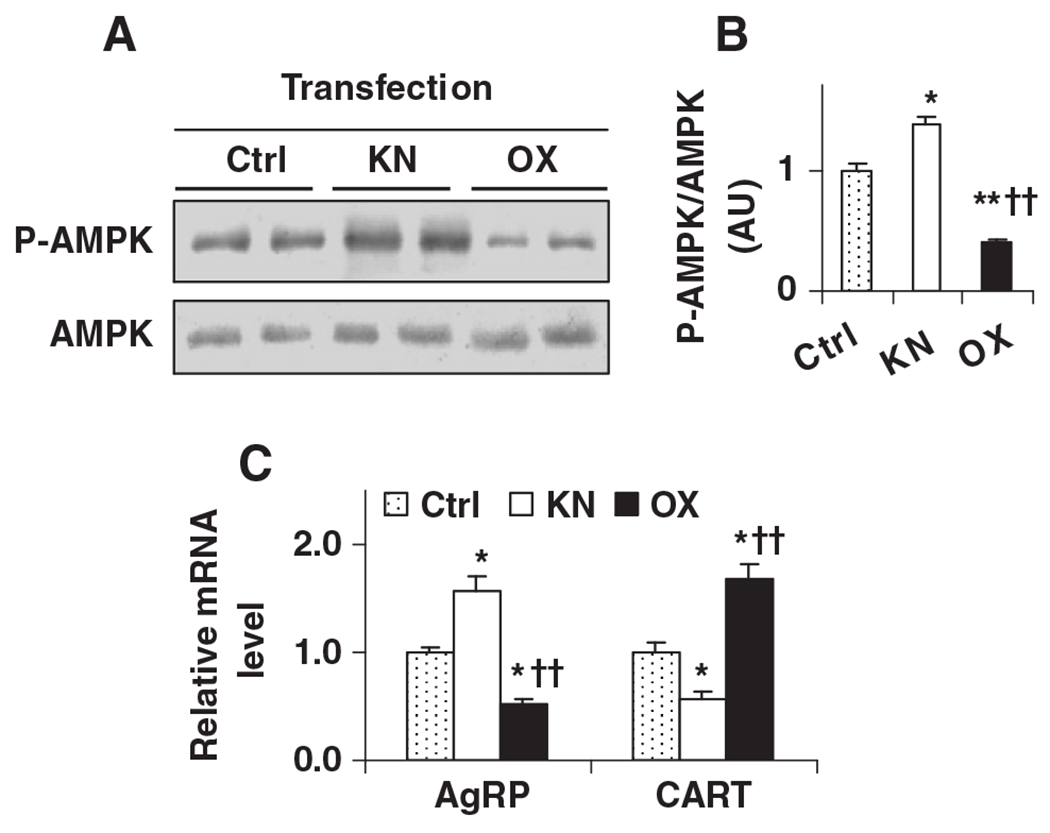
Modulation of neuronal AMPK phosphorylation and neuropeptide expression by altering PFKFB3 expression. N-43/5 cells were transfected with siRNA against PFKFB3 to knock down PFKFB3/iPFK2 or a plasmid containing the cDNA of PFKFB3 to over-express PFKFB3/iPFK2. After transfection for 24 h, cells were harvested to analyze AMPK signaling and neuropeptide expression. (A) Amount of AMPK and phospho-AMPK (Thr172). (B) Ratios of phospho-AMPK/AMPK were calculated using densitometry. AU, arbitrary unit. (C) Total RNA was prepared and subjected to real-time RT-PCR. For B and C, data are means±S.E., n=4–6. *P<.05 and **P<.01 vs. Ctrl; ††P<.01 vs. KN (in B) for the same gene (in C).
4. Discussion
The glucostatic hypothesis was proposed 6 decades ago and used to explain how post-prandial increases in plasma glucose levels cause meal termination [56,57]. Since then, growing evidence has suggested an essential role for neuronal glucose metabolism, but not the levels of plasma glucose, in the control of food intake [58]. Recently, GK in hypothalamic neurons has been considered as a glucose sensor that regulates homeostatic functions, including the control of food intake [29–31], although a direct link between neuronal glucose-sensing and the control of food intake is still lacking [54]. In the present study, two lines of evidence obtained from mice and clonal hypothalamic neurons support a crucial role for PFKFB3/iPFK2 in stimulating neuronal glycolysis and in regulating AMPK phosphorylation and neuropeptide expression.
PFKFB3 is one of the four PFKFB genes that code for different isoforms of 6PFK2/FBP2 [33–41]. It has been previously shown that PFKFB3 is expressed at high abundance in adipose tissue and adipocytes [24], in which iPFK2 (encoded by PFKFB3) links glucose with fatty acid metabolism and the inflammatory response [24,25]. These findings have demonstrated the importance of PFKFB3/iPFK2, not only in the control of glycolysis, but also in the regulation of cell functions. As with adipose tissue, brains of human and rodents also express PFKFB3, whose functions remain to be established. In the hypothalamus, however, the expression pattern of PFKFB3, as well as other PFKFB isoforms, has not been determined. By characterizing the PFKFB expression profile, the present study identified PFKFB3 as the most abundant PFKFB gene in mouse hypothalamus. This enabled us to address a role for PFKFB3/iPFK2 in neuronal glucose-sensing in relation to the control of food intake (see below). Furthermore, compared with other PFKFB genes, PFKFB3 was also expressed at the highest abundance in clonal hypothalamic neurons. This led us to the use of clonal hypothalamic neurons, in order to elucidate a direct role for PFKFB3/iPFK2 in controlling hypothalamic glycolysis, as well as AMPK phosphorylation and neuropeptide expression.
The expression of PFKFB3 in the mouse hypothalamus increased after the transition from fasting to re-feeding. This increase in PFKFB3 expression was due to re-feeding-associated increases in the levels of glucose and/or insulin. In support of this idea, treatment of clonal hypothalamic neurons with glucose and insulin caused an increase in PFKFB3 expression in a pattern similar to that brought about by re-feeding in the mouse hypothalamus. In fact, glucose alone exhibited a stimulatory effect on hypothalamic PFKFB3 expression. However, when combined with insulin, glucose, at either a low or high concentration, caused a significantly greater increase in PFKFB3 expression. These results demonstrate that glucose and insulin work additively, or even synergistically, to stimulate PFKFB3 expression in the hypothalamus. Therefore, insulin has a crucial role in neuronal glucose-sensing, which warrants further investigation in future studies.
Given the property of PFKFB3/iPFK2 in stimulating glycolysis due to activating 6PFK1, as the result of increasing the production of F2,6P2 [24,25], it is postulated here that increased PFKFB3/iPFK2 in the hypothalamus during re-feeding contributed, in large part, to an increase in hypothalamic glycolysis. As substantial evidence, treatment of glucose and insulin increased rates of glycolysis in clonal hypothalamic neurons while stimulating PFFKB3 expression. Furthermore, PFKFB3/iPFK2 knockdown decreased, whereas PFKFB3/iPFK2 overexpression increased, rates of glycolysis in clonal hypothalamic neurons. Thus, PFKFB3/iPFK2 directly controls rates of glycolysis in hypothalamic neurons.
In response to re-feeding, hypothalamic AMPK phosphorylation was decreased. In addition, hypothalamic expression of orexigenic AgRP was decreased whereas hypothalamic expression of anorexigenic CART was increased. These changes have been used by others to explain how signals generated in response to re-feeding suppress food intake [7,8,12]. Relatively little is known, however, about the link among neuronal glucose-sensing, AMPK phosphorylation, and neuropeptide expression. In contrast, relatively much is known about roles for AMPK and neuropeptides in controlling food intake. In the present study, re-feeding-associated changes in hypothalamic AMPK phosphorylation and neuropeptide expression were accompanied by increased PFKFB3 expression. Considering that glucose and insulin stimulate PFKFB3 expression and that PFKFB3/iPFK2 stimulates glycolysis, it is postulated that in the hypothalamus PFKFB3/iPFK2 responds to glucose and insulin to increase glycolysis, and in turn decreases AMPK phosphorylation. The latter effect consequently decreases AgRP expression and increases CART expression. Because associations among hypothalamic events observed in mice were similarly observed in clonal hypothalamic neurons, a role for PFKFB3/iPFK2 in stimulating neuronal glycolysis, decreasing neuronal AMPK phosphorylation, and altering neuropeptide expression was postulated, and is supported by the results obtained from PFKFB3/iPFK2-knockdown and/or PFKFB3/iPFK2-overexpressing neurons. The role proposed here for PFKFB3/iPFK2 provides new insight into the link between glucose-sensing and hypothalamic AMPK in relation to the control of food intake as previously demonstrated [8,15,49].
While PFKFB3/iPFK2 clearly regulates AMPK phosphorylation and neuropeptide expression, it remains to be determined whether or not altering PFKFB3/iPFK2 specifically in the hypothalamus modulates feeding behavior in rodents. In addition, because AMPK in mediobasal hypothalamic neurons appears to have a limited role in regulating food intake [59], PFKFB3/iPFK2 stimulation of glycolysis in relation to AMPK phosphorylation and neuropeptide expression may exist in limited regions of the hypothalamus. Alternatively, PFKFB3/iPFK2 also acts through AMPK-independent signaling cascade(s) to alter neuropeptide expression and regulate food intake.
In summary, the present study demonstrates a crucial role for PFKFB3/iPFK2 in stimulating neuronal glycolysis in relation to the control of food intake. This role of PFKFB3/iPFK2 is evidenced by the fact that PFKFB3 expression is increased in the mouse hypothalamus in response to re-feeding, and in clonal hypothalamic neurons upon treatment with glucose and insulin. Furthermore, altered PFKFB3 in both mouse hypothalamic and clonal hypothalamic neurons correlates with changes in AMPK phosphorylation and neuropeptide expression in a pattern to suppress food intake (with increased PFKFB3/iPFK2) or stimulate feeding (with decreased PFKFB3/iPFK2). Therefore, PFKFB3/iPFK2 is likely an essential glucosensor in hypothalamic neurons.
Acknowledgment
This work was supported, in whole or in part, by ADA grant 1-10-JF-54 and AHA 12BGIA9050003 (to C.W.).
References
- [1].Morley JE, Hernandez EN, Flood JF. Neuropeptide Y increases food intake in mice. Am J Physiol Regul Integr Comp Physiol 1987;253(3):R516–22. [DOI] [PubMed] [Google Scholar]
- [2].Rossi M, Kim MS, Morgan DGA, Small CJ, Edwards CMB, Sunter D, et al. A C-terminal fragment of agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology 1998;139(10):4428–31. [DOI] [PubMed] [Google Scholar]
- [3].Thim L, Kristensen P, Larsen PJ, Wulff BS. CART, a new anorectic peptide. Int J Biochem Cell Biol 1998;30(12):1281–4. [DOI] [PubMed] [Google Scholar]
- [4].Larsen PJ, Vrang N, Petersen PC, Kristensen P. Chronic intracerebroventricular administration of recombinant CART(42-89) peptide inhibits and causes weight loss in lean and obese Zucker (fa/fa) rats. Obes Res 2000;8(8):590–6. [DOI] [PubMed] [Google Scholar]
- [5].Millington GW. The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr Metab 2007;4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem 2004;279(13):12005–8. [DOI] [PubMed] [Google Scholar]
- [7].Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004;2004(428):569–74. [DOI] [PubMed] [Google Scholar]
- [8].Lee K, Li B, Xi X, Suh Y, Martin RJ. Role of neuronal energy status in the regulation of adenosine 5’-monophosphate-activated protein kinase, orexigenic neuropeptides expression, and feeding behavior. Endocrinology 2005;146(1):3–10. [DOI] [PubMed] [Google Scholar]
- [9].Namkoong C, Kim MS, Jang PG, Han SM, Park HS, Koh EH, et al. Enhanced hypothalamic AMP-activated protein kinase activity contributes to hyperphagia in diabetic rats. Diabetes 2005;54(1):63–8. [DOI] [PubMed] [Google Scholar]
- [10].Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 2005;1(1):15–25. [DOI] [PubMed] [Google Scholar]
- [11].Cota D, Proulx K, Seeley RJ. The role of CNS fuel sensing in energy and glucose regulation. Gastroenterology 2007;132(6):2158–68. [DOI] [PubMed] [Google Scholar]
- [12].Mountjoy PD, Bailey SJ, Rutter GA. Inhibition by glucose or leptin of hypothalamic neurons expressing neuropeptide Y requires changes in AMP-activated protein kinase activity. Diabetologia 2007;50(1):168–77. [DOI] [PubMed] [Google Scholar]
- [13].Kim MS, Park JY, Namkoong C, Jang PG, Ryu JW, Song HS, et al. Anti-obesity effects of alpha-lipoic acid mediated by suppression of hypothalamic AMP-activated protein kinase. Nat Med 2004;10(7):727–33. [DOI] [PubMed] [Google Scholar]
- [14].Ahima RS, Lazar MA. Adipokines and the peripheral and neural control of energy balance. Mol Endocrinol 2008;22(5):1023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cheng H, Isoda F, Belsham DD, Mobbs CV. Inhibition of agouti-related peptide expression by glucose in a clonal hypothalamic neuronal cell line is mediated by glycolysis, not oxidative phosphorylation. Endocrinology 2008;149(2):703–10. [DOI] [PubMed] [Google Scholar]
- [16].Pilkis SJ, El-Maghrabi MR, Claus TH. Hormonal regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Biochem 1988;57:755–83. [DOI] [PubMed] [Google Scholar]
- [17].Nordlie RC, Foster JD, Lange AJ. Regulation of glucose production by the liver. Annu Rev Nutr 1999;19:379–406. [DOI] [PubMed] [Google Scholar]
- [18].Wu C, Khan SA, Lange AJ. Regulation of glycolysis-role of insulin. Exp Gerontol 2005;40(11):894–9. [DOI] [PubMed] [Google Scholar]
- [19].Argaud D, Lange AJ, Becker TC, Okar DA, El-Maghrabi MR, Newgard CB, et al. Adenovirus-mediated overexpression of liver 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in gluconeogenic rat hepatoma cells. Paradoxical effect on Fru-2,6-P2 levels. J Biol Chem 1995;270(41):24229–36. [DOI] [PubMed] [Google Scholar]
- [20].Wu C, Okar DA, Newgard CB, Lange AJ. Overexpression of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in mouse liver lowers blood glucose by suppression of hepatic glucose production. J Clin Invest 2001;107(1):91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wu C, Kang JE, Peng L, Li H, Khan SA, Hillard CJ, et al. Enhancing hepatic glycolysis reduces obesity: differential effects on lipogenesis depend on site of glycolytic modulation. Cell Metab 2005;2(2):131–40. [DOI] [PubMed] [Google Scholar]
- [22].Wu C, Khan SA, Peng LJ, Li H, Camela S, Lange AJ. Perturbation of glucose flux in the liver by decreasing fructose-2,6-bisphosphate levels causes hepatic insulin resistance and hyperglycemia. Am J Physiol Endocrinol Metab 2006;291(3):E536–43. [DOI] [PubMed] [Google Scholar]
- [23].Atsumi T, Nishio T, Niwa H, Takeuchi J, Bando H, Shimizu C, et al. Expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase/PFKFB3 isoforms in adipocytes and their potential role in glycolytic regulation. Diabetes 2005;54(12):3349–57. [DOI] [PubMed] [Google Scholar]
- [24].Huo Y, Guo X, Li H, Wang H, Zhang W, Wang Y, et al. Disruption of inducible 6-phosphofructo-2-kinase ameliorates diet-induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response. J Biol Chem 2010;285:3713–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guo X, Xu K, Zhang J, Li H, Zhang W, Wang H, et al. Involvement of inducible 6-phosphofructo-2-kinase in the anti-diabetic effect of PPARγ activation in mice. J Biol Chem 2010;285(31):23711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yang X, Kow L, Funabashi T, Mobbs C. Hypothalamic glucose sensor: similarities to and differences from pancreatic beta-cell mechanisms. Diabetes 1999;48(9):1763–72. [DOI] [PubMed] [Google Scholar]
- [27].Lynch RM, Tompkins LS, Brooks HL, Dunn-Meynell AA, Levin BE. Localization of glucokinase gene expression in the rat brain. Diabetes 2000;49(5):693–700. [DOI] [PubMed] [Google Scholar]
- [28].Levin BE. Glucosensing neurons do more than just sense glucose. Int J Obes Relat Metab Disord 2001;25(Suppl 5):S68–72. [DOI] [PubMed] [Google Scholar]
- [29].Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes 2002;51(7):2056–65. [DOI] [PubMed] [Google Scholar]
- [30].Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes 2004;53(3):549–59. [DOI] [PubMed] [Google Scholar]
- [31].Kang L, Dunn-Meynell AA, Routh VH, Gaspers LD, Nagata Y, Nishimura T, et al. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes 2006;55(2):412–20. [DOI] [PubMed] [Google Scholar]
- [32].Levin BE. Metabolic sensing neurons and the control of energy homeostasis. Physiol Behav 2006;89(4):486–9. [DOI] [PubMed] [Google Scholar]
- [33].Kessler R, Eschrich K. Splice isoforms of ubiquitous 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase in human brain. Brain Res Mol Brain Res 2001;87(2):190–5. [DOI] [PubMed] [Google Scholar]
- [34].Pilkis SJ, Walderhaug M, Murray K, Beth A, Venkataramu SD, Pilkis GJ, et al. 6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase from rat liver: Isolation and. J Biol Chem 1983;258(10):6135–41. [PubMed] [Google Scholar]
- [35].Chikri M, Rousseau G. Rat gene coding for heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: characterization of an unusual promoter region and identification of four mRNAs. Biochemistry 1995;34(27):8876–84. [DOI] [PubMed] [Google Scholar]
- [36].Sakurai T, Johnson J, Uyeda K. Islet fructose 6-phosphate, 2-kinase:fructose 2,6-bisphosphatase: isozymic form, expression, and characterization. Biochem Biophys Res Commun 1996;218(1):159–63. [DOI] [PubMed] [Google Scholar]
- [37].Watanabe G, Sakai A, Furuya E. Novel isoforms of rat brain fructose 6-phosphate 2-kinase/fructose 2,6-bisphosphatase are generated by tissue-specific alternative splicing. J Neurochem 1997;69(1):1–9. [DOI] [PubMed] [Google Scholar]
- [38].Hirata T, Kato M, Okamura N, Fukasawa M, Sakakibara R. Expression of human placental-type 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase in various cells and cell lines. Biochem Biophys Res Comm 1998;242:680–4. [DOI] [PubMed] [Google Scholar]
- [39].Chesney J, Mitchell R, Benigni F, Bacher M, Spiegel L, Al-Abed Y, et al. An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element: role in tumor cell glycolysis and the Warburg effect. Proc Natl Acad Sci USA 1999;96:3047–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Watanabe F, Furuya E. Tissue-specific alternative splicing of rat brain fructose-6 phosphate 2-kinase/fructose 2,6-bisphosphatase. FEBS Lett 1999;458:1–5. [DOI] [PubMed] [Google Scholar]
- [41].Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: head-to-head with a bifunctional enzyme that controls glycolysis. Biochem J 2004;381(3):561–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Depre C, Rider MH, Veitch K, Hue L. Role of fructose-2,6-bisphosphate in the control of heart glycolysis. J Biol Chem 1993;268:13274–9. [PubMed] [Google Scholar]
- [43].Hue L, Rider M. Role of fructose-2,6-bisphosphate in the control of glycolysis in mammalian tissues. J Biochem 1987;245:313–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jin ES, Uyeda K, Kawaguchi T, Burgess SC, Malloy CR, Sherry AD. Increased hepatic fructose 2,6 bisphosphate after an oral glucose load does not affect gluconeogenesis. J Biol Chem 2003;278(31):28427–33. [DOI] [PubMed] [Google Scholar]
- [45].Donthi RV, Ye G, Wu C, McClain DA, Lange AJ, Epstein PN. Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J Biol Chem 2004;279(46):48085–90. [DOI] [PubMed] [Google Scholar]
- [46].Almeida A, Moncada S, Bolanos JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol 2004;6(1):45–51. [DOI] [PubMed] [Google Scholar]
- [47].Calvo MN, Bartrons R, Castano E, Perales JC, Navarro-Sabate A, Manzano A. PFKFB3 gene silencing decreases glycolysis, induces cell-cycle delay and inhibits anchorage-independent growth in HeLa cells. FEBS Lett 2006;580(13):3308–14. [DOI] [PubMed] [Google Scholar]
- [48].Titolo D, Cai F, Belsham DD. Coordinate regulation of neuropeptide Y and agouti-related peptide gene expression by estrogen depends on the ratio of estrogen receptor (ER) alpha to ER{beta} in clonal hypothalamic neurons. Mol Endocrinol 2006;20(9):2080–92. [DOI] [PubMed] [Google Scholar]
- [49].Cai F, Gyulkhandanyan AV, Wheeler MB, Belsham DD. Glucose regulates AMP-activated protein kinase activity and gene expression in clonal, hypothalamic neurons expressing proopiomelanocortin: additive effects of leptin or insulin. J Endocrinol 2007;192(3):605–14. [DOI] [PubMed] [Google Scholar]
- [50].Huo Y, Guo X, Li H, Xu H, Halim V, Zhang W, et al. Targeted overexpression of inducible 6-phosphofructo-2-kinase in adipose tissue increases fat deposition but protects against diet-induced insulin resistance and inflammatory responses. J Biol Chem 2012;287(25):21492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Guo X, Li H, Xu H, Halim V, Zhang W, Wang H, et al. Palmitoleate induces hepatic steatosis but suppresses liver inflammatory response in mice. PLoS One 2012;7(6):e39286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Payne VA, Arden C, Wu C, Lange AJ, Agius L. Dual role of phosphofructokinase-2/fructose bisphosphatase-2 in regulating the compartmentation and expression of glucokinase in hepatocytes. Diabetes 2005;54(7):1949–57. [DOI] [PubMed] [Google Scholar]
- [53].Wu C, Okar DA, Stoeckman AK, Peng L, Herrera AH, Herrera JE, et al. A potential role for fructose-2,6-bisphosphate in the stimulation of hepatic glucokinase gene expression. Endocrinology 2004;145(2):650–8. [DOI] [PubMed] [Google Scholar]
- [54].Levin BE. Neuronal glucose sensing: still a physiological orphan? Cell Metab 2007;6(4):252–4. [DOI] [PubMed] [Google Scholar]
- [55].Kim E, Miller I, Aja S, Landree LE, Pinn M, McFadden J, et al. C75, a fatty acid synthase inhibitor, reduces food intake via hypothalamic AMP-activated protein kinase. J Biol Chem 2004;279(19):19970–6. [DOI] [PubMed] [Google Scholar]
- [56].Mayer J. Glucostatic mechanism of regulation of food intake. N Engl J Med 1953;249(1):13–6. [DOI] [PubMed] [Google Scholar]
- [57].Mayer J. Regulation of energy intake and the body weight: the glucostatic theory and the lipostatic hypothesis. Ann N Y Acad Sci. 1955 Jul 15;63(1):15-43 1955;63 (1):15–43. [DOI] [PubMed] [Google Scholar]
- [58].Mobbs CV, Isoda F, Makimura H, Mastaitis J, Mizuno T, Shu I-W, et al. Impaired glucose signaling as a cause of obesity and the metabolic syndrome: the glucoadipostatic hypothesis. Physiol Behav 2005;85(1):3–23. [DOI] [PubMed] [Google Scholar]
- [59].Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest 2007;117(8):2325–36. [DOI] [PMC free article] [PubMed] [Google Scholar]